

Abstract
Objective
The development of an enzymatic assay for the specific quantification of the C1-oxidation product, i.e. gluconic acid of cellulose active lytic polysaccharide monooxygenases (LPMOs).
Results
In combination with a β-glucosidase, the spectrophotometrical assay can reliably quantify the specific C1- oxidation product of LPMOs acting on cellulose. It is applicable for a pure cellulose model substrate as well as lignocellulosic biomass. The enzymatic assay compares well with the quantification performed by HPAEC-PAD. In addition, we show that simple boiling is not sufficient to inactivate LPMOs and we suggest to apply a metal chelator in addition to boiling or to drastically increase pH for proper inactivation.
Conclusions
We conclude that the versatility of this simple enzymatic assay makes it useful in a wide range of experiments in basic and applied LPMO research and without the need for expensive instrumentation, e.g. HPAEC-PAD.
Keywords: Lytic polysaccharide monooxygenase (LPMO), Biomass degradation, Quantification assay, LPMO inactivation, Cellulose oxidation, Gluconic acid
Introduction
Since their discovery, Lytic polysaccharide monooxygenases (LPMOs) have received considerable interest, due to their ability to boost the degradation of lignocellulose (Vaaje-Kolstad et al. 2010; Horn et al. 2012). LPMOs are mono-copper enzymes with the active site positioned on a relatively flat surface enabling them to interact with crystalline polysaccharides. The catalytic mechanism of LPMOs involves the reduction of the copper from Cu(II) to Cu(I) by an external reducing agent. Hereafter, the copper-active site of the LPMO reacts with either O2 or H2O2 (Walton and Davies 2016; Bissaro et al. 2017) and binds an activated oxygen species that enables the oxidation of the polysaccharide substrate. The oxidative cleavage of the β-1,4 glycosidic bonds can occur at the C1 or the C4 position or both, generating oxidized products, both soluble and insoluble (Quinlan et al. 2011). The soluble products are commonly detected by anion exchange chromatography coupled to a pulsed amperometric detector (HPAEC-PAD) (Westereng et al. 2013). It offers high sensitivity and provides detailed information about the product pattern but its limitations are the absence of commercially available reference compounds, time-consuming sample analysis, and costly instrumentation.
LPMO assays have been developed to circumvent some of these challenges. The Amplex red assay provides a fast measure of LPMO reactivity and is based on the release of H2O2 in the absence of a substrate (Kittl et al. 2012). Recently, a spectrophotometrical assay was developed that exploits peroxidase activity of some LPMOs to oxidize 2,6-dimethoxyphenol (Breslmayr et al. 2018). While both assays can be employed to detect LPMO reactivity, the results cannot be directly correlated to the oxidative activity of LPMOs on polysaccharides. Soluble LPMO products have been analyzed with by carbohydrate gel electrophoresis (PACE) (Quinlan et al. 2011; Frandsen et al. 2016) yielding semi-quantitative results. LPMO activity has also been analyzed using chromogenic polysaccharide hydrogel substrates (Kračun et al. 2015). Assays for quantifying C1-oxidized moieties in the insoluble fraction of the LPMO on pure cellulose model substrates were described, such as a fluorescence-based approach (Vuong et al. 2017), and a colorimetric cation-based assay (Wang et al. 2018). Despite the value of these methods, LPMO activity assays and especially methods that can simplify the quantification of LPMO oxidation, without the necessity of expensive specialized instrumentation, not only on cellulosic model substrates but on complex substrates, such as lignocellulosic biomass are still lacking.
We describe a simple quantification method for cellulose active C1-oxidizing LPMOs, employing a commercially available glycoside hydrolase to hydrolyze the oxidation products to yield gluconic acid and glucose. Subsequently, gluconic acid is quantified spectrophotometrically which is suitable for microplate formats. The release of C1-oxidized products by TtLPMO9E on microcrystalline cellulose (Avicel) and steam pretreated wheat straw (PWS) was quantified and compared to the results by HPEAC-PAD.
Methods
Substrates
The activity of Thielavia terrestris LPMO9E (TtLPMO9E) (previously TtGH61E) was determined on the two substrates; Avicel PH101 (Sigma-Aldrich), and pretreated wheat straw (PWS). The wheat straw was hydrothermally pretreated at 190 °C for 10 min at an initial solid loading of 35%. The heating was done by injection of steam, decreasing the solids level to 15% after 10 min treatment. Using standard NREL methods the composition was determined to be 54% glucan, 4% xylan, 34% lignin, and 6% ash. The substrates were washed five times in MilliQ water and twice in 50 mM citrate buffer pH 6 to remove soluble sugars prior to the incubation with TtLPMO9E.
