

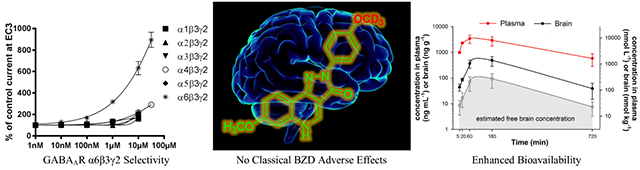
Abstract
Recent reports indicate that α6β2/3γ2 GABAAR selective ligands may be important for the treatment of trigeminal orofacial pain and neuropsychiatric disorders with sensori-motor gating deficits. Based on 3 functionally α6β2/3γ2 GABAAR selective pyrazoloquinolinones, 42 novel analogs were synthesized and their in vitro metabolic stability and cytotoxicity as well as their in vivo pharmacokinetics, basic behavioral pharmacology, and effects on locomotion was investigated. Incorporation of deuterium into the methoxy substituents of the ligands increased their duration of action via improved metabolic stability and bioavailability, while their selectivity for the GABAAR α6 subtype was retained. 8b was identified as the lead compound with a substantially improved pharmacokinetic profile. The ligands allosterically modulated diazepam insensitive α6β2/3γ2 GABAARs and were functionally silent at diazepam sensitive α1β2/3γ2 GABAARs, thus no sedation was detected. In addition, these analogs were not cytotoxic, which render them interesting candidates for treatment of CNS disorders mediated by GABAAR α6β2/3γ2 subtypes.
Graphical Abstract
Introduction
GABAA receptors (GABAARs) are the major inhibitory neurotransmitter receptors in the mammalian brain and the target of many clinically important drugs, which act at GABAAR binding sites. GABAARs are ligand-gated pentameric chloride channels mainly comprised of combinations of 19 different subunits (α1–6, β1–3, γ1–3, δ, ε, π, θ, ρ1–3).1, 2 Classical GABAARs consist of two α, two β and one “tertiary” subunit (γ, δ, ε, θ, or π).1, 3 Due to the vast array of subunit combinations and their diverse regional, cellular and subcellular distribution, a wide-range of pharmacological properties are potentially mediated by these ion channels.2, 4, 5 GABAergic drugs including benzodiazepines (Bzs) and barbiturates modulate GABAARs via allosteric binding sites.6 Many GABAergic drugs on the market today offer little alpha subtype selectivity and thus exhibit undesired side effects (sedation, ataxia, amnesia, tolerance, and dependence), in addition to their therapeutic benefits. Recently, a renewed search for GABAAR subtype selective ligands began7 with the discovery of a new GABAAR αx+βy- ligand binding site.8 The identification of new ligands which specifically interact with this interface might lead to novel therapeutic entities and provided the initial interest in this area of research.9–11
GABAARs containing the α6 subunit (α6βγ2, Figure 1, or α6βδ) were found primarily expressed in the granule cells of the cerebellum,5 where they are located synaptically or extrasynaptically, respectively,12 as well as in the olfactory bulb and the cochlear nucleus.13 Lower expression of α6-GABAARs has recently been reported for the hippocampus where they might play a role in depressive behaviors.14 Recent reports also suggest that α6-containing receptors may play a role in neuropsychiatric disorders with sensori-motor gating deficits (such as tic disorders, certain symptoms of schizophrenia, obsessive compulsive disorder and attention deficit disorders),15, 16 as well as in migraine,17 and trigeminal orofacial pain.18 However, our understanding of the function of α6-containing receptors in the CNS is still in its infancy, and currently it is not possible to indicate with certainty whether α6βγ2, or α6βδ, or both of these receptors, play a major role in the above mentioned disorders. In any case, the synthesis of ligands selective for α6 subunit-containing GABAARs is important to help determine which physiological processes might be mediated by these particular ion channels and if they are a valid target for drug development.
