

Abstract
Genomic instability is a hallmark of many cancers; however, the molecular etiology of chromosomal dysregulation is not well understood. The human T-cell leukemia virus type-1 (HTLV-1) oncoprotein Tax activates NF-κB-signaling and induces DNA-damage and aberrant chromosomal segregation through diverse mechanisms which contribute to viral carcinogenesis. Intriguingly, Stathmin/oncoprotein-18 (Op-18) depolymerizes tubulin and interacts with the p65RelA subunit and functions as a cofactor for NF-κB-dependent transactivation. We thus hypothesized that the dissociation of p65RelA-Stathmin/Op-18 complexes by Tax could lead to the catastrophic destabilization of microtubule (MT) spindle fibers during mitosis and provide a novel mechanistic link between NF-κB-signaling and genomic instability. Here we report that the inhibition of Stathmin expression by the retroviral latency protein, p30II, or knockdown with siRNA-stathmin, dampens Tax-mediated NF-κB transactivation and counters Tax-induced genomic instability and cytotoxicity. The Tax-G148V mutant, defective for NF-κB activation, exhibited reduced p65RelA-Stathmin binding and diminished genomic instability and cytotoxicity. Dominant-negative inhibitors of NF-κB also prevented Tax-induced multinucleation and apoptosis. Moreover, cell clones containing the infectious HTLV-1 ACH.p30II mutant provirus, impaired for p30II production, exhibited increased multinucleation and the accumulation of cytoplasmic tubulin aggregates following nocodozole-treatment. These findings allude to a mechanism whereby NF-κB-signaling regulates tubulin dynamics and mitotic instability through the modulation of p65RelA-Stathmin/Op-18 interactions, and support the notion that p30II enhances the survival of Tax-expressing HTLV-1-transformed cells.
Keywords: HTLV-1, Tax, p30, Viral oncoprotein, ATLL, NF-kappa B, Stathmin, Genomic instability, Microtubule, Tubulin
Graphical Abstract
Introduction
The human T-cell leukemia virus type-1 (HTLV-1) infects and transforms CD4+ T-lymphocytes and causes adult T-cell leukemia/lymphoma (ATLL) -an aggressive, and often-fatal hematological malignancy that generally responds poorly to most anticancer therapies (Johnson et al., 2001; Bangham and Ratner, 2015). One distinguishing pathological feature of acute/lymphoma-stage ATLL is the presence of tumor lymphocytes with atypical pleomorphic or polylobate (“flower-shaped”) nuclei (Johnson et al., 2001; Bangham and Ratner, 2015). The viral transactivator protein Tax induces clastogenic DNA-damage and genomic instability through the generation of reactive oxygen species (ROS) and the inhibition of cellular DNA-damage surveillance and repair components, and by targeting various regulators of the cell-cycle checkpoints (Baydoun et al., 2015; Belgnaoui et al., 2010; Durkin et al., 2008; Haoudi et al., 2003; Chaib-Mezrag et al., 2014; Dayaram et al., 2013; Baydoun et al., 2015; Chaib-Mezrag et al., 2014; Majone and Jeang, 2000; Kinjo et al., 2010; Hutchison et al., 2018; Jin et al., 1998; Majone and Jeang, 2000; Liu et al., 2005; Kehn et al., 2005; Neuveut et al., 1998; Ching et al., 2006; Peloponese et al., 2005; Belgnaoui et al., 2010; Durkin et al., 2008; Haoudi et al., 2003; Dayaram et al., 2013; Zane et al., 2012; Jin et al., 1998; Majone and Jeang, 2000; Liu et al., 2005; Kehn et al., 2005; Neuveut et al., 1998). Further, Tax activates NF-κB-dependent expression of the inducible Nitric oxide synthase (iNOS) which results in nitric oxide (NO) production and double-strand DNA breaks in HTLV-1-infected cells (Baydoun et al., 2015). Chaib-Mezrag et al., 2014 have also reported that Tax-induced dsDNA lesions, associated with the slowed progression of replication forks during DNA-synthesis, were attributed to NF-κB activation and increased intracellular NO levels.
The Tax oncoprotein induces constitutive NF-κB-signaling through the activation of the IκBa-kinase (IκK) complex -consisting of IκKα, IκKβ, and IκKγ subunits, which results in the phosphorylation/polyubiquitination and degradation of the inhibitor IkBα and subsequent nuclear translocation of NF-κB p65RelA/p50 heterodimers that bind to kB-responsive promoter elements (O’Mahony et al., 2004; Sun et al., 1994; Geleziunas et al., 1998; Harhaj et al., 2007; Ho et al., 2015). Tax interacts with the scaffold subunit, IκKγ (also known as the NF-κB essential modulator, or NEMO; Yamaoka et al., 1998; Harhaj et al., 2000), the E3 ubiquitin ligase -Ring finger protein 8 (Ho et al., 2015), and E2 conjugating enzymes Ubc13/Uev1A and Ubc13/Uev2, and induces oligomerization of the IκK complex by generating K63- and Met1-linked polyubiquitin chains (Ho et al., 2015; Shembade et al., 2007; Lavorgna and Harhaj, 2014; Shibata et al., 2017). The assembly of K63-polyubiquitin chains activates TGFβ-activated kinase 1 (TAK1) which stimulates IκK-signaling (Ho et al., 2015; Wu and Sun, 2007). Whereas NF-κB transactivation is essential for the continuous proliferation and survival of HTLV-1-transformed lymphocytes (Harhaj and Giam, 2018; Zhang et al., 2016; Choi and Harhaj, 2014; Lavorgna et al., 2014), the hyperactivation of NF-κB-signaling by Tax induces DNA-damage that can lead to cellular senescence and apoptosis (Baydoun et al., 2015; Chaib-Mezrag et al., 2014; Kinjo et al., 2010; Kuo and Giam, 2006; Ho et al., 2012; Zhi et al., 2011; Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998). We recently demonstrated in Hutchison et al., 2018 that the HTLV-1 latency-maintenance factor p30II (Nicot et al., 2004; Younis et al., 2004; Zhang et al., 2000; Bartoe et al., 2000) inhibits Tax-induced cytotoxicity and cooperates with the viral transactivator to promote oncogenic colony formation in vitro, dependent upon the activation of p53-regulated pro-survival signals, including the TP53-induced glycolysis and apoptosis regulator (TIGAR; Bensaad et al., 2006; Bensaad et al., 2009) which suppresses Tax-induced oxidative stress. p30II interacts with the MYST-family acetyltransferase TIP60 (Awasthi et al., 2005; Romeo et al., 2015) and inhibits lysine K120-acetylation of the p53 protein (Romeo et al., 2018) which differentially regulates the expression of p53-dependent pro-apoptotic genes (Sykes et al., 2006; Tang et al., 2006; Kurash et al., 2008; Dar et al., 2013; Xu et al., 2014). Interestingly, the p53 tumor suppressor is mutated in nearly half of all cancers; however, it is rarely mutated in HTLV-1+ ATLL clinical isolates which frequently contain high levels of wildtype p53 (Zane et al., 2012; bPise-Masison et al., 1998; Tabakin-Fix et al., 2006; Mengle-Gaw and Rabbitts, 1987), suggesting the subversion of p53-regulated target genes may contribute to viral carcinogenesis (Hutchison et al., 2018; Romeo et al., 2018). Although there is currently no commercial antibody available to detect the p30II protein, the alternatively-spliced pX-orfII-p30 mRNA has been detected by RT-PCR in chronically HTLV-1-infected T-cell-lines, primary uncultured ATLL clinical isolates, and PBMCs from asymptomatic carriers (Princler et al., 2003; Berneman et al., 1992; Koralnik et al., 1992; Ciminale et al., 1992). Pique et al., 2000 have further shown that CD8+ cytotoxic T-lymphocytes that specifically target the p30II and p13II peptides can be isolated from HTLV-1+ asymptomatic carriers, as well as HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) and ATLL patients, which suggests these ORF-II products are chronically expressed in vivo.
Stathmin/oncoprotein-18 (Op-18) is one of only a few genes whose expression is negatively regulated by the p53 tumor suppressor/histone deacetylase-1 (HDAC1)/mammalian Sin3a (mSin3a) transcriptional repressor complexes (Murphy et al., 1999; Ahn et al., 1999). The Stathmin protein binds tubulin dimers and increases the catastrophe (i.e., shortening) rate of microtubule (MT) spindle fibers during mitosis (Belmont and Mitchison, 1996; Belmont et al., 1996). Indeed, Holmfeldt et al., 2006 have reported that a hyperactive Q18E mutant, or overexpression of wildtype Stathmin, causes chromosomal instability and aneugenic ploidy changes in leukemic cells. The Stathmin protein is phosphorylated on four serine residues (S16, S25, S38, and S68) by cell-cycle regulators and in response to surface receptor-kinase-signaling; and the hyper-phosphorylation of Stathmin inhibits its ability to destabilize MTs attached to mitotic chromatin (Belmont and Mitchison, 1996; Holmfeldt et al., 2001; Küntziger et al., 2001; Andersen et al., 1997). The catastrophe-promoting activity of Stathmin is dependent upon its N-terminal region which destabilizes MT spindle fibers, independent of binding tubulin dimers associated with the depolymerization of interphase microtubules (Holmfeldt et al., 2001; Larsson et al., 1999). The MT-destabilizing activity of Stathmin/Op-18 is constrained through cytoplasmic interactions with the cyclin-dependent kinase inhibitor (cdk)-inhibitor, p27Kip1, in proliferating cells (Belletti et al., 2010; Schiappacassi et al., 2011; Fabris et al., 2015; Bertron et al., 2014). It is important to note, however, that HTLV-1-transformed tumor lymphocytes contain low levels of p27Kip1 (Kuo and Giam, 2006; Iwanaga et al., 2001) which could lead to unrestrained Stathmin MT-destabilizing activity, altered tubulin dynamics, and catastrophic genomic instability in ATLL cells. Intriguingly, Lu et al., 2014 have reported that Stathmin interacts with the NF-κB subunit p65RelA and acts as a cofactor for NF-κB-signaling by stabilizing the p65RelA protein in aggressive pancreatic tumors. This prompted us to investigate whether HTLV-1 p30II influences p65RelA-Stathmin/Op-18 molecular interactions and NF-κB transcriptional signaling by the Tax oncoprotein.
In the present study, we demonstrate that the repression of Stathmin/Op-18 by the viral latency protein p30II, or small-interfering RNAs targeted against stathmin transcripts (siRNA-stathmin), destabilizes p65RelA and inhibits Tax-induced NF-κB-signaling, genomic instability, and cytotoxicity in HTLV-1-infected cells. These findings suggest that p30II functions as an ancillary factor to promote the survival of Tax-expressing cells which could contribute to viral persistence and the establishment of neoplastic disease (Bartoe et al., 2000; Silverman et al., 2004; Edwards et al., 2011; Valeri et al., 2010; Yamamoto et al., 2008). Importantly, the modulation of tubulin dynamics and destabilization of mitotic MT spindle fibers -as a consequence of p65RelA Stathmin/Op-18 molecular interactions, alludes to a novel mechanistic link between genomic instability and NF-κB inflammatory signaling in certain cancers, such as ATLL, with reduced p27Kip1 expression (Kuo and Giam, 2006; Iwanaga et al., 2001).
Results
The HTLV-1 p30II protein represses Stathmin/Op-18 and inhibits Tax-induced NF-κB transactivation
The 3’ region of the HTLV-1 genome contains a highly-conserved nucleotide sequence, known as pX, that encodes several nonstructural/regulatory products, including the viral transactivator Tax, HBZ and p30II, through alternative mRNA splicing (Fig. 1A; Johnson et al., 2001; Bangham and Ratner, 2015; Bartoe et al., 2000; Koralnik et al., 1992; Nicot et al., 2005; Li et al., 2009; Arnold et al., 2006). The Tax oncoprotein contains zinc-finger, leucine zipper, and PDF-binding domains and modulates viral and host cellular gene expression by activating the cyclic AMP-responsive element binding protein/Activating transcription factor (CREB/ATF), Serum-response factor (p62SRF), and NF-κB transcription pathways (Fig. 1B; Sun et al., 1994; Geleziunas et al., 1998; Harhaj et al., 2000; Wu and Sun, 2007; Zhao and Giam, 1992; Smith and Greene, 1990; aSuzuki et al., 1993; bSuzuki et al., 1993; Xie et al., 2006). The diagram in Fig. 1B shows the sites of amino acid substitution mutations within Tax that impair CREB/ATF-dependent (M47: L319R; L320R; Smith and Greene, 1990) or NF-κB-dependent (M22: T130A; L131S; Smith and Greene, 1990, and G148V; Yamaoka et al., 1996) transactivation. As we have recently shown that the p30II protein activates p53 and inhibits TIP60-mediated K120-acetylation of p53 (Romeo et al., 2018), we tested whether p30II might inhibit the expression of Stathmin/Op-18 (as a result of its negative regulation by p53-containing repressor complexes; Murphy et al., 1999; Ahn et al., 1999). Indeed, the results in Fig. 1C demonstrate that an HTLV-1 p30II-Green fluorescent protein (p30II-GFP) fusion inhibited Stathmin protein expression, as compared to an empty CβS vector control in transfected 293 HEK cells. p30II-GFP was detected by immunoblotting and the relative Actin protein levels are shown for comparison (Fig. 1C). The repression of Stathmin correlated with reduced p65RelA protein levels in Tax-expressing and PHA-stimulated cells (Fig. 1D). These findings are consistent with Stathmin’s role as an essential cofactor for p65RelA (Lu et al., 2014), and suggest p30II might counter the hyperactivation of NF-κB-signaling by the Tax transactivator protein (Ho et al., 2012; Zhi et al., 2011). The full functionality of the Tax construct used in these studies was confirmed by testing its ability to activate CREB/ATF-dependent transcription from the three 21-base-pair repeat Tax-responsive elements (TREs) within the U3 region of the HTLV-1 5’ long terminal repeat (LTR) which drive proviral gene expression and replication (supplementary Fig. S2A; Zhao and Giam, 1992; aSuzuki et al., 1993; Giebler et al., 1997; Kwok et al., 1996; Harrod et al., 1998; Harrod et al., 2000; Geiger et al., 2008). To determine if p30II functionally inhibits Tax-mediated NF-κB transactivation, 293 cells were cotransfected with either a kB-responsive E-Selectin promoter-luciferase reporter plasmid (Fig. 1E; Hong et al., 2007) or HIV-1 κB-LTR (ΔTAR)-luciferase reporter construct, spanning the two kB-responsive elements and three SP1-binding sites and with a deletion of the U-rich trinucleotide bulge of the TAR of the HIV-1LAI promoter (Fig. 1F), and expression constructs for HTLV-1 Tax, p30II-GFP, or an empty CβS vector control. The Tax oncoprotein and p30II-GFP were detected by immunoblotting. These results demonstrate that p30II-GFP markedly inhibited Tax-induced NF-κB transactivation from the E-Selectin and HIV-1 κB-LTR (ΔTAR) promoter-reporter plasmids (Figs. 1E and 1F). For comparison, the data in Fig. 1E are represented as fold transactivation in supplementary Fig. S1A. p30II-GFP also inhibited NF-κB transactivation induced by stimulating the cells with phorbol 12-myristate 13-acetate (PMA; Fig. 1G). As additional controls, we demonstrated that HA-tagged p30II similarly inhibits Tax-dependent NF-κB transactivation and represses Stathmin protein expression, as compared to a GFP negative control (supplementary Figs. S1B–S1D).
Fig. 1.

