

Abstract
A structure-guided design strategy was used to improve the resistance profile of HIV-1 protease inhibitors by optimizing hydrogen bonding and van der Waals interactions with the protease while staying within the substrate envelope. Stereoisomers of 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties were explored as P2′ ligands providing pairs of diastereoisomers epimeric at P2′, which exhibited distinct potency profiles depending on the configuration of the hydroxyl group and size of the P1′ group. While compounds with the 4-(1-hydroxyethyl)benzene P2′ moiety maintained excellent antiviral potency against a panel of multidrug-resistant HIV-1 strains, analogues with the polar 4-(1,2-dihydroxyethyl)benzene moiety were less potent, and only the (R)-epimer incorporating a larger 2-ethylbutyl P1′ group showed improved potency. Crystal structures of protease-inhibitor complexes revealed strong hydrogen bonding interactions of both (R)- and (S-stereoisomers of the hydroxyethyl group with Asp30′. Notably, the (R)-dihydroxyethyl group was involved in a unique pattern of direct hydrogen bonding interactions with the backbone amides of Asp29′ and Asp30′. The SAR data and analysis of crystal structures provide insights for optimizing these promising HIV-1 protease inhibitors.
INTRODUCTION
The HIV-1 protease is a major target for developing antiviral therapies against HIV-1. Drug discovery efforts, aided by structure-based drug design, have led to the development of nine FDA-approved HIV-1 protease inhibitors (PIs).1 All approved HIV-1 PIs are competitive inhibitors, and most contain different dipeptide isosteres as transition state mimetics.2 In addition to extensive hydrophobic interactions, PIs mainly rely on a number of direct and water-mediated hydrogen bonds with the protease for potency. The clinical efficacy of HIV-1 PIs has significantly improved since this drug class was first introduced in the mid-1990s. The development of second-generation PIs with improved potency, tolerability, and pharmacokinetic profiles, and the introduction of low-dose ritonavir as a pharmacokinetic booster in PI-based antiretroviral therapy (ART) has led to improved clinical outcomes. Moreover, PIs allow exceedingly high level of viral inhibition at clinical concentrations due to cooperative dose-response curves with high slopes.3,4 These transition-state mimetic inhibitors represent the most potent anti-HIV-1 drugs.
Despite much success, PI-based therapies are associated with drawbacks that limit their effectiveness, including major side effects, unfavorable pharmacokinetics, and the acquisition of many viable multidrug-resistant (MDR) protease variants.5,6 Drug resistance remains a major issue, as the prevalence of PI resistance mutations increases with duration of ART and is much higher among PI-experienced patients.7,8 Even the most effective PI-based regimens have been reported to select multiple resistance mutations in the protease, resulting in reduced virologic response.7-10 Moreover, the prevalence of transmitted drug resistance continues to increase, which is an added challenge in the treatment of HIV infections.11,12 The emergence of MDR protease variants presents a challenge to structure-based drug design (SBDD), as the target is not a single protein but an ensemble of closely related proteases. The new PIs must maintain activity not only against existing MDR protease variants, but also against future variants that may emerge. These challenges require developing new SBDD strategies to optimize PI potency while avoiding resistance.
The design strategy to maximize interactions in the HIV-1 protease active site, particularly with the protein backbone atoms, has been quite successful leading to the development of the FDA-approved drug darunavir.13,14 Darunavir (DRV, 1) is a highly potent inhibitor of HIV-1 protease with a low pM inhibition constant (Ki) and a high genetic barrier to resistance. The key structural feature that distinguishes DRV from previous generation HIV-1 PIs is the bis-tetrahydrofuran (bis-THF) moiety at the P2 position (Figure 1) that makes strong hydrogen bonding interactions with the backbone NH of Asp29 and Asp30 in the protease active site.1,13,15 The application of the design strategy to maximize interactions with the backbone atoms of HIV-1 protease has recently led to the discovery of DRV analogues that maintain exceptional potency against MDR HIV-1 variants including DRV-resistant strains.16-19
Figure 1.

Structures of HIV protease inhibitors DRV (1), P2′ 4-(1-hydroxymethyl)benzene analogues (2–4) and designed compounds with stereochemically defined P2′ 4-(1-hydroxyethyl)benzene and (12–17) and 4-(1,2-dihydroxyethyl)benzene (22–27) moieties. The canonical nomenclature for inhibitor moiety position is indicated using DRV.
Among the strategies proposed to rationally design PIs against drug-resistant HIV-1 variants, the substrate envelope model provides a more comprehensive framework to incorporate drug resistance considerations into SBDD.20-22 This structure-guided design strategy aims to optimize hydrogen bonding and van der Waals (vdW) interactions with the protease to improve potency against MDR protease variants, while constraining inhibitors within the substrate envelope to avoid resistance.23 We previously used the substrate envelope-guided design strategy to develop a series of highly potent DRV analogues that maintained excellent antiviral potencies against a panel of clinically relevant MDR HIV-1 strains.23 The X-ray cocrystal structures confirmed that the designed PIs optimally fill the substrate envelope and make enhanced vdW contacts and hydrogen bonding interactions with HIV-1 protease.23,24
Among these PIs, compounds 3 and 4 (Figure 1) with the 4-(hydroxymethyl)benzene P2′ moiety were identified as promising lead compounds due to distinct polar interactions of the benzylic hydroxyl group with the protease. Cocrystal structures of PIs 3 and 4 bound to wild-type HIV-1 protease revealed that the P2′ benzylic hydroxyl group is positioned close to the backbone NH of Asp29′ and Asp30′ in the S2′ subsite. The hydroxyl group makes a direct hydrogen bond only with the backbone NH of Asp30′, which is slightly closer than the NH of Asp29′ (Figure S1).23,24 We reasoned that introduction of a small hydrophobic group such as a methyl at the benzylic position could shift the resulting secondary hydroxyl group closer to the backbone NH of Asp29′, allowing polar interactions with both residues, as well as additional vdW contacts in the S2′ subsite. Similarly, a hydroxymethyl group at the benzylic position could serve the same purpose, with the primary hydroxyl group making additional polar interactions in the S2′ subsite of HIV-1 protease (Figure 1). Optimal polar interactions of the P2′ moiety with the backbone NH of Asp29′ and Asp30′ could potentially mimic the strong hydrogen bonding interaction of the P2 bis-THF moiety, further improving potency against MDR HIV-1 variants. Previous efforts to identify an optimal P2′ moiety have only been partially successful, and none of the reported P2′ moieties make direct hydrogen bonding interactions with the backbone atoms of both Asp29′ and Asp30′.16,25-27
On the basis of insights from the structural analysis and modeling, modifications of the P2′ moiety of PIs 3 and 4, in combination with variations at the P1′ group, were explored to enhance inhibitor interactions in the S1′ and S2′ subsites. Here, we describe the substrate envelope-guided design, synthesis, evaluation of biochemical and antiviral potency, and crystal structure analysis of a series of novel HIV-1 PIs. The inhibitors were designed by incorporating stereoisomers of the 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties as novel P2′ ligands to enhance hydrogen bonding interactions in the S2′ subsite of HIV-1 protease. Structure-activity relationship (SAR) studies identified a number of compounds with improved potency profiles compared to DRV and the parent compounds against highly drug-resistant HIV-1 strains representing the spectrum of clinically relevant MDR viruses. We also report high-resolution crystal structures of all new compounds bound to wild-type HIV-1 protease revealing key interactions of the chiral 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties in the protease active site. A unique pattern of hydrogen bonding interactions was observed for compounds incorporating the (R)-4-(1,2-dihydroxyethyl)benzene as P2′ ligand, where both P2 and P2′ moieties make direct hydrogen bonds with the backbone NH of residues Asp29/29′ and Asp30/30′.
CHEMISTRY
The synthesis of HIV-1 PIs with the (S)- and (R)-(1-hydroxyethyl)benzene moieties at the P2′ position is outlined in Scheme 1. We envisioned generating both stereoisomers of the hydroxyethyl group at the P2′ moiety from the corresponding 4-acetylbenzene derivatives by enantioselective reduction of the acetyl group using the Corey–Bakshi–Shibata (CBS) catalyst.28 The 4-acetylbenzenesulfonamide intermediates 8a–c were prepared from the commercially available epoxide 5 in two steps. Ring opening of the chiral epoxide 5 with selected amines provided the amino alcohols 6a–c, which were reacted with 4-acetylbenzenesulfonyl chloride 7 using Na2CO3 as a base under biphasic conditions to afford the 4-acetylbenzenesulfonamide intermediates 8a–c in excellent yield. Reduction of the acetyl group using BH3-THF in the presence of the chiral catalyst (R)-CBS-Me provided the required intermediates with the (S)-(1-hydroxyethyl)benzene moiety 9a–c with excellent enantioselectivity. After purification by flash chromatography, the products were recrystallized from a mixture of EtOAc and hexanes to ensure chiral purity. Removal of the Boc protecting group with TFA and reaction of the resulting amine salts with the bis-THF activated carbonate 11 provided the target compounds 12, 14 and 16 with the (S)-4-(1-hydroxyethyl)benzene moiety at the P2′ position. Inhibitors with the corresponding (R)-4-(1-hydroxyethyl)benzene at the P2′ position were prepared similarly using the (S)-CBS-Me as the catalyst during the stereoselective reduction step and provided the desired inhibitors 13, 15 and 17.
Scheme 1.
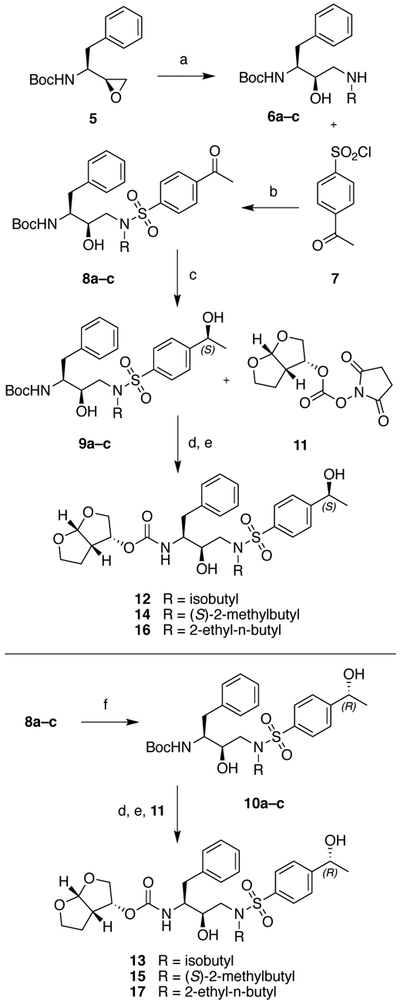
Synthesis of protease inhibitors incorporating (S)- and (R)-4-(1-hydroxyethyl)benzene as P2′ ligands
Reagents and conditions: (a) RNH2, EtOH, 70 °C, 3 h, 79–91%; (b) Na2CO3, EtOAc, H2O, RT, 18 h, 98–100%; (c) R-CBS-Me, BH3-THF (1 M), THF, 0 °C to RT, 3 h, 57–85%; (d) TFA, CH2Cl2, RT, 1 h; (e) DIEA, CH3CN, RT, 24 h, 73–91%; (f) S-CBS-Me, BH3-THF (1 M), THF, 0 °C to RT, 3 h, 47–87%.
The protease inhibitors with the (R)- and (S)-4-(1,2-dihydroxyethyl)benzene moieties at the P2′ position were prepared from the corresponding styrene derivatives by Sharpless asymmetric dihydroxylation using the AD-mix catalyst29,30 as outlined in Scheme 2. Briefly, reactions of amino alcohols 6a–c with 4-vinylbenzenesulfonyl chloride 18 using Na2CO3 as a base under biphasic conditions provided the 4-vinylbenzenesulfonamide intermediates 19a–c. The asymmetric dihydroxylation reaction using AD-mix-β as a chiral catalyst proceeded smoothly and provided the required intermediates with the (R)-4-(1,2-dihydroxyethyl)benzene moiety 20a–c. In all cases the (S)-epimer was not detected by 1H NMR; however, the products were recrystallized to provide enantiomerically pure intermediates 20a–c. Analogous to the compound series with the 4-(1-hydroxyethyl)benzene P2′ moiety, Boc deprotection and the reaction of the resulting amine salts with the bis-THF carbonate 11 provided the target compounds 22, 24 and 26 with the (R)-4-(1,2-dihydroxyethyl)benzene moiety at the P2′ position. Inhibitors with the corresponding (S)-4-(1,2-dihydroxyethyl)benzene moiety at the P2′ position were prepared in a similar fashion using the AD-mix-α as the chiral catalyst in the asymmetric dihydroxylation reaction and provided the target inhibitors 23, 25 and 27.
Scheme 2.

Synthesis of protease inhibitors incorporating (S)- and (R)-4-(1,2-dihydroxyethyl)benzene as P2′ ligands
Reagents and conditions: (a) Na2CO3, EtOAc, H2O, RT, 18 h, 75–94%; (b) AD-mix-β, t-BuOH, H2O, RT, 4 h, 68–87%; (c) TFA, CH2Cl2, RT, 1 h; (d) DIEA, CH3CN, RT, 24 h, 33–79%; (e) AD-mix-α, t-BuOH, H2O, RT, 4 h, 47–86%.