Enzymes
TtLPMO9E was donated by Novozymes A/S (Bagsværd, Denmark). The TtLPMO9E concentration was determined by absorbance at 280 nm using a molar extinction coefficient of 58,120 M−1 cm−1. Novozym 188 was obtained from Novozymes A/S (Bagsværd, Denmark). β-glucosidase from Aspergillus niger and cellobiohydrolase I (CBHI) from Trichoderma longibrachiatum were purchased from Megazyme with a specified activity of 40 U ml−1 and a concentration of 10 mg ml−1, respectively.
Preparation of C1-oxidized cello-oligosaccharide standards for the HPAEC-PAD
C1-oxidized cello-oligosaccharide standards for the HPAEC-PAD were prepared by oxidizing pure cello-oligosaccharides (Megazyme) with iodine as earlier described for the preparation of cellobionic acid (Green et al. 1976; Hildebrand et al. 2016), with the following modifications:
The cello-oligosaccharide was dissolved in a minimum of deionized water and heated at 55 °C for 10 min until dissolved. A solution of iodine in methanol was added to a final molar ratio of 1:2:110 (cello-oligosaccharide:iodine:methanol) and the solution was heated at 40 °C for 15 min. At room temperature 4% (w/v) potassium hydroxide-methanol solution was slowly added to the solution and left for an additional 30 min to a final molar ratio of 1:12 (cello-oligosaccharide:potassium hydroxide). 20 mL deionized water was added to the solution and the methanol was gently boiled off in a rotary evaporator. The pH of the solution was set to pH 5 with citric acid.
All procedures were done with constant stirring.
Inactivation of TtLPMO9E
Samples of Avicel 50 g L−1, 50 mM citrate buffer pH 6, 1 µM TtLPMO, and 2 mM ascorbic acid in a final volume of 200 µl were mixed in Eppendorf tubes. The samples were treated with or without 1 mM EDTA addition, with or without 50 mM NaOH and with or without incubation in a water bath at 100 °C for 20 min. A positive control, not treated with EDTA, NaOH, or heat, was included. The samples were incubated in an Eppendorf ThermoMixer C at 50 °C for 6 h at 1100 rpm. Subsequently, the samples were centrifuged and the supernatant was kept for measuring C1-oxidized sites as described below.
TtLPMO9E activity
Samples of Avicel (50 g L−1) or PWS (20 g L−1), 1 µM TtLPMO9E, 2 mM ascorbic acid in 50 mM citrate buffer pH 6 in a final volume of 200 µl were incubated in an Eppendorf ThermoMixer C at 50 °C. At varying time points (0–24 h), the enzymatic reaction was quenched by the addition of NaOH to a final concentration of 50 mM.
After 24 h, the samples were centrifuged at 1800×g and the supernatant was used for measuring C1-oxidized sites as described below.
The pellet was further hydrolyzed for measuring C1-oxidized sites in the insoluble fraction. The pellet was washed five times in 200 µL 50 mM citrate buffer pH 5, by centrifugation and resuspension, to decrease pH and remove soluble sugars. The pellet was resuspended in 50 mM citrate buffer. Cellobiohydrolase I (CBHI) from Trichoderma longibrachiatum was added to a final concentration of 0.5 g L−1 in a final volume of 200 µL. The samples were incubated in an Eppendorf ThermoMixer C at 50 °C for 24 h at 1100 rpm after which the samples were centrifuged. The supernatant was used for measuring C1-oxidized sites revealed by the CBHI as described below.
Quantification of gluconic acid by HPAEC-PAD
100 µL of the supernatant was diluted in 50 mM citrate buffer pH 5 together with 5 U ml−1 β-glucosidase from Aspergillus niger to a final volume of 200 µL. The samples were incubated at 50 °C for 24 h before the samples were filtered through a nylon filter with a pore size of 0.45 µm and loaded in HPLC vials.
The samples were analyzed by high-performance anion-exchange chromatography (HPAEC) performed on an ICS5000 system, equipped with a pulsed amperometric detector (PAD) (Thermo Scientific) with an analytical CarboPac PA1 column (2 × 250) as described elsewhere (Westereng et al. 2013) with some modifications: The analytes were eluted at 0.25 mL min−1 at 30 °C and the initial conditions were 100% eluent A (0.1 M NaOH). A linear gradient was applied increasing the proportion of eluent B (1 M NaOAc in 0.1 M NaOH) to 90% A:10% B after 10 min. An exponential gradient (curve 6) was applied to reach 83.1% A:16.9% B after 22 min and 0% A:100% B after 23 min. These conditions were kept for 4 min. The column was reconditioned by running initial conditions for 15 min. Gluconic acid was quantified based on external standards of 0–250 mg L−1 gluconic acid in 50 mM citrate pH 5.
Quantification of gluconic acid by the d-Gluconic acid/d-Glucono-δ-lactone assay kit
Gluconic acid was quantified using the assay kit from Megazyme (Bergmeyer and Moellering 1988). The method is based on phosphorylation of gluconic acid to D-gluconate-6-phosphate by ATP in the presence of gluconate kinase with the simultaneous formation of ADP. In the presence of NADP, gluconate-6-phosphate is oxidatively decarboxylated by 6-phosphogluconate dehydrogenase to ribulose-5-phosphate with the stoichiometrically formation of NADPH. The assay was performed according to the microplate procedure described by the manufacturer with the following modifications: 50 µL of the supernatant was mixed with 160 µL MilliQ water, 20 µL solution I (buffer), 20 µL solution II (NADP + ATP), 6 µL solution III (6-PGDH) diluted three times in MilliQ water. The samples were mixed in an Eppendorf ThermoMixer for 4 min at 600 rpm after which the absorbance was measured at 340 mm in microplate reader (Molecular Devices SpectraMax i3). 6 µL solution IV (GCK) diluted three times with MilliQ water was added to the samples and the plate was mixed in an Eppendorf ThermoMixer at 600 rpm for 6 min. The absorbance was measured at 340 nm and the difference was used to quantify gluconic acid based on external standards of 0–250 mg L−1 gluconic acid in 50 mM citrate pH 5. According to the supplier’s information, the range of linearity of the UV-signal is 0.1–5 µg gluconic acid per sample in a sample volume of 1–200 µL. Using the guidelines of the International Conference on Harmonisation (ICH), we calculated the quantitation limit (LOQ) as LOQ = 10Sa/b, where Sa is the standard error of the y-intercept of the regression line of the gluconic acid standard curve and b is the slope of the regression line of the gluconic acid standard curve. Using this approach, we found that LOQ = 0.27 mg gluconic acid L−1.
Quantification of glucose
The hydrolysis yield was determined on an Ultimate 3000 high-performance liquid chromatography equipped with an ultraviolet detector at 210 nm (Thermo Scientific). The separation was performed on a Phenomenex Rezex ROA column at 80 °C with a mobile phase of 5 mM H2SO4 at a flow rate of 0.6 mL min−1. The glucose was quantified based on external standards of 0–10 g L−1 glucose in 50 mM citrate pH 5.
Results and discussion
Hydrolysis of oxidized products
To decrease product complexity, the products of LPMO-catalysis are often hydrolyzed by glycoside hydrolases prior to the quantification by HPAEC-PAD (Cannella et al. 2016; Bissaro et al. 2017; Frommhagen et al. 2017). Here, we assessed the applicability of two commercially available β-glucosidases, Novozym 188 and β-glucosidase from Aspergillus niger to hydrolyze the soluble products generated by TtLPMO9E from a cellulosic substrate to the monomers, glucose and gluconic acid (Fig. 1).
Fig. 1.

HPAEC-PAD chromatograms and elution patterns. a, The supernatant of 50 g L−1 Avicel incubated with 1 µM TtLPMO9E and 2 mM ascorbic acid in 50 mM citrate buffer pH 6 for 6 h at 50 °C. The peaks are assigned based on external standards of C1-oxidized cello-oligosaccharide standards (not shown). The oligosaccharides are abbreviated GlcN and GlcNGlcA with N being the number of glucose units and GlcA being gluconic acid. b, The supernatant from a incubated with Novozym 188. c, The supernatant from a incubated with A. niger β-glucosidase. d, Novozym 188. e, A. niger β-glucosidase. f The supernatant of 50 g L−1 Avicel incubated with 2 mM ascorbic acid for 6 h at 50 °C
The lowest β-glucosidase concentration capable of hydrolyzing the TtLPMO9E products generated in the following experiments to glucose and gluconic acid was determined to be 1.25 U/ml A. niger β-glucosidase corresponding to a specific activity on 4-Nitrophenyl β-D-glucuronide of 1.26 × 10−3 µmol p-nitrophenol min−1 and Novozym 188 corresponding to a specific activity of 1.16 × 10−3 µmol p-nitrophenol min−1 according to the β-glucosidase activity assay described elsewhere (Gunata et al. 1990). These concentrations were used throughout the work presented here. The product profiles were analyzed by HPAEC-PAD (Fig. 1).
Both β-glucosidases hydrolyzed the TtLPMO9E products to the monomers, glucose and gluconic acid (GlcA). As the Novozym 188 contained impurities eluting together with glucose and gluconic acid, the A. niger β-glucosidase was selected for further use.
Inactivation of TtLPMO9E
As TtLPMO9E does not have activity on soluble oligosaccharides (Westereng et al. 2015), the reaction in the supernatant can simply be stopped by filtration. Efficient inactivation of the LPMOs is, however, essential to quantify the activity in the insoluble fraction. Prior to the hydrolysis of the LPMO products by glucoside hydrolases, common procedures to quench the LPMO reaction are 10 min incubations at 100 °C (Cannella et al. 2016; Frommhagen et al. 2017) or by increasing pH (Isaksen et al. 2014; Bissaro et al. 2017). Previous studies have shown that chelating agents, such as EDTA can remove the copper atom from the LPMO active site (Vaaje-Kolstad et al. 2010; Kracher et al. 2017), suggesting that the addition of EDTA could effectively inactivate LPMOs.
The inactivation efficiencies were tested by measuring the activity of TtLPMO9E after boiling for 20 min with and without the addition of EDTA or after the addition of sodium hydroxide, and their activity was compared to the activity of a non-treated TtLPMO9E samples (Fig. 2).
Fig. 2.
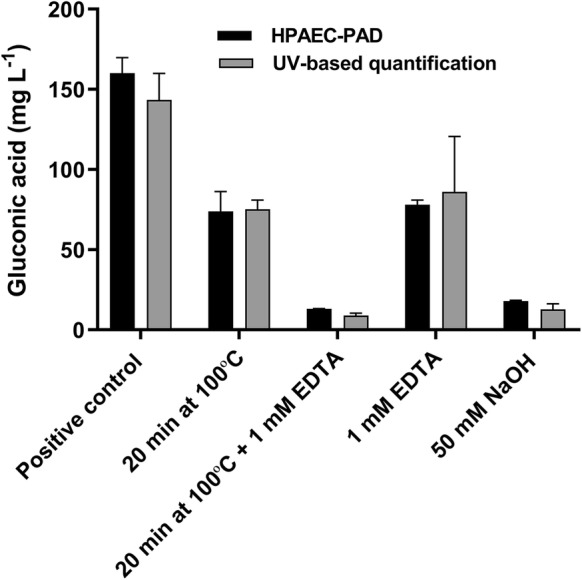
Evaluation of methods to inactivate TtLPMO9E by quantification of gluconic acid. 50 g L−1 Avicel were incubated with 2 mM ascorbic acid and 1 µM TtLPMO9E in 50 mM citrate buffer pH 6 for 6 h at 50 °C. Prior to the incubation, the samples were treated with either, 20 min incubation at 100 °C, 20 min incubation at 100 °C + 1 mM EDTA, 1 mM EDTA, or 50 mM NaOH. A positive control, not treated with NaOH, EDTA, or incubation at 100 °C, was included. Gluconic acid was quantified by HPAEC-PAD (black bars) and by the UV-based gluconic acid kit (grey bars). All incubations were performed in triplicate and the standard deviations of the samples are represented by error bars
TtLPMO9E retained approximately 50% of its activity after boiling for 20 min. This may be due to reversible thermal unfolding as has previously been reported for NcLPMO9C and TaLPMO9A, respectively (Kracher et al. 2017; Singh et al. 2019). These findings suggest that if boiling is used to attempt inactivation of LPMOs, it should be followed by careful evaluation of residual activity. The addition of EDTA decreased the activity by approximately 50%. A combination of boiling and addition of EDTA decreased the activity of TtLPMO9E to less than 10% compared to the positive control not treated with heat or EDTA. A similar decrease in activity was obtained by the addition of 50 mM NaOH to reach pH 11.
Quantification of the LPMO reaction
The activity of TtLPMO9E was measured by quantifying the C1-oxidation products in the soluble and the insoluble fraction of microcrystalline cellulose (Avicel) and pretreated wheat straw (PWS) (Fig. 3). The results should not be interpreted as the absolute quantification of insoluble products, as only a fraction of the insoluble products was hydrolyzed.
Fig. 3.

Quantification of the soluble and insoluble products from microcrystalline cellulose and pretreated wheat straw (PWS). 50 g L−1 Avicel or 20 g L−1 PWS incubated with 2 mM ascorbic acid and 1 µM TtLPMO9E in 50 mM citrate buffer pH 6 at 50 °C from 0 to 24 h. a, Supernatant of Avicel incubated with TtLPMO9E and ascorbic acid incubated with 5 U mL−1A. niger β-glucosidase. b, Pellet of Avicel incubated with TtLPMO9E and ascorbic acid incubated with 0.5 g L−1 T. longibrachiatum CBHI and 5 U mL−1A. niger β-glucosidase. c, Supernatant of PWS incubated with TtLPMO9E and ascorbic acid incubated with 5 U mL−1A. niger β-glucosidase. d, Pellet of PWS incubated with TtLPMO9E and ascorbic acid incubated with 0.5 g L−1 T. longibrachiatum CBHI and 5 U mL−1A. niger β-glucosidase. Gluconic acid detected after 0 h of incubation was subtracted from the results. Gluconic acid was quantified by HPAEC-PAD (black bars) and by the UV-based gluconic acid kit (grey bars). All results are averages of triplicate measurements. The standard deviations of the samples are represented by error bars. e and f, Correlation between the quantification results measured by HPAEC-PAD (y-axis) and gluconic acid kit (x-axis) from a–d. The equation for the best linear regression (slope = 0.9) and the coefficient of determination, r2 (r2 = 0.98) is shown. f: Enlargement of the correlation at gluconic acid concentrations from 0 to 50 mg L−1. For clarity, standard deviations are not shown
The formation of gluconic acid (GlcA) in the soluble fraction of Avicel incubated with TtLPMO9E and ascorbic acid increased up to approximately 175 mg L−1 after 0.5 to 6 h. Between 6 and 24 h, no further changes in GlcA concentration were detected in the soluble fraction. GlcA formation in the insoluble fraction of Avicel increased up to approximately 35 mg L−1 after 24 h. Interestingly, after 6 h of incubation an increase in GlcA was solely detected in the insoluble fraction whereas no increase in GlcA was detected in the soluble fraction.
For the reactions on pretreated wheat straw, the GlcA content in the soluble fraction increased from 0.5 to 6 h up to approximately 175 mg L−1. As with the reaction on Avicel, the GlcA concentration in the soluble fraction remained unchanged between 6 and 24 h. In the insoluble fraction, the formation of GlcA increased steadily up to approximately 60 mg L−1 after 24 h.
The UV-based enzymatic GlcA quantification method produced similar results and detection limits as the HPAEC-PAD when analyzing products from the soluble and insoluble fractions of Avicel as well as PWS. The results of the two methods showed a linear correlation with r2 values within the four data groups varying between 0.986 and 0.996.
Furthermore, dose–response curves of enzyme titration (Fig. 4a) and substrate loading (Fig. 4b) were performed on PWS and the results were fitted to a hyperbolic function with the best fit of [y = 197.97*×/(2.51 + x)] for the enzyme titration data. Likewise, the dose–response curve with increased substrate load was fitted to a hyperbolic Michalies-Menten like function,
Fig. 4.

Dose–response curves of enzyme and substrate titrations. a, Quantification of the soluble products of 20 g L−1 PWS incubated with 2 mM ascorbic acid and varying concentrations of TtLPMO9E in 50 mM citrate buffer pH 6 at 50 °C for 3 h. The dotted line represents the best fit of a hyperbolic function. b, specific activity of TtLPMO9E on PWS measured by quantifying the gluconic acid in the soluble products of varying loads of PWS incubated with 2 mM ascorbic acid and 0.25 µM TtLPMO9E in 50 mM citrate buffer pH 6 at 50 °C in 3 h. The dotted line represents the best fit of a hyperbolic function. All results are averages of triplicate measurements. The standard deviations of the samples are represented by error bars
as described for other interfacial enzymes (Cruys-Bagger et al. 2013) with pVss being the specific activity (product/time/enzyme concentration), pVmax being the maximal specific activity, S0 being the substrate load, and pKM the Michaelis–Menten constant. Using the enzymatic assay, the pKM and the pVmax/E0 were derived from the regression analysis to be 49.80 g L−1 and 0.064 s −1 (3.8 min −1), respectively. The ladder being in line with the results given in a recent review, comparing the apparent enzyme rates of LPMOs, that showed that the apparent catalytic rates (kcat) of LPMOs fall in a relatively small range roughly between 1 and 10 min −1 when not considering H2O2 or light-driven reactions (Bissaro et al. 2018).
Conclusions
We have described a simple, fast, and sensitive UV-based enzymatic assay to quantify specifically the C1-oxidation of cellulose active LPMOs. In recent years, a number of LPMO assays have been developed to decrease the analysis time compared to HPAEC-PAD, as well as eliminating the need for oxidized oligosaccharide standards that are not commercially available, and expensive instrumentation. These methods all have distinct advantages making them suitable for different applications. The method reported in this paper excels by its simplicity and its ability to quantify soluble and insoluble LPMO products on a pure cellulose substrate as well as on a lignocellulosic biomass. The UV-based enzymatic assay is suitable for an auto-analyzer format and, with an analysis time of approximately 30 min per 96 samples in a microtiter plate, compared to approximately 15 min per sample or 24 h per 96 samples in a microtiter plate with HPAEC, the method offers fast measurements (Fig. 5).
Fig. 5.

Schematic representation of the UV-based enzymatic assay to quantify LPMO activity on cellulose. After the LPMO reaction, the plates were centrifuged and the soluble products were collected in the supernatant. β-glucosidase was added to the soluble products and the plates were incubated at 50 °C overnight after which gluconic acid was quantified by HPAEC-PAD (a) and by the UV-based enzymatic assay (b). a: The samples were filtered and gluconic acid was quantified on a CarboPac PA1 column (2 × 250) on a HPAEC-PAD. The peak areas were used for the quantification of gluconic acid based on external standards. The time consumption was 30 min per plate, however this could be optimized for shorter runs. b: Buffer, NADPH + , and 6-Phosphoglucanate dehydrogenase was added to the samples. The samples were mixed for 4 min in a thermomixer at 600 rpm and the absorbance was measured at 340 nm after which gluconate kinase was added to the samples and the plates were mixed for 6 min in a thermomixer at 600 rpm and the absorbance was measured at 340 nm. The difference in absorbance was used for the quantification of gluconic acid based on external standards
Furthermore, we compared methods for TtLPMO9E inactivation and demonstrated that a boiling is not sufficient. Thus, we suggest to apply a metal chelator while boiling or to drastically increase pH for proper inactivation.
The quantification method may be useful in other experiments, such as for comparison of activities of different C1-oxidizing cellulose active LPMOs, for studies of LPMO activities on different substrates and for testing compounds that might influence LPMO activity. Such experiments are technically straightforward by using the quantification approach outlined here, but changing conditions should be followed by calibration against an alternative quantification technique to exclude potential interferences.
Acknowledgements
We would like to sincerely thank Anne Stenbæk and Johan Pelck Olsen from Novozymes A/S for their support and supply of enzyme.
Funding
This work was funded by the Innovation Found, Denmark 7043-00016B (ENACT), the Novo Nordisk Foundation grant NNF16OC0021832 and by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Brazil) via Grants 2015/13684-0 and 2017/01165-4.
Compliance with ethical standards
Conflict of interest
All authors declare that they have no conflicts of interest with the contents of this article.
Research involving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Bergmeyer HU, Moellering H. D-Gluconate (D-Glucono δ-lactone) and D-Gluconate 6-Phosphate. Methods in enzymatic analysis. 3. Cambrigde: VCH Publishers (UK) Ltd; 1988. pp. 220–227. [Google Scholar]
- Bissaro B, Røhr ÅK, Müller G, et al. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2470. [DOI] [PubMed] [Google Scholar]
- Bissaro B, Várnai A, Røhr ÅK, Eijsink VGH. Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass. Microbiol Mol Biol Rev. 2018;82:e00029–e118. doi: 10.1128/MMBR.00029-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslmayr E, Hanžek M, Hanrahan A, et al. A fast and sensitive activity assay for lytic polysaccharide monooxygenase. Biotechnol Biofuels. 2018;11:79. doi: 10.1186/s13068-018-1063-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannella D, Möllers KB, Frigaard N-UU, et al. Light-driven oxidation of polysaccharides by photosynthetic pigments and a metalloenzyme. Nat Commun. 2016;7:1134. doi: 10.1038/ncomms11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruys-Bagger N, Elmerdahl J, Praestgaard E, et al. A steady-state theory for processive cellulases. FEBS J. 2013;280:3952–3961. doi: 10.1111/febs.12397. [DOI] [PubMed] [Google Scholar]
- Frandsen KEH, Simmons TJ, Dupree P, et al. The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases Europe PMC Funders Group. Nat Chem Biol. 2016;12:298–303. doi: 10.1038/nchembio.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommhagen M, Westphal AH, Hilgers R, et al. Quantification of the catalytic performance of C1-cellulose-specific lytic polysaccharide monooxygenases. Appl Microbiol Biotechnol. 2017 doi: 10.1007/s00253-017-8541-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JW, Thompson NS, Pearl IA. Study of the carbohydrate peeling and stopping reactions under the conditions of oxygen-alkali pulping. Wisconsin: Appleton; 1976. [Google Scholar]
- Gunata YZ, Bayonove CL, Cordonnier RE, et al. Hydrolysis of grape monoterpenyl glycosides by Candida molischiana and Candida wickerhamii β-glucosidases. J Sci Food Agric. 1990;50:499–506. doi: 10.1002/jsfa.2740500408. [DOI] [Google Scholar]
- Hildebrand A, Addison JB, Kasuga T, Fan Z. Cellobionic acid inhibition of cellobiohydrolase I and cellobiose dehydrogenase. Biochem Eng J. 2016;109:236–242. doi: 10.1016/j.bej.2016.01.024. [DOI] [Google Scholar]
- Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels. 2012;5:45. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksen T, Westereng B, Aachmann FL, et al. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J Biol Chem. 2014;289:2632–2642. doi: 10.1074/jbc.M113.530196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittl R, Kracher D, Burgstaller D, et al. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels. 2012;5:79. doi: 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracher D, Andlar M, Furtmüller PG, Ludwig R. Active-site copper reduction promotes substrate binding of fungal lytic polysaccharide monooxygenase and reduces stability. J Biol Chem. 2017;293:1676. doi: 10.1074/jbc.RA117.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kračun SK, Schückel J, Westereng B, et al. A new generation of versatile chromogenic substrates for high-throughput analysis of biomass-degrading enzymes. Biotechnol Biofuels. 2015;8:70. doi: 10.1186/s13068-015-0250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan RJ, Sweeney MD, Lo Leggio L, et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Blossom BM, Russo DA, et al. Thermal unfolding and refolding of a lytic polysaccharide monooxygenase from Thermoascus aurantiacus. RSC Adv. 2019;9:29734–29742. doi: 10.1039/C9RA05920B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaaje-Kolstad G, Westereng B, Horn SJ, et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science. 2010;330:219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- Vuong TV, Liu B, Sandgren M, Master ER. Microplate-based detection of lytic polysaccharide monooxygenase activity by fluorescence-labeling of insoluble oxidized products. Biomacromol. 2017;18:610–616. doi: 10.1021/acs.biomac.6b01790. [DOI] [PubMed] [Google Scholar]
- Walton PH, Davies GJ. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr Opin Chem Biol. 2016;31:195–207. doi: 10.1016/j.cbpa.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Wang D, Li J, Wong ACY, et al. A colorimetric assay to rapidly determine the activities of lytic polysaccharide monooxygenases. Biotechnol Biofuels. 2018;11:215. doi: 10.1186/s13068-018-1211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westereng B, Agger JW, Horn SJ, et al. Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J Chromatogr A. 2013;1271:144–152. doi: 10.1016/J.CHROMA.2012.11.048. [DOI] [PubMed] [Google Scholar]
- Westereng B, Cannella D, Agger JW, et al. Enzymatic cellulose oxidation is linked to lignin by long-range electron transfer. Nat Publ Gr. 2015 doi: 10.1038/srep18561. [DOI] [PMC free article] [PubMed] [Google Scholar]