HTLV-1 p30II represses the p65RelA-binding cofactor, Stathmin, and inhibits Tax-induced NF-κB transactivation. (A) Diagram of the HTLV-1 proviral genome and its products. The conserved pX nucleotide sequence is indicated and the protein coding regions are represented by shaded boxes (p30II, tax, and hbz are in bold). The alternatively-spliced mRNAs are represented by dotted lines. The antisense hbz gene product is transcribed from the 3’ long terminal repeat (LTR). (B) A schematic of the HTLV-1 transactivator protein Tax and its functional domains. ZF, zinc-finger motif; LZ, leucine zipper; NES, nuclear export signal. The sites of the M22 (T130A; L131S), G148V, and M47 (L319R; L320S) amino acid substitution mutations are indicated (Smith and Greene, 1990; Yamaoka et al., 1996). (C) 293 HEK cells were transfected with increasing amounts (0.12, 0.25, and 0.5 mg) of an HTLV-1 p30II-GFP expression construct or CβS empty vector control, and the expression of Stathmin and p30II-GFP was detected by SDS-PAGE and immunoblotting. The relative levels of Actin are shown as a protein loading control. (D) The NF-κB p65RelA subunit is destabilized by HTLV-1 p30II-GFP and correlates with the repression of Stathmin. 293 HEK cells were cotransfected with 0.25 mg of RcCMVHTLV-1 Tax and/or pEGFP-N3-HTLV-1 p30II-GFP or a CβS empty vector, and the expression of the Stathmin, p65RelA, p30II-GFP, Tax, and Actin proteins was detected by immunoblotting. Alternatively, the cells were stimulated with 10 ng/ml PHA for 14 hrs, either alone or in combination with HTLV-1 p30II-GFP expression. UT, untransfected cells. (E and F) The effects of HTLV-1 p30II and p30II-mediated Stathmin repression upon Tax-dependent NF-κB transactivation were determined by cotransfecting 293 HEK cells with 0.25 mg of either a kB-responsive E-Selectin promoter-luciferase reporter plasmid (E), or an HIV-1 κB-LTR (ΔTAR)-luciferase reporter plasmid which contains nucleotides 345–531 of the HIV-1LAI promoter, spanning the two kB-responsive elements (F), together with expression constructs for HTLV-1 Tax or p30II-GFP or a CβS empty vector control. Relative luciferase activities were measured and normalized for equivalent total cellular protein levels. The Tax, p30II-GFP, and Actin proteins were detected by immunoblotting. (G) The general inhibition of NF-κB transactivation by the HTLV-1 latency factor p30II was demonstrated by cotransfecting 293 cells with an ESelectin promoter-luciferase reporter plasmid and increasing amounts (0.25 and 0.5 mg) of either a pEGFP-N3-HTLV-1 p30II-GFP expression construct or CβS empty vector, and then stimulating the cells with 100 ng/ml PMA for 14 hrs. Relative luciferase activities were quantified and normalized for equivalent total cellular protein levels. The p30II-GFP and Actin proteins were detected by immunoblotting. UT, untransfected cells. All the data is representative of at least three independent experiments. The data in E, F, and G represent the mean of the experiments ± standard deviation (error bars).
The NF-κB-defective transactivation mutants, Tax-M22 and Tax-G148V (Smith and Greene, 1990; Yamaoka et al., 1996), were impaired for the activation of kB-responsive transcription from the E-Selectin promoter, as compared to wildtype Tax or the Tax-M47 mutant (Fig. 2A; Smith and Greene, 1990). The co-expression of p30II-GFP also inhibited NF-κB transactivation by the Tax-M47 mutant (Fig. 2A). The wildtype Tax and the Tax-M22, Tax-M47, and Tax-G148V mutant proteins, as well as p30II-GFP were detected by immunoblotting (Fig. 2A, lower panels). The NF-κB-dependence of Tax transactivation from the E-Selectin gene promoter was further demonstrated by cotransfecting the cells with a “super repressor” mutant of IkBα, IkBα-S32A/S36A, defective for Ser32/Ser36-phosphorylation and degradation (supplementary Fig. S2B; DiDonato et al., 1996), or with dominant-negative deletion mutants of the IκKβ subunit:IκKβΔ9 and IκKβD34 (supplementary Fig. S2C; Sylla et al., 1998). The IkBα-S32A/S36A,IκKβΔ9 (FLAG-tagged) and IκKβD34 (FLAG-tagged) mutants, and HTLV-1 Tax proteins were detected by immunoblotting (Figs. S2B and S2C). We also demonstrated that p30II-GFP inhibits Tax-induced NF-κB transactivation in Jurkat T-lymphocytes cotransfected with an ESelectin-luciferase reporter plasmid and various expression constructs for Tax, p30II-GFP, HBZ, the Tax-M22, Tax-M47, or Tax-G148V mutants, or an empty CβS vector control (Fig. 2B). The HTLV-1 antisense protein HBZ did not significantly activate NF-κB-dependent transcription (Fig. 2B). Both the Tax-M22 and Tax-G148V mutants were impaired for NF-κB transactivation, whereas Tax-M47 induced kB-responsive transactivation at levels comparable to wildtype Tax (Fig. 2B). To determine whether the repression of Stathmin/Op-18 directly inhibits Tax-induced NF-κB-signaling, 293 cells were repeatedly cotransfected with siRNA oligonucleotides targeted against stathmin transcripts (siRNA-stathmin) or a non-specific RNA (nsRNA) as negative control. The siRNA-stathmin oligonucleotides, #1 and #2, which bind different nucleotide sequences, inhibited Stathmin expression and destabilized the p65RelA protein, as compared to the nsRNA control (Figs. 2C and 2D). We observed that siRNA-stathmin oligo #2 was consistently more effective at inhibiting Stathmin and, therefore, this siRNA molecule was used for most of the experiments throughout this study. The co-expression of p30II-GFP inhibited Tax-induced NF-κB transactivation from the E-Selectin promoter (Fig. 2E). The siRNA-stathmin oligonucleotide (#2) also potently inhibited Tax-induced NF-κB-signaling, relative to the nsRNA negative control, in cotransfected cells (Fig. 2E). The relative expression of Stathmin was detected by immunoblotting (Fig. 2E, lower panels) and quantified by densitometry (Fig. 2F). These findings demonstrate that HTLV-1 p30II represses Stathmin/Op-18 and destabilizes the p65RelA subunit, and suggest p30II could cooperate with the viral transactivator by dampening Tax-induced NF-κB-signaling (Hutchison et al., 2018; Ho et al., 2012; Zhi et al., 2011).
Fig. 2.

The HTLV-1 p30II protein and siRNA-knockdown of Stathmin inhibit the NF-κB activating functions of Tax. (A) 293 HEK cells were cotransfected with 0.25 mg of an E-Selectin promoter-luciferase reporter plasmid and RcCMV expression constructs (0.25 mg) for wildtype HTLV-1 Tax or the Tax substitution mutants, Tax-M22, Tax-G148V, or Tax-M47 (Smith and Greene, 1990; Yamaoka et al., 1996). The co-expression of increasing amounts (0.25 and 0.5 mg) of p30II-GFP inhibited NF-κB transactivation by the Tax-M47 mutant, which is impaired for CREB-dependent transcriptional activation from the HTLV-1 promoter (Smith and Greene, 1990). Relative luciferase activities were measured and normalized for equivalent total cellular protein levels. The data represent the mean ± standard deviation (error bars) from three independent experiments. The expression of wildtype Tax, Tax-M22, Tax-M47, Tax-G148V, and p30II-GFP was detected by SDS-PAGE and immunoblotting (lower panels). Relative Actin protein levels are provided for comparison. (B) Jurkat T-lymphocytes were cotransfected with 0.25 mg of the E-Selectin-luc reporter plasmid and various combinations of the expression constructs for wildtype HTLV-1 Tax, Tax-M22, Tax-M47, or Tax-G148V mutants, HTLV-1 HBZ, p30II-GFP, or a CβS empty vector control. Relative luciferase activities were measured and normalized for equivalent total cellular proteins. The data represent the mean ± standard deviation from three independent experiments. (C) 293 HEK cells were repeatedly transfected with siRNAs targeted against stathmin transcripts (siRNA-stathmin #1 and #2) or a non-specific RNA (nsRNA) control in increasing amounts (50 and 100 ng), and the expression of Stathmin and p65RelA was assessed by SDS-PAGE and immunoblotting. Relative Actin protein levels are shown for comparison. (D) The relative expression of Stathmin and p65RelA in C was quantified by densitometry analysis of the immunoblot bands. (E) The effects of targeted siRNA-knockdown of Stathmin expression upon Tax-dependent NF-κB transactivation were determined by cotransfecting 293 HEK cells with E-selectin-luc and expression constructs (0.25 mg) for Tax and/or p30II-GFP, or Tax in the presence of increasing amounts (50 and 100 ng) of siRNA-stathmin or nsRNA as a negative control. Relative luciferase activities were measured and normalized for equivalent total cellular proteins. The relative expression of Stathmin and Tubulin was detected by immunoblotting (lower panels) and (F) the bands were quantified by densitometry. UT, untransfected cells. All the data is representative of at least three independent experiments. The data in D, E, and F represent the experimental mean ± standard deviation (error bars).
Stathmin/Op-18 and the NF-κB subunit p65RelA interact in HTLV-1-transformed lymphoma T-cells
We next investigated whether p65RelA and Stathmin/Op-18 interact or exist as part of a stable complex by performing coimmunoprecipitations. The results in Figs. 3A and 3B show that NF-κB-signaling -induced by stimulating 293 HEK cells with PMA, led to increased expression of the p65RelA protein. The relative input levels of Actin are shown for comparison. Stathmin/Op-18 was coimmunoprecipitated in p65RelA-containing complexes using Protein-G-agarose and Anti-Stathmin/Op-18 or Anti-p65RelA antibodies, and extracts prepared from PMA-treated cells (Figs. 3A and 3B). The HTLV-1 p30II-GFP inhibited NF-κB-dependent transactivation induced by PMA-stimulation, as well as by the Tax oncoprotein (Figs. 1E–1G). We therefore tested whether p30II-GFP might interfere with the formation of p65RelA-Stathmin immune-complexes in Tax-expressing cells. The results in Figs. 3C and 3D demonstrate that p30II-GFP, either alone or in combination with Tax, inhibited the co-immunoprecipitation of p65RelA with Stathmin using a monoclonal Anti-p65RelA antibody (Fig. 3C, lower panel). The input levels of Tax, p30II-GFP, Stathmin, and Actin are shown in the upper panels of Fig. 3C. We then investigated whether p65RelA and Stathmin stably interact in HTLV-1- transformed ATLL tumor cell-lines that express high levels of the Tax oncoprotein. Indeed, the NF-κB p65RelA subunit was coimmunoprecipitated in Stathmin-containing complexes using an Anti-p65RelA antibody and extracts prepared from the HTLV-1-transformed lymphoma T-cell-lines, MJG11 and SLB1 (Fig. 3E, lower panels, and Fig. 3F). Consistent with the constitutive activation of NF-κB-signaling in HTLV-1-transformed cells (Harhaj et al., 2000; Wu and Sun, 2007; Harhaj and Giam, 2018), the MJG11 and SLB1 lymphocytes exhibited more p65RelA-Stathmin immune-complex formation than hu-PBMCs as determined by densitometry quantitation (Figs. 3E and 3F). As a negative control, the monoclonal Anti-p65RelA antibody was heat-inactivated prior to use in the coimmunoprecipitation reactions (Fig. 3E, bottom panel, and Fig. 3F). We further demonstrated that the knockdown of Stathmin, by transfecting HTLV-1-transformed SLB1 lymphocytes with siRNA-stathmin oligonucleotides (#1 or #2) which bind to different sequences, concomitantly resulted in the destabilization of the NF-κB p65RelA subunit in HTLV-1-transformed cells (Figs. 3G–3J).
Fig. 3.

Stathmin interacts with the NF-κB p65RelA subunit in HTLV-1-transformed ATLL cells. (A) p65RelA-Stathmin protein complexes were immunoprecipitated from precleared whole-cell lysates prepared from PMA-stimulated 293 HEK cells using Protein-G-agarose (20 ml of a 50% slurry) and monoclonal Anti-p65RelA and rabbit polyclonal Anti-Stathmin/Op-18 antibodies. The relative input levels of Stathmin, p65RelA, and Actin are shown in the upper panels. The precipitated bound p65RelA and Stathmin proteins were resolved by SDS-PAGE and detected by immunoblotting (lower panels). UT, untreated cells. (B) The relative levels of the immunoprecipitated Stathmin (left) and p65RelA (right) proteins in A were quantified by densitometry analysis of the immunoblot bands. (C) The formation of HTLV-1 Tax-induced p65RelA-Stathmin immune-complexes is markedly diminished in cotransfected cells expressing increasing amounts (2.5 and 5.0 mg) of p30II-GFP. The relative input levels of HTLV-1 Tax, p30II-GFP, Stathmin, and Actin are shown (upper panels). The Stathmin/Op-18 protein, complexed with p65RelA, was co-immunoprecipitated using Protein-G-agarose and an Anti-p65RelA antibody and detected by immunoblotting (lower panel). (D) The relative levels of the immunoprecipitated Stathmin protein in C was quantified by densitometry. (E) p65RelA-Stathmin protein complexes were immunoprecipitated from precleared extracts prepared from cultured/activated hu-PBMCs and the HTLV-1-transformed ATLL T-cell-lines, MJG11 and SLB1, using a monoclonal Anti-p65RelA antibody. As a negative control, the Anti-p65RelA antibody was heat-inactivated prior to its use in immunoprecipitation reactions. The relative input levels of Stathmin, p65RelA, Tax, and Actin are shown in the upper panels. The precipitated Stathmin protein was resolved by SDS-PAGE and detected by immunoblotting using a rabbit polyclonal Anti-Stathmin/Op-18 antibody (lower panels). A non-specific band (ns) is also indicated. (F) The relative levels of immunoprecipitated Stathmin in E was quantified by densitometry analysis of the immunoblot bands. (G-J) siRNA-inhibition of Stathmin expression destabilizes the NF-κB p65RelA subunit. HTLV-1-transformed SLB1 lymphoma T-cells were repeatedly transfected with 180 ng of a siRNA-stathmin oligonucleotide (#1 in G, or #2 in I) or non-specific RNA (nsRNA) as a negative control, and the Stathmin, p65RelA, and HTLV-1 Tax proteins were detected by SDS-PAGE and immunoblotting. (H and J) The relative expression of Stathmin, p65RelA and Tax in G (H) and I (J) was quantified by densitometry. All the data is representative of at least three independent experiments. The data in B, D, F, H and J represent the mean ± standard deviation (error bars).
The expression of Stathmin is negatively regulated by p53/HDAC1/mSin3a repressor complexes (Murphy et al., 1999; Ahn et al., 1999), and our studies have demonstrated that the viral latency protein p30II activates p53 (Romeo et al., 2018) and represses Stathmin/Op-18 (Figs. 1C, 1D, 4A, and 4B). Therefore, to determine whether inhibiting p53-dependent transcriptional activity might interfere with the ability of p30II-GFP to repress stathmin gene expression, 293 cells were cotransfected with various expression constructs for HTLV-1 p30II-GFP, wildtype p53, or a dominant-negative DNA-binding-defective p53 mutant, p53-R175H (Hermeking et al., 1997), or an empty CβS vector control. The expression of the Stathmin, p30II-GFP, p53 (wildtype and the p53-R175H mutant), and Actin proteins was detected by immunoblotting and quantified by densitometry. p30II-GFP significantly inhibited the expression of Stathmin, as compared to untransfected cells or the empty CβS vector (Figs. 4A and 4B). The overexpression of wildtype p53 also inhibited Stathmin protein expression (Figs. 4A and 4B), whereas the dominant-negative DNA-binding mutant, p53-R175H, resulted in higher levels of Stathmin and countered the ability of p30II GFP to repress Stathmin (Figs. 4A and 4B). These findings confirm that the repression of Stathmin expression by p30II is p53-dependent (Murphy et al., 1999; Ahn et al., 1999) and can be countered by inhibiting p53 transcriptional activation. It was observed that the p53-R175H mutant inhibited Tax-induced NF-κB transactivation from the E-Selectin promoter (Fig. 4C). One possible explanation for this unexpected result is that the p53-R175H mutant could interfere with p65RelA transcriptional interactions through directly binding p65RelA (Jeong et al., 2004; Jeong et al., 2005; bPise-Masison et al., 2000), or it might hinder Tax from interacting with the IκK-γ subunit of the IκK complex (Yamaoka et al., 1998; Harhaj et al., 2000; aPise-Masison et al., 1998; Tabakin-Fix et al., 2006; bPise-Masison et al., 1998). These data demonstrate that p65RelA interacts with Stathmin/Op-18 associated with constitutive NF-κB activation in Tax-expressing HTLV-1-transformed lymphoma cells. Moreover, the viral p30II protein represses Stathmin/Op-18 in a p53-dependent manner and inhibits Tax-mediated NF-κB transactivation.
Fig. 4.

A dominant-negative DNA-binding mutant of p53 counters the repression of Stathmin by HTLV-1 p30II. (A) 293 HEK cells were cotransfected with 0.25 mg of pEGFP-N3-HTLV-1 p30II-GFP in combination with increasing amounts (0.25 and 0.5 mg) of pCEP4 expression constructs for wildtype p53 or a dominant-negative DNA-binding mutant, p53-R175H (Hermeking et al., 1997), or a CβS empty vector control. The relative expression of Stathmin, p53, and HTLV-1 p30II-GFP was detected by SDS-PAGE and immunoblotting. The Actin protein levels are shown for comparison. (B) The relative expression of Stathmin and Actin in A was quantified by densitometry analysis. (C) 293 cells were cotransfected with 0.25 mg of an ESelectin promoter-luciferase reporter plasmid and various expression constructs for HTLV-1 Tax, p30II-GFP, or the dominant-negative p53-R175H mutant, or CβS empty vector. Relative luciferase activities were measured and normalized for equivalent total cellular protein levels. UT, untransfected cells. All the data are representative of at least three independent experiments. The data in B and C represent the experimental mean ± standard deviation (error bars).