RESULTS AND DISCUSSION
Our goal was to improve potency of PIs against MDR HIV-1 variants by optimizing hydrogen bonding interactions in the S2′ subsite of HIV-1 protease. Specifically, we explored the possibility of additional hydrogen bonding interactions between the hydroxyl group of the P2′ moiety and the backbone amide NH of Asp29′ and Asp30′ on the basis of an analysis of 3- and 4-bound HIV-1 protease structures and molecular modeling. Addition of a methyl group to the hydroxymethyl substituent was expected to shift the hydroxyl group closer to the backbone NH of Asp29′, while increasing vdW interactions with residues in the S2′ subsite. Similarly, replacement of the hydroxymethyl group with a more polar 1,2-dihydroxyethyl was intended to enhance polar interactions; particularly the second hydroxyl group was expected to replace the water-mediated interactions between the P2′ moiety and the Asp30′ side chain. However, due to the free rotation of the hydroxyethyl and dihydroxyethyl groups, modeling did not provide clear distinction which of the two stereoisomers would provide optimal polar and vdW interactions. Thus, both (R)- and (S)-stereoisomers of 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties were explored as P2′ ligands in combination with three P1′ groups of varying size and hydrophobicity.
Enzyme inhibition assays.
The binding affinity of HIV-1 PIs has reached a level where assessing the inhibition of wild-type protease using standard FRET-based assays is quite difficult. Accurate measurement of inhibition constants in the low pM range remains a challenge even with the recently reported highly sensitive fluorogenic assay.31 Due to these limitations, two drug-resistant protease variants, I84V and I50V/A71V were selected to assess the potency of new PIs. I84V is a common drug resistance mutation that reduces susceptibility to all FDA-approved PIs, while I50V/A71V mutations arise in response to therapy with APV and DRV-based regimens, resulting in reduced susceptibility to these PIs. The enzyme inhibition constants (Ki) were determined against the I84V and I50V/A71V protease variants using the highly sensitive FRET assay31; DRV was used as a control in all assays (Table 1).
Table 1.
Protease inhibitory activity of PIs 12–17 and 22–27 against drug-resistant variants
| Inhibitor | Structure | Ki (nM) | |
|---|---|---|---|
| I84V | I50V/A71V | ||
| 12 |  |
0.048 ± 0.004 | 0.057 ± 0.008 |
| 13 |  |
0.109 ± 0.007 | 0.092 ± 0.008 |
| 14 |  |
0.111 ± 0.008 | 0.093 ± 0.013 |
| 15 |  |
0.095 ± 0.005 | 0.132 ± 0.014 |
| 16 |  |
0.020 ± 0.002 | 0.102 ± 0.010 |
| 17 |  |
0.028 ± 0.003 | 0.107 ± 0.014 |
| 22 |  |
0.098 ± 0.008 | 0.090 ± 0.008 |
| 23 |  |
0.283 ± 0.024 | NT |
| 24 |  |
0.216 ± 0.011 | NT |
| 25 |  |
0.504 ± 0.039 | NT |
| 26 |  |
0.057 ± 0.004 | 0.074 ± 0.006 |
| 27 |  |
0.134 ± 0.007 | 0.220 ± 0.011 |
| 3 |  |
0.010 ± 0.003 | 0.080 ± 0.006 |
| 4 |  |
0.005 ± 0.002 | 0.055 ± 0.004 |
| DRV | 0.025 ± 0.006 | 0.075 ± 0.006 | |
NT: not tested
DRV retained excellent potency against the I84V and I50V/A71V protease variants (Ki = 25 pM and 75 pM, respectively) compared to that reported for wild-type protease (Ki = 5–16 pM).13,23,31 Compound 3, which incorporates a 4-(hydroxymethyl)benzene moiety as P2′ ligand and (S)-2-methylbutyl group at the P1′ position, showed better potency against the I84V protease but was equipotent to DRV against the I50V/A71V variant. Analogue 4 with a larger 2-ethylbutyl group at the P1′ position was more active than DRV against both the I84V and I50V/A71V protease variants. The corresponding analogue 2 with the isobutyl P1′ group was previously reported which was equipotent to DRV against wild-type protease but exhibited relatively lower antiviral potency, presumably due to reduced hydrophobicity.13,32 Thus, as we have previously shown, PIs with 4-(hydroxymethyl)benzene as the P2′ moiety in combination with larger, more hydrophobic (S)-2-methylbutyl and 2-ethylbutyl P1′ groups maintained potency against drug-resistant protease variants.
Replacement of the 4-(hydroxymethyl)benzene P2′ moiety of PIs 2–4 with (S)- and (R)-4-(1-hydroxyethyl)benzene resulted in pairs of diastereoisomers epimeric at P2′. The P2′ hydroxyethyl analogues 12–17 showed excellent inhibitory potencies against the I84V and I50V/A71V protease variants with Ki values ranging between 20 to 132 pM. In each case, both diastereoisomers were relatively less potent than the corresponding parent hydroxymethyl compound, particularly against the I84V protease variant. However, no major difference in inhibitory potency was observed between P2′ epimeric compounds. Only compounds with the isobutyl P1′ group showed minor differences in inhibitory potencies between the P2′ epimers, as diastereoisomer 12 with (S)-configuration of the hydroxyethyl group was slightly more potent than the corresponding (R)-epimer 13. While this work was underway, Ghosh et al. reported the synthesis of PIs 12 and 13 using a much longer reaction sequence; both compounds were tested against wild-type HIV-1 protease, and only compound 13 was evaluated for antiviral activity.33
Diastereoisomers 14 and 15 with the (S)-2-methylbutyl P1′ group showed similar potencies against the I84V and I50V/A71V protease variants, and both were less potent than the parent 3. Similarly, no difference in inhibitory potency was observed between diastereoisomers 16 and 17 incorporating the 2-ethylbutyl P1′ group. However, analogues 16 and 17 were more potent against the I84V protease (Ki = 20 and 28 pM, respectively) and exhibited overall potency profiles comparable to that of DRV. Together, these data demonstrate that minor modifications to the P1′ and P2′ moieties result in distinct potency profiles.
In contrast to the 4-(1-hydroxyethyl)benzene P2′ series, compounds with the 4-(1,2-dihydroxyethyl)benzene P2′ moiety showed clear differences in inhibitory potency between the P2′-epimers. Compounds incorporating the (R)-4-(1,2-dihydroxyethyl)benzene P2′ moiety were 2–3-fold more potent than the corresponding (S)-epimers. Diastereoisomer 22, with the isobutyl group at the P1′ position and (R)-configuration of the dihydroxyethyl group, exhibited excellent inhibitory potency against the I84V and I50V/A71V proteases, while the corresponding (R)-epimer 23 was about 3-fold less active against the I84V protease. A similar trend was observed for diastereoisomers 24 and 25 incorporating the (S)-2-methylbutyl P1′ group, though both compounds were 2-fold less active against the I84V protease than the corresponding isobutyl P1′ analogues 23 and 24. The analogue 26 incorporating the larger 2-ethylbutyl P1′ group and (R)-configuration of the dihydroxyethyl group was the most potent in this series, with enzyme potency against the I50V/A71V protease variant comparable to that of DRV. Again, the corresponding P2′ (S)-epimer 27 was 2–3-fold less potent against the protease variants tested. Thus, shape of the hydrophobic group at P1′ and the configuration of the dihydroxyethyl group at P2′ affected inhibitor potency against drug-resistant protease variants.
Antiviral assays.
The potency and resistance profiles of PIs were evaluated using a cell-based antiviral assay against wild-type HIV-1 and three representative MDR variants selected from a panel of clinically relevant multi-PI resistant recombinant clones (Table 2, Table S1).34 Relative to the wild-type HIV-1 strain NL4-3 (with a near consensus protease sequence), the MDR variants contain 19, 20 and 24 amino acid substitutions in the protease and are named SLK19, VSL20, and KY24, respectively. All three variants show high-level resistance and cross-resistance to multiple PIs including DRV, as determined by the Monogram Biosciences (South San Francisco, CA) PhenoSense assay.34 Accordingly, in antiviral assays, the MDR variants exhibited increasingly high-level resistance to DRV: compared to wild-type HIV-1, DRV was 6-fold less potent against SLK19, 58-fold less potent against VSL20, and 210-fold less potent against KY24. Thus, the three selected MDR HIV-1 strains represent clinically relevant MDR viruses with a wide spectrum of DRV resistance.
Table 2.
Antiviral potency of PIs 12–17 and 22–27 against WT HIV-1 and drug-resistant variants
| Inhibitor | cLogPa | Antiviral EC50 (nM) (fold change)b | |||
|---|---|---|---|---|---|
| WT | SLK19 | VSL20 | KY24 | ||
| 12 | 2.885 | 4.7 | 31 (7) | 117 (25) | 352 (75) |
| 13 | 2.885 | 7.5 | 100 (13) | 148 (20) | 360 (48) |
| 14 | 3.414 | 4.6 | 49 (11) | 281 (61) | 490 (107) |
| 15 | 3.414 | 4.2 | 91 (22) | 291 (69) | 342 (81) |
| 16 | 3.943 | 4.4 | 26 (6) | 112 (25) | 196 (45) |
| 17 | 3.943 | 5.1 | 32 (6) | 420 (82) | 345 (68) |
| 22 | 1.673 | 176 | NT | NT | NT |
| 23 | 1.673 | 189 | NT | NT | NT |
| 24 | 2.202 | 80 | NT | NT | NT |
| 25 | 2.202 | 92 | NT | NT | NT |
| 26 | 2.731 | 40 | 116 (3) | 100 (3) | 507 (13) |
| 27 | 2.731 | 41 | 151 (4) | 417 (10) | 1154 (28) |
| 3 | 3.105 | 5.7 | 52 (9) | 310 (54) | 1087 (191) |
| 4 | 3.634 | 4.2 | 36 (9) | 426 (101) | 495 (118) |
| DRV | 2.387 | 5.5 | 33 (6) | 320 (58) | 1157 (210) |
cLogP values were calculated using ChemDraw 18.
The multidrug-resistant HIV-1variants SLK19, VSL20 and KY24 contain the following amino acid substitutions in HIV-1 protease compared to the WT NL4-3 strain: SLK19 (L10I, V11I, I13V, K14R, A22V, E35D, M36I, N37D, G48M, F53L, I54V, I62V, L63P, A71V, T74S, V82A, I84V, T91S, Q92K); VSL20 (L10I, K20R, L33F, E34A, E35D, M36I, N37D, K43T, M46I, G48V, I50V, I54T, I62V, L63P, A71V, I72V, V77I, V82A, I85V, I93L); KY24 (L10V, T12V, I13V, I15V, K20M, V32I, L33F, K43T, M46I, I47V, I54M, D60E, Q61N, I62V, L63P, C67Y, H69K, A71I, I72L, G73S, V77I, V82A, L89V, L90M). NT: not tested.
The 4-(hydroxymethyl)benzene P2′ analogues 3 and 4 were equipotent to DRV against wild-type HIV-1 with EC50 values of 5.7 and 4.2 nM, respectively. The two analogues retained potent activity against the SLK19 variant but were significantly less potent against the VSL20 variant, with EC50 values similar to that of DRV. Compared to DRV and compound 3, both of which showed similar potencies against the three MDR variants, analogue 4 retained 2-fold better potency against the KY24 variant. The slightly improved resistance profile of compound 4 is in agreement with previous results and indicates that optimizing vdW interactions in the S1′ pocket can improve potency against MDR viruses.23
All compounds incorporating the 4-(1-hydroxyethyl)benzene P2′ moieties (12–17) potently inhibited wild-type HIV-1 with EC50 values similar to that of DRV and parent compounds 3 and 4. However, the three MDR HIV-1 variants showed distinct susceptibilities to each of these compounds, depending on the P1′ group and the configuration of the hydroxyethyl group at the P2′ moiety. Compound 12, with the isobutyl P1′ group and the (S)-4-(1-hydroxyethyl)benzene P2′ moiety, was equipotent to DRV against SLK19 but exhibited 2–3-fold better potency against the highly resistant MDR variants VSL20 and KY24, resulting in an overall improved resistance profile. The corresponding epimer 13, incorporating the (R)-4-(1-hydroxyethyl)benzene P2′ moiety, while showing a 3-fold lower potency against SLK19, retained similar potency as 12 against VSL20 and KY24. Diastereoisomers 14 and 15 with the (S)-2-methylbutyl P1′ group exhibited similar potency profiles as the corresponding analogues 12 and 13, except for a 2-fold loss in potency against VSL20. Further improvement in overall potency profile was realized with analogue 16 incorporating a larger 2-ethylbutyl P1′ group and the (S)-4-(1-hydroxyethyl)benzene P2′ moiety, which maintained similar potency against SLK19 as DRV but demonstrated about 3- and 6-fold increase in potency against VSL20 and KY24, respectively. Moreover, compared to the parent compound 4, analogue 16 demonstrated 4- and 2-fold improved potency against VSL20 and KY24. Compared to 16, the (R)-epimer 17 maintained similar potency against SLK19 but exhibited lower potency against VSL20 and KY24. While all compounds with the 4-(1-hydroxyethyl)benzene as P2′ ligands maintained excellent potency against MDR HIV-1 variants, each exhibited distinct resistance profile depending on the configuration of the hydroxyethyl group and size of the P1′ group.