Figure 1:

The GABAAR α6β3γ2 Subtypea
aThe GABAAR α6β3γ2 subtype is a pentameric ligand-gated chloride channel comprised of two α6, two β3 and one γ2 subunits. PQs, like 8b, mediate their effects at the α6+β3- interface (PQ Site) as positive allosteric modulators and act at the DI-Bz site (α6+γ2- interface) as null modulators. The PQ site, the DI-Bz binding site and the endogenous ligand (GABA) binding sites at the β3+α6- interfaces are displayed. Image modified from Jacob et al., Nature Reviews Neuroscience, 2008.
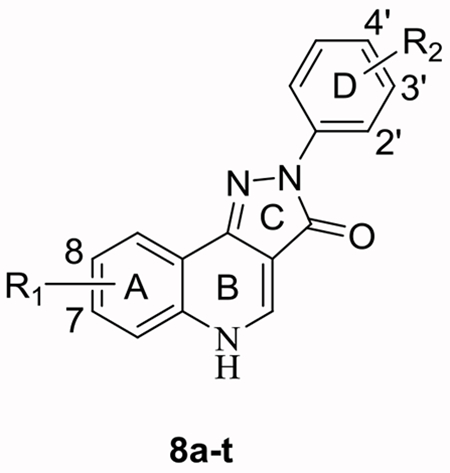
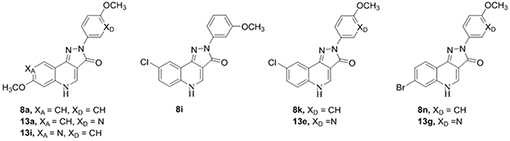
The Ciba-Geigy Corporation reported pyrazoloquinolinones (PQs), CGS 8216 and CGS 9896, as the first nonbenzodiazepine ligands to interact with the Bz site of GABAARs.19, 20 These compounds exhibited Bz site agonist (positive allosteric modulatory) and antagonist (null modulatory) properties at GABAARs, but their receptor binding selectivity between diazepam sensitive (DS, αβγ GABAARs containing α1, α2, α3, or α5 subunits) and diazepam insensitive (DI, αβγ GABAARs containing α4 or α6 subunits) GABAAR subtypes was rather poor.21 Incorporation of a 7-methoxy substituent on the “A-ring” (see Figure 2) of PQs by the Skolnick group resulted in improved DI affinity.22 Deconstruction of the CGS-related compounds via application of the pharmacophore/receptor model for GABAAR subtypes was used to incorporate structural modifications of the “A-ring” and “D-ring” (Figure 2) of PQs. 23, 24 These modifications resulted in the synthesis of PZ-II-029 (8a) and several other ligands which bound tightly to the Bz/GABAAR α6 subtype with a similar affinity as imidazobenzodiazepines.25 The size and position of substituents on the “D-ring” affected the affinity towards the Bz/GABAAR α6 subtype.26, 27 More recently, structural analogues including 8a were found not only to bind to α6β2/3γ2 GABAARs, but also to potentiate these receptors, representing the first functionally selective α6β3γ2 subtype ligands reported to date.10 Prototypes of these ligands mediated their physiological effects at the recently discovered α6+β3- interface8 of GABAARs as positive allosteric modulators, while in addition acting as antagonists (null modulators) at the Bz site (α+γ2- interface) of α1-6βγ2 receptors.10, 11 In a parallel study, the Ernst group synthesized very similar analogs LAU 159 (8i)28 and LAU 463 (8n).29 These compounds exhibited a functional selectivity at α6β2/3γ2 GABAARs comparable to 8a, within the series of α1-6β2/3γ2 receptors. Compounds 8a, 8i and 8n thus represent the three lead compounds described in this study (see Figure 2).29 Since all these PQs exert their positive allosteric modulation via the α6+β3- interface8 of GABAARs, they also are able to modulate α1-6β2/3δ receptors or any receptors containing an α1-6β2/3 interface.30 But the modulation of α1β3δ, α4β3δ, or α6β3δ receptors by all three lead compounds (8a, 8i, 8n) is much weaker than that of α6β3γ2 receptors,29 and was thus not investigated in the present study.
