

Abstract
The use of DNA‐based nanomaterials in biomedical applications is continuing to grow, yet more emphasis is being put on the need for guaranteed structural stability of DNA nanostructures in physiological conditions. Various methods have been developed to stabilize DNA origami against low concentrations of divalent cations and the presence of nucleases. However, existing strategies typically require the complete encapsulation of nanostructures, which makes accessing the encased DNA strands difficult, or chemical modification, such as covalent crosslinking of DNA strands. We present a stabilization method involving the synthesis of DNA brick nanostructures with dendritic oligonucleotides attached to the outer surface. We find that nanostructures assembled from DNA brick motifs remain stable against denaturation without any chemical modifications. Furthermore, densely coating the outer surface of DNA brick nanostructures with dendritic oligonucleotides prevents nuclease digestion.
Keywords: dendrimers, DNA bricks, DNA nanotechnology, nanostructures, self-assembly
Brick by brick: DNA brick nanostructures with binding domains of 13 nt or longer are stable in solutions with low divalent salt concentration. Coating these DNA bricks with dendritic oligonucleotides increases their resistance to DNAse I, enhancing their biocompatible stability.
DNA nanotechnology enables the synthesis of rationally designed DNA nanostructures of arbitrary geometric configuration,1 and such molecular‐scale nanodevices can be used in biomedical applications, driving the field towards programmable and customizable nanomedicine.2, 3 Examples of DNA nanocages,4, 5 capsules,6 and carriers7 have been studied as delivery vehicles or diagnostic devices for drug delivery, cancer treatment, and immunotherapy.2, 7 Nevertheless, more recent literature highlights the importance of the structural stability of DNA‐based materials under physiological conditions when using them in vitro and/or in vivo.8, 9, 10, 11 This is due to two main factors involved in degradation of DNA nanostructures upon exposure to biological conditions: i) denaturation caused by low divalent cation concentration (physiological salt concentration approximately 0.04–0.8 mm MgCl2), and ii) digestion caused by the presence of nucleases.8
Multiple strategies have been developed to chemically or physically prevent the DNA nanostructures from falling apart in cellular media (for example, 10 % FBS),12, 13, 14, 15, 16, 17, 18, 19, 20 and in general, encapsulation of DNA nanostructures with different coating moieties prolongs the survival time the longest published.12, 14, 17, 18, 19 For example, while most bare DNA origami falls apart easily under physiological conditions, PEG‐oligolysines,12 lipid molecules,17 or cationic polymers18, 19 can be applied as a coating material to extend the half‐life of DNA origami by an order of up to approximately 100.12 Such an encasing strategy, however, often covers the entire outer surface of DNA origami and therefore in theory, makes it difficult to access the DNA strands post‐coating. Ideally, one should be able to access the DNA strands and nanostructures for binding without having to penetrate through a thick layer of overlaid material.
In this work, we aim to structurally stabilize DNA nanostructures with only oligonucleotide strands such that both biocompatible stability and DNA accessibility remain viable. And in doing so, we present two findings: i) nanostructures consisting of certain DNA brick motifs21, 22 remain structurally stable at low divalent salt concentrations (for example, 1×PBS), and ii) functionalizing the outer surface of DNA brick nanostructures with dendritic oligonucleotides prevents the digestion of nanostructures from nucleases due to putative steric hindrance (Figure 1). As a result, this strategy suggests that neither chemical protectants nor covalent base‐pair interlocking is necessary to enhance the stability of DNA brick nanostructures.
Figure 1.
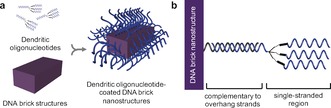
DNA nanostructures are constructed by a) attaching dendritic oligonucleotides to the outer surface of DNA brick nanostructures via b) hybridization to the complementary, protruding overhang strands.