The introduction of a more polar 4-(1,2-dihydroxyethyl)benzene moiety as the P2′ ligand resulted in significantly reduced potency against wild-type HIV-1, despite compounds 22–27 showing pM inhibitory activities in biochemical assays. The observed loss of potency in cellular assays is likely due to reduced hydrophobicity, as indicated by lower calculated partition coefficient (LogP) values compared to DRV (Table 1). The cellular potency of HIV-1 protease inhibitors has been shown to strongly correlate with the hydrophobicity descriptor LogP, suggesting that membrane transport is a key factor affecting antiviral potency.35
The observed antiviral potencies of the 4-(1,2-dihydroxyethyl)benzene containing compounds indeed correlate with their cLogP values (Figure S2). In all cases both diastereoisomers exhibited similar antiviral potencies against wild-type HIV-1. Compounds 22 and 23 incorporating the isobutyl P1′ group showed significantly lower potency than DRV. Replacement of the isobutyl P1′ group with a slightly more hydrophobic (S)-2-methylbutyl group, providing analogues 24 and 25, resulted in further improvement in antiviral potency. Compounds 26 and 27 incorporating a larger 2-ethylbutyl P1′ group further increased hydrophobicity and resulted in a 2-fold improvement in potency compared to the (S)-2-methylbuty P1′ analogues 24 and 25. However, both diastereoisomers 26 and 27 were 8-fold less active than DRV against wild-type HIV-1 despite 26 exhibiting similar potency in enzyme inhibition assays. Compared to DRV the P2′ (R)-epimer 26 maintained better potency against MDR variants VSL20 and KY24 and exhibited much lower fold potency losses. The corresponding (R)-epimer 27, though less active against SLK19, showed similar potency as DRV against VSL20 and KY24. Thus, the combination of a more hydrophobic 2-ethylbutyl P1′ group and the (R)-4-(1,2-dihydroxyethyl)benzene P2′ moiety provided compounds with improved potency profiles.
Analysis of protease-inhibitor complexes.
To explore molecular interactions of the chiral 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene P2′ moieties, we determined crystal structures of PIs 12–17 and 22–27 bound to wild-type HIV-1 protease of the NL4–3 strain. The cocrystal structures of PIs 2–4 were also determined with the same protease enzyme, as the previously reported cocrystal structures of PIs 3 and 4 were with the wild-type protease of SF-2 sequence, which is slightly different from NL4-3. The two variants differ by four amino acids which caused minor structural differences at the distal loops, but the active sites were nearly identical, including inhibitor binding and crystallographic waters. The crystallographic data collection and refinement statistics are summarized in Table S2. All 15 high-resolution (1.86–2.03 Å) cocrystal structures were solved in the same P212121 space group with one protease homodimer in the asymmetric unit, and only one orientation of the bound inhibitor in the protease active site, which was crucial for direct comparison and analysis.
The overall binding conformations of all PIs incorporating the 4-(hydroxymethyl)benzene (2–4), 4-(1-hydroxyethyl)benzene (12–17), and 4-(1,2-dihydroxyethyl)benzene (22–27) P2′ moieties are similar to that of DRV (Figure 2A). However, clear differences in contacts were observed in the binding of the P2′ moieties depending on the substituent at the 4-position of the benzene ring and the configuration of the hydroxyl group (Figure 2B, 2D). Compared to DRV, all PIs with modified P2′ ligands maintain similar hydrogen bonding interactions with the protease, except in the S2′ subsite (Figures 3 and 4). The cocrystal structures of PIs 2–4 in complex with protease showed minor changes in the conformation of the 4-(hydroxymethyl)benzene moiety compared to the 4-aminobenzene of DRV in the S2′ subsite (Figure S3). In the DRV-bound protease structure, the amino group of the 4-aminobenzene P2′ moiety is involved in hydrogen bonding interactions with the main chain carbonyl of Asp30′ and water-mediated interactions with the side-chain carboxylate of Asp30′ (Figure S1). The primary hydroxyl group of the 4-(hydroxymethyl)benzene P2′ moiety in compounds 2–4 is oriented toward Asp29′ and Asp30′ backbone and makes a direct hydrogen bond with the backbone NH of Asp30′. Moreover, the hydroxyl group interacts with the backbone NH of Asp29′ and the side chain of Asp30′ through water-mediated hydrogen bonds. This network of hydrogen bonding interactions in the S2′ subsite likely underlie the improved potency of compounds 2–4 compared to DRV (Figure S1).
Figure 2.

Comparison of binding modes of protease inhibitors with 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene P2′ moieties in the active site of wild-type HIV-1 protease. The two protease monomers are in cyan (denoted as non-prime) and magenta (denoted as prime). Superposition of protease complexes with DRV (1), parent compounds (2–4), and new analogues (12–17 and 22–27). The inhibitors are shown as sticks and HIV protease dimers are shown as ribbons. (B) Zoomed-in active site of superimposed complexes. The inhibitors bind to wild-type HIV protease in similar conformations except minor variations in the S2′ subsite. (C) Fit of inhibitors within the substrate envelope. The substrate envelope is in blue space filling representation, and the superimposed inhibitors are displayed as sticks. There is minimal protrusion of inhibitors outside the active site. (D) Variations in the binding mode of the inhibitors’ P2¢ moieties.
Figure 3.

Crystal structures of wild-type HIV-1 protease in complex with inhibitors (A) 16, (B) 17, (C) 26, and (D) 27. Both (R)- and (R)-stereoisomers of the P2′ 4-(1-hydroxyethyl)benzene moiety make direct hydrogen bonding interactions with the backbone NH of Asp30′ in the S2′ subsite (A and B). The (R)-stereoisomer of the P2′ 4-(1,2-dihydroxyethyl)benzene moiety makes hydrogen bonding interactions with backbone NH of Asp29′ and Asp30′ (C). The (S)-stereoisomer of the P2′ 4-(1,2-dihydroxyethyl)benzene moiety makes hydrogen bonding interactions with the backbone NH and side chain carboxylate group of Asp30′ (D).
Figure 4.

Comparison of binding interactions of representative PIs with modified P2′ moieties in the S2′ subsite of HIV-1 protease. Binding interactions of (A) DRV (PDB 6DGX), (B) parent compound 4 (PDB 6OXQ) with the 4-(hydroxymethyl)benzene P2′ moiety, (C) inhibitor 16 with (S)-4-(1-hydroxyethyl)benzene P2′ moiety, (D) inhibitor 17 with the (R)-configuration of the P2′ moiety, (E) inhibitor 26 with (R)-4-(1,2-dihydroxyethyl)benzene P2′ moiety, and (F) inhibitor 27 with the (S)-configuration of the P2′ moiety.
The PIs 12–17 with the 4-(1-hydroxyethyl)benzene moiety at the P2′ position were designed to better position the hydroxyl group between the backbone NH of Asp29′ and Asp30′. In addition, the methyl group was expected to make vdW interactions with the hydrophobic residues around the S2′ subsite. Similar to the parent compounds, PIs 12–17 fit well within the substrate envelope (Figure 2C). For both the (S)- and (R)-4-(1-hydroxyethyl)benzene P2′ moieties only a portion of the hydroxyethyl substituent protrudes outside the envelope. The structures of PIs 12, 14 and 16 bound to protease superimpose very well with the corresponding parent compound structures with only subtle changes in the position and orientation of the (S)-4-(1-hydroxyethyl)benzene P2′ moiety. The differences in the conformation of the P2′ moiety are more evident for PIs 13, 15 and 17 incorporating the (R)-4-(1-hydroxyethyl)benzene, which is shifted upward toward the Gly48 backbone with a slight change in the orientation of the benzene ring compared to the position in the corresponding epimers, likely to maintain polar interactions between the hydroxyl group and the backbone NH of Asp30′ (Figure S3).
Despite clear differences in the overall binding conformations of (S)- and (R)-4-(1-hydroxyethyl)benzene P2′ moieties, the secondary hydroxyl group is oriented in the same direction of Asp29′ and Asp30′ backbone (Figure 4). Instead, the orientation of the methyl group is altered. As a result, regardless of the configuration of the hydroxyl group, the PIs incorporating the 4-(1-hydroxyethyl)benzene moiety (12–17) make identical direct and water-mediated hydrogen bonding interactions in the protease active site as the parent compounds 2–4 (Figure 4). In both configurations, the hydroxyl group is positioned between the backbone NH of Asp29′ and Asp30′. However, in all structures the hydroxyl group makes a direct hydrogen bond only with the backbone NH of Asp30′, which is closer (3.0–3.2 Å) than the NH of Asp29′ (3.6–3.8 Å). The distance between the hydroxyl group and the backbone NH of Asp30′ is slightly shorter (3.0–3.1 Å) for compounds with the (S)-4-(1-hydroxyethyl)benzene P2′ moiety than the corresponding (R)-epimers (3.2 Å) (Table S3). The hydroxyl group also interacts with the side chain of Asp30′, mostly through water-mediated hydrogen bonds but directly in case of compound 14. In the 14-protease complex structure the side chain of Asp30′ is shifted toward the P2′ moiety, resulting in a strong, direct hydrogen bond between the hydroxyl and carboxylate groups. Moreover, the hydroxyl group makes water-mediated interactions with the backbone NH of Asp29′, which are not observed in DRV-protease complex. Thus, the hydroxyl group at the P2′ moiety is involved in a network of direct and water-mediated interactions in the protease active site (Figure 4).
Cocrystal structures were analyzed for vdW contacts, and compounds with the modified P2′ moieties showed enhanced vdW interactions with protease residues in the S2′ subsite compared to the corresponding parent PIs 2–4 (Figure 5). To evaluate the changes in vdW interactions, total vdW energies per residue were calculated for each protease-inhibitor complex and the corresponding values for parent compound subtracted (Figure S5). The additional methyl group is oriented toward Ile47 in complexes with PIs 12, 14 and 16 incorporating the (S)-4-(1-hydroxyethyl)benzene P2′ moiety and toward Val32 and Leu76 in the corresponding (R)-epimers 13, 15 and 17. Compounds with the same P1′ group show minor differences in vdW interactions with residues in the S2′ subsite depending on the configuration of the P2′ hydroxyl group. The methyl group of analogue 12 with the (S)-configuration of the hydroxyl group makes additional vdW interactions with the methylene portion of Asp30′ side chain compared to the parent compound 2. Surprisingly, there is no increase in vdW contacts with Ile47 even though the methyl group of compound 12 is oriented toward this residue. The methyl group of the P2′ (R)-epimer 13 forms vdW contacts with Ile47 and Leu76. The corresponding analogues with the (S)-2-methylbutyl and 2-ethylbutyl P1′ groups showed largely similar interactions as observed for 12 and 13, respectively, but a slight shift in the benzene ring caused minor differences in the vdW contacts. As previously shown for parent compounds 3 and 4, larger (S)-2-methyl butyl and 2-ethylbutyl P1′ groups result in increased vdW contacts in the S1′ subsite, compared to the corresponding isobutyl group. Compounds with the same P1′ group show similar vdW contacts in the S1′ subsite. Thus, enhanced vdW interaction of PIs 12–17 in the S2′ subsite result from the additional methyl group and altered position of the modified P2′ moiety.
Figure 5.

Packing of inhibitors (A) 16, (B) 17, (C) 26, and (D) 27 in the S2′ subsite of HIV-1 protease. The protease residues are colored blue to red for increasing van der Waals (vdW) contact potentials with the inhibitor mapped onto the surface of cocrystal structures. Inhibitors and key residues are shown as sticks.
While compounds with the (S)- and (R)-4-(1-hydroxyethyl)benzene P2′ moieties show minor changes in vdW contacts in the S2′ subsite, both diastereoisomers make identical hydrogen bonding interactions with the protease because the orientation of the hydroxyl group is similar in both configurations. The largely similar overall binding interactions of P2′ epimeric compounds correlates with the similar enzyme inhibitory potencies observed between diastereoisomers. PIs with the (S)-4-(1-hydroxyethyl)benzene P2′ moiety showed improved antiviral potency against MDR variants likely due to stronger polar interactions with the backbone atoms without relying on contacts with side chains that could mutate to cause resistance. Overall, the cocrystal structures provided insights into the binding of these new PIs with modified P2′ moieties, revealing the structural basis for the observed inhibitory potencies.
The cocrystal structures of PIs 22–27 incorporating the 4-(1,2-dihydroxyethyl)benzene in complex with protease revealed a larger shift in the position of the P2′ moiety compared to DRV (Figure S3). The benzene ring is moved toward the flaps, likely to accommodate the larger substituent at the 4-position. With the (R)-configuration of the dihydroxyethyl group, the primary hydroxyl group in PIs 22, 24 and 26 is oriented toward the Asp29′ backbone and the S3′ subsite. As a result, these PIs fit very well within the substrate envelope (Figure 2C). In contrast, the primary hydroxyl group in the corresponding (S)-epimer compounds 23, 25 and 27 is oriented in the opposite direction, toward the Asp30′ and Leu76 side chains, and lie largely outside the substrate envelope. The cocrystal structures of PIs 22–27 also showed subtle variations in the binding of the secondary hydroxyl group of the (R)- and (S)-4-(1,2-dihydroxyethyl)benzene P2′ moieties. Despite differences in the binding of the dihydroxyethyl stereoisomers, similar conformations of the benzene ring were observed for both (R)- and (S)-4-(1,2-dihydroxyethyl)benzene P2′ moieties.
