

Abstract
Arrhythmogenic cardiomyopathy (ACM) encompasses a group of inherited cardiomyopathies including arrhythmogenic right ventricular cardiomyopathy (ARVC) whose molecular disease mechanism is associated with dysregulation of the canonical WNT signalling pathway. Recent evidence indicates that ARVC and ACM caused by pathogenic variants in the FLNC gene encoding filamin C, a major cardiac structural protein, may have different molecular mechanisms of pathogenesis. We sought to identify dysregulated biological pathways in FLNC-associated ACM.
RNA was extracted from seven paraffin-embedded left ventricular tissue samples from deceased ACM patients carrying FLNC variants and sequenced.
Transcript levels of 623 genes were upregulated and 486 genes were reduced in ACM in comparison to control samples. The cell adhesion pathway and ILK signalling were among the prominent dysregulated pathways in ACM. Consistent with these findings, transcript levels of cell adhesion genes JAM2, NEO1, VCAM1 and PTPRC were upregulated in ACM samples. Moreover, several actin-associated genes, including FLNC, VCL, PARVB and MYL7, were suppressed, suggesting dysregulation of the actin cytoskeleton. Analysis of the transcriptome for dysregulated biological pathways predicted activation of inflammation and apoptosis and suppression of oxidative phosphorylation and MTORC1 signalling in ACM.
Our data suggests dysregulated cell adhesion and ILK signalling as novel putative pathogenic mechanisms of ACM caused by FLNC variants which are distinct from the postulated disease mechanism of classic ARVC caused by desmosomal gene mutations. This knowledge could help in the design of future gene therapy strategies which would target specific components of these pathways and potentially lead to novel treatments for ACM.
Keywords: Arrhythmogenic cardiomyopathy, RNA sequencing, Filamin C, Focal adhesion pathway, Integrin linked kinase pathway
1. Introduction
Arrhythmogenic cardiomyopathy (ACM) is an important cause of sudden cardiac death (SCD) in those under 35 years of age [1]. ACM is characterised by global or regional ventricular dysfunction associated with fibro-fatty replacement of the myocardium, conduction defects, and arrhythmias [2].
Arrhythmogenic right ventricular cardiomyopathy (ARVC), a major subtype of ACM, is considered a disease of the desmosome, commonly caused by mutations in genes coding for major desmosomal proteins [2,3]. Canonical WNT signalling has been implicated in the pathogenesis of classic ARVC caused by mutations in desmosomal genes [4]. However, the role of this signalling pathway in other subtypes of ACM is unknown.
Pathogenic variants in the FLNC gene encoding filamin C, also known as actin binding like protein γ-filamin, a major structural protein, have been recognised to cause several cardiomyopathies associated with SCD including ACM [5–8]. We recently studied the clinical and molecular “signature” of FLNC-associated disease in pedigrees with ACM as well as in fixed cardiac tissue specimens from deceased ACM cases with FLNC pathogenic variants [9]. In that study immunohistochemistry analysis provided early evidence that the pathogenic mechanism related to genetic variants in FLNC may be different to typical ARVC [9]. However, currently the disease mechanisms related to FLNC are mostly unknown as is the molecular pathogenesis of ACM caused by FLNC variants. The purpose of this study was to gain insights into the molecular pathogenesis of ACM caused by FLNC variants.
2. Methods
2.1. Sample selection
Fixed cardiac tissue from six deceased ACM cases with FLNC variants was obtained through a collaborative research project between Spanish pathology centres and hospitals as previously described [9]. All cases were sudden cardiac death victims with a diagnosis of ARVC or left dominant arrhythmogenic cardiomyopathy at post mortem. [9]. FLNC variants include five pathogenic and one variant of unknown significance according to the American College of Medical Genetics (ACMG) variant classification [10]. In addition, one ACM case carrying the novel c.7252–1G>A splicing variant in FLNC (also classified as pathogenic) was identified from sudden death archive cases in the UK. All cases were negative for pathogenic variants in other known ACM genes. A list of FLNC variants present in samples used in this study is provided in Appendix Table A.1.