Recently, the Yin lab developed a strategy to assemble DNA nanostructures of different sizes and shapes using short, synthetic oligonucleotide strands.21, 22 This assembly of DNA bricks, which consist of four short binding domains arranged so that the bricks can interlock, does not require a scaffold. And while the first generation of bricks were 32 nucleotides (nt) long, consisting of four 8 nt binding domains,21 more recent developments investigated brick strands with longer binding domains (52 nt bricks with four 13 nt domains; 74 nt bricks with two 18 nt and two 19 nt domains).22
We discovered that DNA brick nanostructures with binding domains of 13 nt or longer remain stable even when they are placed in 1×PBS with no divalent salt. First, the stability of DNA brick nanostructures was tested by removing cations from solution. Three separate sets, each consisting of approximately two hundred strands of 32, 52, or 74 nt bricks, were assembled in 10 mm or 40 mm MgCl2 (note that assembly salt conditions were chosen based on previous literature;22 see the Supporting Information for a detailed protocol). All sets of samples were divided into two subsets for comparison: i) maintaining the overall MgCl2 concentration at 10 mm or 40 mm as control, and ii) replacing the buffer with 1×PBS via incubating the assembled structures in 1×PBS at 37 °C for 1 hour, then using filtration to remove any excess divalent salt in solution. To note, 3D DNA origami with a binding length of 8 nt was included for comparison (see the Supporting Information for design and assembly conditions). Gel electrophoresis results show the dissociation of structures in 1× PBS for 32 nt brick nanostructures and 3D DNA origami (lanes 5 and 14, respectively, in Figure 2 a), while 52 and 74 nt brick nanostructures remained stable, implying the significance of binding‐domain length when designing DNA nanostructures (lanes 8 and 11, respectively, in Figure 2 a). This experiment was repeated with a longer incubation time (24 hours; Supporting Information, Figure S2) and differently sized brick nanostructures (Supporting Information, Figure S3), and results indicate that all 52 nt brick nanostructures remain stable regardless of their overall size. Transmission electron microscopy (TEM) data also verified the structural stability of the DNA brick nanostructures in 1×PBS (Figure 1 b) and demonstrated that they retain their structure even after 4 days of storage at room temperature (Supporting Information, Figure S4). Though it is expected that longer complementary binding lengths lead to stronger interlocking between base‐pairs, all results emphasize the importance of binding‐domain length as a contributing factor in stabilizing DNA nanostructures. This finding also helps explain why most 3D DNA origami structures with average binding‐domain lengths of 7–10 base‐pairs degrade when the global divalent salt concentration is low (0–0.8 mm MgCl2).12, 13, 14, 15, 16, 17, 18, 19, 20
Figure 2.

Structural stability test against low salt denaturation. a) 32, 52, and 74 nt brick nanostructures and 3D origami were assembled at either 10 or 40 mm MgCl2 (lanes 4, 7, 10, and 13). Assembled samples were incubated at 37 °C in 1×PBS for 1 hour, then spin‐filtered to completely remove any remaining salt in solution (lanes 5, 8, 11, and 14). All samples were characterized via agarose gel electrophoresis. b) TEM images were taken to confirm the preservation of structures. Scale bars indicate 50 nm in length.
DNA brick nanostructures that remain stable even in 1×PBS were still susceptible to nucleases. After confirming that DNA brick nanostructures were stable against low salt denaturation without the requirement of any additional stabilization techniques, the same 52 nt brick nanostructures were tested against nuclease digestion (Supporting Information, Figure S5). We followed a DNase I titration assay used in previous literature,8 and brick nanostructures were incubated with varying concentrations of DNase I at 37 °C for 1 hour, then characterized via gel electrophoresis. A decrease in band intensity as well as a slight shift in band position were found (Figure 3 and Supporting Information, Figure S5), suggesting that 52 nt bricks fall apart at a DNase I concentration above approximately 5 U mL−1. The nuclease resistance of DNA brick nanostructures is not high enough when compared to that of chemically stabilized or encapsulated DNA origami,12, 15 and therefore an additional method is required to stabilize DNA brick nanostructures against nuclease digestion.