The HTLV-1 p30II protein and dominant-negative inhibitors of NF-κB dampen kB transcriptional signaling and inhibit MT- and genomic-instability
To determine whether p30II influences NF-κB-signaling in the context of the full-length HTLV-1 provirus, HT1080 clones containing the infectious HTLV-1 ACH.wildtype or ACH.p30II mutant provirus, defective for p30II production (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998), were cotransfected with a tk-renilla-luciferase plasmid and increasing amounts of a κB-responsive ESelectin-firefly-luciferase (Fig. 5A) or HIV-1 κB-LTR (ΔTAR)-firefly-luciferase reporter plasmid (Fig. 5B). Dual-luciferase assays were performed and the relative values of κB-responsive firefly-luciferase activities were normalized to equivalent renilla-luciferase levels. The results in Figs. 5A and 5B revealed that NF-κB-dependent transactivation was greater in the HT1080 clones expressing the HTLV-1 ACH.p30II mutant provirus, compared to wildtype ACH. There were no significant differences observed in the S36-phosphorylation and degradation of the inhibitor IκB-α in the HTLV-1 ACH.wt and ACH.p30II mutant proviral clones over a time-course of cycloheximide-treatment (supplementary Figs. S3A and S3C). However, the ACH.p30II mutant exhibited significantly higher levels of p65RelA than ACH.wt, consistent with the proposed model (Figs. S3A and S3B). Despite the significant activation of NF-κB-dependent transcription by the ACH.p30II mutant (Figs. 5A and 5B) – interestingly, the majority of the p65RelA protein in the cycloheximide-treated HT1080/HTLV-1 ACH.p30II cells was localized in the cytoplasm (Figs. S3D and S3E), although we do not rule out the possibility that the nuclear p65RelA protein may have become degraded over the 2-hrs cycloheximide-treatment in these studies. The mutation in the ACH.p30II provirus also results in an in-frame insertion of eight amino acids (YLEEESRG) after the activation domain of the antisense HBZ protein (Robek et al., 1998; Clerc et al., 2008); however, this insertion is not predicted or likely to disrupt HBZ (see Materials and methods). Nevertheless, we cannot exclude the possibility that the ACH.p30II mutant provirus could have impaired HBZ functions and, as Zhi et al., 2011 have shown HBZ also inhibits Tax-induced NF-κB transactivation, the inactivation of both p30II and HBZ could produce a combined effect upon NF-κB-signaling. These findings are in agreement with the results in Figs. 1E and 1F, and demonstrate that the viral latency protein p30II dampens Tax-induced NF-κB-signaling in the context of the full-length HTLV-1 provirus.
Fig. 5.

Increased Stathmin levels and NF-κB-signaling are associated with MT- and genomic-instability in cell clones containing the HTLV-1 ACH.p30II mutant provirus. (A and B) Transiently-amplified HT1080 clones expressing the infectious HTLV-1 ACH.wildtype (wt) or ACH.p30II mutant provirus, defective for p30II production (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998), were cotransfected with 0.25 mg of a tk-renilla-luciferase plasmid and increasing amounts (0.1, 0.25, 0.5, and 0.75 mg) of an ESelectin promoter-firefly-luciferase (A) or HIV-1 κB-LTR (ΔTAR)-firefly-luciferase reporter plasmid (B). Dual luciferase assays were carried out and the relative NF-κB-dependent firefly-luciferase activities were quantified and normalized for equivalent renilla-luciferase levels. The data in A and B represent the mean ± standard deviation (error bars) from three independent experiments. (C) The HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were treated with nocodozole (400 ng/ml) for 24 hrs and then labeled with BrdU for 6 hrs, permeabilized, fixed and stained using an Anti-BrdU-FITC-conjugated antibody and 7-aminoactinomycin D (7-AAD, BD-Pharmingen), and subsequently analyzed by flow-cytometry. The total cellular genomic DNA-contents were analyzed using the ModFit LT 3.0 cell-cycle algorithm to determine the relative percentages of diploid cells in the G1, S, and G2-phases, as well as the total percentages of aneuploid cells (bar graph at top and cell-cycle profiles in the bottom panels). The data are representative of three independent experiments. (D and E) The relative percentages of multinucleate cells per field in the untreated HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones, ± nocodozole-treatment, were determined by staining the samples with a monoclonal Anti-Tubulin primary antibody and rhodamine-red-conjugated secondary antibody (red) and 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI; blue). Scale bar, 10 mm. The numbers of multinucleate cells, as compared to the total numbers of cells (D and E), in triplicate visual fields were counted using confocal immunofluorescence-microscopy. (F) The relative levels of the Stathmin protein expressed in the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones, compared to the parental HT1080 cell-line, were detected by immunoblotting. Actin protein levels are shown for comparison. (G-I) The HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones were transfected with 0.5 mg of pEGFP-N3-HTLV-1 p30II-GFP, or expression constructs for the dominant-negative NF-κB- signaling mutants: IkBα-S32A/S36A, IκKβΔ9, or IκKbD34, together with pcDNA3.1-GFP. The HT1080 cells were also transfected with pcDNA3.1-GFP. The samples were then fixed, permeabilized, and immunostained using an Anti-Tubulin primary antibody, rhodamine-redconjugated secondary antibody (red), and DAPI-nuclear stain (blue). The relative percentages of GFP-positive (i.e., transfected) cells with cytoplasmic tubulin aggregates (arrows) were quantified in-triplicate using confocal microscopy. Representative micrographs are shown in H and I. Scale bar, 10 mm. (J) The levels of acetylated Alpha-Tubulin in HT1080 cells or the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones, ± nocodozole-treatment (400 ng/ml for 24 hrs), were assessed by SDS-PAGE and immunoblotting using a monoclonal Anti-Acetyl-Tubulin antibody. The relative expression of total Alpha-Tubulin was detected by immunoblotting and is provided for reference (lower panels). The relative levels of acetylatedtubulin were quantified by densitometry and normalized for total tubulin and are indicated below the immunoblots. All the data in D-J are representative of at least three independent experiments. The data in D and G represent the experimental mean ± standard deviation (error bars).
Stathmin/Op-18 destabilizes mitotic MT spindle fibers and has been shown to induce aberrant chromosomal segregation (Holmfeldt et al., 2010; Houghtaling et al., 2009; Andersen et al., 1997), which suggests p65RelA-Stathmin molecular interactions could promote altered MT dynamics and genomic instability in HTLV-1-infected cells. To address this intriguing possibility, the HT1080 clones containing either the HTLV-1 ACH.wt or ACH.p30II mutant proviruses were labeled with bromodeoxyuridine (BrdU), in the absence or presence of nocodozole-treatment, and flow-cytometry-based cell-cycle analyses were performed. The relative percentages of cells in the G1, S, or G2-phases, or with aneuploidy were then quantified using the ModFit LT 3.0 algorithm. Interestingly, the ACH.p30II mutant clones exhibited significant aneuploidy and bypassed nocodozole-induced metaphase-phase arrest -with an increased percentage of treated cells in the S-phase (Fig. 5C). To further validate these results, the HTLV-1 ACH.wt and ACH.p30II proviral clones were treated with nocodozole and then stained with DAPI and a primary antibody against Alpha-Tubulin and a rhodamine red-conjugated secondary antibody, and the samples were analyzed by immunofluorescence-confocal microscopy. The data in Figs. 5D and 5E show that the ACH.p30II mutant clones exhibited greater genomic instability and an increased percentage of multinucleate cells, even in the absence of nocodozole-treatment (see the representative micrographs in Fig. 5E). By contrast, the ACH.wt clones contained reduced levels of Stathmin relative to the ACH.p30II mutant (Fig. 5F). We therefore sought to determine if the ACH.p30II mutant exhibits more MT-instability than the ACH.wt clones. For these experiments, the parental HT1080 cells, or HT1080/HTLV-1 ACH.wt or ACH.p30II mutant proviral clones were transfected with p30II-GFP, or pcDNA3.1-GFP and various expression constructs for the IkBα-S32A/S36A (DiDonato et al., 1996), IκKβD9, or IκKβD34 mutants (Sylla et al., 1998) to inhibit NF-κB-signaling. The relative percentages of GFP-positive (i.e., transfected) cells that contained cytoplasmic tubulin aggregates were then quantified using confocal microscopy. As HTLV-1-infected cells contain reduced levels of p27Kip1 (Kuo and Giam, 2006; Iwanaga et al., 2001). Therefore, as a rationale for these experiments, we hypothesized this might lead to unstrained Stathmin activity and result in the depolymerization of tubulin and the increased accumulation of cytoplasmic tubulin aggregates. These results demonstrate that the ACH.p30II mutant clones exhibited aberrant MT dynamics and contained significantly more cytoplasmic tubulin aggregates than the ACH.wt clones (Figs. 5G–5I; arrows in Figs. 5H and 5I). The suppression of NF-κB transcriptional signaling in the ACH.p30II mutant clones, either through co-expression of p30II-GFP or dominant-negative inhibitors of NF-κB, abrogated the MT dynamics and accumulation of cytoplasmic tubulin aggregates in these cells (Figs. 5G–5I). The representative micrographs in Fig. 5I show the suppression of altered MT dynamics in the GFP-positive cotransfected cells expressing the IκKβD34 mutant, as well as tubulin aggregates (arrows) in the surrounding GFP-negative ACH.p30II mutant cells. The acetylation of Alpha-Tubulin on lysine K40 has been associated with the stabilization of long-lived microtubules (Portran et al., 2017; Howes et al., 2014; Janke and Montagnac, 2017). We therefore examined the acetylation status of Alpha-Tubulin in HT1080 cells and the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones, either in the absence or presence of nocodozole-treatment. The results in Fig. 5J demonstrate that the acetylation of Alpha-Tubulin was diminished in the ACH.p30II mutant clones, compared to ACH.wt (see quantitation below the immunoblots). The relative levels of total Alpha-Tubulin are provided for reference (Fig. 5J, lower panels). To determine whether Tax-induced NF-κB-signaling is responsible for the genomic-instability and altered MT dynamics observed in HTLV-1-infected cells, we quantified the relative percentages of multinucleation in HT1080 cells expressing the Tax oncoprotein or the Tax-G148V mutant, defective for NF-κB transactivation (Yamaoka et al., 1996), either in the absence or presence of nocodozole-treatment. As shown in Fig. 6A, wildtype Tax induced pronounced genomic-instability and multinucleation, even in the absence of nocodozole, compared to untransfected cells or the Tax-G148V mutant. We then cotransfected the cells with pcDNA3.1-GFP and expression constructs for wildtype Tax or the Tax-G148V mutant, and quantified the relative percentages of GFP-positive (i.e., transfected) cells that contained cytoplasmic tubulin aggregates. The results in 6B–6D demonstrate that wildtype Tax expression altered the MT dynamics and led to more cytoplasmic tubulin aggregates (arrows) than the Tax-G148V mutant. Also, cells with the Tax-G148V mutant had more acetylated-Alpha-Tubulin than either wildtype Tax-expressing or untransfected cells (Fig. 6E). We also performed co-immunoprecipitations to detect the p65RelA-Stathmin/Op-18 complexes in transfected cells expressing the Tax oncoprotein or Tax-G148V mutant. These experiments revealed that the Tax-G148V mutant was associated with diminished p65RelA-Stathmin/Op-18 interactions as compared to wildtype Tax (Figs. 6F and 6G). Importantly, these findings allude to a novel link between NF-κB-signaling and the catastrophic genomic-instability mediated by p65RelA-Stathmin/Op-18 molecular interactions in Tax-expressing HTLV-1-infected cells.
Fig. 6.

The NF-κB transactivation-defective mutant, Tax-G148V, exhibits reduced genomic instability and is impaired for p65RelA-Stathmin molecular interactions. (A) HT1080 cells were transfected with 0.5 mg of the expression constructs for wildtype HTLV-1 Tax or the Tax-G148V mutant, defective for NF-κB transactivation (Yamaoka et al., 1996), and confocal immunofluorescence-microscopy was performed to determine the relative percentages of multinucleate cells per field. Certain samples were also treated with nocodozole (400 ng/ml for 24 hrs). The samples were fixed, permeabilized, and then stained using an Anti-Tubulin primary antibody and rhodamine red-conjugated secondary antibody and DAPI. The data represent the mean ± standard deviation (error bars) from three independent experiments. (B-D) HT1080 cells were cotransfected with 0.5 mg of pcDNA3.1-GFP and the expression constructs for either wildtype HTLV-1 Tax or the Tax-G148V mutant, and the relative percentages of GFP-positive cells with cytoplasmic tubulin aggregates (arrows in C and D) were quantified in-triplicate using confocal immunofluorescence-microscopy. Representative micrographs are shown in C and D. Scale bar, 10 mm. The data in B represent the mean ± standard deviation (error bars) from three independent experiments. (E) The relative levels of acetylated tubulin in nocodozole-treated, untransfected HT1080 cells or transfected cells expressing wildtype HTLV-1 Tax or the Tax-G148V mutant were determined by SDS-PAGE and immunoblotting. Total Alpha-Tubulin protein levels are shown for comparison. (F) 293 HEK cells were transfected with the expression constructs for wildtype HTLV-1 Tax or the Tax-G148V mutant and the Stathmin protein complexed with p65RelA was co-immunoprecipitated from precleared extracts using Protein G-agarose and a monoclonal Anti-p65RelA antibody. The relative input levels of Stathmin, p65RelA, Tax, and Actin are shown in the upper panels. The precipitated Stathmin protein was detected by immunoblotting (lower panel). (G) The relative levels of immunoprecipitated Stathmin in F were quantified by densitometry analysis of the immunoblot bands. All the data in E, F and G are representative of at least three independent experiments; and the data in G represent the mean ± standard deviation (error bars).
Inhibition of NF-κB-signaling by HTLV-1 p30II or siRNA-stathmin counters Tax-induced cytotoxicity
The hyperactivation of NF-κB-signaling by the HTLV-1 transactivator Tax induces cellular senescence (Ho et al., 2012; Zhi et al., 2011); and the NF-κB-dependent expression of iNOS could cause oxidative stress and contribute to Tax-induced apoptosis (Baydoun et al., 2015; Chaib-Mezrag et al., 2014; Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998; Hall et al., 1998; Chen et al., 1997; Yamada, 1996). Zhi et al., 2011 have reported that Tax-induced senescence in HeLa cells is mitigated by the antisense HBZ protein which interacts with the p65RelA subunit, prevents its DNA-binding, and inhibits NF-κB-signaling. We investigated whether the pX-encoded proteins p30II and/or HBZ could prevent Tax-induced cellular senescence by cotransfecting HeLa cells with various expression constructs for HTLV-1 Tax, p30II-GFP, HBZ (Myc), or an empty CβS vector control. The Tax, p30II-GFP, and HBZ (Myc-tagged) proteins were detected by immunoblotting (Fig. 7A, lower panels). The expression of senescence-associated Beta-galactosidase (SA-β-Gal) was visualized by staining the cultures with an X-Gal solution and the relative numbers of senescent (blue) cells per well were quantified by microscopy. The results in Fig. 7A revealed that p30II-GFP was more effective at suppressing Tax-induced cellular senescence under these experimental conditions than the antisense HBZ protein. Representative micrographs are provided in Fig. 7B. Furthermore, we also demonstrated that p30II-GFP could prevent Tax-induced senescence in cotransfected HT1080 cells (Fig. 7C).
Fig. 7.

The cytotoxicity induced by the HTLV-1 transactivator Tax is countered by the viral latency protein p30II or siRNA-knockdown of Stathmin. (A and B) HeLa cells were cotransfected with 0.25 mg of the expression constructs for HTLV-1 Tax, HBZ, or p30II-GFP, or a CβS empty vector control and the expression of senescence-associated Beta-galactosidase (SA- b-Gal) was detected by staining the cells with an X-Gal solution. The relative numbers of senescent SA-β-Gal-positive (blue) cells per well were quantified in-triplicate using color and DIC phase-contrast microscopy. Representative merged images are shown in B. Scale bar, 10 mm. The expression of the HTLV-1 Tax, HBZ (Myc-tagged), p30II-GFP, and Actin proteins was detected by SDS-PAGE and immunoblotting (lower panels in A). A non-specific band (ns) is also indicated. (C) HT1080 fibrosarcoma cells were cotransfected as in A with 0.25 mg of pEGFP-N3-HTLV-1 p30II-GFP, RcCMV-HTLV-1 Tax, and/or a CβS empty vector control and the numbers of senescent (SA-β-Gal-positive) cells per well were quantified by microscopy. The data in A and C represent the mean ± standard deviation (error bars) from three independent experiments. (D and E) Jurkat T-lymphocytes were cotransfected with 0.25 mg of the various expression constructs for HTLV-1 Tax, HBZ, or p30II-GFP, or a CβS empty vector, and the samples were subsequently stained with a Click-iT Alexa Fluor 594 TUNEL kit (Invitrogen, Carlsbad, CA). The relative percentages of TUNEL-positive apoptotic cells per field were quantified in-triplicate using confocal microscopy. As a positive control for TUNEL-staining, the cells were permeabilized and treated with 0.15 U/ml of RNAse-free DNAse I (Qiagen). DAPI nuclear-staining and DIC phase-contrast are included in the representative merged images in E. (F-H) 293 cells were cotransfected with 0.25 mg of the expression constructs for wildtype HTLV-1 Tax, the Tax-G148V mutant, or p30II (HA-tagged) or a CβS empty vector, and the samples were then stained with Annexin V-FITC and propidium iodide (PI) to determine the relative percentages of apoptotic (i.e., Annexin V-FITC and/or PI-positive) cells per well by confocal microscopy. Alternatively, 293 HEK cells were cotransfected with RcCMV-HTLV-1 Tax and either siRNA-stathmin oligonucleotides (#1 and #2) or a nsRNA control (in G and H). As a positive control for apoptosis, the cells were treated with staurosporine (12 nM) for 4 hrs in F and H. Representative micrographs are shown in H. DIC phase-contrast is provided in the merged image for the Tax-G148V mutant which exhibited reduced apoptosis. Scale bar, 20 mm. All the data is representative of at least three independent experiments. The data in D, F, and G represent the experimental mean ± standard deviation (error bars).