The hydrogen bonding patterns of compounds incorporating the 4-(1,2-dihydroxyethyl)benzene as P2′ ligands varied considerably depending on the configuration of the secondary hydroxyl group (Figure 4). In the cocrystal structures of PIs 22, 24, and 26 with the (R)-configuration of the dihydroxyethyl group, the secondary hydroxyl group makes a strong, direct hydrogen bond with the backbone NH of Asp30′. Another weaker but direct hydrogen bond connects the secondary hydroxyl group with the side chain carboxylate of Asp30′, replacing the water-mediated interactions observed for compounds with the 4-(hydroxymethyl)- and 4-(1-hydroxyethyl)-benzene P2′ moieties. The primary hydroxyl group of the (R)-dihydroxyethyl moiety is oriented toward the protease backbone and forms a direct hydrogen bond with the backbone NH of Asp29′, displacing a water molecule observed in the parent compound structures (Figure S4). The primary hydroxyl group is also involved in weaker (3.3–3.5 Å), direct hydrogen bonding interactions with the side chain carboxylate of Asp29′. The unique pattern of polar interactions of the (R)-dihydroxyethyl group in the S2′ subsite, particularly involving the backbone NH of Asp29′ and Asp30′, mimics the interactions of the bis-THF moiety in the S2 subsite.
The cocrystal structures of PIs 23, 25, and 27 with the (S)-configuration of the P2′ dihydroxyethyl group also revealed a unique network of hydrogen bonds between the hydroxyl groups and the protease. The secondary hydroxyl group forms two direct hydrogen bonds, one with the backbone NH of Asp30′ and another with the side chain carboxylate of the same residue. The secondary hydroxyl group is also positioned close to the backbone NH of Asp29′ (3.5–3.6 Å) and makes water-mediated interactions with both the backbone NH and side chain carboxylate of this residue. The primary hydroxyl group is oriented toward the side chain of Asp30′ and is involved in a direct hydrogen bonding interaction with the carboxyl group, replacing the water molecule observed in complexes with compounds incorporating the 4-(hydroxymethyl)- and 4-(1-hydroxyethyl)-benzene P2′ moieties. In this orientation, the dihydroxyethyl group forms an additional direct hydrogen bond with the protease replacing a water-mediated interaction.
In addition, PIs 22–27 incorporating the 4-(1,2-dihydroxyethyl)benzene moieties as P2′ ligands form enhanced vdW contacts with the protease compared to the corresponding parent compounds 2–4. The altered position of the P2′ moiety places the benzene ring closer to Gly48′ in the flaps, resulting in an overall increase in vdW contacts for all PIs in this series. Minor changes in vdW interactions were observed between P2′ epimeric compounds, with additional subtle variations resulting from changes at the P1′ position (Figure S5). In general, compounds with the (R)-configuration of the modified P2′ moiety (22, 24, and 26) make enhanced vdW interactions with Asp29′, Pro81, and residues 47′–50′ in the flap region, but slightly reduced interactions with Val32 and Ile84 (Figure 5). The corresponding (S)-epimers (23, 25, and 27) formed more vdW contacts with a different set of residues around the active site, including Asp30′, Lys45′, Ile47, and Leu76′, and weaker contacts with Asp30 and Ile50. However, few exceptions to these general trends were observed for compounds 24 and 25 that incorporate the (S)-2-methylbutyl group at the P1′ position, likely due to the asymmetric shape of the P1′ group (Figure S5). Overall, for PIs incorporating the 4-(1,2-dihydroxyethyl)benzene P2′ moieties, the increase in vdW interactions with specific protease residues mainly depend on the configuration of the dihydroxyethyl group.
The cocrystal structures of PIs with the 4-(1,2-dihydroxyethyl)benzene P2′ moieties revealed unique hydrogen bonding patterns and vdW contacts that vary significantly, depending on the configuration of the modified P2′ moiety. The improved potency profiles of PIs with the (R)-4-(1,2-dihydroxyethyl)benzene P2′ moiety compared to the corresponding (S)-epimers likely result from enhanced backbone interactions with residues Asp29′ and Asp30′, as well as increased vdW contacts with the side chains of Asp29′ and residues in the flap region. Notably, these PIs also fit very well in the substrate envelope. Compound 26, with the 2-ethylbutyl P1′ group, is a promising lead for exploring further modifications, particularly in combination with more hydrophobic P1 and P2 groups, to further improve potency against MDR variants.
CONCLUSIONS
New HIV-1 protease inhibitors were designed by incorporating stereoisomers of the 4-(1-hydroxyethyl)benzene and 4-(1,2-dihydroxyethyl)benzene moieties as P2′ ligands to enhance hydrogen bonding interactions in the S2′ subsite. Compounds with the polar dihydroxyethyl group at the P2′ moiety were generally less potent than DRV while inhibitors with the hydroxyethyl group exhibited improved antiviral potency against MDR variants. Crystal structures of protease-inhibitor complexes show that all PIs make enhanced hydrogen bonding and vdW interactions with the protease. The reduced cellular potency of PIs with the dihydroxyethyl group is likely due to their relatively low hydrophobicity, which can be counterbalanced with increased hydrophobicity at other positions such as P1. Overall, the PI potency profile was determined by the polar substituent at the 4-position of the P2′ benzene ring, configuration of the secondary hydroxyl group, and the size of the P1′ group. Exploration of both stereoisomers of P2′ moiety revealed unique polar and vdW interactions, including a network of direct and water-mediated hydrogen bonding with the backbone and side chain atoms of Asp29′ and Asp30′. Notably, PIs with the (R)-4-(1,2-dihydroxyethyl)benzene moiety make hydrogen bonding interactions with the backbone NH of Asp29′ and Asp30′ in the S2′ subsite, mimicking the polar interactions of the P2 bis-THF moiety, and show improved potency and resistance profiles compared to the corresponding (S)-epimers. In general, compounds that make increased hydrogen bonding interactions with the backbone and vdW contacts with invariant residues, and fit better within the substrate envelope maintain better potency against highly resistant MDR HIV-1 strains. These SAR data and structural insights may allow further optimization of these promising inhibitors.
EXPERIMENTAL SECTION
General.
All reactions were performed in oven-dried round- bottomed fitted with rubber septa under argon atmosphere unless otherwise noted. All reagents and solvents, including anhydrous solvents, were purchased from commercial sources and used as received. Flash column chromatography was performed on an automated Teledyne ISCO CombiFlash Rf+ system equipped with a UV-Vis detector using disposable Redisep Gold high performance silica gel columns, or manually using silica gel (230–400 mesh, EMD Millipore). Thin- layer chromatography (TLC) was performed using silica gel (60 F- 254) coated aluminum plates (EMD Millipore), and spots were visualized by exposure to ultraviolet light (UV), exposure to iodine adsorbed on silica gel, and/or staining with alcohol solutions of phosphomolybdic acid (PMA) and ninhydrin followed by brief heating. 1H NMR and 13C NMR spectra were acquired on Varian Mercury 400 MHz and Bruker Avance III HD 500 MHz NMR instruments. Chemical shifts are reported in ppm (δ scale) with the residual solvent signal used as reference, and coupling constant (J) values are reported in hertz (Hz). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, dd = doublet of triplet, t = triplet, m = multiplet, br s = broad singlet), coupling constant in Hz, and integration. High-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Orbitrap Velos Pro mass spectrometer coupled with a Thermo Scientific Accela 1250 UPLC and an autosampler using electrospray ionization (ESI) in the positive mode. The purity of final compounds was determined by analytical HPLC and was found to be ≥95% pure. HPLC was performed on a Agilent 1200 system equipped with a multiple wavelength detector and a manual injector under the following conditions: column, Phenomenex Hypersil-BDS-5u-C18 (5 μm, 4.6 mm × 250 mm, 130 Å); solvent A, H2O containing 0.1% trifluoroacetic acid (TFA); solvent B, CH3CN containing 0.1% TFA; gradient, 20% B to 100% B over 15 min followed by 100% B over 5 min; injection volume, 20 μL; flow rate, 1 mL/min. The wavelengths of detection were 254nm and 280 nm. Retention times and purity data for each target compound are provided in the Experimental Section.
Synthesis of Protease Inhibitors
tert-Butyl ((2S,3R)-3-hydroxy-4-((4-((S)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (9a).
A solution of tert-butyl ((2S,3R)-4-((4-acetyl-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate 8a (1.00 g, 1.93 mmol) in anhydrous tetrahydrofuran (40 mL) was cooled to 0 °C and the (R)-2-methyl-CBS-oxazaborolidine (1.07 g, 3.86 mmol) catalyst was added. After stirring the reaction mixture for 15 minutes, borane solution (1 M in THF) (2.90 mL, 2.90 mmol) was added dropwise over an hour. Stirring was continued at 0 °C until the reaction was complete, as indicated by TLC. The reaction was quenched with acetone/methanol mixture (1:1, 20 mL) and the solvent was removed under reduced pressure. The crude product was purified by automated flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–100% EtOAc/hexanes), to give compound 9a as a white solid. Recrystallization from a mixture of ethyl acetate/hexanes (1:1) provided a white solid (0.77 g, 77%). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2 H), 7.51 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 5.00–4.95 (m, 1 H), 4.63 (d, J = 7.5 Hz, 1 H), 3.92 (s, 1 H), 3.82–3.71 (m, 2 H), 3.13–3.05 (m, 2 H), 3.02–2.80 (m, 4 H), 2.02 (d, J = 3.5 Hz, 1 H), 1.89–1.81 (m, 1 H), 1.51 (d, J = 6.5 Hz, 3 H), 1.34 (s, 9 H), 0.91 (d, J = 6.5 Hz, 3 H), 0.87 (d, J = 7.0 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.20, 151.19, 137.93, 137.24, 129.67, 128.60, 127.72, 126.57, 126.22, 79.87, 72.91, 69.72, 58.76, 54.84, 53.81, 35.58, 28.37, 27.30, 25.54, 20.23, 19.99 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C27H41N2O6S, 521.2680; found 521.2676.
tert-Butyl ((2S,3R)-3-hydroxy-4-((4-((S)-1-hydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (9b).
The same procedure was used as described above for compound 9a. Compound 8b (1.00 g, 1.88 mmol) was treated with (R)-2-methyl-CBS-oxazaborolidine (1.04 g, 3.75 mmol) and borane solution (1 M in THF) (2.82 mL, 2.82 mmol) to give compound 9b as a white solid (yield after recrystallization: 0.57 g, 57%). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2 H), 7.51 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 5.00–4.95 (m, 1 H), 4.61 (d, J = 6.5 Hz, 1 H), 3.87 (s, 1 H), 3.80–3.71 (m, 2 H), 3.13–2.89 (m, 5 H), 2.82 (dd, J = 13.0, 7.0 Hz, 1 H), 2.00 (d, J = 3.7 Hz, 1 H), 1.64–1.58 (m, 1 H), 1.51 (d, J = 6.5 Hz, 3 H), 1.50–1.44 (m, 1 H, overlapping), 1.34 (s, 9 H), 1.11–1.02 (m, 1 H), 0.86–0.83 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.18, 151.11, 137.85, 137.26, 129.72, 128.64, 127.78, 126.61, 126.22, 79.88, 72.83, 69.80, 57.39, 54.74, 53.80, 35.59, 33.50, 28.40, 26.56, 25.58, 17.02, 11.17 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C28H43N2O6S, 535.2836; found 535.2832.
tert-Butyl ((2S,3R)-4-((N-(2-ethylbutyl)-4-((S)-1-hydroxyethyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (9c).
The same procedure was used as described above for compound 9a. Compound 8c (1.00 g, 1.83 mmol) was treated with (R)-2-methyl-CBS-oxazaborolidine (1.01 g, 3.66 mmol) and borane solution (1 M in THF) (2.74 mL, 2.74 mmol) to give compound 9c as a white solid (yield after recrystallization: 0.85 g, 85%). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2 H), 7.52 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 5.00–4.96 (m, 1 H), 4.58 (d, J = 5.9 Hz, 1 H), 3.92 (s, 1 H), 3.75 (m, 2 H), 3.12–2.98 (m, 4 H), 2.92–2.86 (m, 2 H), 2.01 (d, J = 3.7 Hz, 1 H), 1.51 (d, J = 6.5 Hz, 3 H), 1.49–1.37 (m, 2 H), 1.34 (s, 9 H), 1.34–1.24 (m, 3 H, overlapping), 0.83–0.79 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.16, 151.13, 137.83, 137.14, 129.71, 128.63, 127.81, 126.60, 126.24, 79.86, 73.06, 69.80, 54.91, 54.69, 53.81, 39.09, 35.77, 28.40, 25.58, 23.18, 22.86, 10.70, 10.42 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H45N2O6S, 549.2993; found 549.2989.
tert-Butyl ((2S,3R)-3-hydroxy-4-((4-((R)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (10a).
The same procedure was used as described above for compound 9a. Compound 8a (1.00 g, 1.93 mmol) was treated with (S)-2-methyl-CBS-oxazaborolidine (1.22 g, 4.40 mmol) and borane solution (1 M in THF) (3.3 mL, 3.30 mmol) to give compound 10a as a white solid (yield after crystallization: 0.6 g, 60%). 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 8.4 Hz, 2 H), 7.50 (d, J = 8.3 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.24–7.19 (m, 3 H), 4.98–4.93 (m, 1 H), 4.67 (d, J = 7.8 Hz, 1 H), 3.94 (s, 1 H), 3.84–3.70 (m, 2 H), 3.12–3.04 (m, 2 H), 3.01–2.80 (m, 4 H), 2.35 (s, 1 H), 1.89–1.81 (m, 1 H), 1.49 (d, J = 6.5 Hz, 3 H), 1.33 (s, 9 H), 0.90 (d, J = 7.0 Hz, 3 H), 0.86 (d, J = 6.5 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.20, 151.23, 137.94, 137.19, 129.66, 128.59, 127.69, 126.55, 126.21, 79.85, 72.91, 69.67, 58.74, 54.83, 53.80, 35.57, 28.36, 27.28, 25.54, 20.22, 19.98 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C27H41N2O6S, 521.2680; found 521.2682.
tert-Butyl ((2S,3R)-3-hydroxy-4-((4-((R)-1-hydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (10b).
The same procedure was used as described above for compound 9a. Compound 8b (1.00 g, 1.88 mmol) was treated with (S)-2-methyl-CBS-oxazaborolidine (1.04 g, 3.75 mmol) and borane solution (1M in THF) (2.82 mL, 2.82 mmol) to give 10b as a white solid (yield after recrystallization: 0.76 g, 76%). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.4 Hz, 2 H), 7.52 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 5.00–4.96 (m, 1 H), 4.61 (d, J = 6.7 Hz, 1 H), 3.87 (s, 1 H), 3.82–3.71 (m, 2 H), 3.13–2.99 (m, 5 H), 2.82 (d, J = 13.0, 7.0 Hz, 1 H), 1.95 (d, J = 3.5 Hz, 1 H), 1.64–1.57 (m, 1 H), 1.51 (d, J = 6.5 Hz, 3 H), 1.51–1.44 (m, 1 H, overlapping), 1.35 (s, 9 H), 1.11–1.02 (m, 1 H), 0.86–0.83 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.18, 151.09, 137.85, 137.28, 129.72, 128.65, 127.80, 126.61, 126.22, 79.88, 72.82, 69.82, 57.41, 54.73, 53.82, 35.60, 33.51, 28.41, 26.56, 25.60, 17.02, 11.17 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C28H43N2O6S, 535.2836; found 535.2835.
tert-Butyl ((2S,3R)-4-((N-(2-ethylbutyl)-4-((R)-1-hydroxyethyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (10c).
The same procedure was used as described above for compound 9a. Compound 8c (1.00 g, 1.83 mmol) was treated with (S)-2-methyl-CBS-oxazaborolidine (1.01 g, 3.66 mmol) and borane solution (1 M in THF) (2.74 mL, 2.74 mmol) to give compound 10c as a white solid (yield after recrystallization: 0.71 g, 69%). 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 2 H), 7.52 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 5.00–4.96 (m, 1 H), 4.58 (d, J = 5.5 Hz, 1 H), 3.93 (s, 1 H), 3.79–3.71 (m, 2 H), 3.12–2.99 (m, 4 H), 2.92–2.85 (m, 2 H), 1.96 (d, J = 3.4 Hz, 1 H), 1.51 (d, J = 6.5 Hz, 3 H), 1.48–1.37 (m, 2 H), 1.34 (s, 9 H), 1.34–1.18 (m, 3 H, overlapping), 0.83–0.79 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.15, 151.09, 137.83, 137.15, 129.72, 128.64, 127.82, 126.60, 126.24, 79.86, 73.07, 69.83, 54.93, 54.68, 53.83, 39.10, 35.78, 28.40, 25.60, 23.18, 22.86, 10.70, 10.42 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H45N2O6S, 549.2993; found 549.2992.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((S)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (12).
A solution of compound 9a (0.45 g, 0.864 mmol) in anhydrous dichloromethane (6 mL) was treated with trifluoroacetic acid (5 mL). After stirring the reaction mixture at room temperature for 1 h, solvents were evaporated under reduced pressure and the residue was dried under high vacuum. A solution of the resulting amine salt in anhydrous acetonitrile (15 mL) was cooled to 0 °C and treated with diisopropylethylamine (0.34 g, 2.59 mmol) followed by bis-THF activated carbonate 11 (0.23 g, 0.864 mmol). After 15 min, the reaction mixture was allowed to warm to room temperature and stirred for 24 h. The solvents were removed under reduced pressure and the residue was purified by automated flash chromatography using a silica gel column (RediSep Gold, 24 g, gradient elution with 0–10% methanol/dichloromethane) to give the target compound 12 (0.44 g, 88%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.2 Hz, 2 H), 7.54 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.19 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.02–4.96 (m, 2 H), 4.90 (d, J = 8.4 Hz, 1 H), 3.96–3.83 (m, 4 H), 3.70–3.65 (m, 2 H), 3.60 (br s, 1 H), 3.19–3.14 (m, 1 H), 3.08 (dd, J = 14.0, 3.6 Hz, 1 H), 3.03–2.97 (m, 2 H), 2.92–2.77 (m, 3 H), 2.06 (br s, 1 H), 1.87–1.81 (m, 1 H), 1.68–1.57 (m, 1 H), 1.52 (d, J = 6.5 Hz, 3 H, overlapping), 1.51–1.47 (m, 1 H), 0.94 (d, J = 6.6 Hz, 3 H), 0.89 (d, J = 6.6 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.60, 151.33, 137.63, 136.92, 129.48, 128.72, 127.74, 126.77, 126.34, 109.44, 73.59, 72.95, 70.98, 69.74, 59.09, 55.21, 53.96, 45.44, 35.79, 27.46, 25.99, 25.59, 20.29, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H41N2O8S, 577.2578; found 577.2575. Anal. HPLC: tR 11.22 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((R)-1-hydroxyethyl)-N-isobutylphenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (13).
The same procedure was used as described above for compound 12. Compound 10a (0.40 g, 0.768 mmol) was treated with trifluoroacetic acid (5 mL), and the resulting deprotected amine was treated with diisopropylethylamine (0.30 g, 2.304 mmol) and bis-THF activated carbonate 11 (0.21 g, 0.768 mmol) to give compound 13 (0.40 g, 90%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.3 Hz, 2 H), 7.53 (d, J = 8.3 Hz, 2 H), 7.29–7.26 (m, 2 H), 7.22–7.19 (m, 3 H), 5.63 (d, J = 5.2 Hz, 1 H), 5.01–4.95 (m, 3 H), 3.94–3.81 (m, 4 H), 3.69–3.60 (m, 3 H), 3.17–3.12 (m, 1 H), 3.08 (dd, J = 14.0, 3.6 Hz, 1 H), 3.05–2.96 (m, 2 H), 2.90–2.76 (m, 3 H), 2.27 (br s, 1 H), 1.89–1.81 (m, 1 H), 1.66–1.58 (m, 1 H), 1.50 (d, J = 6.5 Hz, 3 H, overlapping), 1.50–1.44 (m, 1 H), 0.93 (d, J = 6.6 Hz, 3 H), 0.89 (d, J = 6.6 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.59, 151.34, 137.62, 136.89, 129.48, 128.73, 127.76, 126.78, 126.32, 109.44, 73.61, 72.95, 71.01, 69.75, 59.14, 55.21, 54.01, 45.43, 35.83, 27.47, 26.00, 25.62, 20.29, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H41N2O8S, 577.2578; found 577.2581. Anal. HPLC: tR 11.37 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((S)-1-hydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (14).
The same procedure was used as described above for compound 12. Compound 9b (0.43 g, 0.804 mmol) was treated with trifluoroacetic acid (5 mL), and the resulting deprotected amine was treated with diisopropylethylamine (0.31 g, 2.41 mmol) and bis-THF activated carbonate 11 (0.22 g, 0.804 mmol) to give compound 14 (0.41 g, 86%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.3 Hz, 2 H), 7.53 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.03–4.96 (m, 2 H), 4.89 (d, J = 8.9 Hz, 1 H), 3.94 (dd, J = 9.5, 6.0 Hz, 1 H), 3.91–3.83 (m, 3 H), 3.71–3.65 (m, 2 H), 3.56 (br s, 1 H), 3.17–2.98 (m, 4 H), 2.92–2.87 (m, 1 H), 2.84–2.80 (m, 2 H), 2.10 (br s, 1 H), 1.69–1.59 (m, 2 H), 1.54–1.46 (m, 2 H), 1.51 (d, J = 6.5 Hz, 3 H, overlapping), 1.14–1.05 (m, 1 H), 0.88–0.85 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 155.61, 151.35, 137.60, 136.93, 129.52, 128.74, 127.77, 126.79, 126.33, 109.44, 73.62, 72.83, 70.98, 69.73, 57.60, 55.20, 53.90, 45.46, 35.81, 33.63, 26.54, 25.98, 25.57, 17.05, 11.18 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C30H43N2O8S, 591.2735; found 591.2732. Anal. HPLC: tR 11.95 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-hydroxy-4-((4-((R)-1-hydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (15).
The same procedure was used as described above for compound 12. Compound 10b (0.40 g, 0.748 mmol) was treated with trifluoroacetic acid (5 mL), and the resulting deprotected amine was treated with diisopropylethylamine (0.29 mg, 2.24 mmol) and bis-THF activated carbonate 11 (0.20 g, 0.748 mmol) to give compound 15 (0.40 g, 91%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2 H), 7.54 (d, J = 8.3 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.23–7.20 (m, 3 H), 5.65 (d, J = 5.2 Hz, 1 H), 5.03–4.96 (m, 2 H), 4.86 (d, J = 9.0 Hz, 1 H), 3.94 (dd, J = 9.5, 6.0 Hz, 1 H), 3.91–3.84 (m, 3 H), 3.72–3.64 (m, 2 H), 3.55 (br s, 1 H), 3.14 (dd, J = 15.0, 8.5 Hz, 1 H), 3.10–3.04 (m, 2 H), 2.99 (dd, J = 15.0, 3.0 Hz, 1 H), 2.92–2.87 (m, 1 H), 2.85–2.80 (m, 2 H), 2.05 (br s, 1 H), 1.70–1.55 (m, 2 H), 1.54–1.47 (m, 2 H), 1.52 (d, J = 6.5 Hz, 3 H, overlapping), 1.14–1.05 (m, 1 H), 0.88–0.85 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 155.61, 151.38, 137.59, 136.90, 129.52, 128.74, 127.78, 126.80, 126.31, 109.45, 73.62, 72.83, 71.01, 69.73, 57.64, 55.19, 53.93, 45.46, 35.85, 33.64, 26.56, 25.99, 25.59, 17.05, 11.18 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C30H43N2O8S, 591.2735; found 591.2734. Anal. HPLC: tR 11.87 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((N-(2-ethylbutyl)-4-((S)-1-hydroxyethyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (16).
The same procedure was used as described above for compound 12. Compound 9c (0.40 g, 0.729 mmol) was treated with trifluoroacetic acid (5 mL), and the resulting deprotected amine was treated with diisopropylethylamine (0.28 g, 2.19 mmol) and bis-THF activated carbonate 11 (0.22 g, 0.801 mmol) to give compound 16 (0.36 g, 82%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2 H), 7.54 (d, J = 8.3 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.02–4.97 (m, 2 H), 4.84 (d, J = 9.1 Hz, 1 H), 3.94 (d, J = 9.5, 6.0 Hz, 1 H), 3.88–3.80 (m, 3 H), 3.70–3.64 (m, 3 H), 3.17–3.06 (m, 3 H), 2.98 (dd, J = 15.0, 3.0 Hz, 1 H), 2.92–2.85 (m, 2 H), 2.81 (dd, J = 14.0, 9.0 Hz, 1 H), 2.02 (br s, 1 H), 1.68–1.60 (m, 1 H), 1.55–1.42 (m, 3 H), 1.52 (d, J = 6.5 Hz, 3H, overlapping), 1.35–1.25 (m, 3 H), 0.85–0.81 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.58, 151.34, 137.57, 136.69, 129.50, 128.74, 127.79, 126.78, 126.35, 109.43, 73.60, 73.19, 70.91, 69.75, 55.11, 53.95, 45.44, 39.26, 35.93, 25.97, 25.59, 23.16, 22.85, 10.77, 10.41ppm. HRMS (ESI) m/z: [M + H]+ calcd for C31H45N2O8S, 605.2891; found 605.2890. Anal. HPLC: tR 12.56 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((N-(2-ethylbutyl)-4-((R)-1-hydroxyethyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (17).
The same procedure was used as described above for compound 12. Compound 10c (0.40 g, 0.729 mmol) was treated with trifluoroacetic acid (5 mL), and the resulting deprotected amine was treated with diisopropylethylamine (0.28 g, 2.19 mmol) and bis-THF activated carbonate 11 (0.28 g, 0.801 mmol) to give compound 17 (0.32 g, 73%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.3 Hz, 2 H), 7.54 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.23–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.02–4.97 (m, 2 H), 4.84 (d, J = 8.5 Hz, 1H), 4.93 (dd, J = 10.0, 6.5 Hz, 1H), 3.87–3.80 (m, 3 H), 3.71– 3.64 (m, 3 H), 3.16–3.06 (m, 3 H), 2.99–2.96 (dd, J = 15.5, 2.5 Hz, 1 H), 2.92–2.86 (m, 2 H), 2.80 (dd, J = 14.0, 9.2 Hz, 1 H), 2.06 (br s, 1 H), 1.66–1.60 (m, 1 H), 1.53–1.40 (m, 3 H), 1.52 (d, J = 6.5 Hz, 3H, overlapping), 1.35–1.25 (m, 3 H), 0.85–0.81 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.57, 151.46, 137.65, 136.56, 129.45, 128.65, 127.71, 126.68, 126.29, 109.40, 73.51, 73.18, 70.95, 69.72, 69.59, 55.16, 54.97, 53.81, 45.45, 39.12, 35.90, 25.94, 25.52, 23.07, 22.81, 10.67, 10.40 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C31H45N2O8S, 605.2891; found 605.2891. Anal. HPLC: tR 12.61 min, purity 100%.
tert-Butyl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (20a).
t-Butanol (7.5 mL) and water (7.5 mL) were charged into a round-bottom flask and AD-mix-β (2.80 g) was added. The mixture was stirred at room temperature until two clear phases appeared. The mixture was cooled to 0 °C and intermediate 19a (0.86 g, 1.71 mmol) was added in one portion. The resulting reaction mixture was stirred at 4 °C for 48 h. Reaction was quenched by adding sodium sulfite (3.00 g) and the mixture was allowed to warm to room temperature. Ethyl acetate (10 mL) was added, phases were separated, and the aqueous layer was extracted with ethyl acetate (3 × 10 mL). The combined organic portions were dried (Na2SO4), filtered, and concentrated. The residue was purified by automated flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 20–100% ethyl acetate/hexanes) to give compound 20a as a white solid (0.80 g, 87%). 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2 H), 7.52 (d, J = 8.3 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.24–7.20 (m, 3 H), 4.88 (d, J = 6.0 Hz, 1 H), 4.65 (d, J = 7.4 Hz, 1 H), 3.93 (s, 1 H), 3.83–3.70 (m, 3 H), 3.62 (dd, J = 11.0, 8.0 Hz, 1 H), 3.14–3.07 (m, 2 H), 3.01–2.82 (m, 5 H), 2.31 (s, 1 H), 1.89–1.81 (m, 1 H), 1.34 (s, 9 H), 0.90 (d, J = 6.5 Hz, 3 H), 0.86 (d, J = 6.5 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.27, 145.97, 137.93, 137.89, 129.67, 128.64, 127.68, 126.97, 126.62, 79.99, 74.00, 72.88, 67.88, 58.70, 54.89, 53.73, 35.58, 28.39, 27.30, 20.24, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C27H41N2O7S, 537.2629; found 537.2627.
tert-Butyl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (20b).
The same procedure was used as described above for compound 20a. Compound 19b (1.22 g, 2.36 mmol) was treated with AD-mix-β (2.80 g) and the reaction was quenched with solid sodium sulfite (3.00 g) to give compound 20b as a white solid (0.88 g, 68%). 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 8.5 Hz, 2 H), 7.48 (d, J = 8.0 Hz, 2 H), 7.29–7.25 (m, 2 H), 7.22–7.18 (m, 3 H), 4.84–4.82 (m, 1 H), 4.74 (d, J = 7.4 Hz, 1 H), 3.97 (s, 1 H), 3.80–3.69 (m, 4 H), 3.58–3.55 (m, 1 H), 3.14–3.04 (m, 3 H), 3.02–2.93 (m, 2 H), 2.90–2.79 (m, 2 H), 1.64–1.57 (m, 1 H), 1.47–1.39 (m, 1 H), 1.32 (s, 9 H), 1.08–0.99 (m, 1 H), 0.84–0.81 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.23, 146.24, 137.77, 137.59, 129.60, 128.58, 127.55, 126.96, 126.57, 79.97, 73.94, 72.64, 67.80, 57.09, 54.78, 53.46, 35.60, 33.37, 28.33, 26.55, 16.95, 11.14 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C28H43N2O7S, 551.2785; found 551.2786.
tert-Butyl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-(2-ethylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (20c).
The same procedure was used as described above for compound 20a. Compound 19c (1.30 g, 2.45 mmol) was treated with AD-mix-β (3.50 g) and the reaction was quenched with sodium sulfite (4.00 g) to give compound 20c as a white solid (1.02 g, 74%). 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.0 Hz, 2 H), 7.53 (d, J = 8.0 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 4.90 (d, J = 7.5 Hz, 1 H), 4.58 (d, J = 7.0 Hz, 1 H), 3.92 (s, 1 H), 3.82 (d, J = 11.0 Hz, 1 H), 3.77–3.71 (m, 2 H), 3.64 (t, J = 10.0 Hz, 1 H), 3.13–2.98 (m, 4 H), 2.92–2.81 (m, 3 H), 2.15 (br s, 1 H), 1.48–1.38 (m, 2 H), 1.36–1.23 (m, 3 H), 1.34 (s, 9 H, overlapping), 0.83–0.79 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.23, 146.25, 137.75, 137.52, 129.61, 128.59, 127.59, 126.98, 126.58, 79.98, 73.95, 72.88, 67.80, 54.74, 54.61, 53.47, 38.95, 35.77, 28.34, 23.06, 22.81, 10.61, 10.41 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H45N2O7S, 565.2942; found 565.2939.
tert-Butyl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (21a).
The same procedure was used as described above for compound 20a. Compound 19a (0.65 g, 1.30 mmol) was treated with AD-mix-α (2.10 g) and the reaction was quenched with sodium sulfite (2.5 g) to give compound 21a as a white solid (0.33 g, 47%). 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 2 H), 7.52 (d, J = 8.5 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.21 (m, 3 H), 4.91–4.88 (m, 1 H), 4.63 (d, J = 7.5 Hz, 1 H), 3.92 (s, 1 H), 3.83–3.70 (m, 3 H), 3.66–3.61 (m, 1 H), 3.14–3.06 (m, 2 H), 3.02–2.81 (m, 5 H), 2.13 (t, J = 5.4 Hz, 1 H), 1.89–1.81 (m, 1 H), 1.34 (s, 9 H), 0.90 (d, J = 6.5 Hz, 3 H), 0.86 (d, J = 6.5 Hz, 3 H) ppm.13C NMR (125 MHz, CDCl3) δ 156.29, 145.86, 138.01, 137.90, 129.69, 128.65, 127.72, 126.97, 126.62, 79.96, 74.02, 72.90, 67.90, 58.75, 54.88, 53.80, 35.61, 28.39, 27.32, 20.25, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C27H41N2O7S, 537.2629; found 537.2631.
tert-Butyl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (21b).
The same procedure was used as described above for compound 20a. Compound 19b (1.00 g, 1.94 mmol) was treated with AD-mix-α (2.80 g) and the reaction was quenched with solid sodium sulfite (3.00 g) to give compound 21b as a white solid (0.92 g, 86%). 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 2 H), 7.52 (d, J = 8.5 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 4.91–4.88 (m, 1 H), 4.62 (d, J = 7.1 Hz, 1 H), 3.87 (br s, 1 H), 3.83–3.71 (m, 3 H), 3.66–3.61 (m, H), 3.13–2.87 (m, 5 H), 2.85–2.80 (m, 2 H), 2.16 (t, J = 5.5 Hz, 1 H), 1.65–1.58 (m, 1 H), 1.51–1.43 (m, 1 H), 1.35 (s, 9 H), 1.10–1.02 (m, 1 H), 0.86–0.83 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.22, 145.86, 137.92, 137.80, 129.70, 128.66, 127.73, 126.96, 126.64, 79.96, 74.02, 72.77, 67.90, 57.32, 54.78, 53.71, 35.63, 33.49, 28.40, 26.56, 17.01, 11.18 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C28H43N2O7S, 551.2785; found 551.2787.
tert-Butyl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-(2-ethylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (21c).
The same procedure was used as described above for compound 20a. Compound 19c (1.50 g, 2.45 mmol) was treated with AD-mix-α (3.80 g) and the reaction was quenched with sodium sulfite (4.00 g) to give compound 21c as a white solid (0.95 g, 60%). 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 2 H), 7.53 (d, J = 8.5 Hz, 2 H), 7.31–7.28 (m, 2 H), 7.25–7.20 (m, 3 H), 4.92–4.89 (m, 1 H), 4.58 (d, J = 7.0 Hz, 1 H), 3.93 (s, 1 H), 3.84–3.80 (m, 1 H), 3.77–3.71 (m, 2 H), 3.66–3.62 (m, 1 H), 3.13–2.98 (m, 4 H), 2.93–2.77 (m, 3 H), 2.16–2.08 (m, 1 H), 1.48–1.37 (m, 2 H), 1.36–1.20 (m, 3 H), 1.34 (s, 9 H, overlapping), 0.83–0.79 (m, 6 H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.23, 146.13, 137.77, 137.61, 129.65, 128.63, 127.65, 126.99, 126.62, 79.99, 73.98, 72.94, 67.85, 54.72, 53.57, 39.01, 35.79, 28.37, 23.11, 22.83, 10.65, 10.42 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H45N2O7S, 565.2942; found 565.2942.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (22).
A solution of compound 20a (0.78 g, 1.45 mmol) in anhydrous dichloromethane (5 mL) was treated with a solution of 4 N HCl in 1,4-dioxane (10 mL). After stirring the reaction mixture at room temperature for 2.5 h, solvents were evaporated under reduced pressure, and the residue was triturated with diethyl ether and dried under high vacuum. A solution of the deprotected amine salt in anhydrous acetonitrile (15 mL) was cooled to 0 °C and treated with diisopropylethylamine (0.75 g, 5.813 mmol) followed by bis-THF activated carbonate 11 (0.39 g, 1.45 mmol). The resulting reaction mixture was stirred at room temperature for 24 h. Solvents were evaporated under reduced pressure, and the residue was purified by automated flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–20% methanol/dichloromethane) to give compound 22 (0.28 g, 33%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2 H), 7.55 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 4.99 (q, J = 7.0 Hz, 1 H), 4.93–4.89 (m, 2 H), 3.94 (dd, J = 9.6, 6.3 Hz, 1 H), 3.89–3.81 (m, 4 H), 3.69–3.62 (m, 3 H), 3.56 (br s, 1 H), 3.16 (dd, J = 15.5, 8.5 Hz, 1 H), 3.08 (dd, J = 13.5, 3.0 Hz, 1 H), 3.02–2.97 (m, 3 H), 2.91–2.83 (m, 2 H), 2.79 (dd, J = 13.5, 9.0 Hz, 1 H), 1.88–1.81 (m, 1 H), 1.68–1.60 (m, 1 H), 1.49–1.46 (m, 1 H), 0.94 (d, J = 6.6 Hz, 3 H), 0.89 (d, J = 6.6 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.61, 146.12, 137.59, 129.48, 128.74, 127.68, 127.08, 126.79, 109.44, 73.93, 73.62, 72.89, 71.04, 69.78, 67.83, 59.04, 55.24, 53.92, 45.45, 35.80, 27.46, 26.00, 20.28, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H41N2O9S, 593.2527; found 593.2528. Anal. HPLC: tR 9.69 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (23).
The same procedure was used as described above for compound 22. Compound 21a (0.35 g, 0.652 mmol) was treated with 4 N HCl in 1,4-dioxane (5 mL), and the resulting amine salt was treated with diisopropylethylamine (0.34 g, 2.61 mmol) and bis-THF activated carbonate 11 (0.18 g, 0.652 mmol) to give compound 23 (0.34 g, 77%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 8.3 Hz, 2 H), 7.55 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.23–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 4.99 (q, J = 6.0 Hz, 1 H), 4.92–4.87 (m, 2 H), 3.93 (dd, J = 9.6, 6.2 Hz, 1 H), 3.88–3.81 (m, 4 H), 3.70–3.62 (m, 3 H), 3.54 (br s, 1 H), 3.17–3.07 (m, 2 H), 3.02–2.97 (m, 3 H), 2.92–2.74 (m, 3 H), 2.15 (br s, 1 H), 1.88–1.82 (m, 1 H), 1.69–1.60 (m, 1 H), 1.52–1.48 (m, 1 H), 0.94 (d, J = 6.7 Hz, 3 H), 0.90 (d, J = 6.6 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.60, 146.15, 137.56, 129.48, 128.75, 127.70, 127.5, 126.80, 109.46, 73.91, 73.63, 72.89, 71.10, 69.79, 67.85, 59.12, 55.22, 53.99, 45.43, 35.87, 27.47, 26.02, 20.28, 20.00 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C29H41N2O9S, 593.2527; found 593.2530. Anal. HPLC: tR 9.85 min, purity 98%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (24).