Figure 2:

Lead Ligands of this Worka
aThe lead ligands with the “A-ring” and “D-ring” displayed.
Due to the possibility of O-demethylation in vivo in this series, as well as their limited solubility in water,31 the bioavailability and half-life of these α6β3γ2 GABAAR selective ligands were of concern. One way to enhance the bioavailability of compounds is via selective replacement of hydrogen atoms with deuterium at key metabolic positions on the ligand (see Figure 3). Deuteration has been shown to positively affect the metabolic stability while retaining the pharmacologic profile of active compounds.32 Consequently, the primary deuterium kinetic isotope (D vs H) effect (DIE) was employed here to enhance metabolic stability.33 The kinetic DIE results from a difference in the zero-point energy of C-D and C-H bonds due to the lower vibrational frequency of C-D bonds. Consequently, C-D bond cleavage requires a higher activation energy, followed by a concomitant slower metabolic rate than a C-H bond. Since the lead compounds contained methoxy groups, it was assumed that those would be primary metabolic sites for O-demethylation by cytochrome P450 enzymes.34 Thus, one could take advantage of the stability of the C-D bond to reduce O-demethylation of deuterated analogs.35 Herein, the first synthesis of novel deuterated GABAAR α6 subtype selective ligands is reported, as well as the concomitant improved metabolic profile with respect to their parent OCH3 containing counterparts. Moreover, it was found that their improved metabolic stability resulted in improved bioavailability in vivo and ultimately the achievement of higher brain concentrations via both oral and intraperitoneal (IP) administration, a circumstance that is very important in regard to drug development.
Figure 3:

General Strategy of this Work to Improve the Bioavailability of the Ligands
Another approach to improve ligand bioavailability of CNS-acting drugs is to improve the solubility and lipophilicity since these modifications can impact membrane permeability and enhance their bioavailability and ability to reach the desired biological target.36, 37 Therefore, nitrogen atoms were incorporated into the “A-ring” or “D-ring” of the lead ligands (8a, 8n and 8i) to improve the water solubility (see Figure 3), while still maintaining a reasonable cLogP. In addition to their metabolic stability, GABAAR-α6 subtype selective efficacy and the pharmacokinetic profile of these N-hetero-modified ligands were also studied.
To expand the structure-activity relationship (SAR) library of the GABAAR-α6 subtype selective ligands, a series of 42 structural analogs related to 8a, 8n and 8i were synthesized. GABAAR-α6 subtype efficacy of some of these novel ligands was measured to analyze the effect of deuteration and structural modifications. Furthermore, a binding affinity screen over a panel of 46 receptors (including the hERG) was conducted to determine any possible off-target interactions of these novel compounds. In addition, the effects of these compounds on the rotarod test and on HEPG2 (liver) and HEK293 (kidney) cell lines were examined to screen motor activity and hepato and renal toxicities, respectively. Finally, basic in vivo behavioral tests on the elevated plus maze, grip strength and cued water-maze training were run on these ligands to assess the occurrence of anxiolysis, muscle strength and basic sensorimotor activity, respectively.
Results
Chemistry.