To determine if p30II cooperates with the viral transactivator by preventing Tax-induced cellular apoptosis, we cotransfected Jurkat T-lymphocytes and performed fluorescent terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assays. As a positive control, certain samples were permeabilized and treated with ribonuclease-free deoxyribonuclease I (RNAse-free DNAse I). The results in Figs. 7D and 7E demonstrate that the Tax oncoprotein and, to a lesser extent, the antisense HBZ protein, induced programmed cell-death as determined by TUNEL-staining. Both p30II (HA-tagged) and HBZ effectively countered Tax-induced apoptosis in cotransfected cells (Figs. 7D and 7E). We then compared the relative levels of apoptosis in 293 HEK cells expressing wildtype Tax or the Tax-G148V mutant, and/or p30II (HA-tagged), or an empty CβS vector by staining the cells with Annexin V-FITC and propidium iodide (PI). Staurosporine-treated cells were included as a positive control for apoptosis. As shown in Figs. 7F and 7H, the Tax oncoprotein induced more apoptosis (i.e., percentages of Annexin V-FITC and/or PI-positive cells) than the NF-κB-defective Tax-G148V mutant or p30II expression construct. The p30II protein countered the induction of cellular apoptosis by the Tax transactivator (Figs. 7F and 7H). To determine whether inhibiting Stathmin could prevent Tax-induced cytotoxicity, 293 cells were cotransfected with siRNA-stathmin oligonucleotides (#1 and #2) or nsRNA as a negative control, and a Tax expression construct, and then stained with Annexin V-FITC/PI and subsequently analyzed by confocal-microscopy. These results demonstrate that the siRNA-knockdown of Stathmin inhibited Tax-induced apoptosis, as compared to the nsRNA control (Figs. 7G and 7H).
These results have shown that NF-κB-signaling contributes to HTLV-1 Tax-induced genomic instability and altered MT dynamics, mediated through p65RelA-Stathmin/Op-18 interactions, and suggest that p30II cooperates with the viral transactivator by dampening Tax-induced NF-κB transactivation and genotoxicity by repressing Stathmin/Op-18.
Discussion
In the present study, we have demonstrated that genomic instability induced by the HTLV-1 Tax oncoprotein is modulated by NF-κB-signaling, p65RelA-Stathmin/Op-18 interactions, and Stathmin-mediated MT-destabilization. The Tax protein activates the IκK signalosome through binding to the scaffold subunit, NEMO or IκK-γ, via lysine K63-linked polyubiquitin chains (Sun et al., 1994; Geleziunas et al., 1998; Harhaj et al., 2007; Ho et al., 2015; Yamaoka et al., 1998; Harhaj et al., 2000; Shembade et al., 2007; Wu and Sun, 2007) and induces constitutive NF-κB-signaling which is essential for the survival and proliferation of HTLV-1-transformed tumor cells (Harhaj and Giam, 2018; Zhang et al., 2016; Choi and Harhaj, 2014; Lavorgna et al., 2014). However, hyperactivation of the NF-κB pathway by Tax has been shown to induce oxidative stress and cumulative DNA-damage (Baydoun et al., 2015; Belgnaoui et al., 2010; Durkin et al., 2008; Haoudi et al., 2003; Chaib-Mezrag et al., 2014; Kinjo et al., 2010; Hutchison et al., 2018; Takahashi et al., 2013; Los et al., 1998), resulting in cellular senescence and cytotoxicity (Kinjo et al., 2010; Hutchison et al., 2018; Kuo and Giam, 2006; Ho et al., 2012; Zhi et al., 2011; Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998) which can be countered through its cooperation with other pX-encoded viral proteins (HBZ or p30II) that inhibit NF-κB transactivation (Hutchison et al., 2018; Zhi et al., 2011; see Figs. 1A, 1E, 1F, 2B, 5A, 5B, 7A–7F, 7H). Moreover, the Tax oncoprotein induces a plethora of chromosomal abnormalities and causes double-strand DNA-breaks and genomic instability as a consequence of its effects upon cellular metabolism, DNA damage-repair components, regulators of the cell-cycle checkpoints, and centrosome amplification (Baydoun et al., 2015; Belgnaoui et al., 2010; Durkin et al., 2008; Haoudi et al., 2003; Chaib-Mezrag et al., 2014; Dayaram et al., 2013; Jin et al., 1998; Majone and Jeang, 2000; Liu et al., 2005; Kehn et al., 2005; Neuveut et al., 1998; Ching et al., 2006; Peloponese et al., 2005).
Stathmin/Op-18 gene expression is negatively regulated through the recruitment of p53 tumor suppressor/HDAC1/mSin3a transcriptional repressor complexes to p53-binding sites within its promoter (Murphy et al., 1999; Ahn et al., 1999). Intriguingly, in contrast to other cancers, the p53 tumor suppressor is rarely mutated in HTLV-1-transformed ATLL clinical isolates (Zane et al., 2012; bPise-Masison et al., 1998; Tabakin-Fix et al., 2006; Mengle-Gaw and Rabbitts, 1987) – suggesting that p53-regulated target genes may contribute to viral pathogenesis. The HTLV-1 latency-maintenance factor p30II interacts with the MYST-family acetyltransferase TIP60 and inhibits lysine K120-acetylation of p53 (Awasthi et al., 2005; Romeo et al., 2015; Romeo et al., 2018) which differentially regulates the expression of p53-dependent proapoptotic genes (Sykes et al., 2006; Tang et al., 2006; Kurash et al., 2008; Dar et al., 2013; Xu et al., 2014). We recently demonstrated that p30II activates p53 and induces the expression of p53-regulated pro-survival signals, including the TIGAR, which is required for its cooperation with cellular (e.g., c-Myc) and viral (Tax and HBZ) oncoproteins (Hutchison et al., 2018; Romeo et al., 2018). Others have reported that Stathmin/Op-18 binds to the NF-κB subunit p65RelA and functions as a cofactor by regulating the stability of p65RelA in aggressive pancreatic cancers (Lu et al., 2014). Here we show that the HTLV-1 p30II protein suppresses the expression of Stathmin and destabilizes p65RelA, and inhibits Tax-induced NF-κB transactivation (Figs. 1C, 1D, 1E, 1F). The targeted knockdown of Stathmin with a siRNA-stathmin oligonucleotide also destabilized p65RelA and inhibited Tax-mediated NF-κB transactivation (Figs. 2C–2F). Further, the siRNA-knockdown of Stathmin destabilized the p65RelA subunit in transfected, HTLV-1-transformed SLB1 lymphoma T-cells (Figs. 3G–3J). The HT1080 clones containing the infectious HTLV-1 ACH.p30II mutant provirus, defective for p30II production (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998), exhibited increased NF-κB-dependent transcriptional activity, multinucleation, and genomic instability either in the absence or presence of nocodozole-treatment, as compared to wildtype ACH (Figs. 5A–5E). The ACH.p30II mutant clones also contained higher Stathmin expression (Fig. 5F) and more cytoplasmic tubulin aggregates, as a result of MT-destabilization (Figs. 5G–5I) and reduced levels of acetylated-Alpha-Tubulin (Fig. 5J). These findings are consistent with the observation that the cycloheximide-treated ACH.p30II mutant cells had significantly higher p65RelA protein levels than the ACH.wt clone (supplementary Figs. S3A, S3B, S3D and S3E). We cannot rule out the possibility that the antisense HBZ protein, which has been shown to inhibit Tax-induced NF-κB transactivation through directly binding p65RelA (Zhi et al., 2011; Zhao et al., 2009), might be functionally impaired in the ACH.p30II mutant provirus and, together with p30II-inactivation, could have a combined effect upon NF-κB transactivation.
Consistent with the negative regulation of stathmin gene expression by p53 (Murphy et al., 1999; Ahn et al., 1999), a dominant-negative DNA-binding mutant of p53, p53-R175H (Hermeking et al., 1997), countered the repression of Stathmin by p30II-GFP (Figs. 4A and 4B), yet inhibited Tax-induced NF-κB transactivation from the E-selectin promoter (Fig. 4C). Several studies have reported that Tax promotes an interaction between p65RelA and p53, and NF-κB transactivation is required for the inhibition of p53 apoptotic functions by the viral transactivator protein (Jeong et al., 2004; Jeong et al., 2005; aPise-Masison et al., 2000; bPise-Masison et al., 2000; Jung et al., 2008). It is therefore possible that the p53-R175H mutant could interfere with p65RelA transcriptional interactions (Jeong et al., 2004; Jeong et al., 2005; bPise-Masison et al., 2000) or, alternatively, might sterically hinder Tax from binding the IκK-γ subunit of the IκK complex through direct p53-Tax interactions (Yamaoka et al., 1998; Harhaj et al., 2000; aPise-Masison et al., 1998; Tabakin-Fix et al., 2006; bPise-Masison et al., 1998).
The Tax-G148V mutant, defective for NF-κB transactivation (Yamaoka et al., 1996), exhibited reduced multinucleation and genomic instability as compared to wildtype Tax, either in the absence or presence of nocodozole-treatment (Fig. 6A). This mutant also had reduced MT-destabilization and fewer cytoplasmic tubulin aggregates (Figs. 6B–6D) that correlated with increased levels of acetylated-Alpha-Tubulin and diminished p65RelA-Stathmin interactions, as determined by co-immunoprecipitations (Figs. 6E–6G). Moreover, the repression of Stathmin by HTLV-1 p30II or siRNA-stathmin-knockdown countered the cellular senescence, cytotoxicity, and apoptosis induced by the Tax oncoprotein (Figs. 7A–7H; Kuo and Giam, 2006; Ho et al., 2012; Zhi et al., 2011; Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998; Hall et al., 1998; Chen et al., 1997; Yamada, 1996). The Tax-G148V mutant did not exhibit significant apoptosis as compared to wildtype Tax (Figs. 7F and 7H).
The modulation of p65RelA-Stathmin/Op-18 interactions by viral and/or cellular oncoproteins has broad implications to impact NF-κB functions and MT dynamics associated with cellular transformation, migration, and neoplastic disease progression. In addition to its effects upon cytoskeletal dynamics and cell migration, NF-κB activation can also cause DNA-damage and genomic instability. Baydoun et al., 2015 have demonstrated that Tax-induced constitutive activation of NF-κB-signaling results in the increased expression of iNOS and production of NO which causes double-strand DNA-breaks in HTLV-1-infected cells. The NF-κB transcription pathway regulates the expression of Polo-like kinase 4 (PLK4) through direct binding to κB-responsive elements within the plk4 gene promoter and influences the assembly of centrosomes during the cell cycle (Ledoux et al., 2013). Also, Ben-Abdallah et al., 2012 have demonstrated that the intracellular pathogen, Cryptococcus neoformans, inhibits the proliferation and viability of macrophages through the activation of NF-κB-signaling which resulted in disruption of the cell-cycle and pronounced aneuploidy.
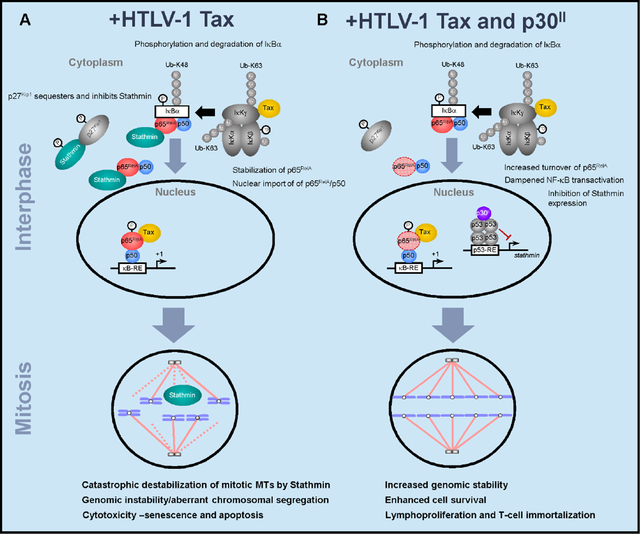
In conclusion, these studies have revealed a novel link between NF-κB-signaling and genomic instability mediated through p65RelA-Stathmin/Op-18 molecular interactions (Lu et al., 2014) and concomitant MT-destabilization. The hyperactivation of NF-κB transcriptional signaling promotes Tax-induced aberrant chromosomal segregation, multinucleation (Belgnaoui et al., 2010; Dayaram et al., 2013; Jin et al., 1998; Majone and Jeang, 2000; Ching et al., 2006; Peloponese et al., 2005; Vernin et al., 2014; Lemoine and Marriott, 2002; Tsukasaki et al., 2001), and cytotoxicity (Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998; Hall et al., 1998; Chen et al., 1997; Yamada, 1996) which are countered by the viral latency protein p30II and the repression of stathmin gene expression in HTLV-1-infected cells (Figs. 8A and 8B). While the cdk-inhibitor, p27Kip1, has beenshown to interact with cytoplasmic Stathmin and stabilizes MT dynamics by restraining the tubulin-depolymerizing activity of Stathmin (Belletti et al., 2010; Fabris et al., 2015; Berton et al., 2014; Fig. 8A), Tax-expressing HTLV-1-transformed cells have low levels of p27Kip1 (Kuo and Giam, 2006; Iwanaga et al., 2001) which could lead to unchecked Stathmin activity, MT-destabilization, and defective spindle functions during mitosis. Moreover, the Tax oncoprotein targets the human mitotic arrest-defective 1 (hMAD1) regulator of the M-phase checkpoint and induces aberrant multinucleation under conditions of mitotic spindle damage (Jin et al., 1998). We recently reported that the HTLV-1 latency-maintenance factor p30II cooperates with Tax and enhances its oncogenic potential in colony-formation assays through the activation of p53-regulated pro-survival signals, including the TIGAR (Hutchison et al., 2018). Our present studies demonstrate that p30II represses Stathmin -a p65RelA-binding cofactor and MT-destabilizing factor that is negatively regulated by p53/HDAC1/mSin3a transcription complexes (Murphy et al., 1999; Ahn et al., 1999), and suppresses Tax-induced NF-κB-signaling, genomic instability and apoptosis in HTLV-1-infected cells (Figs. 8A and 8B). Importantly, these findings have shed new light upon the relationship between p65RelA-Stathmin/Op-18 interactions and genomic instability in human cancers with reduced p27Kip1 expression, and support the ancillary function of p30II in enhancing the survival of Tax-expressing HTLV-1+ cells by dampening the genotoxic effects of NF-κB-signaling during viral carcinogenesis (Hutchison et al., 2018).
Fig. 8.

Model of NF-κB-signaling and p65RelA-Stathmin molecular interactions as mediators of Tax-induced genomic instability and cytotoxicity in HTLV-1-infected cells. (A) The high-level expression of Tax has been shown to hyperactivate NF-κB transcriptional signaling and induce cellular senescence and cytotoxicity (Ho et al., 2012). Tax activates both the canonical and non-canonical IκK signaling pathways in the cytoplasm which leads to the phosphorylation/polyubiquitination and degradation of the inhibitor IkBα -concomitant with the nuclear translocation of NF-κB p65RelA/p50 heterodimers. Consequently, the nuclear translocation of p65RelA could dissociate the molecular interactions between p65RelA and Stathmin/Op-18. As these cells progress into mitosis, free Stathmin could destabilize the MT spindle fibers associated with aberrant chromosomal segregation, catastrophic genomic instability, and cytotoxicity. (B) The viral latency-maintenance factor p30II activates p53 and represses stathmin gene expression (Romeo et al., 2018; Murphy et al., 1999; Ahn et al., 1999). The reduced levels of Stathmin destabilize the p65RelA protein (Lu et al., 2014) and, thereby, dampen Tax-induced NF-κB-signaling -resulting in less genomic instability and the enhanced survival of HTLV-1-infected cells.