In total, paraffin embedded left ventricular (LV) tissue samples were available from seven patients for RNA extraction and sequencing. Paraffin fixed LV samples from four age-matched individuals, who died from non-cardiac conditions, were used as controls.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the relevant NHS Health Research Authority Ethics Committees (REC ID: 15/LO/0549) in the UK, CEIC Hospital Virgen de la Arrixaca, – CEIC Hospital Universitario y Politécnico La Fe in Spain and with the 1964 Helsinki declaration and its later amendments. Human samples and data on sudden death cases were collected with informed consent from first-degree family members.
2.2. RNA sequencing
RNA from LV tissue was extracted and sequenced by Qiagen as a commercial service project. Detailed methodology is provided in the Appendix (Supplementary methods).
2.3. Pathway analysis
Advanced bioinformatic data analysis and interpretation was completed in-house using WebGestalt, g:Profiler (g:GOSt) and Qiagen Ingenuity Pathway Analysis (IPA) as described in detail in the Supplementary material, Appendix.
2.4. Quantitative polymerase chain reaction (qPCR)
Comprehensive validation of changes in the transcript levels of the target genes, detected by RNA-seq, was not feasible due to limited amount of tissue samples available. Therefore, changes in the transcript levels of selected genes related to the predicted dysregulated pathways were investigated by qPCR. In particular, Taqman assays (Thermo Fisher Scientific) were used for five genes: FLNC, Hs00155124_m1; VCL, Hs00419715_m1; PARVB, Hs00203381_m1; THBS4, Hs00170261_m1; MYL7, Hs01085598_g1 (Supplementary methods, Appendix Table A.2).
3. Results
3.1. RNA sequencing quality
On average 53.1 million reads were obtained per sample. It is estimated that 40 million reads per sample for total RNA-seq is sufficient for detecting differential expression of genes [11]. Approximately, 26.4% of the sequence reads were mapped to the reference human genome assembly GRCh37 (hg19) and were used in subsequent analysis.
3.2. Differentially expressed genes (DEGs)
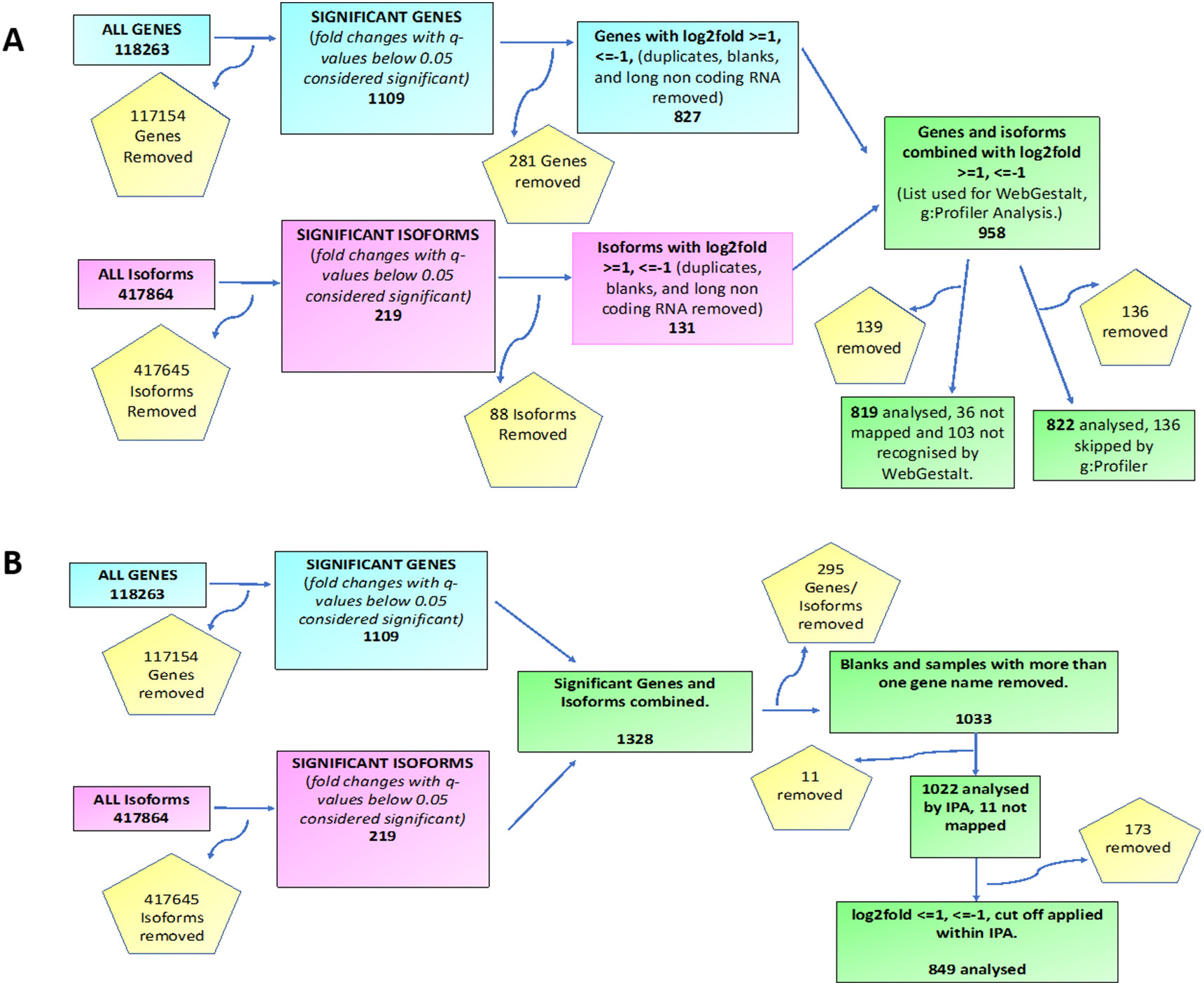