The synthesis of 8a25 and 8i,28 but not that of 8n has been reported before. Since we improved the route of their synthesis as well as their purification, we also report this here. The synthesis of analogs of the α6β2/3γ2-subtype selective ligand 8a,11 which contained deuterium on the methoxy groups of the “A-ring” and/or “D-ring” of the quinolinone nucleus was undertaken. Analogs of 8i and 8n, which also contained deuterium on the methoxy groups of the “D-ring” of the PQ nucleus, were also prepared. To permit an extension of the SARs of possible α6β2/3γ2-selective PQ compounds to be performed, additional deuterated compounds were synthesized, in which the OCD3 group in the “D-ring” was moved to alternative positions, or in which the 8-Cl of 8i or 8k in the “A-ring” was replaced by 8-OCD3 (Table 1). The compounds (8a-t) in Table 1 were generated in a convergent fashion following the steps outlined in Scheme 1. First, the deuterated methoxy phenyl hydrazines (4a-c) were synthesized starting from the appropriate hydroxyanilines (1a-c). Amide protection of the amine functionality was achieved using acetic anhydride. N-Acetylation was accomplished over O-acetylation via strict stoichiometric (1.05 eq.) control of the amount of acetic anhydride. This was followed by installation of the OCD3 groups via alkylation of the hydroxyphenylacetamides (2a-c) using iodomethane-d3 and base to yield the deuterated methoxy phenyl acetamides (3a-c) in high yield. Aryl hydrazines (4a-c) were obtained via a one-pot procedure, which involved the deprotection of the amine with hydrochloric acid, diazonium salt formation with sodium nitrite and finally reduction of the diazonium salts to yield the desired hydrazines using tin (II) chloride.38 As reported by Frahn and Illman,39 control of the temperature (−20°C) of the diazotization and reduction steps was key to avoid elimination of nitrogen from the decomposition of the diazonium salt, especially in the cases of the para- and ortho-methoxy substituted series.39
Table 1:
Ligands Synthesized via the Chemistry in Scheme 1
| ||
|---|---|---|
| Compound | R1 | R2 |
| 8a | 7-OCH3 | 4’-OCH3 |
| 8ba | 7-OCH3 | 4’-OCD3 |
| 8ca | 7-OCD3 | 4’-OCH3 |
| 8da | 7-OCD3 | 4’-OCD3 |
| 8ea | 7-OCD3 | 3’-OCD3 |
| 8fa | 7-OCD3 | 2’-OCD3 |
| 8ga | 7-OCH3 | 3’-OCD3 |
| 8ha | 7-OCH3 | 2’-OCD3 |
| 8i | 8-Cl | 3’-OCH3 |
| 8jb | 8-Cl | 3’-OCD3 |
| 8kb | 8-Cl | 4’-OCH3 |
| 8lb | 8-Cl | 4’-OCD3 |
| 8mb | 8-Cl | 2’-OCD3 |
| 8n | 7-Br | 4’-OCH3 |
| 8oc | 7-Br | 4’-OCD3 |
| 8pc | 7-Br | 3’-OCD3 |
| 8qc | 7-Br | 2’-OCD3 |
| 8r | 8-OCD3 | 4’-OCD3 |
| 8s | 8-OCD3 | 3’-OCD3 |
| 8t | 8-OCD3 | 2’-OCD3 |
Scheme 1:

Synthesis of Deuterated Analogs
The hydrolysis of methoxyphenylacetamides (3a and 3b) yielded deuterated anisidines (5a and 5b) in excellent yields. The 4-hydroxyquinoline derivatives (6a-e) were then prepared in a modified “one pot” procedure following previous reports (6c40 and 6e41) which involved condensation of the substituted anilines (5a-e) with diethyl ethoxymethylenemalonate and this was followed by cyclo-acylation to the quinolone core using the Gould-Jacobs reaction.42 The application of para-substituted anilines typically resulted in higher yields than meta-substituted anilines since the cyclo-acylation with para-anilines resulted in only one 4-hydroxyquinoline regioisomer with the substituent in the 6-position of the final compound. The cyclo-acylation with meta-anilines resulted in the formation of the 5-substituted 4-hydroxyquinolines in addition to the desired 7-substituted 4-hydroxyquinolines in a 1:5 ratio. The undesired 5-substituted 4-hydroxyquinolines were easily removed during the isolation process by using ethanol to selectively dissolve the undesired isomer. The 4-chloroquinolines (7a-e) were obtained by chlorination of the respective 4-hydroxyquinolines (6a-e) by generation of the catalytic Vilsmeier reagent in situ using oxalyl chloride and a catalytic amount of N,N-dimethylformamide43 nin high yields (80-90%). Typically, the hydrochloride salts of the 4-chloroquinolines were formed in the chlorination reactions and the free bases were generated by quenching the reaction mixtures into aqueous potassium carbonate solution during the isolation of 4-chloroquinolines (7a-e). Utilization of the catalytic Vilsmeier reagent simplified the purification and improved the yield and safety of this chlorination reaction over the reported phosphorus oxychloride (7c)41 and thionyl chloride (7e)40 methods.
Finally, PQs (8a-t) were obtained by the convergent coupling of phenylhydrazines (4a-e) with appropriate chloroquinolines (7a-e) by simple modification of the previous work of Ciba-Geigy19, 20 in mixed xylenes at 135-140°C. A similar procedure was reported in the deconstruction approach to PQs by He et al.26 In all cases, excess triethylamine (1.2 eq.) was used to scavenge the hydrogen chloride formed from the nucleophilic aromatic substitution (SNAr) of chlorine by the primary amino group of the respective phenylhydrazines. Following the SNAr reaction, thermal cyclization via the secondary amine of the phenylhydrazine occurred with subsequent elimination of ethanol to form the PQ core. Upon completion of the cyclization, the reaction mixtures were quenched into ethanol and filtered to yield the pure PQs (8a-t) in 40 - 60% yields. This improved procedure required no further purification in contrast to the previously reported methods.40, 44 In cases where phenylhydrazine hydrochloride salts were employed, the amount of triethylamine was adjusted to accommodate for the additional hydrogen chloride.
To increase the solubility and lipophilicity of the lead compounds, the “A-ring” and “D-ring” N-hetero PQs (13a-i) in Table 2 were synthesized in a convergent fashion following the steps depicted in Scheme 2 and Scheme 3. Initially, to install the “N-hetero” methoxypyridyl groups into the “D-ring” of PQs (13a-h), pyridylhydrazines (12a-b) were synthesized. The 2-methoxy-d3-5-nitropyridine (10) was generated in 87% yield by reacting 2-chloro-5-nitropyridine (9) in deuterated methanol (CD3OD) as the solvent in the presence of potassium t-butoxide. Reduction of the nitro group with iron metal in aqueous hydrogen chloride yielded 5-amino-2-methoxy-d3-pyridine (11a) in 95% yield. The aminopyridines (11a-b) were converted into their respective pyridylhydrazines (12a-b) using the same procedure via diazonium salt formation and tin reduction, as described above for aryl hydrazines (4a-c). A similar procedure for the synthesis of 12b has been reported.45 Finally, reaction of the pyridylhydrazines (12a-b) with chloroquinolines (7a-e) yielded PQs (13a-h) in moderate yields (40-75%) using the same condensation process as described above.
Table 2:
Scheme 2:

Synthesis of “D-Ring” N-Hetero Analogs
Scheme 3:

Synthesis of “A-Ring” N-Hetero Analogs
The “A-ring” substituted N-hetero PQ (13i) provided a unique challenge (Scheme 3). The poor electron density of the amino pyridine ring (11c) disfavored the Gould-Jacobs cyclization as compared to the previous syntheses that were described. The lower yields (15%) to form 4-hydroxynaphthyridine (6f) was not desirable. In addition, chlorination of 4-hydroxynaphthyridine (6f) using the catalytic Vilsmeier reagent was also not as selective as in the 4-hydroxyquinoline series. Cleavage of the pyridyl methoxy group under the acidic reaction conditions to a pyridone appeared to be the problem, which resulted in degradation. Despite, degradation of the product which resulted in a low overall yield (40%) of 4-chloronapthridine (7f), as mentioned, the pure product was isolated. Lastly, the reaction of 4-methoxyphenylhydrazine hydrochloride (4d) with 4-chloronapthyridine (7f) to form PQ (13i) proceeded in 69% yield.