Materials and methods
Cell-lines
The following cell-lines and culture conditions were used for these studies: 293 human embryonic kidney (HEK) cells (CRL-1573; ATCC, Manassas, VA) were cultured in Eagle’s Minimum Essential Medium (EMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Biowest, Riverside, MO), 100 U/ml penicillin, 100 mg/ml streptomycinsulfate, and 20 mg/ml gentamycin-sulfate (Life Technologies, Waltham, MA), in a humidified incubator at 37°C under 5% CO2. HeLa cells (CCL-2; ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% heat-inactivated FBS and antibiotics, at 37°C and 5% CO2. All cell-lines were negative for mycoplasma contamination. The HTLV-1-transformed MJG11 T-cell-line (CRL-8294; ATCC) was cultured in RPMI-1640 medium, supplemented with 20% FBS and antibiotics, at 37°C under 10% CO2. The HTLV-1-transformed SLB1 lymphoma T-cell-line (kindly provided by P. Green, The Ohio State University-Comprehensive Cancer Center; Arnold et al., 2008) was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM), supplemented with 10% FBS and antibiotics, in a humidified incubator at 37°C under 10% CO2. The Jurkat E6.1 T-cell-line (TIB-152; ATCC) was grown in RPMI-1640 medium, supplemented with 10% FBS and antibiotics, at 37°C under 5% CO2. Primary human PBMCs (hu-PBMCs) were isolated from de-identified whole-blood samples obtained from the SMU Memorial Health Center under a protocol approved by the SMU Institutional Review Board and which abides by the Declaration of Helsinki principles. Buffy-coat hu-PBMCs were isolated by mixing 2 ml of blood with 2 ml of sterile phosphate-buffered saline (PBS), pH 7.4 and layering the mixture over 3 ml of Lymphocyte separation medium (MP Biomedicals, Aurora, OH) in a 15 ml conical tube. The samples were centrifuged at 30 min at 400 x g at room temp. Buffy-coat lymphocytes were aspirated and rinsed 2X in RPMI-1640 medium and centrifuged for 7 min at 260 x g. The cell pellets were then resuspended in 20 ml of RPMI-1640 medium, supplemented with 20% FBS, antibiotics, 50 U/ml recombinant human interleukin-2 (hu-IL-2; Roche Applied Science, Indianapolis, IN), and 10 ng/ml phytohemagglutinin (PHA; Sigma-Aldrich, St Louis, MO) and cultured at 37°C under 10% CO2. After 24 hrs, the cells were pelleted by centrifugation for 7 min at 260 x g to remove the PHA, rinsed 2X with serum-free RPMI-1640 medium, and then resuspended and cultured in complete medium with antibiotics and hu-IL-2 as described. The transiently-amplified HT1080 cell clones, expressing the infectious HTLV-1 ACH.wt or ACH.p30II mutant proviruses, were generated by transfecting 2×105 cells in 6-well tissue-culture plates with plasmids that contain the full-length ACH.wt or ACH.p30II proviral nucleotide sequences (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998). The ACH.p30II mutant contains a premature stop codon in the p30II nucleotide coding sequence; however, this mutation also results in an in-frame insertion of eight amino acids (YLEEESRG) after residue G56 of the antisense HBZ protein at the end of the activation domain (Robek et al., 1998). The insertion is a repeat of an LEEE repeat domain that immediately follows both LXXLL motifs that are involved in HBZ interactions with p300 (Clerc et al., 2008). It is unlikely to be detrimental to the function of the activation domain since the full activity of the activation domain only requires amino acids 5–53. The insertion is also well upstream of basic regions 1 and 2 (Clerc et al., 2008); and secondary structure prediction programs suggest the insertion is located within an unstructured region of the protein. The transfected cultures were then seeded in 96-well microtiter plates and the virus-producing HT1080/HTLV-1 ACH clones were screened by quantifying the amounts of extracellular HTLV-1 p19Gag core antigen released into the culture supernatants by performing Anti-HTLV-1 p19Gag enzyme-linked immunosorbent assays (ELISAs; Zeptometrix, Buffalo, NY). The supernatants were filtered using 0.22 mm cellulose acetate syringe filters (Sartorius, Goettingen, Germany). The extracellular p19Gag levels were measured relative to a HTLV-1 p19Gag protein standard using a Berthold Tristar microtiter plate-reader in Absorbance mode (Berthold Technologies, Oak Ridge, TN). The virus-producing HT1080/HTLV-1 ACH.wt and ACH.p30II clones were expanded and repeatedly passaged and the continuous production of infectious HTLV-1 particles was confirmed by performing Anti-HTLV-1 p19Gag ELISAs (Hutchison et al., 2018; Romeo et al., 2018). The HT1080/HTLV-1 ACH proviral clones and parental HT1080 human fibrosarcoma cell-line (CCL-121; ATCC) were cultured in EMEM containing high-glucose (4.5 g/L) and supplemented with 10% FBS and antibiotics, and incubated at 37°C under 5% CO2.
Plasmids and antibodies
The RcCMV-wildtype HTLV-1 Tax (Harrod et al., 1998; Harrod et al., 2000; Adya and Giam, 1995), RcCMV-Tax-M22 and Tax-M47 mutants -impaired for NF-κB- and CREB-dependent transactivation, respectively (Smith and Greene, 1990), and the NF-κB-defective Tax-G148V mutant (Yamaoka et al., 1996) were kindly provided by C.Z. Giam (The Uniformed Services University of the Health Sciences). The pEGFP-N3-HTLV-1 p30II-GFP and pMH-HTLV-1 p30II (HA-tagged) expression constructs were described in Nicot et al., 2004 and were generously provided by G. Franchini (NCI/NIH). The pGL2-Basic-HTLV-1 TRE-luciferase reporter plasmid (Giebler et al., 1997) was provided by J. Nyborg (Colorado State University); and the pcDNA-HBZ-MycHis expression construct (Clerc et al., 2008; Hiven et al., 2005) was provided by I. Lemasson (East Carolina University). The NF-κB-responsive E-Selectin promoter-luciferase reporter plasmid has been described in Hong et al., 2007. The pCV63-HIV-1 κB-LTR (ΔTAR)-luciferase reporter construct contains nucleotides 345–531 of the HIV-1LAI promoter (spanning the two κB-responsive elements and three SP1-binding sites with a deletion of the U-rich trinucleotide bulge of the TAR) which were inserted into pGL2-Basic (Promega, Madison, WI). pcDNA3.1-GFP was described in Nicot and Harrod, 2000. The pCDNA3-FLAG-IκKbΔ9 mutant and IκKβΔ34 mutant expression constructs were described in Sylla et al., 1998. The pRc-IkBα-S32A/S36A construct which expresses a “super repressor” mutant of IkBα, defective for Ser32/Ser36-phosphorylation and degradation (DiDonato et al., 1996), was kindly provided by P. West (Texas A&M University). pCEP-wildtype p53 and the pCEP-p53-R175H mutant expression constructs were described in Hermeking et al., 1997 and were provided by B. Vogelstein (Johns Hopkins University).
The following antibodies were used in this study: mouse monoclonal Anti-HTLV-1 Tax (1A3), rabbit polyclonal Anti-Stathmin/Op-18 (FL-149), mouse monoclonal Anti-NF-κB p65RelA (F-6), rabbit polyclonal Anti-IkBα (C-15), mouse monoclonal Anti-p53 (DO-2), mouse monoclonal Anti-Alpha-Tubulin (TU-02), mouse monoclonal Anti-Acetylated-Alpha-Tubulin (6-11B-1), rabbit polyclonal Anti-GFP (FL), mouse monoclonal Anti-c-Myc (9E10), and goat polyclonal Anti-Actin (C-11). All were purchased from Santa Cruz Biotechnology (Dallas, TX). The other antibodies used were: rabbit monoclonal Anti-IkB-a (E130; Abcam, Cambridge, MA), rabbit monoclonal Anti-phospho-S36-IkB-a (EPR6235(2); Abcam), mouse monoclonal Anti-FLAG M2 (Sigma-Aldrich), fluorescein isothiocyanate (FITC)-conjugated Anti-Bromodeoxyuridine (FITC-Anti-BrdU; BDPharmingen, San Diego, CA), and polyclonal Anti-HTLV-1 p19Gag (Zeptometrix). The fluorescent rhodamine red-conjugated Anti-Mouse IgG (H+L) secondary antibody and horseradish peroxidase (HRP)-conjugated secondary antibodies (Anti-Mouse IgG [H+L]-HRP; Anti-Rabbit IgG [H+L]-HRP; Anti-Goat IgG [H+L]-HRP) used for chemiluminescence-imaging and immunoblotting were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Immunoblotting and co-immunoprecipitations
Protein expression was detected by Western blotting. In brief, the transfected cells were harvested and pelleted by centrifugation for 7 min at 260 x g at 4°C, washed 2X with PBS, pH 7.4, centrifuged, and resuspended in 1X Reporter Lysis Buffer (Promega). The cells were lysed by repeated freeze-thawing over dry-ice and passaging through a 27-gauge tuberculin syringe needle. The samples were then centrifuged for 2 min at 5,000 x g at 4°C, mixed with 2X Laemmli Sample Buffer containing 2-mercaptoethanol (Biorad Laboratories, Hercules, CA), heat-denatured at 95°C for 3 min, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12.5% polyacrylamide gel with a 4% stacking layer. The proteins were transferred onto a Protran BA83 0.2 mm nitrocellulose membrane (Whatman, Maidstone, UK) using a model TE 77 PWR semi-dry blotting unit (Amersham Biosciences, Little Chalfont, UK). The membranes were blocked for 1 hr with gentle agitation in Blocking buffer (3% w/v bovine serum albumin and 0.5% v/v Tween-20 in PBS, pH 7.4) and then incubated for 2 hrs in primary antibodies (diluted 1:1000 or 1:2000 in Blotto buffer: 50 mM Tris-Cl, pH 8.0, 2 mM CaCl2, 80 mM NaCl, 0.2% v/v IGEPALCA630, 0.02% w/v sodium azide, and 5% w/v nonfat dry milk) with gentle shaking. The membranes were washed 2X with Blotto buffer for 10 min and incubated for 1 hr with various HRP-conjugated secondary antibodies (diluted 1:500). The blots were finally washed 2X with Blotto buffer and once with TMN solution (100 mM NaCl, 5 mM MgCl2, 100 mM Tris-Cl, pH 9.5) for 10 min, and developed by chemiluminescence-based detection using Pierce ECL Western Blot Reagent (Thermo Scientific, Rockford, IL) and a ChemiDoc Touch imaging system (BioRad Laboratories). Co-immunoprecipitations were performed by lysing the cells in 500 ml of RIPA buffer (0.15 M NaCl, 50 mM Tris-Cl, pH 7.4, 0.5% sodium deoxycholate, 0.5% Nonidet P-40, 0.1% SDS), containing 50 ng/ml each of the protease inhibitors: pepstatin, leupeptin, chymostatin, bestatin, and antipain-dihydrochloride (Roche Applied Science), by repeated freeze-thawing and either syringe-shearing or sonication over an ice-bath using a microtip probe and Misonix S-4000 model instrument set at 70% amplitude (for the HTLV-1-infected MJG11 and SLB1 ATLL cell-lines). Cell debris was removed by centrifuging the samples for 5 min at 5,000 x g at 4°C. Two hundred and fifty microliters from each sample were used in immunoprecipitation reactions with 20 ml of a 50% slurry of Protein-G-agarose and 3–5 ml of the primary antibodies overnight at 4°C with gentle rotation. The agarose-immune complexes were then pelleted by centrifugation for 5 min at 5,000 x g at 4°C, washed 2X with RIPA buffer, and resuspended in 30 ml of 2X Laemmli Sample Buffer with 2-mercaptoethanol. The samples were incubated in a heat-block at 95°C for 5 min, briefly centrifuged, and the bound proteins were resolved and detected by SDS-PAGE and immunoblotting. The quantitation of immunoblot bands by densitometry was performed using Image Lab 5.2.1 software (BioRad Laboratories).
siRNA-knockdown of Stathmin expression
To specifically inhibit Stathmin expression in HTLV-1-transformed SLB1 T-lymphoblasts or 293 HEK cells, the cells were repeatedly transfected with either a 2’-O-methyl-uridine-modified siRNA-stathmin (#1) oligonucleotide with 5’ and 3’ terminal phosphorothioate linkages and the sequence: 5’-mUCCAGUUCUUUCACCUGGAUAUC-3’, or a bridged nucleic acid (BNA) siRNA-stathmin (#2) oligonucleotide with phosphorothioate linkages at the 5’ and 3’ ends with the sequence: 5’-AAUCAGCUCAAAGCCUGGCCU-3’ (Biosynthesis, Lewisville, TX), using the HiPerFect transfection reagent (Qiagen, Germantown, MD) as per the manufacturer’s recommended protocol. Alternatively, the cells were transfected with a 2’-O-methyl-uridine-modified non-specific RNA (nsRNA) oligonucleotide with the sequence: 5’-UUACCGAGACCGUACGUAU-3’ (Biosynthesis), as a negative control. The targeted siRNA-knockdown of Stathmin protein expression was confirmed by SDS-PAGE and immunoblotting using a rabbit polyclonal Anti-Stathmin/Op-18 primary antibody (Santa Cruz Biotechnology).
NF-κB-dependent transactivation and luciferase reporter gene assays
To determine whether the inhibition of stathmin expression by HTLV-1 p30II influences Tax-dependent NF-κB transcriptional activation, 293 HEK cells were cotransfected with either an E-Selectin promoter-luciferase reporter plasmid or HIV-1 κB-LTRLAI (ΔTAR) promoter-luciferase reporter plasmid (0.25 mg), and RcCMVHTLV-1 Tax and an HTLV-1 p30II-GFP expression construct or empty CβS vector control. Alternatively, the cells were transfected with expression constructs for the Tax-M22, Tax-M47, or Tax-G148V transactivation mutants (Smith and Greene, 1990; Yamaoka et al., 1996), or the dominant-negative p53 DNA-binding mutant, p53-R175H (Hermeking et al., 1997), dominant-negative IκKβ mutants,IκKβΔ9 and IκKβΔ34 (Sylla et al., 1998), or a phosphorylation/degradation-defective IkBα “super-repressor” mutant, IkBα-S32A/S36A (DiDonato et al., 1996). For some experiments, 293 cells were cotransfected with an E-Selectin promoter-luciferase reporter plasmid and pEGFP-N3-HTLV-1 p30II-GFP and then stimulated with 100 ng/ml of phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) to induce NF-κB-dependent transactivation. Certain samples were also repeatedly transfected with siRNA-stathmin to knockdown Stathmin/OP-18 expression or an nsRNA oligonucleotide as a negative control. The CREB-dependent transactivation of the HTLV-1 promoter by Tax was demonstrated by cotransfecting 293 HEK cells with 0.25 mg of a pGL2-Basic-HTLV-1 TRE-luciferase reporter plasmid (Giebler et al., 1997). The cells were harvested by scraping, pelleted by centrifugation for 7 min at 260 x g at 4°C, rinsed with PBS, pH 7.4, and resuspended in 80 ml of Reporter Lysis Buffer (Promega). The cells were lysed by rapid freeze-thawing and passaging through a 27-gauge syringe needle. Then, the samples were centrifuged for 2 min at 5,000 x g at 4°C and the protein concentrations were determined using the Bradford microassay and spectrophotometric analysis at 595 nm. Luciferase assays (Promega) were performed and read on a Berthold Lumat LB 9507 luminometer (Berthold Technologies), and the values were normalized to equivalent amounts of total cellular protein. To compare the relative levels of NF-κB-dependent transactivation between the HT1080/HTLV-1 ACH.wt and HT1080/HTLV-1 ACH.p30II mutant proviral clones (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998), the cells were cotransfected with E-Selectin promoter-firefly-luciferase (or HIV-1 κB-LTR (ΔTAR)-firefly-luciferase) and tk-renilla-luciferase reporter plasmids and dual-luciferase assays were performed using a Dual-Glo kit as recommended by the manufacturer’s protocol (Promega). The kB-dependent E-Selectin promoter-driven firefly-luciferase readouts were normalized to equivalent tk-renillaluciferase activities.
IkB-a Ser36-phosphorylation and degradation and the preparation of nuclear extracts
To assess the phosphorylation and degradation of the inhibitor IkB-a as well as the expression of NF-κB p65RelA in the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones, 7×105 cells were plated in 60 mm2 tissue-culture dishes and, after 48 hrs, the cultures were treated with the protein synthesis-inhibitor, cycloheximide (50 mg/ml; Sigma-Aldrich). The cells were then harvested by scraping and centrifugation, washed 2X with PBS, and protein extracts were prepared over a time-course of 0, 0.5, 1, 2, 4, and 6 hrs. The relative S36-phosphorylation and degradation of IkB-α and the expression of p65RelA were detected by SDS-PAGE and immunoblotting and quantified by densitometry analysis.