Analysis of RNA-seq data led to identification of 623 upregulated genes, 486 downregulated genes, 92 upregulated isoforms and 127 downregulated isoforms, all with a q value <0.05, in ACM hearts as compared to controls (Fig. 1A). The DEGs were targets of a number of transcriptional regulators, including STAT1, IRF family of transcription factors, CEBPB and CTNNB1, which were predicted, based on their target transcript levels, to be among the most activated transcriptional regulators, whereas TRIM24 and MYCN were predicted to be among the most suppressed transcriptional regulators (Fig. 1B and C).
Fig. 1.

Dysregulated gene expression in ACM cardiac tissue samples carrying FLNC variants. A). Volcano plots showing differentially expressed genes (q < 0.05) in ACM samples compared to controls. Z score plots showing transcriptional regulators targeting B). upregulated or C). downregulated genes. D). Gene set enrichment analysis plot of differentially expressed genes showing enrichment of genes involved in cell adhesion. E). Heatmap of dysregulated genes in the cell adhesion pathway.
The DEGs identified were filtered as shown in Appendix Fig. A.1A and B to produce a gene list for Gene Ontology (GO) terms and pathway analysis by different software programmes. In accord with the known interactions of FLNC with the actin cytoskeleton in striated muscles and its perceived functions in localization of signalling receptors and the structural organization of Z-discs [12], the most enriched Gene Ontology terms found by both WebGestalt and g:GOSt included ‘cell adhesion’ (p = 5.70 × 10−23), ‘extracellular matrix organization’ (p = 4.91 × 10−22) and ‘circulatory system development’ (p = 1.24 × 10−12) (Fig. 2 and Appendix Table A.3). Gene set enrichment analysis (GSEA) also identified enrichment of genes involved in cell adhesion among the DEGs (Fig. 1D). Heat map of the dysregulated genes involved in the cell adhesion pathway is shown in Fig. 1E. Thus, the mutant FLNC proteins appear to have an impact on the molecular composition of the extracellular matrix (ECM) and the proteins involved in cell adhesion or the regulation of cell adhesion.
Fig. 2.