Figure 3.
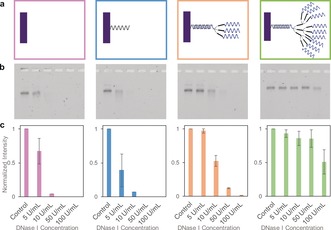
Structural stability test against nuclease digestion. a) DNA brick nanostructures with four different surface densities were studied: bare (pink), single (blue), triple (orange), and nonuple (green). All assembled DNA brick nanostructures were incubated at 37 °C for 1 hour with varying DNase I concentrations (0 to 100 U mL−1). b) Incubated samples were characterized via gel electrophoresis (Lane 1: control; Lane 2: 5 U/L of DNAse 1; Lane 3: 0 U/L of DNAse 1; Lane 4: 50 U/L of DNAse 1; Lane 5: 100 U/L of DNAse 1). c) Gel band intensities from each gel were quantified via a gel‐imaging software, then normalized such that the intensity of the control is always 1. Data from three separate gel results were averaged, and standard deviations were calculated..
We envisioned that densely functionalizing the outer surface of DNA brick nanostructures with additional oligonucleotide strands would stabilize them against enzymes. This was mainly inspired by spherical nucleic acids (SNAs),23 which are formed by organizing nucleic acids radially around a nanoparticle core. SNAs can enter cells without transfection reagents, and once inside the cell, the nucleic acid components of SNAs resist nuclease degradation, leading to longer intracellular lifetimes.23 Because bare DNA brick nanostructures alone were not stable against nucleases, we hypothesized that introducing surrounding oligonucleotides in high density would prevent enzyme accessibility and therefore stabilize them against digestion.
Because only a limited number of overhang strands can be designed into a given DNA brick nanostructure, the surface density could be increased by attaching dendritic oligonucleotides. To do this, 52 nt DNA brick nanostructures were carefully designed to have approximately 100 protruding overhang strands to which dendritic oligonucleotides can be attached (Figure 1 b). Dendritic oligonucleotides were synthesized by incorporating a trebler phosphoramidite, a branching reagent that can be incorporated in regular DNA synthesis protocols. Integrating one trebler modifier enables each single‐stranded DNA to branch into three separate single‐stranded arms (3×), while including two repeated trebler moieties enables the dendritic oligonucleotide to have nine single stranded arms (9×). 3× or 9× dendrimers can hybridize with the overhang strands to systematically increase the total number of available single‐stranded oligonucleotides on the outermost surface.
DNA brick nanostructures were tested against nuclease digestion at four different surface oligonucleotide densities: i) bare, with no protruding strands from the brick nanostructures, ii) single, with approximately 100 protruding overhang DNA strands, iii) triple, with 3× dendrimers attached, and iv) nonuple, with 9× dendrimers attached (Figure 3 a). All samples were exposed to different concentrations of DNase I, ranging from 0 to 100 U mL−1. These concentrations were chosen based on previous literature12 and a DNase I titration assay was used to characterize nuclease resistance. Gel analysis shows that DNA brick nanostructures have an increased resistance to DNase I at higher oligonucleotide density on the outer surface. Some samples show the formation of multimeric (for example, dimer) structures, however, this phenomenon is found in most synthesized brick structures (including bare ones)22, and therefore we do not believe the multimeric structures themselves dramatically increase the overall structural stability. To calculate the number of oligonucleotides available on the outer surface, fluorescence measurements were performed (Supporting Information, Figures S7 and S8). The estimated numbers of available DNA strands, relatively close to the expected numbers with some error, indirectly validate the accessibility of DNA sequences at the outer surface of DNA brick nanostructures.