The subcellular localization of the NF-κB p65RelA subunit in the HT1080/HTLV-1 ACH.wt and ACH.p30II mutant proviral clones was determined by preparing nuclear and cytoplasmic extracts from cycloheximide-treated cells. In brief, 2×106 cells were plated in 100 mm2 tissue-culture dishes and, after 48 hours, the cultures were treated with cycloheximide (50 mg/ml) for 2-hrs. The cell monolayers were then washed with PBS and harvested by scraping in PBS, followed by centrifugation for 7 min at 260 x g at 4°C. The cell pellets were resuspended in 500 ml of Lysis buffer (10 mM Tris-Cl, pH 7.4, 3 mM CaCl2, 2 mM MgCl2) and an equal volume of Lysis buffer + IGEPAL CA-630 (1% v/v; Sigma-Aldrich) was added, and the samples were gently lysed by repeated passage through a 27-gauge syringe needle. The nuclei were then collected by centrifugation for 10 min at 1,000 x g at 4°C and resuspended and stored in 200 ml aliquots of Nuclear freezing buffer (50 mM Tris-Cl, pH 8.3, 5 mM MgCl2, 0.1 mM EDTA, 40% v/v glycerol). Nuclear extracts were prepared through the addition of 20 ml of 400 mM KCl, followed by rapid freeze-thawing over dry-ice. The samples were clarified by centrifugation for 2 min at 5,000 x g at 4°C prior to use for SDS-PAGE and immunoblotting. The nitrocellulose membranes were stained using a Ponceau S (Sigma-Aldrich) solution (0.1% w/v in 5% acetic acid) to demonstrate equivalent loading between the nuclear and cytoplasmic fractions.
Quantification of multinucleation and tubulin aggregates
The effects of Tax-induced NF-κB-signaling and p30II expression upon genomic instability in HTLV-1-infected cells (Belgnaoui et al., 2010; Dayaram et al., 2013; Jin et al., 1998; Majone and Jeang, 2000; Ching et al., 2006; Peloponese et al., 2005; Vernin et al., 2014; Lemoine and Marriott, 2002; Tsukasaki et al., 2001) were determined by treating HT1080 human fibrosarcoma cells, or the HT1080/HTLV-1 ACH.wt and HT1080/HTLV-1 ACH.p30II mutant proviral clones (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998) with the microtubuledepolymerizing agent, nocodozole (400 ng/ml; Sigma-Aldrich), for 24 hrs to induce metaphase (M-phase)-arrest. The cells were plated and treated on sterile glass coverslips in 35 mm2 tissue-culture dishes and then incubated at 37°C and 5% CO2. The samples were fixed in 0.2% glutaraldehyde-1% formaldehyde in PBS for 15 min, washed 2X with PBS, and then incubated in Blocking buffer with gentle shaking. The slides were immunostained using a monoclonal Anti-Tubulin primary antibody (diluted 1:2000 in Blotto buffer) and a rhodamine red-conjugated Anti-Mouse IgG (H+L) secondary antibody (Jackson ImmunoResearch Laboratories; diluted 1:200), in combination with 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI; Invitrogen, Carlsbad, CA) nuclear-staining. We performed confocal immunofluorescence-microscopy to determine the relative percentages of multinucleate cells (i.e., which had bypassed nocodozole-induced M-phase arrest) per field by counting triplicate visual fields. To determine whether the wildtype Tax protein differentially induces genomic instability, as compared to the NF-κB transactivation mutant, Tax-G148V (Yamaoka et al., 1996), 1×105 HT1080 cells were plated on glass coverslips and transfected with RcCMV expression constructs for wildtype HTLV-1 Tax or the Tax-G148V mutant. Certain samples were also treated with nocodozole to induce M-phase arrest. The slides were then fixed, blocked and immunostained, and confocal microscopy was performed as described to quantify the relative percentages of multinucleate cells per field. Alternatively, flow cytometry was performed on cultures labeled with BrdU and immunostained using an Anti-BrdU antibody conjugated with fluorescein isothiocyanate (FITC; BD-Pharmingen) to determine the effects of p30II upon Tax-induced genomic instability. The transiently-amplified HT1080 fibrosarcoma clones, containing the HTLV-1 ACH.wt or ACH.p30II mutant proviruses, were seeded at a density of 2×105 cells/well and cotransfected with a pLenti-6.2/V5-DESTHTLV-1 p30II (HA-tagged) expression construct or empty CβS vector control in 6-well tissue culture plates. After 72 hrs, the cells were labeled for 6 hrs with 10 mM BrdU in EMEM supplemented with 20% FBS. The cells were subsequently permeabilized and stained using the FITC-Anti-BrdU antibody; and the total genomic DNA content was quantified by staining the cultures with 7-aminoactinomycin D (7-AAD; BD-Pharmingen). Flow cytometry was performed on a Becton Dickinson FACSCaliber instrument (Becton Dickinson, Franklin Lakes, NJ) and the data were analyzed using ModFit LT 3.0 software (Verity Software House, Topsham, ME). The samples were gated during acquisition to exclude sub-G1 cellular debris and apoptotic fragments.
To determine the effects of Tax-dependent NF-κB-signaling and the repression of Stathmin expression by p30II upon tubulin dynamics and the MT network, the parental HT1080 cell-line (1×105 cells) and the HT1080/HTLV-1 ACH.wt and HT1080/HTLV-1 ACH.p30II mutant proviral clones (Hutchison et al., 2018; Bartoe et al., 2000; Romeo et al., 2018; Kimata et al., 1994; Robek et al., 1998) were plated on sterile glass coverslips in 35 mm2 tissue-culture dishes and then cotransfected with pcDNA3.1-GFP or pEGFP-N3-HTLV-1 p30II-GFP and/or various expression constructs for the dominant-negative IκKb deletion mutants: IκKβΔ9 and IκKbD34 (Sylla et al., 1998), or the IkBα “super repressor” mutant, IkBα-S32A/S36A (DiDonato et al., 1996). Following 72 hrs, the samples were fixed with 0.2% glutaraldehyde-1% formaldehyde in PBS for 15 min at room temperature, washed 2X with PBS, and incubated in Blocking buffer for 1 hr with gentle agitation. The samples were then immunostained with a monoclonal Anti-Tubulin primary antibody and a rhodaminered-conjugated Anti-Mouse IgG (H+L) secondary antibody, together with DAPI nuclear-stain. Confocal immunofluorescence-microscopy was performed and the relative percentages of GFP-positive (i.e., transfected) cells with cytoplasmic tubulin aggregates were quantified by counting each slide in-triplicate. Further, to determine how Tax-dependent NF-κB signaling affects tubulin polymer dynamics, HT1080 cells were cotransfected with pcDNA3.1-GFP and an expression construct for wildtype HTLV-1 Tax or the NF-κB transactivation-defective mutant, Tax-G148V (Yamaoka et al., 1996), and the relative percentages of GFP-positive cells with cytoplasmic tubulin aggregates were quantified as described using confocal immunofluorescence-microscopy. The relative fluorescence-intensities of the DAPI, GFP, and Tubulin-specific (red) signals were measured using the Zen 2.5D analysis tool (Carl Zeiss Microscopy, Jena, Germany).
Measuring cellular senescence and apoptosis
Apoptosis assays were performed by cotransfecting 293 HEK cells plated on 8-well glass chamber slides with 0.25 mg of RcCMV- wildtype HTLV-1 Tax or the Tax-G148V NF-κB-defective transactivation mutant (Yamaoka et al., 1996), pMH-HTLV-1 p30II (HA-tagged; Nicot et al., 2004), or a CβS empty vector in various combinations. To determine the effects of inhibiting Stathmin expression upon Tax-induced programmed cell-death (Nicot and Harrod, 2000; Takahashi et al., 2013; Los et al., 1998; Hall et al., 1998; Chen et al., 1997; Yamada, 1996), certain samples were also cotransfected with the RcCMV-HTLV-1 Tax construct and 50 ng of a siRNA-stathmin (#1 and #2) oligonucleotide or nsRNA as a negative control. After 72 hrs, the cells were stained with Annexin V conjugated to FITC (Annexin V-FITC) and propidium iodide (PI; BD-Pharmingen), and the relative percentages of apoptotic (i.e., Annexin V-FITC and/or PI-positive) cells per well were determined using confocal fluorescence-microscopy and by counting in-triplicate. Staurosporine-treated cells (12 nM for 4 hrs; Sigma-Aldrich) were included as a positive control for apoptosis. For the fluorescence-based, terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) experiments, 2×105 Jurkat T-cells were resuspended in 500 ml of Gene Pulser Electroporation buffer (Bio-rad Laboratories, Hercules, CA) and then cotransfected by electroporation in sterile cuvettes with a 0.4 cm gap width using a Gene Pulser Xcell instrument (Bio-rad Laboratories) with 0.25 mg of the RcCMV-HTLV-1 Tax, pEGFP-N3-HTLV-1 p30II-GFP, and pcDNAHBZ-MycHis expression constructs or a CβS empty vector control. Two pulses of 10 sec each were administered with instrument settings of 250 volts, 1,500 mF capacitance, and 1,000 ohms resistance. The cells were cultured in 12-well tissue-culture plates in RPMI-1640 medium, supplemented with 20% FBS and antibiotics. After 72 hrs, the cells were washed 2X with PBS and subsequently transferred to poly-lysine/concanavalin A (Sigma-Aldrich)-treated glass slides, permeabilized, and labeled using a Click-iT Alexa Fluor 594 TUNEL kit (Invitrogen) as per the manufacturer’s suggested protocol. As a positive control for TUNEL, certain samples were treated with 0.15 U/ml of ribonuclease-free deoxyribonuclease I (RNAse-free DNAse I; Qiagen). The cell nuclei were visualized by staining with DAPI. The relative percentages of fluorescent TUNEL-positive apoptotic cells per field were quantified by counting triplicate visual fields using confocal microscopy. The total numbers of cells were determined by DAPI nuclear-staining and DIC phase-contrast microscopy.
To determine the effects of HTLV-1 p30II and HBZ upon Tax-induced cellular senescence (Kuo and Giam, 2006; Ho et al., 2012), HeLa cells were cotransfected with various combinations of the expression constructs pRcCMV-HTLV-1 Tax, pEGFPN3-HTLV-1 p30II-GFP, pcDNA-HBZMycHis, or an empty CβS vector control, and then cultured for four days in complete medium at 37°C under 5% CO2. The cells were then serum-starved (0.05% FBS) for 24 hrs and stained using X-Gal (Sigma-Aldrich) to detect senescence-associated Beta-galactosidase (SA-β-Gal) expression. The cells were rinsed with PBS, pH 7.4, and incubated in a fixative solution of 2% formaldehyde and 0.2% glutaraldehyde in PBS for 15 min at room temperature. The samples were then washed 2X with PBS and stained with the X-Gal-Staining Solution (37.2 mM citric acid/sodium phosphate, 140 mM NaCl, 2 mM MgCl2, 0.5 mM potassium ferrocyanide, 0.5 mM potassium ferricyanide, 1 mg/ml X-Gal, and 2% N-N-dimethylformamide, pH 6.0) at 37°C overnight in a dry incubator without CO2. The numbers of SA-β-Gal-positive (blue) senescent cells were quantified by counting triplicate visual fields using a DIC filter on a Zeiss AxioImager Z2 fluorescence-microscope equipped with AxioCam MRc color and HRm monochromatic cameras and a Plan-Apochromat 20x/0.8 objective lens. The expression of Tax, HBZ (Myc-tagged), and p30II-GFP was confirmed by SDS-PAGE and immunoblotting with appropriate primary antibodies. The p30II-GFP fusion was visualized in the transfected cells by direct fluorescence-microscopy.
Microscopy
Confocal fluorescence-microscopy to visualize tubulin aggregates, multinucleation, Annexin V-FITC/PI or Alexa Fluor 594 TUNEL-staining in HTLV-1 Tax-expressing cells was performed on a Zeiss LSM800 instrument with an Airyscan super-resolution detector and stage CO2 incubator (Carl Zeiss Microscopy). All images were taken using either Plan-Apochromat 40x/1.3 or Plan-Apochromat 63x/1.4 oil immersion objectives and Zeiss ZEN system software. The expression of HTLV-1 p30II-GFP in cotransfected cells was visualized on an inverted Nikon Eclipse TE2000-U microscope and D-Eclipse C1 confocal system (Nikon Instruments, Melville, NY) equipped with 633 nm and 543 nm He/Ne and 488 nm Ar lasers using a Plan Apo 20x/0.75 objective lens.
Statistical analysis
The statistical significance of experimental data sets was determined using unpaired two-tailed Student’s t-tests (alpha = 0.05) and calculated P-values using the Shapiro-Wilk normality test and Graphpad Prism 7.03 software. The P-values were defined as: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), <0.0001 (****). Error bars represent the SEM from at least three independent experiments.
Supplementary Material
Supplemental Fig. S1. The HA-tagged HTLV-1 p30II protein represses Stathmin expression and inhibits Tax-dependent NF-κB transactivation similar to p30II-GFP. (A) The data from Fig. 1E which shows Tax-dependent transactivation of the E-selectin promoter-luciferase reporter plasmid and inhibition by p30II-GFP are represented as ‘Fold transactivation’ for comparison. (B) 293 HEK cells were cotransfected with the E-selectin-luciferase reporter construct and an RcCMV-HTLV-1 Tax expression construct, and then transduced with lentiviral expression vectors for GFP or HTLV-1 p30II (HA). Relative luciferase activities were measured and normalized for equivalent total cellular proteins. (C) The effects of pLenti-GFP and pLenti-HTLV-1 p30II (HA) upon Stathmin expression were determined by transducing 293 cells with the lentiviral constructs and then performing SDS-PAGE and immunoblot analyses. The relative Stathmin protein levels were quantified by densitometry and normalized to Actin. (D) The expression of HTLV-1 p30II (HA) and GFP in lentiviral transduced cultures was visualized by confocal microscopy either by immunostaining the samples with an Anti-HA tag monoclonal antibody (Abcam) and rhodamine red-conjugated secondary antibody (red signal), or by direct-fluorescence for GFP (green). DAPI nuclear-staining is provided for reference. Untransduced (UT) cells were included as negative controls for the Anti-HA antibody and GFP fluorescence detection (n=3). Scale bar, 20 mm.
Supplemental Fig. S2. Transactivation of the E-selectin gene promoter by HTLV-1 Tax is dependent upon NF-κB-signaling. (A) The functionality of the Tax protein used in these studies and its ability to transactivate the CREB-dependent Tax-responsive elements (TREs) within the HTLV-1 promoter was demonstrated by cotransfecting 293 HEK cells with 0.25 mg of an HTLV-1 TRE-luciferase reporter plasmid (Giebler et al., 1997) and increasing amounts (0.25 and 0.5 mg) of RcCMV-HTLV-1 Tax or a pcDNA3.1-GFP control. Relative luciferase activities were measured and normalized for equivalent total cellular protein levels. The expression of the HTLV-1 Tax, GFP, and Actin proteins was detected by SDS-PAGE and immunoblotting. (B and C) Dominant-negative mutants of IkBα or the IκKb subunit inhibit Tax-induced NF-κB transactivation. 293 cells were cotransfected with an E-selectin promoter-luciferase reporter plasmid and either RcCMV-HTLV-1 Tax alone, or together with a phosphorylation/degradation-defective IkBα “super repressor” mutant, IkBα-S32A/S36A (DiDonato et al., 1996) in B. The HTLV-1 Tax, IkBα-S32A/S36A mutant, and Actin proteins were detected by immunoblotting. In C, 293 cells were cotransfected with the E-Selectin-luc reporter plasmid and RcCMV-HTLV-1 Tax alone, or with expression constructs (0.25 and 0.5 mg) for the dominant-negative IκKβ subunit deletion mutants, IκKβΔ9 or IκKbD34 (Sylla et al., 1998). Relative luciferase activities were then measured and normalized for equivalent total cellular proteins. The HTLV-1 Tax, IκKβΔ9 (FLAG), IκKβΔ34 (FLAG), and Actin proteins were detected by SDS-PAGE and immunoblotting. For A-C, the data represent the mean ± standard deviation (error bars) from three independent experiments.