Map displaying the most enriched GO terms for the FLNC variant carriers vs control group using WebGestalt [25]. The enriched GO terms are shown in the context of the ontology tree; the stronger the red colour in the GO term boxes, the more significant the enrichment is. The GO terms in the white boxes are not significantly enriched but are included in order to provide information about the parentage of the enriched terms. gNum, Number of genes in the gene set also in the category. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
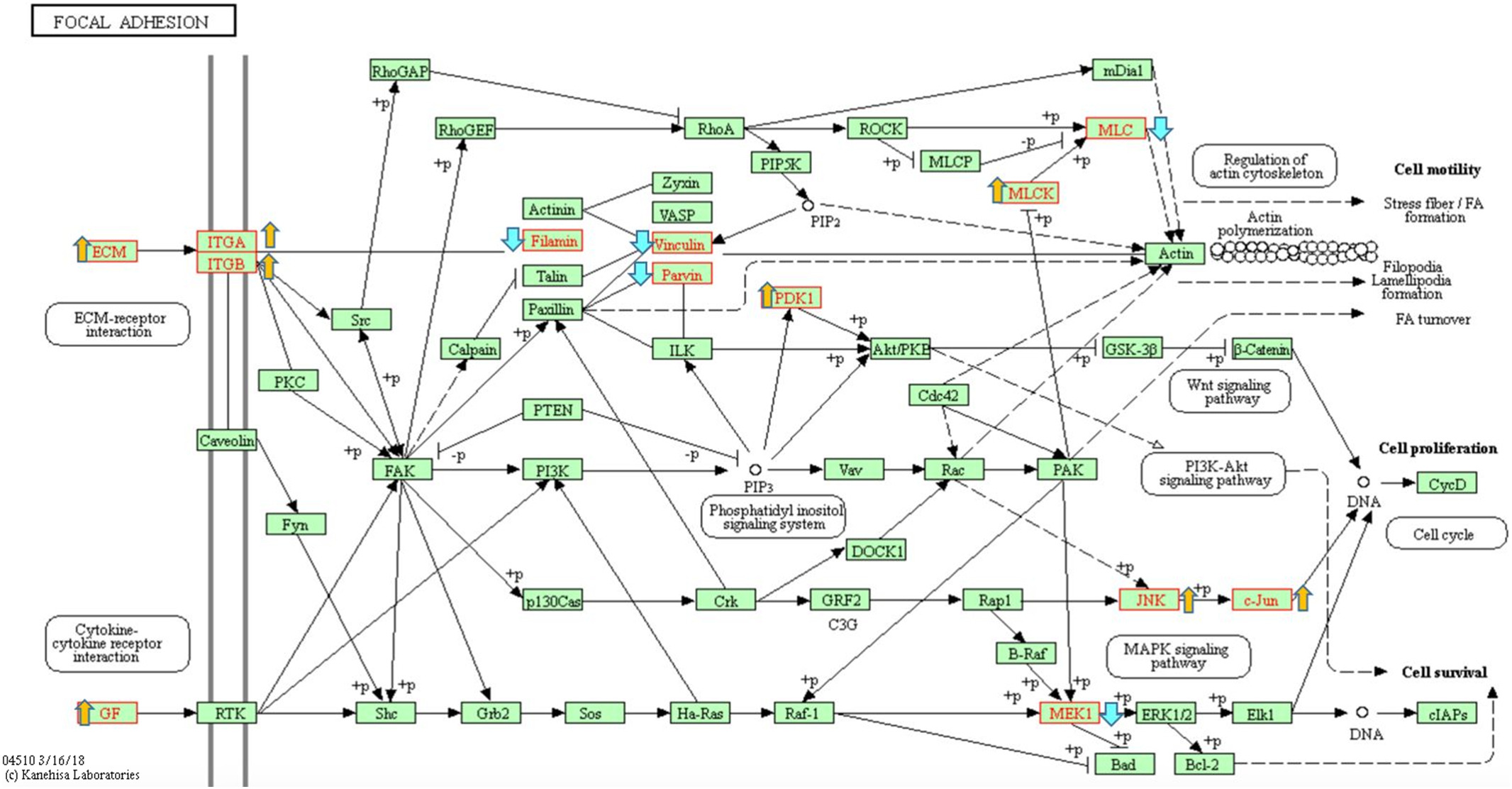
WebGestalt was also used to identify the most enriched KEGG pathways, as shown in Appendix Table A.4. The most dysregulated KEGG pathway, which contained FLNC and 23 other dysregulated genes in the dataset, was the “focal adhesion” pathway (p = 5.67 × 10−5). The ‘protein digestion and absorption’ and ‘ECM-receptor interaction’ KEGG pathways were also dysregulated (p = 6.78 × 10−6 and p = 9.17 × 10−6 respectively).