Lastly, in vitro studies were carried out on DNA brick nanostructures incubated in 10 % FBS cellular media for different lengths of times. To compare the effect of dendritic brushes, bare and nonuple DNA brick nanostructures were tested in 10 % FBS (Supporting Information, Figure S9). Results reveal a shorter survival time for bare DNA brick nanostructures while nonuple brick nanostructures survived up to 30 hours without significant degradation (Figure 4 a and Supporting Information, Figure S10). Cellular uptake studies were also performed by incubating nonuple DNA brick nanostructures in HEK293T cells. Nonuple DNA brick nanostructures were fluorescently labeled via hybridizing complementary Cy5‐ssDNA to the single‐stranded region of the dendritic oligonucleotides. In situ imaging results show a clear difference in fluorescence between Cy5‐ssDNA only versus Cy5‐DNA brick nanostructures (Figure 4 b), as well as successful uptake of the DNA brick nanostructures inside the cells.
Figure 4.
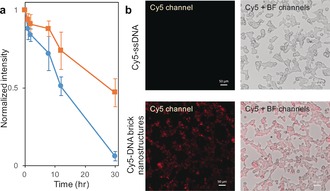
Testing stability in cellular media (10 % FBS) and conducting cellular uptake. a) Bare (blue circles) and nonuple (orange squares) DNA brick nanostructures were assembled, then incubated in 10 % FBS at 37 °C for varying lengths of time (0, 1, 2, 8, 12, 30 hours). Normalized band intensities, from three separately conducted gel electrophoresis experiments, were averaged and plotted against time. b) Nonuple DNA brick nanostructures (200 nm) were incubated with complementary Cy5‐ssDNA (2 μm) to fluorescently label the structures. HEK293T cells were incubated with 200 nm fluorescently labeled nonuple DNA brick nanostructures, and as control, a separate set of HEK293T cells were incubated with 2 μm Cy5‐ssDNA. Both sets were characterized via an inverted fluorescence microscopy using the Cy5 and bright‐field channels.
In conclusion, DNA brick nanostructures with binding‐domain lengths of 13 nt or longer are stable against denaturation in low divalent salt concentration and attaching dendritic oligonucleotides to the outer surface of DNA brick nanostructures stabilizes them against nuclease digestion. Dendritic‐oligonucleotide‐coated DNA brick nanostructures do not require chemical base‐pair interlocking techniques or encapsulation methods yet still display structural stability in cellular media as well as accessibility of DNA sequences at the surface. As a result, DNA brick nanostructures offer a promising alternative to DNA origami. Furthermore, unlike spherical nucleic acids in which DNA strands are coated in an isotropic manner, these DNA brick nanostructures can become asymmetrically functionalized with precise location control, which will have important implications as research efforts shift to the use of multi‐functionalized nanomaterials.24, 25
Conflict of interest
P.Y. is a co‐inventor in DNA brick patents. P.Y. is also a co‐founder of Ultivue Inc. and NuProbe Global.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the National Institutes of Health (1DP1GM133052), the Office of Naval Research (N00014‐18‐1‐2549), the National Science Foundation (1540214), and the Wyss Institute's Immune Materials Platform. Y. K. thanks Dr. Ayush Verma, Prof. William M. Shih, Dr. Zhao Zhao, Dr. Frances Anastassacos for helpful discussions, and Dr. Nikhil Gopalkrishnan for insight in DNA origami.