Supplemental Fig. S3. The HT1080/HTLV-1 ACH.wt proviral clone contains reduced NF-κB p65RelA levels relative to the ACH.p30II mutant. (A) The HT1080/HTLV-1 ACH.wt and ACH.p30II clones were treated with the protein synthesis-inhibitor, cycloheximide (50 mg/ml; Sigma-Aldrich), and the S36-phosphorylation and degradation of IkB-α and expression of the NF-κB p65RelA subunit were detected by SDS-PAGE and immunoblotting. A representative Ponceau S-stained nitrocellulose membrane is shown for comparison. (B and C) The relative levels of p65RelA, S36-phosphorylated IkB-a, and total IkB-a were quantified by densitometry analysis of the immunoblot bands. (D) Nuclear (Nuc) and cytoplasmic (Cyt) extracts were prepared from the HT1080/HTLV-1 ACH.wt and ACH.p30II clones that were treated with cycloheximide for 2 hrs, and the relative levels of p65RelA were detected by immunoblotting. A nonspecific band (ns) is also indicated. A representative Ponceau S-stained membrane is provided for comparison. (E) The expression of p65RelA was quantified by densitometry. All the data is representative of three independent experiments. The data in B, C, and E represent the experimental mean ± standard deviation (error bars).
NF-κB-signaling by the HTLV-1 Tax oncoprotein induces genomic instability and apoptosis
Tax dissociates molecular interactions between the NF-κB p65RelA subunit and Stathmin
The HTLV-1 latency protein p30II represses Stathmin and counters Tax-induced cytotoxicity
Repression of Stathmin destabilizes p65RelA and inhibits Tax-induced NF-κB transactivation
p30II promotes the survival of HTLV-1+ cells by inhibiting Tax-induced mitotic catastrophe
Acknowledgements
The authors thank B. Vogelstein for the pCEP4-wildtype p53 and pCEP4-p53-R175H expression constructs, I. Lemasson for the pcDNA-HBZ-MycHis plasmid, and P. Green for the HTLV-1+ SLB1 lymphoma T-cell-line. We also thank P. West for kindly providing the pRc-IkBα-S32A/S36A “super repressor” mutant of IkBα. AM, TH, LY, KS, KN, RB, JP, and MR performed the experiments, contributed to data analysis, and read the manuscript. RH conceived the project, designed and performed experiments, analyzed all results, and wrote the manuscript. CH, LR, and CVL contributed to data analysis and commented on the manuscript. This work was supported by National Cancer Institute/National Institutes of Health grants 1R15CA202265–01A1 and 1R15CA158945–01A1 to RH. TH is a recipient of a 2015 American Society for Microbiology-Eugene & Millicent Goldschmidt Graduate Student Fellowship. CVL is Research Director of the Belgian Fund for Scientific Research (FRS-FNRS, Belgium) and acknowledges support from the Televie Program of the FRS-FNRS. The laboratory of CVL is part of the ULB-Cancer Research Centre (U-CRC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adya N, Giam CZ, 1995. Distinct regions in human T-cell lymphotropic virus type I tax mediate interactions with activator protein CREB and basal transcription factors. J. Virol 69, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Murphy M, Kratowicz S, Wang A, Levine AJ, George DL, 1999. Down-regulation of the Stathmin/Op18 and FKBP25 following p53 induction. Oncogene 18, 5954–5958. [DOI] [PubMed] [Google Scholar]
- Andersen SS, Ashford AJ, Tournebize R, Gavet O, Sobel A, Hyman AA, Karsenti E, 1997. Mitotic chromatin regulates phosphorylation of Stathmin/Op18. Nature 389, 640–643. [DOI] [PubMed] [Google Scholar]
- Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL, 2006. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood 107, 3976–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL, 2008. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 112, 3788–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, Motloch P, Takemoto S, Taguchi H, Cole MD, Lüscher B, Dittrich O, Tagami H, Nakatani Y, McGee M, Girard AM, Gaughan L, Robson CN, Monnat RJ Jr., Harrod R, 2005. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol. Cell. Biol 25, 6178–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangham CR, Ratner L, 2015. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr. Opin. Virol 14, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoe JT, Albrecht B, Collins ND, Ratner L, Green PL, Lairmore MD, 2000. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J. Virol 74, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydoun HH, Cherian MA, Green P, Ratner L, 2015. Inducible nitric oxide synthase mediates DNA double strand breaks in Human T-Cell Leukemia Virus Type 1-induced leukemia/lymphoma. Retrovirology 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui SM, Fryear KA, Nyalwidhe JO, Guo X, Semmes OJ, 2010. The viral oncoprotein tax sequesters DNA damage response factors by tethering MDC1 to chromatin. J. Biol. Chem 285, 32897–32905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belletti B, Pellizzari I, Berton S, Fabris L, Wolf K, Lovat F, Schiappacassi M, D’Andrea S, Nicoloso MS, Lovisa S, Sonego M, Defilippi P, Vecchione A, Colombatti A, Friedl P, Baldassarre G, 2010. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol. Cell. Biol 30, 2229–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont L, Mitchison T, Deacon HW, 1996. Catastrophic revelations about Op18/stathmin. Trends Biochem. Sci 21, 197–198. [PubMed] [Google Scholar]
- Belmont LD, Mitchison TJ, 1996. Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84, 623–631. [DOI] [PubMed] [Google Scholar]
- Ben-Abdallah M, Sturny-Leclère A, Avé P, Louise A, Moyrand F, Weih F, Janbon G, Mémet S, 2012. Fungal-induced cell cycle impairment, chromosome instability and apoptosis via differential activation of NF-κB. PLoS Pathog 8, e1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH, 2009. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28, 3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH, 2006. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120. [DOI] [PubMed] [Google Scholar]
- Berneman ZN, Gartenhaus RB, Reitz MS Jr., Blattner WA, Manns A, Hanchard B, Ikehara O, Gallo RC, Klotman ME, 1992. Expression of alternatively spliced human T-lymphotropic virus type I pX mRNA in infected cell lines and in primary uncultured cells from patients with adult T-cell leukemia/lymphoma and healthy carriers. Proc. Natl. Acad. Sci., USA 89, 3005–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton S, Pellizzari I, Fabris L, D’Andrea S, Segatto I, Canonieri V, Marconi D, Schiappacassi M, Benevol S, Gattei V, Colombatti A, Belletti B, Baldassarre G, 2014. Genetic characterization of p27(kip1) and stathmin in controlling cell proliferation in vivo. Cell Cycle 13, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaib-Mezrag H, Lemaçon D, Fontaine H, Bellon M, Bai XT, Drac M, Coquelle A, Nicot C, 2014. Tax impairs DNA replication forks and increases DNA breaks in specific oncogenic genome regions. Mol. Cancer 13, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zachar V, Zdravkovic M, Guo M, Ebbesen P, Liu X, 1997. Role of the Fas/Fas ligand pathway in apoptotic cell death induced by the human T cell lymphotropic virus type I Tax transactivator. J. Gen. Virol 78, 3277–3285. [DOI] [PubMed] [Google Scholar]
- Ching YP, Chan SF, Jeang KT, Jin DY, 2006. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat. Cell Biol 8, 717–724. [DOI] [PubMed] [Google Scholar]
- Choi YB, Harhaj EW, 2014. HTLV-1 tax stabilizes MCL-1 via TRAF6-dependent K63-linked polyubiquitination to promote cell survival and transformation. PLoS Pathog 10, e1004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciminale V, Pavlakis GN, Derse D, Cunningham CP, Felber BK, 1992. Complex splicing in the human T-cell leukemia virus (HTLV) family of retroviruses: novel mRNAs and proteins produced by HTLV type I. J. Virol 66, 1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc I, Polakowski N, André-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I, 2008. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J. Biol. Chem 283, 23903–23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A, Shibata E, Dutta A, 2013. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell. Biol 33, 3309–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayaram T, Lemoine FJ, Donehower LA, Marriott SJ, 2013. Activation of WIP1 phosphatase by HTLV-1 Tax mitigates the cellular response to DNA damage. PLoS One 8, e55989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M, 1996. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol. Cell. Biol 16, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SS, Guo X, Fryear KA, Mihaylova VT, Gupta SK, Belgnaoui SM, Haoudi A, Kupfer GM, Semmes OJ, 2008. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem 283, 36311–36320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, Franchini G, 2011. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses 3, 861–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabris L, Berton S, Pellizzari I, Segatto I, D’Andrea S, Armenia J, Bomben R, Schiappacassi M, Gattei V, Philips MR, Vecchione A, Belletti B, Baldassarre G, 2015. p27kip1 controls H-Ras/MAPK activation and cell cycle entry via modulation of MT stability. Proc. Natl. Acad. Sci., USA 112, 13916–13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger TR, Sharma N, Kim YM, Nyborg JK, 2008. The human T-cell leukemia virus type 1 tax protein confers CBP/p300 recruitment and transcriptional activation properties to phosphorylated CREB. Mol. Cell. Biol 28, 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geleziunas R, Ferrell S, Lin X, Mu Y, Cunningham ET Jr., Grant M, Connelly MA, Hambor JE, Marcu KB, Greene WC, 1998. Human T-cell leukemia virus type 1 Tax induction of NF-kappaB involves activation of the IkappaB kinase alpha (IKKalpha) and IKKbeta cellular kinases. Mol. Cell. Biol 18, 5157–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebler HA, Loring JE, van Orden K, Colgin MA, Garrus JE, Escudero KW, Brauweiler A, Nyborg JK, 1997. Anchoring of CREB-binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol. Cell. Biol 17, 5156–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AP, Irvine J, Blyth K, Cameron ER, Onions DE, Campbell ME, 1998. Tumours derived from HTLV-I tax transgenic mice are characterized by enhanced levels of apoptosis and oncogene expression. J. Pathol 186, 209–214. [DOI] [PubMed] [Google Scholar]
- Haoudi A, Daniels RC, Wong E, Kupfer G, Semmes OJ, 2003. Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem 278, 37736–37744. [DOI] [PubMed] [Google Scholar]
- Harhaj EW, Giam CZ, 2018. NF-κB signaling mechanisms in HTLV-1-induced adult T-cell leukemia/lymphoma. FEBS J doi: 10.1111/febs.14492 [epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj EW, Good L, Xiao G, Uhlik M, Cvijic ME, Rivera-Walsh I, Sun SC, 2000. Somatic mutagenesis studies of NF-kappa B signaling in human T cells: evidence for an essential role of IKK gamma in NF-kappa B activation by T-cell costimulatory signals and HTLV-I Tax protein. Oncogene 19, 1448–1456. [DOI] [PubMed] [Google Scholar]
- Harhaj NS, Sun SC, Harhaj EW, 2007. Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa B kinase. J. Biol. Chem 282, 4185–4192. [DOI] [PubMed] [Google Scholar]
- Harrod R, Kuo YL, Tang Y, Yao Y, Vassilev A, Nakatani Y, Giam CZ, 2000. p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J. Biol. Chem 275, 11852–11857. [DOI] [PubMed] [Google Scholar]
- Harrod R, Tang Y, Nicot C, Lu HS, Vassilev A, Nakatani Y, Giam CZ, 1998. An exposed KID-like domain in human T-cell lymphotropic virus type 1 Tax is responsible for the recruitment of coactivators CBP/p300. Mol. Cell. Biol 18, 5052–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B, 1997. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1, 3–11. [DOI] [PubMed] [Google Scholar]
- Hiven P, Frédéric M, Arpin-André C, Basbous J, Gay B, Thébault S, Mesnard JM, 2005. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J. Cell Sci 118, 1355–1362. [DOI] [PubMed] [Google Scholar]
- Ho YK, Zhi H, Bowlin T, Dorjbal B, Philip S, Zahoor MA, Shih HM, Semmes OJ, Schaefer B, Glover JN, Giam CZ, 2015. HTLV-1 Tax stimulates ubiquitin E3 ligase, ring finger protein 8, to assemble lysine 63-linked polyubiquitin chains for TAK1 and IKK activation. PLoS Pathog 11, e1005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YK, Zhi H, DeBiaso D, Philip S, Shih HM, Giam CZ, 2012. HTLV-1 tax-induced rapid senescence is driven by the transcriptional activity of NF-κB and depends on chronically activated IKKa and p65/RelA. J. Virol 86, 9474–9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt P, Brännström K, Stenmark S, Gullberg M, 2006. Aneugenic activity of Op18/stathmin is potentiated by the somatic Q18 E mutation in leukemic cells. Mol. Biol. Cell 17, 2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt P, Larsson N, Segerman B, Howell B, Morabito J, Cassimeris L, Gullberg M, 2001. The catastrophe-promoting activity of ectopic Op18/Stathmin is required for disruption of mitotic spindles but not Interphase microtubules. Mol. Biol. Cell 12, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt P, Sellin ME, Gullberg M, 2010. Upregulated Op18/stathmin activity causes chromosomal instability through a mechanism that evades the spindle assembly checkpoint. Exp. Cell Res 316, 2017–2026. [DOI] [PubMed] [Google Scholar]
- Hong S, Wang LC, Gao X, Kuo YL, Liu B, Merling R, Kung HJ, Shih HM, Giam CZ, 2007. Heptad repeats regulate protein phosphatase 2a recruitment to I-kappaB kinase gamma/NF-kappaB essential modulator and are targeted by human T-lymphotropic virus type 1 tax. J. Biol. Chem 282, 12119–12126. [DOI] [PubMed] [Google Scholar]
- Houghtaling BR, Yang G, Matov A, Danuser G, Kapoor TM, 2009. Op18 reveals the contribution of nonkinetochore microtubules to the dynamic organization of the vertebrate meiotic spindle. Proc. Natl. Acad. Sci., USA 106, 15338–15343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes SC, Alushin GM, Shida T, Nachury MV, Nogales E, 2014. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 25, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison T, Malu A, Yapindi L, Bergeson R, Peck K, Romeo M, Harrod C, Pope J, Smitherman L, Gwinn W, Ratner L, Yates C, Harrod R, 2018. The TP53-Induced Glycolysis and Apoptosis Regulator mediates cooperation between HTLV-1 p30II and the retroviral oncoproteins Tax and HBZ and is highly expressed in an in vivo xenograft model of HTLV-1-induced lymphoma. Virology 520, 39–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga R, Ohtani K, Hayashi T, Nakamura M, 2001. Molecular mechanism of cell cycle progression induced by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene 20, 2055–2067. [DOI] [PubMed] [Google Scholar]
- Janke C, Montagnac G, 2017. Causes and consequences of microtubule acetylation. Curr. Biol 27, R1287–R1292. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN, 2005. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 24, 6719–6728. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Radonovich M, Brady JN, Pise-Masison CA, 2004. HTLV-I Tax induces a novel interaction between p65/RelA and p53 that results in inhibition of p53 transcriptional activity. Blood 104, 1490–1497. [DOI] [PubMed] [Google Scholar]
- Jin DY, Spencer F, Jeang KT, 1998. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell 93, 81–91. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Harrod R, Franchini G, 2001. Molecular biology and pathogenesis of the human T-cell leukaemia/lymphotropic virus type-1 (HTLV-1). Int. J. Exp. Pathol 82, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KJ, Dasgupta A, Huang K, Jeong SJ, Pise-Masison C, Gurova KV, Brady JN, 2008. Small-molecule inhibitor which reactivates p53 in human T-cell leukemia virus type 1-transformed cells. J. Virol 82, 8537–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehn K, de la Fuente C, Strouss K, Berro R, Jiang H, Brady J, Mahieux R, Pumfery A, Bottazzi ME, Kashanchi F, 2005. The HTLV-I Tax oncoprotein targets the retinoblastoma protein for proteasomal degradation. Oncogene 24, 525–540. [DOI] [PubMed] [Google Scholar]
- Kimata JT, Wong FH, Wang JJ, Ratner L, 1994. Construction and characterization of infectious human T-cell leukemia virus type 1 molecular clones. Virology 204, 656–664. [DOI] [PubMed] [Google Scholar]
- Kinjo T, Ham-Terhune J, Peloponese JM Jr., Jeang KT, 2010. Induction of reactive oxygen species by human T-cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J. Virol 84, 5431–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralnik IJ, Gessain A, Klotman ME, Lo Monico A, Berneman ZN, Franchini G, 1992. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc. Natl. Acad. Sci., USA 89, 8813–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küntziger T, Gavet O, Sobel A, Bornens M, 2001. Differential effect of two stathmin/Op18 phosphorylation mutants on Xenopus embryo development. J. Biol. Chem 276, 22979–22984. [DOI] [PubMed] [Google Scholar]
- Kuo YL, Giam CZ, 2006. Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. EMBO J 25, 1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F, 2008. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 29, 392–400. [DOI] [PubMed] [Google Scholar]
- Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih H, Connor LM, Marriott SJ, Goodman RH, 1996. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature 380, 642–646. [DOI] [PubMed] [Google Scholar]
- Larsson N, Segerman B, Gradin HM, Wandzioch E, Cassimeris L, Gullberg M, 1999. Mutations of oncoproteins 18/stathmin identify tubulin-directed regulatory activities distinct from tubulin association. Mol. Cell. Biol 19, 2242–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavorgna A, Harhaj EW, 2014. Regulation of HTLV-1 tax stability, cellular trafficking and NF-κB activation by the ubiquitin-proteasome pathway. Viruses 6, 3925–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavorgna A, Matsuoka M, Harhaj EW, 2014. A critical role for IL-17RB signaling in HTLV-1 tax-induced NF-κB activation and T-cell transformation. PLoS Pathog 10, e1004418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux AC, Sellier H, Gillies K, Iannetti A, James J, Perkins ND, 2013. NFkB regulates expression of Polo-like kinase 4. Cell Cycle 12, 3052–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Marriott SJ, 2002. Genomic instability driven by the human T-cell leukemia virus type I (HTLV-I) oncoprotein, Tax. Oncogene 21, 7230–7234. [DOI] [PubMed] [Google Scholar]
- Li M, Kesic M, Yin H, Yu L, Green PL, 2009. Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J. Virol 83, 3788–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hong S, Tang Z, Yu H, Giam CZ, 2005. HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc. Natl. Acad. Sci., USA 102, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M, Khazaie K, Schulze-Osthoff K, Baeuerle PA, Schirrmacher V, Chlichlia K, 1998. Human T cell leukemia virus-I (HTLV-I) Tax-mediated apoptosis in activated T cells requires an enhanced intracellular prooxidant state. J. Immunol 161, 3050–3055. [PubMed] [Google Scholar]
- Lu Y, Liu C, Cheng H, Xu Y, Jiang J, Xu J, Long J, Liu L, Yu X, 2014. Stathmin, interacting with NF-κB, promotes tumor growth and predicts poor prognosis of pancreatic cancer. Curr. Mol. Med 14, 328–339. [DOI] [PubMed] [Google Scholar]
- Majone F, Jeang KT, 2000. Clastogenic effect of the human T-cell leukemia virus type I Tax oncoprotein correlates with unstabilized DNA breaks. J. Biol. Chem 275, 32906–32910. [DOI] [PubMed] [Google Scholar]
- Mengle-Gaw L, Rabbitts TH, 1987. A human chromosome 8 region with abnormalities in B cell, HTLV-I+ T cell and c-myc amplified tumours. EMBO J 6, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, George DL, 1999. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev 13, 2490–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuveut C, Low KG, Maldarelli F, Schmitt I, Majone F, Grassmann R, Jeang KT, 1998. Human T-cell leukemia virus type 1 Tax and cell cycle progression: role of cyclin D-cdk and p110Rb. Mol. Cell. Biol 18, 3620–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C, Dundr M, Johnson JM, Fullen JR, Alonzo N, Fukumoto R, Princler GL, Derse D, Misteli T, Franchini G, 2004. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med 10, 197–201. [DOI] [PubMed] [Google Scholar]
- Nicot C, Harrod R, 2000. Distinct p300-responsive mechanisms promote caspase-dependent apoptosis by human T-cell lymphotropic virus type 1 Tax protein. Mol. Cell. Biol 20, 8580–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C, Harrod RL, Ciminale V, Franchini G, 2005. Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene 24, 6026–6034. [DOI] [PubMed] [Google Scholar]
- O’Mahony AM, Montano M, Van Beneden K, Chen LF, Greene WC, 2004. Human T-cell lymphotropic virus type 1 tax induction of biologically active NF-kappaB requires IkappaB kinase-1-mediated phosphorylation of RelA/p65. J. Biol. Chem 279, 18137–18145. [DOI] [PubMed] [Google Scholar]
- Peloponese JM Jr., Haller K, Miyazato A, Jeang KT, 2005. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoproteins. Proc. Natl. Acad. Sci., USA 102, 18974–18979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique C, Ureta-Vidal A, Gessain A, Chancerel B, Gout O, Tamouza R, Agis F, Dokhélar MC, 2000. Evidence for the chronic in vivo production of human T cell leukemia virus type I Rof and Tof proteins from cytotoxic T lymphocytes directed against viral peptides. J. Exp. Med 191, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a.Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN, 1998. Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J. Virol 72, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a.Pise-Masison CA, Mahieux R, Jiang H, Ashcroft M, Radonovich M, Duvall J, Guillerm C, Brady JN, 2000. Inactivation of p53 by human T-cell lymphotropic virus type 1 Tax requires activation of the NF-kappaB pathway and is dependent on p53 phosphorylation. Mol. Cell. Biol 20, 3377–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Pise-Masison CA, Mahieux R, Radonovich M, Jiang H, Duvall J, Guillerm C, Brady JN, 2000. Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res. Hum. Retroviruses 16, 1669–1675. [DOI] [PubMed] [Google Scholar]
- b.Pise-Masison CA, Radonovich M, Sakaguchi K, Appella E, Brady JN, 1998. Phosphorylation of p53: a novel pathway for p53 inactivation in human T-cell lymphotropic virus type 1-transformed cells. J. Virol 72, 6348–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portran D, Schaedel L, Xu Z, Théry M, Nachury MV, 2017. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol 19, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princler GL, Julias JG, Hughes SH, Derse D, 2003. Roles of viral and cellular proteins in the expression of alternatively spliced HTLV-1 pX mRNAs. Virology 317, 136–145. [DOI] [PubMed] [Google Scholar]
- Robek MD, Wong FH, Ratner L, 1998. Human T-cell leukemia virus type 1 pX-I and pX-II open reading frames are dispensable for the immortalization of primary lymphocytes. J. Virol 72, 4458–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo M, Hutchison T, Malu A, White A, Kim J, Gardner R, Smith K, Nelson K, Bergeson R, McKee R, Harrod C, Ratner L, Lüscher B, Martinez E, Harrod R, 2018. The human T-cell leukemia virus type-1 p30II protein activates p53 and induces the TIGAR and suppresses oncogene-induced oxidative stress during viral carcinogenesis. Virology 518, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, Harrod CK, Barnett B, Ratner L, Lairmore MD, Martinez E, Lüscher B, Robson CN, Henriksson M, Harrod R, 2015. Acetylation of the c-MYC oncoprotein is required for cooperation with the HTLV-1 p30(II) accessory protein and the induction of oncogenic cellular transformation by p30(II)/c-MYC. Virology 476, 271–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiappacassi M, Lovisa S, Lovat F, Fabris L, Colombatti A, Belletti B, Baldassarre G, 2011. Role of T198 modification in the regulation of p27(Kip1) protein stability and function. PLoS One 6, e17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Yamamoto M, Akira S, Harhaj EW, 2007. The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-kappaB activation. J. Virol 81, 13735–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Tokunaga F, Goto E, Komatsu G, Gohda J, Saeki Y, Tanaka K, Takahashi H, Sawasaki T, Inoue S, Oshiumi H, Seya T, Nakano H, Tanaka Y, Iwai K, Inoue JI, 2017. HTLV-1 Tax induces formation of the active macromolecular IKK complex by generating Lys63- and Met1-linked hybrid polyubiquitin chains. PLoS Pathog 13, e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman LR, Phipps AJ, Montgomery A, Ratner L, Lairmore MD, 2004. Human T-cell lymphotropic virus type 1 open reading frame II-encoded p30II is required for in vivo replication: evidence of in vivo reversion. J. Virol 78, 3837–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Greene WC, 1990. Identification of HTLV-I tax trans-activator mutants exhibiting novel transcriptional phenotypes. Genes Dev 4, 1875–1885. [DOI] [PubMed] [Google Scholar]
- Sun EC, Elwood J, Beraud C, Greene WC, 1994. Human T-cell leukemia virus type I Tax activation of NF-kappa B/Rel involves phosphorylation and degradation of I kappa B alpha and RelA (p65)-mediated induction of the c-rel gene. Mol. Cell. Biol 14, 7377–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a.Suzuki T, Fujisawa JI, Toita M, Yoshida M, 1993. The trans-activator tax of human T-cell leukemia virus type 1 (HTLV-1) interacts with cAMP-responsive element (CRE) binding and CRE modulator proteins that bind to the 21-base-pair enhancer of HTLV-1. Proc. Natl. Acad. Sci., USA 90, 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- b.Suzuki T, Hirai H, Fujisawa J, Fujita T, Yoshida M, 1993. A trans-activator Tax of human T-cell leukemia virus type 1 binds to NF-kappa B p50 and serum response factor (SRF) and associates with enhancer DNAs of the NF-kappa B site and CArG box. Oncogene 8, 2391–2397. [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB, 2006. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylla BS, Hung SC, Davidson DM, Hatzivassiliou E, Malinin NL, Wallach D, Gilmore TD, Kieff E, Mosialos G, 1998. Epstein-Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-κB through a pathway that includes the NF-κB-inducing kinase and the IkB kinases IKKa and IKKb. Proc. Natl. Acad. Sci., USA 95, 10106–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabakin-Fix Y, Azran I, Schavinky-Khrapunsky Y, Levy O, Aboud M, 2006. Functional inactivation of p53 by human T-cell leukemia virus type 1 Tax protein: mechanisms and clinical implications. Carcinogenesis 27, 673–681. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Higuchi M, Makokha GN, Matsuki H, Yoshita M, Tanaka Y, Fujii M, 2013. HTLV-1 Tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood 122, 715–725. [DOI] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W, 2006. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. [DOI] [PubMed] [Google Scholar]
- Tsukasaki K, Krebs J, Nagai K, Tomonaga M, Koeffler HP, Bartram CR, Jauch A, 2001. Comparative genomic hybridization analysis in adult T-cell leukemia/lymphoma: correlation with clinical course. Blood 97, 3875–3881. [DOI] [PubMed] [Google Scholar]
- Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, Nicot C, Cecchinato V, Ruscetti F, Franchini G, 2010. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood 116, 3809–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernin C, Thenoz M, Pinatel C, Gessain A, Gout O, Delfau-Larue MH, Nazaret N, Legras-Lachuer C, Wattel E, Mortreux F, 2014. HTLV-1 bZIP factor HBZ promotes cell proliferation and genetic instability by activating OncomiRs. Cancer Res 74, 6082–6093. [DOI] [PubMed] [Google Scholar]
- Wu X, Sun SC, 2007. Retroviral oncoprotein Tax deregulates NF-kappaB by activating Tak1 and mediating the physical association of Tak1-IKK. EMBO Rep 8, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Yamamoto B, Haoudi A, Semmes OJ, Green PL, 2006. PDZ binding motif of HTLV-1 Tax promotes virus-mediated T-cell proliferation in vitro and persistence in vivo. Blood 107, 1980–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Liao R, Li N, Xiang R, Sun P, 2014. Phosphorylation of Tip60 by p38a regulates p53-mediated PUMA induction and apoptosis in response to DNA damage. Oncotarget 5, 12555–12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, 1996. The human T-cell leukemia virus type I (HTLV-I) tax protein induces apoptosis. Nihon Rinsho 54, 1855–1859. [PubMed] [Google Scholar]
- Yamamoto B, Li M, Kesic M, Younis I, Lairmore MD, Green PL, 2008. Human T-cell leukemia virus type 2 post-transcriptional control protein p28 is required for viral infectivity and persistence in vivo. Retrovirology 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A, 1998. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93, 1231–1240. [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Inoue H, Sakurai M, Sugiyama T, Hazama M, Yamada T, Hatanaka M, 1996. Constitutive activation of NF-κB is essential for transformation of rat fibroblasts by the human T-cell leukemia virus type I Tax protein. EMBO J 15, 873–887. [PMC free article] [PubMed] [Google Scholar]
- Younis I, Khair L, Dundr M, Lairmore MD, Franchini G, Green PL, 2004. Repression of human T-cell leukemia virus type 1 and 2 replication by a viral mRNA-encoded posttranscriptional regulator. J. Virol 78, 11077–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zane L, Yasunaga J, Mitagami Y, Yedavalli V, Tang SW, Chen CY, Ratner L, Lu X, Jeang KT, 2012. Wip1 and p53 contribute to HTLV-1 Tax-induced tumorigenesis. Retrovirology 9, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chen L, Cai SH, Cheng H, 2016. Identification of TBK1 and IKKe, the non-canonical IkB kinases, as crucial pro-survival factors in HTLV-1-transformed T lymphocytes. Leuk. Res 46, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Nisbet JW, Bartoe JT, Ding W, Lairmore MD, 2000. Human T-lymphotropic virus type 1 p30(II) functions as a transcription factor and differentially modulates CREB-responsive promoters. J. Virol 74, 11270–11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LJ, Giam CZ, 1992. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc. Natl. Acad. Sci., USA 89, 7070–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Yasunaga J, Satou Y, Nakao M, Takahashi M, Fujii M, Matsuoka M, 2009. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-κB. Blood 113, 2755–2764. [DOI] [PubMed] [Google Scholar]
- Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ, 2011. NF-κB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog 7, e1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1. The HA-tagged HTLV-1 p30II protein represses Stathmin expression and inhibits Tax-dependent NF-κB transactivation similar to p30II-GFP. (A) The data from Fig. 1E which shows Tax-dependent transactivation of the E-selectin promoter-luciferase reporter plasmid and inhibition by p30II-GFP are represented as ‘Fold transactivation’ for comparison. (B) 293 HEK cells were cotransfected with the E-selectin-luciferase reporter construct and an RcCMV-HTLV-1 Tax expression construct, and then transduced with lentiviral expression vectors for GFP or HTLV-1 p30II (HA). Relative luciferase activities were measured and normalized for equivalent total cellular proteins. (C) The effects of pLenti-GFP and pLenti-HTLV-1 p30II (HA) upon Stathmin expression were determined by transducing 293 cells with the lentiviral constructs and then performing SDS-PAGE and immunoblot analyses. The relative Stathmin protein levels were quantified by densitometry and normalized to Actin. (D) The expression of HTLV-1 p30II (HA) and GFP in lentiviral transduced cultures was visualized by confocal microscopy either by immunostaining the samples with an Anti-HA tag monoclonal antibody (Abcam) and rhodamine red-conjugated secondary antibody (red signal), or by direct-fluorescence for GFP (green). DAPI nuclear-staining is provided for reference. Untransduced (UT) cells were included as negative controls for the Anti-HA antibody and GFP fluorescence detection (n=3). Scale bar, 20 mm.
Supplemental Fig. S2. Transactivation of the E-selectin gene promoter by HTLV-1 Tax is dependent upon NF-κB-signaling. (A) The functionality of the Tax protein used in these studies and its ability to transactivate the CREB-dependent Tax-responsive elements (TREs) within the HTLV-1 promoter was demonstrated by cotransfecting 293 HEK cells with 0.25 mg of an HTLV-1 TRE-luciferase reporter plasmid (Giebler et al., 1997) and increasing amounts (0.25 and 0.5 mg) of RcCMV-HTLV-1 Tax or a pcDNA3.1-GFP control. Relative luciferase activities were measured and normalized for equivalent total cellular protein levels. The expression of the HTLV-1 Tax, GFP, and Actin proteins was detected by SDS-PAGE and immunoblotting. (B and C) Dominant-negative mutants of IkBα or the IκKb subunit inhibit Tax-induced NF-κB transactivation. 293 cells were cotransfected with an E-selectin promoter-luciferase reporter plasmid and either RcCMV-HTLV-1 Tax alone, or together with a phosphorylation/degradation-defective IkBα “super repressor” mutant, IkBα-S32A/S36A (DiDonato et al., 1996) in B. The HTLV-1 Tax, IkBα-S32A/S36A mutant, and Actin proteins were detected by immunoblotting. In C, 293 cells were cotransfected with the E-Selectin-luc reporter plasmid and RcCMV-HTLV-1 Tax alone, or with expression constructs (0.25 and 0.5 mg) for the dominant-negative IκKβ subunit deletion mutants, IκKβΔ9 or IκKbD34 (Sylla et al., 1998). Relative luciferase activities were then measured and normalized for equivalent total cellular proteins. The HTLV-1 Tax, IκKβΔ9 (FLAG), IκKβΔ34 (FLAG), and Actin proteins were detected by SDS-PAGE and immunoblotting. For A-C, the data represent the mean ± standard deviation (error bars) from three independent experiments.
Supplemental Fig. S3. The HT1080/HTLV-1 ACH.wt proviral clone contains reduced NF-κB p65RelA levels relative to the ACH.p30II mutant. (A) The HT1080/HTLV-1 ACH.wt and ACH.p30II clones were treated with the protein synthesis-inhibitor, cycloheximide (50 mg/ml; Sigma-Aldrich), and the S36-phosphorylation and degradation of IkB-α and expression of the NF-κB p65RelA subunit were detected by SDS-PAGE and immunoblotting. A representative Ponceau S-stained nitrocellulose membrane is shown for comparison. (B and C) The relative levels of p65RelA, S36-phosphorylated IkB-a, and total IkB-a were quantified by densitometry analysis of the immunoblot bands. (D) Nuclear (Nuc) and cytoplasmic (Cyt) extracts were prepared from the HT1080/HTLV-1 ACH.wt and ACH.p30II clones that were treated with cycloheximide for 2 hrs, and the relative levels of p65RelA were detected by immunoblotting. A nonspecific band (ns) is also indicated. A representative Ponceau S-stained membrane is provided for comparison. (E) The expression of p65RelA was quantified by densitometry. All the data is representative of three independent experiments. The data in B, C, and E represent the experimental mean ± standard deviation (error bars).