In the focal adhesion KEGG Pathway (https://www.genome.jp/kegg-bin/show_pathway?map04510 [13] - Appendix Fig. A.2), 18 genes were upregulated and six genes were downregulated in the cardiac tissue from FLNC variant carriers compared to control cardiac tissue. The pathway contains several abbreviations for terms, which group multiple genes under the same category, including, ECM and GF (growth factor).
To identify enriched pathways within the dataset, after filtering for significance (q < 0.05), genes and isoforms were combined and uploaded to IPA (Qiagen). This identified a number of dysregulated pathways, including those involved in inflammation and ILK signalling among the major dysregulated pathways (Fig. 3A). The corresponding heat map of DEGs in the ILK pathway is presented in Fig. 3B and a Circos map showing the DEGs in the main dysregulated pathways, namely those involved in inflammation and ILK signalling, is shown in Fig. 3C.
Fig. 3.

Biological pathways disrupted in ACM cardiac tissue samples carrying FLNC variants identified by IPA analysis. A). Histogram of dysregulated canonical pathways. B). Heatmap of DEGs in the ILK pathway. C). Circos plot showing differentially expressed genes in the ILK signalling and inflammation pathways.
The two most significantly enriched canonical pathways identified by IPA (Appendix Fig. A.3) are the ‘integrin linked kinase pathway’ (ILK pathway, Fig. 4) which contains FLNC (p = 2.17 × 10−5) and the ‘hepatic fibrosis pathway’ (p = 7.75 × 10−7). It is worth noting that the canonical WNT signalling pathway does not appear to be dysregulated in FLNC cardiac samples.
Fig. 4.

The ILK Signalling pathway generated using IPA. The genes with an outer pink border are present in this dataset. Those coloured in green are downregulated in the FLNC cardiac specimens and those that are coloured in red/pink are upregulated compared to the control group.
3.3. Confirmation analysis with qPCR
Changes in the transcript levels of selected DEGs were further validated by qPCR on RNA extracted from the same paraffin cardiac samples used in the RNA sequencing process. Five genes in the ILK pathway, FLNC, VCL, PARVB, THBS4 and MYL7, were confirmed to show the same pattern of dysregulation as found by RNA sequencing at a statistically significant level (p < 0.0001 and p < 0.05) (Appendix Fig. A.5A). In the cell adhesion pathway, two genes, JAM2, NEO1, showed statistically significant increased transcription levels compared to controls (Appendix Fig. A.5B). Two additional genes, VCAM1 and PTPRC, showed undetectable transcript levels in control tissue samples but had detectable transcript levels in FLNC cardiac samples (Appendix Fig. A.5B).
4. Discussion
We defined transcriptomic profiles in 7 LV samples from patients with ACM and 4 controls and identified DEGs in ACM samples associated with pathogenic variants in the FLNC gene. The notable findings were dysregulated pathways pertaining to cell adhesion, extracellular matrix protein, and inflammation. The findings were replicated across multiple bioinformatics platforms and were partially validated by qPCR of selected target genes pertaining to the dysregulated pathways. Overall, the most significantly enriched pathways were: the focal adhesion pathway, the protein digestion and absorption pathway, and ECM receptor interaction pathway.
The above three KEGG pathways are intrinsically linked and there is a great degree of overlap with a large number of genes present in all three. For example, the protein digestion and absorption pathway and ECM receptor interaction pathway contain genes that overlap with those in the focal adhesion pathway but the former did not contain FLNC. In addition, ECM organization involves proteolysis which links this pathway to the protein digestion and absorption pathway. Similarly, the ECM receptor interaction pathway is also related to the focal adhesion pathway, as focal adhesions form a connection between the actin cytoskeleton of the cell and the extracellular matrix [14]. Given that the protein digestion and absorption pathways are not cardiac specific and the ECM receptor interaction pathway did not directly involve FLNC, these two pathways were not investigated further.