Y. Kim, P. Yin, Angew. Chem. Int. Ed. 2020, 59, 700.
References
- 1. Seeman N. C., Annu. Rev. Biochem. 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu Q., Li H., Wang L., Gu H., Fan C., Chem. Rev. 2019, 119, 6459–6506. [DOI] [PubMed] [Google Scholar]
- 3. Seeman N. C., Slieman H. F., Nat. Rev. Mater. 2018, 3, 17068. [Google Scholar]
- 4. Douglas S. M., Bachelet I., Church G. M., Science 2012, 335, 831–834. [DOI] [PubMed] [Google Scholar]
- 5. Li S., Jiang Q., Liu S., Zhang Y., Tian Y., Song C., Wang J., Zou Y., Anderson G. J., Han J.-Y., Chang Y., Liu Y., Zhang C., Chen L., Zhou G., Nie G., Yan H., Ding B., Zhao Y., Nat. Biotechnol. 2018, 36, 258–264. [DOI] [PubMed] [Google Scholar]
- 6. Ijäs H., Hakaste I., Shen B., Kostiainen M. A., Linko V., ACS Nano 2019, 13, 5959–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Madhanagopal B. R., Zhang S., Demirel E., Wady H., Chandrasekaran A. R., Trends Biochem. Sci. 2018, 43, 997–1013. [DOI] [PubMed] [Google Scholar]
- 8. Hahn J., Wickham S. F. J., Shih W. M., Perrault S. D., ACS Nano 2014, 8, 8765–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramakrishnan S., Ijas H., Linko V., Keller A., Comput. Struct. Biotechnol. J. 2018, 16, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kielar C., Xin Y., Shen B., Kostiainen M. A., Grundmeier G., Linko V., Keller A., Angew. Chem. Int. Ed. 2018, 57, 9470–9474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9614–9618. [Google Scholar]
- 11. Bila H., Kurisinkal E. E., Bastings M. M. C., Biomater. Sci. 2019, 7, 532–541. [DOI] [PubMed] [Google Scholar]
- 12. Ponnuswamy N., Bastings M. M. C., Nathwani B., Ryu J., Chou L. Y. T., Vinther M., Li W. A., Anastassacos F. M., Mooney D. J., Shih W. M., Nat. Commun. 2017, 8, 15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahmadi Y., Llano E. D., Barisic I., Nanoscale 2018, 10, 7494–7504. [DOI] [PubMed] [Google Scholar]
- 14. Perrault S. D., Shih W. M., ACS Nano 2014, 8, 5132–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bastings M. M. C., Anastassacos F. M., Ponnuswamy N., Leifer F. G., Cuneo G., Lin C., Shih W. M., Nano Lett. 2018, 18, 3557–3564. [DOI] [PubMed] [Google Scholar]
- 16. Gerling T., Kube M., Kick B., Dietz H., Sci. Adv. 2018, 4, eaau1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mikkilä J., Eskelinen A.-P., Niemelä E. H., Linko V., Frilander M. J., Törmä P., Nano Lett. 2014, 14, 2196–2200. [DOI] [PubMed] [Google Scholar]
- 18. Auvinen H., Zhang H., Kopilow A., Niemela E. H., Nummelin S., Adv. Healthcare Mater. 2017, 6, 1700692. [DOI] [PubMed] [Google Scholar]
- 19. Agarwal N. P., Matthies M., Gur F. N., Osada K., Schmidt T. L., Angew. Chem. Int. Ed. 2017, 56, 5460–5464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5552–5556. [Google Scholar]
- 20. Cassinelli V., Oberleitner B., Sobotta J., Nickels P., Grossi G., Kempter S., Angew. Chem. Int. Ed. 2015, 54, 7795–7798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7905–7909. [Google Scholar]
- 21. Ke Y., Ong L. L., Shih W. M., Yin P., Science 2012, 338, 1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong L. L., Hanikel N., Yaghi O. K., Grun C., Strauss M. T., Bron P., Lai-Kee-Him J., Schueder F., Wang B., Wang P., Kishi J. Y., Myhryold C. A., Zhu A., Jungmann R., Bellot G., Ke Y., Yin P., Nature 2017, 552, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cutler J. I., Auyeung E., Mirkin C. A., J. Am. Chem. Soc. 2012, 134, 1376–1391. [DOI] [PubMed] [Google Scholar]
- 24. Chang M., Yang C.-S., Huang D.-M., ACS Nano 2011, 5, 6156–61163. [DOI] [PubMed] [Google Scholar]
- 25. Wang S., Qin L., Yamankurt G., Skakuj K., Huang Z., Chen P., Dominguez D., Lee A., Zhang B., Mirkin C. A., Proc. Natl. Acad. Sci. USA 2019, 116, 10473–10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary