

Amplicon sequencing of the 16S rRNA gene is commonly performed for the assessment and comparison of microbiomes. Here, we introduce GenePiper, an open-source R Shiny application that provides an easy-to-use interface, a wide range of analytical methods, and optimized graphical outputs for offline microbiome data analyses.
ABSTRACT
Amplicon sequencing of the 16S rRNA gene is commonly performed for the assessment and comparison of microbiomes. Here, we introduce GenePiper, an open-source R Shiny application that provides an easy-to-use interface, a wide range of analytical methods, and optimized graphical outputs for offline microbiome data analyses.
ANNOUNCEMENT
The profiling of microbiomes by high-throughput amplicon sequencing has become a standard approach in many research disciplines. Many bioinformatics tools have been developed to analyze such data, in standalone or online applications (1–5). However, most of these tools are command line based and have a steep learning curve for general users. Many of the packages require numerous sources of dependencies with different compatibilities, which complicates their installation and maintenance, whereas Web applications have data transfer and computation time constraints, especially for huge data sets. The uploading of data onto online servers also raises data security concerns. To address these issues, we developed GenePiper, an open-source R Shiny application, based on a virtual environment, that provides a united offline system for microbiome data analyses.
GenePiper is an open-source R Shiny application built in a virtual Linux environment. It depends on VirtualBox (Oracle) and Vagrant (HashiCorp, USA), which are available on Windows, Mac-OS X, and Linux platforms. Users download the GenePiper Vagrant configuration file, which Vagrant uses to set up the R Shiny (6) server, with all of the applications, within a virtual environment on the local computer. The main interface of GenePiper is accessed locally through a Web browser (such as Chrome, Firefox, or Safari).
GenePiper requires three input files, namely, an operational taxonomic unit (OTU) table, a taxonomy table, and a sample data table (Fig. 1). It is optional to provide a phylogenetic tree for UniFrac distance calculations (7). These files are loaded into GenePiper via the data import module. GenePiper constructs a “phyloseq-class” data structure with the loaded data and stores it in RDS format in the virtual environment. These RDS data are saved and can be recalled by a unique data label in subsequent analytical modules. Alternatively, a phyloseq-class data object stored in RDS format can be imported into GenePiper for analysis.
FIG 1.
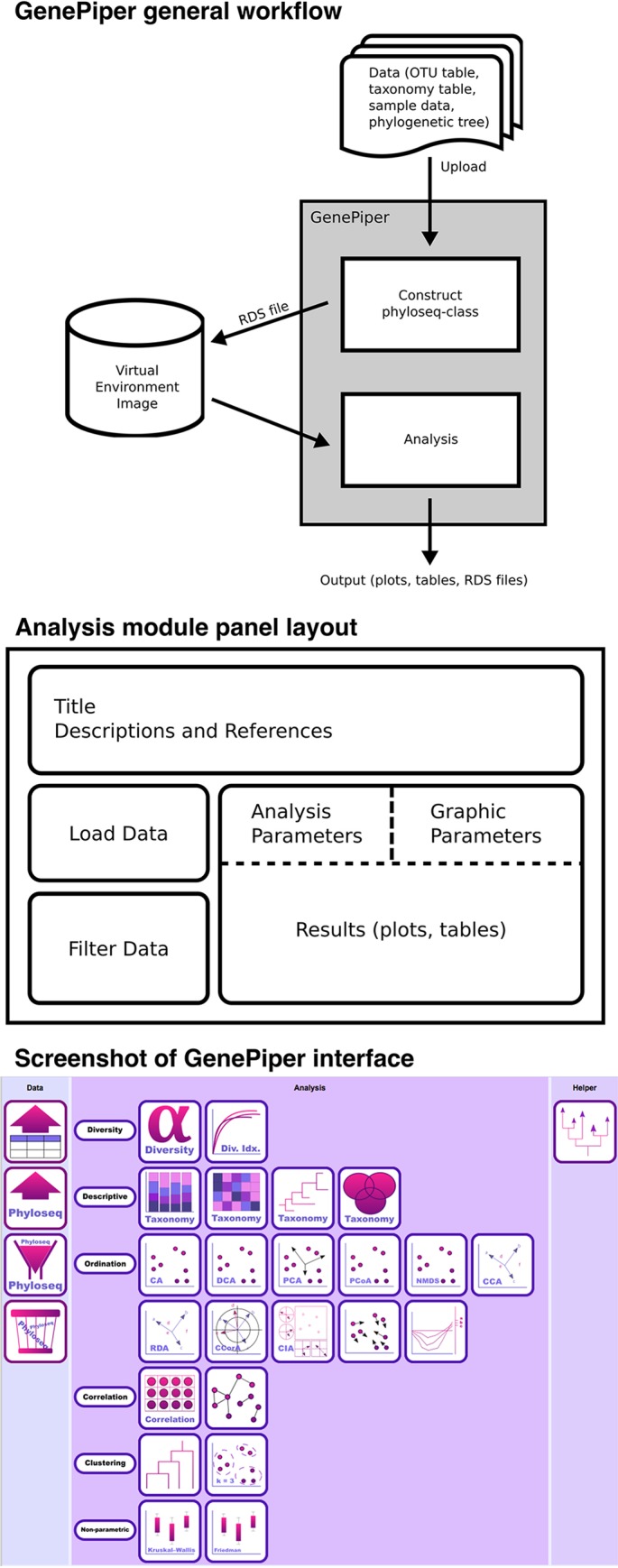
(Top) GenePiper general workflow. Data loaded into GenePiper are formatted into a phyloseq-class structure and stored in the virtual environment as an RDS file. Downstream analytical modules recall this RDS file for analysis. (Middle) Analysis module panel layout. All of the analytical modules in GenePiper share the same layout, with a top panel that shows the title, description, and references, a bottom-left panel for loading and filtering data, and a bottom-right panel for setting up the analysis parameters and displaying the results. (Bottom) Screenshot of the GenePiper interface.
GenePiper complements existing packages, with easy access to many popular and well-documented analytical methods, including phyloseq (3), vegan (8), phangorn (9), ape (10), VennDiagram (11), Hmisc (12), SpiecEasi (13), SparCC (14), and many others. The analytical modules are categorized into six broad groups, i.e., diversity analysis, descriptive analysis, ordination, clustering, correlation analysis, and nonparametric statistical tests. GenePiper generates figures mainly using the ggplot R package (15) and provides full control of the graphical parameters. Users may explore their microbiome data with options for visualization including a diversity index curve, taxonomic bar chart and heatmap, phylogenetic tree, Venn diagram, scatterplot ordination such as correspondence analysis, detrended correspondence analysis, principal component analysis, principal coordinate analysis, nonmetric multidimensional scaling, canonical correspondence analysis, redundancy analysis, canonical correlation analysis, coinertia analysis, Procrustes analysis, principal response curve, correlation plot, correlation network plot, clustering dendrogram, and boxplot with nonparametric test.
In summary, GenePiper is an integrated data-mining application in which the routine analytical pipeline can be operated easily using a graphical user interface. GenePiper allows researchers to efficiently test-run different parameter combinations for optimization and for generation of results for publication.
Data availability.
GenePiper is available at https://github.com/raytonghk/GenePiper. A step-by-step overview tutorial is available at https://github.com/raytonghk/genepiper/wiki/01.-Introduction.
ACKNOWLEDGMENT
Work by Y.C. is supported by the University Research Committee of the University of Hong Kong Seed Fund for Basic Research.
REFERENCES
- 1.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kandlikar GS, Gold ZJ, Cowen MC, Meyer RS, Freise AC, Kraft NJ, Moberg-Parker J, Sprague J, Kushner DJ, Curd EE. 2018. ranacapa: an R package and Shiny Web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Res 7:1734. doi: 10.12688/f1000research.16680.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMurdie PJ, Holmes S. 2015. Shiny-phyloseq: Web application for interactive microbiome analysis with provenance tracking. Bioinformatics 31:282–283. doi: 10.1093/bioinformatics/btu616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lahti L, Shetty S. 2017. Tools for microbiome analysis in R. Microbiome package version 1.9.14. https://github.com/microbiome/microbiome/.
- 6.Chang W, Cheng J, Allaire JJ, Xie Y, McPherson J. 2017. Shiny: Web application framework for R. https://cran.r-project.org/web/packages/shiny/index.html.
- 7.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oksanen J, Blanchet FG, Friendly M, Kidt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MH, Szoecs E, Wagner H. 2019. Vegan: community ecology package. R package version 2.5-6. https://cran.r-project.org/web/packages/vegan/index.html.
- 9.Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 11.Chen H, Boutros PC. 2011. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrell FE Jr, Dupont C. 2019. Hmisc: Harrell miscellaneous. R package version 4.3-0. http://biostat.mc.vanderbilt.edu/Hmisc.
- 13.Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. 2015. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol 11:e1004226. doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman J, Alm EJ. 2012. Inferring correlation networks from genomic survey data. PLoS Comput Biol 8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
GenePiper is available at https://github.com/raytonghk/GenePiper. A step-by-step overview tutorial is available at https://github.com/raytonghk/genepiper/wiki/01.-Introduction.