The IPA software package was used to confirm the KEGG pathway analysis performed by WebGestalt and identified the integrin linked kinase (ILK) canonical pathway and the hepatic fibrosis canonical pathway as most statistically significant. The ILK signalling pathway is involved in the regulation of cell adhesion, migration, differentiation and apoptotic cell death [15] and substantially overlaps with the KEGG focal adhesion pathway. In particular, the ILK pathway contains both FLNC and six other genes which are also present in the KEGG focal adhesion pathway: PARVB, ITGB8, JUN, MAPK10, MYL7 and VCL. The dysregulated hepatic fibrosis canonical pathway suggests involvement of genes that are likely to be commonly involved in the induction of fibrosis in both cardiac and liver tissue. The hepatic fibrosis pathway, however, did not relate to GO terms identified by WebGestalt and g:GOSt and, therefore, was not further investigated.
Analysis of the dysregulated genes in the focal adhesion pathway showed a possible link to the regulation of the actin cytoskeleton. Focal adhesions form between the actin cytoskeleton and extracellular matrix and have the ability to act as signalling transduction platforms [16]. Actin filaments are connected to transmembrane receptors of the integrin family via junctional plaque proteins. In addition, there are a number of proteins present in the focal adhesion pathway, for example paxillin, filamin and vinculin, which interact with the tails of the integrin subunits either directly or indirectly. These interactions have both a structural component, linking them to the actin cytoskeleton, and a role in signal transduction [17].
Comparison of the ILK pathway to the focal adhesion pathway reveals important similarities including the presence of FLNC. ILK has been located in focal adhesions and fibrillar adhesions [18] and is known to interact with multiple focal adhesion proteins, including PINCH, alpha-parvin and paxillin [18]. Additionally the proteins in the ILK pathway are involved in cell adhesion, cytoskeleton organization and actin reorganization similarly to those in the focal adhesion pathway (Appendix Fig. A.2 and Fig. 4).
Previous studies have identified the KEGG focal adhesion and ILK canonical pathways as vital for cardiac function [19,20]. It is important to highlight that the dysregulated pathways in ACM LV samples, annotated differently in different bioinformatics platforms, all converge to highlight dysregulated cell adhesion, involved in actin cytoskeleton and ECM organization, as a mechanism in the pathogenesis of ACM associated with the FLNC variants. The mechanism seems to differ from those involved in the pathogenesis of ACM caused by mutations in genes coding for desmosome proteins.
The focal adhesion pathway also included GSK3b and CTNNB1 genes, which are members of the canonical WNT signalling pathway. However, transcript levels of these two genes were unchanged in ACM heart samples associated with FLNC variants as compared to controls. This is consistent with previous immunohistochemistry analysis which detected no apparent reduction of the immunoreactive signal for GSK3b in LV specimens from the same ACM cases [9]. Activation of the ILK/focal adhesion pathways is implicated in activation of β-catenin which, in turn, binds to the Wnt1 promoter region to upregulate gene transcription leading to positive feedback of the WNT/β-catenin pathway [21]. Transcript levels of the canonical WNT pathway target genes were upregulated in the ACM hearts. However, whether the upregulation was a consequence of activation of the ILK pathway or independent of it, or secondary to advanced heart failure, remains unclear. Overall, the finding suggests involvement of multiple pathways, particularly those involved in cell adhesion in the pathogenesis of ACM caused by FLNC variants.
It is also worth noting that the Alpha-parvin/ILK/Pinch1 (PARVA/ILK/LIMS1) complex, which is involved in linking focal adhesions to the cytoskeleton, has been previously associated with dilated cardiomyopathy DCM [22]. Although our dataset shows no dysregulation of this complex, we did detect a downregulation of Parvin-beta (PARVB), a paralog of PARVA, in cardiac samples with FLNC variants compared to control samples. Parvin-beta has been found to localise to focal adhesions as a cytoplasmic adaptor protein playing part in the connection of integrin receptors to the cytoplasm [23]. Parvin-beta could, therefore, be having a similar effect on focal adhesion formation as alpha-parvin, ultimately affecting the structure of the cytoskeleton, and thereby contributing to this unique LV ACM/DCM cross-over phenotype.
Interestingly, a recent study reported an association of novel missense variants in ILK with arrhythmogenic cardiomyopathy [24]. In that study, Brodelh et al. presented evidence that in vitro loss of focal adhesion location in mutant cells may be due to disruption of the ILK-PINCH complex. These findings are in line with data presented in this study that suggest a dysregulation of the ILK pathway in ACM cardiac tissue.
Given the limited quantities of extracted RNA we were able to validate by qPCR changes in transcript levels of a small number of selected DEGs in the ILK and cell adhesion pathways including actin-associated genes. We also confirmed suppression of FLNC in LV samples carrying variants in this gene. This appears to be consistent with recent findings that showed reduced immunoreactive signal for filamin C in the same tissue samples [9]. This data strengthens earlier observations that ACM caused by FLNC variants is associated with a predominant left ventricle disease pattern [8,9].
4.1. Limitations
This study has a number of limitations including the material source being paraffin fixed tissue and the low yield of intact RNA in such tissues compared to fresh or frozen cardiac tissues, which hampered extensive confirmation of the RNA-seq and bioinformatics findings. The relatively small quantity of extracted RNA limited qPCR confirmation experiments to 5 and 4 genes from the focal adhesion and adhesion pathways, respectively. In addition, the small amount of cardiac tissue available precluded the validation of the results at the protein level by Western blotting analysis. The study was also limited by the small number of samples available, in part because of the challenges in collecting human heart samples and in part because of genetic heterogeneity of human ACM and uncommon nature of the FLNC variants.
Given that the heart samples were patients with expressed phenotypes including heart failure, it is difficult to discern the primary changes resulting directly from FLNC variants from those secondary to cardiac dysfunction, heart failure, and other cardiac pathologies.
5. Conclusion
This is the first study that reports on transcriptomic profiles of cardiac tissue samples from patients with ACM associated with FLNC variants. The findings were notable for dysregulated ILK and the focal adhesion pathways, potentially affecting the actin cytoskeleton organization and leading to impaired function and dysregulated related signalling pathways. The findings implicate disruption to these pathways as putative disease mechanisms in ACM caused by the FLNC variants. Our data suggests the presence of different pathogenic mechanisms between ARVC caused by mutant desmosome proteins, which involves the canonical WNT signalling pathway, and ACM caused by FLNC variants, which may involve disruption of cell adhesion pathways. As this is the first study reporting on the transcriptomic profile of FLNC-associated ACM, these findings need to be confirmed by future studies which will focus on the interactions between mutant filamin C and specific components of the identified pathways.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was funded by Fondation Leducq Transatlantic Networks of Excellence Program grant (no 14CVD03) (A.J.M. and W.J.M.) and the NIHR University College London Hospitals Biomedical Research Centre, UK. Support was provided by grants from National Institutes of Health, National Heart, Lung and Blood Institute (NHLBI, R01HL088498 and 1R01HL132401); the Ewing Halsell Foundation; George and Mary Josephine Hamman Foundation; and TexGen Fund from Greater Houston Community Foundation (A.J.M). In Spain, the study was funded by Plan Estatal de I+D+I 2013-2016 - European Regional Development Fund (FEDER) “A way of making Europe”, Instituto de Salud Carlos III, Spain [PI14/01477, PI18/01582 and La Fe Biobank PT17/0015/0043 (E.Z.) and PI14/01676, PI18/01231 and the BioBank “Biobanco en Red de la Región de Murcia” (PT17/0015/0038) (J.R.G)].
Abbreviations:
- ACM
arrhythmogenic cardiomyopathy
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- DEGs
differentially expressed genes
- DCM
dilated cardiomyopathy
- ECM
extracellular matrix
- FLNC
filamin C
- GO
gene ontology
- GSEA
gene set enrichment analysis
- ILK
integrin linked kinase
- IPA
ingenuity pathway analysis
- KEGG
Kyoto encyclopedia of genes and genomes
- LV
left ventricle
- MTORC1
mechanistic target of rapamycin complex 1
- SCD
sudden cardiac death
Footnotes
Declaration of competing interest
None.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijcard.2019.12.002.
References
- [1].Sen-Chowdhry S, Morgan RD, Chambers JC, Mckenna WJ, Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment, Annu. Rev. Med 61 (2010) 233–253. [DOI] [PubMed] [Google Scholar]
- [2].Corrado D, Basso C, Pilichou K, Thiene G, Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia, Heart. 97 (2011) 530–539. [DOI] [PubMed] [Google Scholar]
- [3].Sen-Chowdhry S, Syrris P, Mckenna WJ, Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy, J Am Coll Cardiol. 50 (2007) 1813–1821. [DOI] [PubMed] [Google Scholar]
- [4].Que D, Yang P, Song X, Liu L, Traditional vs. genetic pathogenesis of arrhythmogenic right ventricular cardiomyopathy, Europace. 17 (2015) 1770–1776. [DOI] [PubMed] [Google Scholar]
- [5].Valdes-Mas R, Gutierrez-Fernandez A, Gomez J, Coto E, Astudillo A, Puente DA, et al. , Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy, Nat. Commun 5 (2014) 5326. [DOI] [PubMed] [Google Scholar]
- [6].Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, et al. , Mutations in FLNC are associated with familial restrictive cardiomyopathy, Hum. Mutat 37 (2016) 269–279. [DOI] [PubMed] [Google Scholar]
- [7].Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, et al. , FLNC gene splice mutations cause dilated cardiomyopathy, JACC: Basic to Translational Science. 1 (2016) 344–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, et al. , Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures, JACC Clin Electrophysiol. 4 (2018) 504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hall CL, Akhtar MM, Sabater-Molina M, Futema M, Asimaki A, Protonotarios A, et al. , Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype, Int J Cardiol. (2019) 10.1016/j.ijcard.2019.09.048. [DOI] [PubMed] [Google Scholar]
- [10].Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. , Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet Med. 17 (2015) 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sheng Q, Vickers K, Zhao S, Wang J, Samuels DC, Koues O, et al. , Multiperspective quality control of Illumina RNA sequencing data analysis, Brief Funct Genomics. 16 (2017) 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fürst DO, Goldfarb LG, Kley RA, Vorgerd M, Olivé M, Van PF Der Ven Filamin, C-related myopathies: pathology and mechanisms, Acta Neuropathol. 125 (2013) 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kanehisa M, Goto S, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Res. 28 (2000) 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Burridge K, Focal adhesions: a personal perspective on a half century of progress, FEBS J. 284 (2017) 3355–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saito A, Hayashi T, Okuno S, Nishi T, Chan PH, Oxidative stress affects the integrin-linked kinase signaling pathway after transient focal cerebral ischemia, Stroke. 35 (2004) 2560–2565. [DOI] [PubMed] [Google Scholar]
- [16].Liu B, Xu L, Yu X, Jiao X, Yan J, Li W, et al. , Genistein inhibited estradiol-induced vascular endothelial cell injury by downregulating the FAK/focal adhesion pathway, Cell. Physiol. Biochem 49 (2018) 2277–2292. [DOI] [PubMed] [Google Scholar]
- [17].Turner Paxillin CE, Focal adhesion signalling, Nat. Cell Biol 2 (2000) E231–E236. [DOI] [PubMed] [Google Scholar]
- [18].Wu C, Dedhar S, Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes, J. Cell Biol 155 (2001) 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hannigan GE, Coles JG, Dedhar S, Integrin-linked kinase at the heart of cardiac contractility, repair, and disease, Circ. Res 100 (2007) 1408–1414. [DOI] [PubMed] [Google Scholar]
- [20].Hirth S, Buhler A, Buhrdel JB, Rudeck S, Dahme T, Rottbauer W, et al. , Paxillin and focal adhesion kinase (FAK) regulate cardiac contractility in the zebrafish heart, PLoS One 11 (2016)(e0150323). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Du J, Zu Y, Li J, Du S, Xu Y, Zhang L, et al. , Extracellular matrix stiffness dictates Wnt expression through integrin pathway, Sci. Rep 6 (2016)(20395). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sopko N, Qin Y, Finan A, Dadabayev A, Chigurupati S, Qin J, et al. , Significance of thymosin beta4 and implication of PINCH-1-ILK-alpha-parvin (PIP) complex in human dilated cardiomyopathy, PLoS One 6 (2011) e20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stiegler AL, Draheim KM, Li X, Chayen NE, Calderwood DA, Boggon TJ, Structural basis for paxillin binding and focal adhesion targeting of beta-parvin, J. Biol. Chem 287 (2012) 32566–32577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brodehl A, Rezazadeh S, Williams T, Munsie NM, Liedtke D, Oh T, et al. , Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy, Transl. Res 208 (2019) 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang J, Vasaikar S, Shi Z, Greer M, Zhang B, WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit, Nucleic Acids Res. 45 (2017) W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.