The same procedure was used as described above for compound 22. Compound 20b (0.52 g, 0.944 mmol) was treated with 4 N HCl in 1,4-dioxane (8 mL), and the resulting amine salt was treated with disopropylethylamine (0.49 g, 3.776 mmol) and bis-THF activated carbonate 11 (0.26 g, 0.944 mmol) to give compound 24 (0.40 g, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.1 Hz, 2 H), 7.55 (d, J = 8.1 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.1 Hz, 1 H), 5.01 (q, J = 6.5 Hz, 1 H), 4.91–4.87 (m, 2 H), 3.94 (dd, J = 9.5, 6.5 Hz, 1 H), 3.86–3.80 (m, 4 H), 3.70–3.62 (m, 3 H), 3.52 (br s, 1 H), 3.17–2.97 (m, 4 H), 3.91–2.97 (m, 4 H), 2.16 (br s, 1 H), 1.69–1.47 (m, 4 H) 1.14–1.05 (m, 1 H), 0.88–0.86 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.63, 146.40, 137.66, 137.34, 129.44, 128.65, 127.60, 127.05, 126.70, 109.43, 73.90, 73.51, 72.77, 71.13, 69.77, 67.73, 57.32, 55.28, 53.59, 45.53, 35.81, 33.45, 26.61, 25.97, 17.02, 11.23 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C30H43N2O9S, 607.2684; found 607.2687. Anal. HPLC: tR 10.53 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-((S)-2-methylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (25).
The same procedure was used as described above for compound 22. Compound 21b (0.45 g, 0.817 mmol) was treated with 4 N HCl in 1,4-dioxane (8 mL), and the resulting amine salt was treated with diisopropylethylamine (0.42 g, 3.27 mmol) and bis-THF activated carbonate 11 (0.22 g, 0.817 mmol) to give compound 25 (0.39 g, 79%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2 H), 7.55 (d, J = 8.3 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.23–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 4.99 (q, J = 6.0 Hz 1 H), 4.91–4.87 (m, 2 H), 3.93 (dd, J = 9.5, 6.0 Hz, 1 H), 3.87–3.81 (m, 4 H), 3.71–3.62 (m, 3 H), 3.50 (br s, 1 H), 3.15–2.97 (m, 5 H), 2.92–2.78 (m, 3 H), 1.70–1.45 (m, 4 H), 1.14–1.05 (m, 1 H), 0.88–0.85 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.63, 146.44, 137.66, 137.28, 129.45, 128.65, 127.62, 127.03, 126.70, 109.44, 73.88, 73.51, 72.78, 71.19, 69.78, 67.74, 57.38, 55.28, 53.63, 45.52, 35.86, 33.46, 26.63, 25.98, 17.02, 11.23 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C30H43N2O9S, 607.2684; found 607.2687. Anal. HPLC: tR 10.39 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((R)-1,2-dihydroxyethyl)-N-(2-ethylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (26).
The same procedure was used as described above for compound 22. Compound 20c (0.51 g, 0.903 mmol) was treated with 4 N HCl in 1,4-dioxane (8 mL), and the resulting amine salt was treated with diisopropylethylamine (0.47 g, 3.61 mmol) and bis-THF activated carbonate 11 (0.24 g, 0.903 mmol) to give compound 26 (0.31 g, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 8.3 Hz, 2 H), 7.55 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.00 (q, J = 6.5 Hz, 1 H), 4.90 (dd, J = 7.8, 3.4 Hz, 1 H), 4.86 (d, J = 9.0 Hz, 1 H), 3.93 (dd, J = 9.7, 6.3 Hz, 1 H), 3.87–3.78 (m, 4 H), 3.69–3.62 (m, 4 H), 3.16–3.04 (m, 3 H), 2.98 (d, J = 15.0 Hz, 1 H), 2.91–2.87 (m, 2 H), 2.80 (dd, J = 14.1, 9.3 Hz, 1 H), 1.68–1.60 (m, 1 H), 1.50–1.39 (m, 3 H), 1.35–1.25 (m, 3 H), 0.85–0.81 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.62, 146.38, 137.64, 137.22, 129.45, 128.67, 127.65, 127.08, 126.72, 109.43, 73.92, 73.54, 73.11, 71.05, 69.77, 67.76, 55.23, 54.85, 53.68, 45.51, 39.05, 35.91, 25.96, 23.06, 22.84, 10.65, 10.45 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C31H45N2O9S, 621.2840; found 621.284. Anal. HPLC: tR 11.11 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-((S)-1,2-dihydroxyethyl)-N-(2-ethylbutyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (27).
The same procedure was used as described above for compound 22. Compound 21c (0.50 g, 0.885 mmol) was treated with 4 N HCl in dioxane (10 mL), and the resulting amine salt was treated with diisopropylethylamine (0.46 g, 3.54 mmol) and bis-THF activated carbonate 11 (0.24 g, 0.885 mmol) to give compound 27 (0.31 g, 57%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.3 Hz, 2 H), 7.56 (d, J = 8.2 Hz, 2 H), 7.30–7.27 (m, 2 H), 7.22–7.20 (m, 3 H), 5.64 (d, J = 5.2 Hz, 1 H), 5.01–4.99 (q, J = 6.0 Hz, 1 H), 4.91 (d, J = 7.0 Hz, 1 H), 4.83 (d, J = 9.0 Hz, 1 H), 3.92 (dd, J = 9.6, 6.2 Hz, 1 H), 3.88–3.80 (m, 4 H), 3.70–3.60 (m, 4 H), 3.15–3.05 (m, 3 H), 2.99 (d, J = 15.0, 2.0 Hz, 1 H), 2.92–2.89 (m, 2 H), 2.80 (dd, J = 14.1, 9.2 Hz, 1 H), 2.12 (br s, 1 H), 1.69–1.61 (m, 1 H), 1.51–1.39 (m, 3 H), 1.35–1.28 (m, 3 H), 0.85–0.81 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3) δ 155.59, 146.18, 137.52, 137.28, 129.49, 128.75, 127.74, 127.06, 126.80, 109.45, 73.92, 73.63, 73.13, 71.05, 69.78, 67.84, 55.12, 53.95, 45.44, 39.24, 36.00, 26.00, 23.14, 22.87, 10.73, 10.45 ppm. HRMS (ESI) m/z: [M + H]+ calcd for C31H45N2O9S, 621.2840; found 621.2847. Anal. HPLC: tR 11.15 min, purity 99%.
Antiviral Assays.
293T and TZM-BL36 cells (NIH AIDS Research and Reference Reagent Program) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum in the presence of penicillin and streptomycin at 37 °C with 5% CO2. To determine the concentration of drugs achieving 50% inhibition of infection compared with the drug-free control, 4.5×106 293T cells were seeded onto a 10-cm plate 24 h before transfection. Cells were transfected with 8 μg of either the wild-type plasmid, infectious molecular clone pNL-CH derived from the pNL4-3 clone of HIV-1, or each PI-resistant HIV-1 variant34 using FuGENE 6 transfection reagent (Roche). The culture supernatant of 293T cells transfected with wild-type or PI-resistant HIV-1 variant was removed 18 h after transfection and the cells were washed with 1 × PBS. The 293T cells were collected and transferred to wells of a 24-well plate. Briefly, each drug was serially diluted in the culture medium and the dilutions were added to the wells of a 24-well plate. The 293T cells (0.5 × 106 per well) collected from the transfection were added to wells containing various concentrations of drug. The culture supernatant containing virus particles was harvested 18 h after the 293T cells were reseeded in the presence of drug. This supernatant was filtered through a 0.45-μm-pore-size membrane (Millipore) to remove cell debris then used to infect 2 × 104 TZM-BL cells in a 96-well plate following a procedure previously described.37 The culture supernatant was removed from each well 48 h post-infection, and the cells were washed with 1 × PBS. For the luciferase assay, infected TZM-BL cells were lysed in 1× reporter lysis buffer (Promega) and the cells were kept at −80 °C. After one freeze-thaw cycle, the cell lysates were transferred into a 96-well assay plate (Costar), and luciferase activity was measured using a luminometer (Promega). The culture supernatant harvested from 293T cells reseeded in the absence of drugs was used as a drug-free control. EC50 was determined based on a dose-response curve generated using GraphPad Prism (version 7.0).
Protease Gene Construction.
Protease gene construction was carried out as previously described.38,39 The NL4-3 strain has four naturally occurring polymorphisms in the protease relative to the SF2 strain.40 In short, the protease variant genes (I84V, I50V/A71V) were constructed using QuikChange site-directed mutagenesis (Genewiz) onto NL4-3 wild-type protease on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. A Q7K mutation was included to prevent autoproteolysis.41
Protein Expression and Purification.
The expression, isolation, and purification of WT and mutant HIV-1 proteases used for the kinetic assays and crystallization were carried out as previously described.38,39 Briefly, the gene encoding the HIV protease was subcloned into the heat-inducible pXC35 expression vector (ATCC) and transformed into E. coli TAP-106 cells. Cells grown in 6 L of Terrific Broth were lysed with a cell disruptor and the protein was purified from inclusion bodies.42 The inclusion body centrifugation pellet was dissolved in 50% acetic acid followed by another round of centrifugation to remove impurities. Size exclusion chromatography was used to separate high molecular weight proteins from the desired protease. This was carried out on a 2.1 L Sephadex G-75 superfine column (Millipore Sigma) equilibrated with 50% acetic acid. The cleanest fractions of HIV protease were refolded into a 10-fold dilution of 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Folded protein was concentrated down to 0.7–2 mg/mL and stored. This stored protease was used in Km and Ki assays. For crystallography, a final purification was performed with a Pharmacia Superdex 75 FPLC column equilibrated with 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Protease fractions purified from the size exclusion column were concentrated to 1–2 mg/mL using an Amicon Ultra-15 10-kDa device (Millipore) for crystallization.
Enzyme Inhibition Assays.
The Km and Ki Assays were carried out as previously described.31,43 In the Km assay, a 10-amino acid substrate containing the natural MA/CA cleavage site with an EDANS/DABCYL FRET pair was dissolved in 8% DMSO at 40 nM and 6% DMSO at 30 nM. The 30 nM of substrate was 4/5th serially diluted from 30 nM to 6 nM, including a 0 nM control. HIV protease was diluted to 120 nM and, using a PerkinElmer Envision plate reader, 5 μL was added to the 96-well plate to obtain a final concentration of 10 nM. The fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts, for approximately 23 minutes. FRET inner filter effect correction was applied as previously described.44 Corrected data were analyzed with Prism7.
To determine the enzyme inhibition constant (Ki), in a 96-well plate, each inhibitor was 2/3 serially diluted from 3 nM to 52 pM, including a 0 pM control, and incubated with 0.35 nM protein for 1 hour. A 10-amino acid substrate containing an optimized protease cleavage site with an EDANS/DABCYL FRET pair was dissolved in 4% DMSO at 120 μM. Using the Envision plate reader, 5 μL of the 120 μM substrate was added to the 96-well plate to a final concentration of 10 μM. The fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts, for approximately 60 min. Data was analyzed with Prism7.
Protein Crystallization.
The condition reliably producing cocrystals of NL4-3 WT protease bound to PIs was discovered and optimized as previously described.45 Briefly, all cocrystals were grown at room temperature by hanging drop vapor diffusion method in a 24-well VDX hanging-drop trays (Hampton Research) with a protease concentration of 1.0–1.7 mg/mL with 3-fold molar excess of inhibitors. Crystallization drops 1 μL protein-inhibitor solution and 2 μL reservoir solution consisting of 22–26% (w/v) ammonium sulfate with 0.1 M bis-Tris-methane buffer at pH 5.5. Drops were micro-seeded with a cat whisker. Diffraction quality crystals were obtained within 1 week. As data were collected at 100 K, cryogenic conditions contained the precipitant solution supplemented with 25% glycerol.
X-Ray Data Collection and Structure Solution.
X-ray diffraction data were collected and solved as previously described.39,45,46 Diffraction quality crystals were flash frozen under a cryostream when mounting the crystal at our in-house Rigaku_Saturn944 X-ray system. The cocrystal diffraction intensities were indexed, integrated, and scaled using HKL3000.47 Structures were solved using molecular replacement with PHASER.48 Model building and refinement were performed using Coot49 and Phenix.50 Ligands were designed in Maestro and the output sdf files were used in the Phenix program eLBOW51 to generate cif files containing atomic positions and constraints necessary for ligand refinement. Iterative rounds of crystallographic refinement were carried out until convergence was achieved. To limit bias throughout the refinement process, five percent of the data were reserved for the free R-value calculation.52 MolProbity53 was applied to evaluate the final structures before deposition in the PDB. Structure analysis, superposition and figure generation was done using PyMOL.54 X-ray data collection and crystallographic refinement statistics are presented in the Supporting Information (Table S2).
Molecular Modeling.
Molecular modeling was carried out using MacroModel (Schrödinger, LLC, New York, NY).55 Briefly, inhibitors were modeled into the active site of wild-type HIV-1 protease using the protease-3 and protease-4 co-complex structures (PDB code: 3O9B and 3O9G).23 Structures were prepared using the Protein Preparation tool in Maestro 11 (Schrödinger, LLC, New York, NY).55 2D chemical structures were modified with the appropriate changes using the Build tool in Maestro. Once modeled, molecular energy minimizations were performed for each inhibitor–protease complex using the PRCG method with 2500 maximum iterations and 0.05 gradient convergence threshold. PDB files of modeled complexes were generated in Maestro for structural analysis.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institute of General Medical Sciences of the NIH (P01 GM109767). We thank Kaneka USA for generous gifts of chiral epoxides, Ellen A. Nalivaika for providing the HIV-1 protease variants, the AIDS Research and Reference Reagent Program (NIAD, NIH) for reference protease inhibitors, and Drs. William Royer, Brian Kelch and members of the Schiffer and Miller laboratories for helpful discussions.
Funding Sources
This work was supported by a grant from the National Institute of General Medical Sciences of the NIH (P01 GM109767).
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type 1
- AIDS
acquired immunodeficiency syndrome
- bis-THF
bis-tetrahydrofuran
- Boc
tert-butoxycarbonyl
- DRV
darunavir
- FRET
fluorescence resonance energy transfer
- MDR
multidrug-resistant
- PIs
protease inhibitors
- SAR
structure-activity relationships
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Synthesis and analytical data of intermediate compounds, Figures S1-S5, Tables S1-S3 (PDF) Molecular formula strings (CSV)
Accessions Codes
The PDB accession codes for X-ray crystal structures of WT HIV-1 protease in complex with DRV and inhibitors 2–4, 12–17 and 22–27 are 6DGX, 6OXO, 6OXP, 6OXQ, 6OXR, 6OXS, 6OXT, 6OXU, 6OXV, 6OXW, 6OXX, 6OXY, 6OXZ, 6OY0, 6OY1, 6OY2. Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- 1.Ghosh AK; Osswald HL; Prato G Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali A; Bandaranayake RM; Cai Y; King NM; Kolli M; Mittal S; Murzycki JF; Nalam MN; Nalivaika EA; Ozen A; Prabu-Jeyabalan MM; Thayer K; Schiffer CA Molecular basis for drug resistance in HIV-1 protease. Viruses 2010, 2, 2509–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen L; Peterson S; Sedaghat AR; McMahon MA; Callender M; Zhang H; Zhou Y; Pitt E; Anderson KS; Acosta EP; Siliciano RF Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med 2008, 14, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampah ME; Shen L; Jilek BL; Siliciano RF Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 7613–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cihlar T; Fordyce M Current status and prospects of HIV treatment. Curr. Opin. Virol 2016, 18, 50–56. [DOI] [PubMed] [Google Scholar]
- 6.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services; Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf (Accessed May 20, 2019). [Google Scholar]
- 7.El Bouzidi K; White E; Mbisa JL; Sabin CA; Phillips AN; Mackie N; Pozniak AL; Tostevin A; Pillay D; Dunn DT HIV-1 drug resistance mutations emerging on darunavir therapy in PI-naive and -experienced patients in the UK. J. Antimicrob. Chemother 2016, 71, 3487–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stockdale AJ; Saunders MJ; Boyd MA; Bonnett LJ; Johnston V; Wandeler G; Schoffelen AF; Ciaffi L; Stafford K; Collier AC; Paton NI; Geretti AM Effectiveness of protease inhibitor/nucleos(t)ide reverse transcriptase inhibitor-based second-line antiretroviral therapy for the treatment of human immunodeficiency virus type 1 infection in sub-Saharan Africa: a systematic review and meta-analysis. Clin. Infect. Dis 2018, 66, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafer RW Human immunodeficiency virus type 1 drug resistance mutations update. J. Infect. Dis 2017, 216, S843–S846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wensing AM; Calvez V; Gunthard HF; Johnson VA; Paredes R; Pillay D; Shafer RW; Richman DD 2017 Update of the drug resistance mutations in HIV-1. Top. Antivir. Med 2017, 24, 132–133. [PMC free article] [PubMed] [Google Scholar]
- 11.Gunthard HF; Calvez V; Paredes R; Pillay D; Shafer RW; Wensing AM; Jacobsen DM; Richman DD Human immunodeficiency virus drug resistance: 2018 recommendations of the international antiviral society-USA panel. Clin. Infect. Dis 2019, 68, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saag MS; Benson CA; Gandhi RT; Hoy JF; Landovitz RJ; Mugavero MJ; Sax PE; Smith DM; Thompson MA; Buchbinder SP; Del Rio C; Eron JJ Jr.; Fatkenheuer G; Gunthard HF; Molina JM; Jacobsen DM; Volberding PA Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the international antiviral society-USA panel. JAMA 2018, 320, 379–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh AK; Chapsal BD; Weber IT; Mitsuya H Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 14.Surleraux DL; Tahri A; Verschueren WG; Pille GM; de Kock HA; Jonckers TH; Peeters A; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu-Jeyabalan M; Schiffer CA; Wigerinck PB Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem 2005, 48, 1813–1822. [DOI] [PubMed] [Google Scholar]
- 15.Tie Y; Boross PI; Wang YF; Gaddis L; Hussain AK; Leshchenko S; Ghosh AK; Louis JM; Harrison RW; Weber IT High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh AK; Rao KV; Nyalapatla PR; Osswald HL; Martyr CD; Aoki M; Hayashi H; Agniswamy J; Wang YF; Bulut H; Das D; Weber IT; Mitsuya H Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants. J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK; P RN; Kovela S; Rao KV; Brindisi M; Osswald HL; Amano M; Aoki M; Agniswamy J; Wang YF; Weber IT; Mitsuya H Design and synthesis of highly potent HIV-1 protease inhibitors containing tricyclic fused ring systems as novel P2 ligands: structure-activity studies, biological and X-ray structural analysis. J. Med. Chem 2018, 61, 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK; Rao KV; Nyalapatla PR; Kovela S; Brindisi M; Osswald HL; Sekhara Reddy B; Agniswamy J; Wang YF; Aoki M; Hattori SI; Weber IT; Mitsuya H Design of highly potent, dual-acting and central-nervous-system-penetrating HIV-1 protease inhibitors with excellent potency against multidrug-resistant HIV-1 variants. ChemMedChem 2018, 13, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki M; Hayashi H; Rao KV; Das D; Higashi-Kuwata N; Bulut H; Aoki-Ogata H; Takamatsu Y; Yedidi RS; Davis DA; Hattori SI; Nishida N; Hasegawa K; Takamune N; Nyalapatla PR; Osswald HL; Jono H; Saito H; Yarchoan R; Misumi S; Ghosh AK; Mitsuya H A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife 2017, 6, e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King NM; Prabu-Jeyabalan M; Nalivaika EA; Schiffer CA Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem. Biol 2004, 11, 1333–1338. [DOI] [PubMed] [Google Scholar]
- 21.Altman MD; Ali A; Reddy GS; Nalam MN; Anjum SG; Cao H; Chellappan S; Kairys V; Fernandes MX; Gilson MK; Schiffer CA; Rana TM; Tidor B HIV-1 protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J. Am. Chem. Soc 2008, 130, 6099–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurt Yilmaz N; Swanstrom R; Schiffer CA Improving viral protease inhibitors to counter drug resistance. Trends Microbiol. 2016, 24, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nalam MN; Ali A; Reddy GS; Cao H; Anjum SG; Altman MD; Yilmaz NK; Tidor B; Rana TM; Schiffer CA Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem. Biol 2013, 20, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulsen JL; Leidner F; Ragland DA; Kurt Yilmaz N; Schiffer CA Interdependence of inhibitor recognition in HIV-1 protease. J. Chem. Theory. Comput 2017, 13, 2300–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surleraux DL; de Kock HA; Verschueren WG; Pille GM; Maes LJ; Peeters A; Vendeville S; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu-Jeyabalan M; Schiffer CA; Wigerinck PB Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J. Med. Chem 2005, 48, 1965–1973. [DOI] [PubMed] [Google Scholar]
- 26.Amano M; Koh Y; Das D; Li J; Leschenko S; Wang YF; Boross PI; Weber IT; Ghosh AK; Mitsuya H A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother 2007, 51, 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windsor IW; Palte MJ; Lukesh JC 3rd; Gold B; Forest KT; Raines RT Sub-picomolar inhibition of HIV-1 protease with a boronic acid. J. Am. Chem. Soc 2018, 140, 14015–14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corey EJ; Bakshi RK; Shibata S Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc 1987, 109, 5551–5553. [Google Scholar]
- 29.Sharpless KB; Amberg W; Bennani YL; Crispino GA; Hartung J; Jeong KS; Kwong HL; Morikawa K; Wang ZM The osmium-catalyzed asymmetric dihydroxylation: a new ligand class and a process improvement. J. Org. Chem 1992, 57, 2768–2771. [Google Scholar]
- 30.Szermerski M; Melesina J; Wichapong K; Loppenberg M; Jose J; Sippl W; Holl R Synthesis, biological evaluation and molecular docking studies of benzyloxyacetohydroxamic acids as LpxC inhibitors. Bioorg. Med. Chem 2014, 22, 1016–1628. [DOI] [PubMed] [Google Scholar]
- 31.Windsor IW; Raines RT Fluorogenic assay for inhibitors of HIV-1 protease with sub-picomolar affinity. Sci. Rep 2015, 5, 11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh AK, Ed. Aspartic Acid Proteases as Therapeutic Targets. Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- 33.Ghosh AK; Fyvie WS; Brindisi M; Steffey M; Agniswamy J; Wang YF; Aoki M; Amano M; Weber IT; Mitsuya H Design, synthesis, biological evaluation, and X-ray studies of HIV-1 protease inhibitors with modified P2' ligands of darunavir. ChemMedChem 2017, 12, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varghese V; Mitsuya Y; Fessel WJ; Liu TF; Melikian GL; Katzenstein DA; Schiffer CA; Holmes SP; Shafer RW Prototypical recombinant multi-protease-inhibitor-resistant infectious molecular clones of human immunodeficiency virus type 1. Antimicrob. Agents Chemother 2013, 57, 4290–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller JF; Andrews CW; Brieger M; Furfine ES; Hale MR; Hanlon MH; Hazen RJ; Kaldor I; McLean EW; Reynolds D; Sammond DM; Spaltenstein A; Tung R; Turner EM; Xu RX; Sherrill RG Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett 2006, 16, 1788–1794. [DOI] [PubMed] [Google Scholar]
- 36.Wei X; Decker JM; Liu H; Zhang Z; Arani RB; Kilby JM; Saag MS; Wu X; Shaw GM; Kappes JC Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother 2002, 46, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SK; Harris J; Swanstrom R A strongly transdominant mutation in the human immunodeficiency virus type 1 gag gene defines an Achilles heel in the virus life cycle. J. Virol 2009, 83, 8536–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.King NM; Melnick L; Prabu-Jeyabalan M; Nalivaika EA; Yang SS; Gao Y; Nie X; Zepp C; Heefner DL; Schiffer CA Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 2002, 11, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozen A; Lin KH; Kurt Yilmaz N; Schiffer CA Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrie KA; Perez EE; Lamers SL; Farmerie WG; Dunn BM; Sleasman JW; Goodenow MM Natural variation in HIV-1 protease, Gag p7 and p6, and protease cleavage sites within gag/pol polyproteins: amino acid substitutions in the absence of protease inhibitors in mothers and children infected by human immunodeficiency virus type 1. Virology 1996, 219, 407–416. [DOI] [PubMed] [Google Scholar]
- 41.Rose JR; Salto R; Craik CS Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem 1993, 268, 11939–11945. [PubMed] [Google Scholar]
- 42.Hui JO; Tomasselli AG; Reardon IM; Lull JM; Brunner DP; Tomich CS; Heinrikson RL Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J. Protein Chem 1993, 12, 323–327. [DOI] [PubMed] [Google Scholar]
- 43.Matayoshi ED; Wang GT; Krafft GA; Erickson J Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 1990, 247, 954–958. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y; Kati W; Chen CM; Tripathi R; Molla A; Kohlbrenner W Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem 1999, 267, 331–335. [DOI] [PubMed] [Google Scholar]
- 45.Lockbaum GJ; Leidner F; Rusere LN; Henes M; Kosovrasti K; Nachum GS; Nalivaika EA; Ali A; Yilmaz NK; Schiffer CA Structural adaptation of darunavir analogues against primary mutations in HIV-1 protease. ACS Infect. Dis 2019, 5, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soumana DI; Kurt Yilmaz N; Ali A; Prachanronarong KL; Schiffer CA Molecular and dynamic mechanism underlying drug resistance in genotype 3 hepatitis C NS3/4A protease. J. Am. Chem. Soc 2016, 138, 11850–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 48.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P; Cowtan K Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004, D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moriarty NW; Grosse-Kunstleve RW; Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 2009, 65, 1074–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunger AT Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 53.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB; Snoeyink J; Richardson JS; Richardson DC MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl. Acids Res 2007, 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.PyMOL: The PyMOL Molecular Graphics System, Version 2.1, Schrödinger, LLC. [Google Scholar]
- 55.Schrödinger. Schrödinger Release 2018–3; Schrödinger, LLC, New York, NY, United States: 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.