The synthesis of 14 additional structural analogs (14a-n) of the parent compounds were also pursued to further expand the SAR library of GABAAR-α6 subtype selective ligands (see (S) Table S4 and Scheme S1). These analogs incorporated substituent modifications of the “A-ring” and “D-ring” of the PQ core which included: bromo-, chloro-, fluoro-, methoxy-, methyl-, nitro-, trifluoromethyl-, and trifluoromethoxy- groups (see ((S) Experimental).
Metabolic Stability with Human Liver (HLM) and Mouse Liver (MLM) Microsomes.
Since it was assumed that the α6β3γ2 selectivity of deuterated ligands would not be significantly different from that of non-deuterated ligands, and since the generation of compounds with an increased metabolic stability was the main aim of this study, the compounds synthesized were first investigated for their metabolic stability. The deuterated analogs displayed improved metabolic stability in both HLM and MLM, as compared to their respective nondeuterated parent compounds in all cases (Table 3). The non-deuterated parent ligands 8a and 8i both exhibited similar HLM-mediated metabolic half-lives of 3.6 and 3.4 hours, respectively, while 8n displayed the most rapid metabolic rate of 1.7 hours in the presence of HLM. In MLM, 8a exhibited a half-life of 3.2 hours and 8n displayed a half-life of 1.9 hours, while 8i displayed the most rapid metabolic rate in MLM with a half-life of 1.5 hours. Among the 8a series, deuteration of the “A-ring” methoxy group (8c) exhibited a three-fold increase in stability in the presence of HLM and a four-fold increase of stability in MLM with respect to the parent ligand. Deuteration of the “D-ring” methoxy group (8b) exhibited a 2.5-fold increased half-life in HLM and a three-fold increase in MLM, as compared to 8a. These results indicated a slightly faster metabolism of the “A-ring” methoxy group over the “D-ring” methoxy group in the 8a parent series within experimental error. The comparison of metabolic stability in the presence of HLM of “D-ring” meta-OCD3 isomers between “A-ring” OCD3 ligand 8e and “A-ring” OCH3 ligand 8g further supports the evidence of faster metabolism of the “A-ring” versus the “D-ring” methoxy group. Additionally, a similar trend was observed for the “D-ring” ortho-OCD3 isomers between “A-ring” OCD3 ligand 8f and “A-ring” OCH3 ligand 8h. The % percent remaining after 1 hour in the presence of HLM and MLM for each of the three deuterated ligands (8b, 8c, and 8d) was virtually identical between 91.5 and 93.1%, within experimental error. In the case of 8d, deuteration of both methoxy groups did not have an additive effect on the metabolic stability in comparison with the mono-OCD3 analogs 8b and 8c. Thus, from a cost and ease of synthesis point of view the mono-OCD3 substituted analogs were pursued versus the di-OCD3 substituted analog 8d for further studies. In the 8i series, the deuterated analog 8j exhibited a three-fold increase in HLM-mediated half-life and a two-fold increase in MLM-mediated half-life. Similarly, the deuterated analog 8o of the parent 8n exhibited a two-fold increase in HLM-mediated half-life and a 20 % increase in MLM-mediated half-life. In summary, deuteration of the methoxy substituent(s) resulted in the desired improvement in metabolic stability in the presence of both HLM and MLM for the compounds.
Table 3:
Metabolic Stability in the Presence of HLM and MLMa
| Compound | Half-life (HLM) (hr) | % left after 1 hr. (HLM) | Half-life (MLM) (hr) | % left after 1 hr. (MLM) |
|---|---|---|---|---|
| 8ap | 3.6 ± 0.6 | 86.10 ± 0.71 | 3.2 ± 0.1 | 80.35 ± 0.10 |
| 8bb,p | 8.7 ± 0.6 | 91.69 ± 0.09 | 10.5 ± 0.9 | 92.80 ± 0.07 |
| 8cb,p | 11.1 ± 3.6 | 92.00 ± 0.21 | 14.3 ± 2.8 | 93.17 ± 0.11 |
| 8db,p | 13.0 ± 3.0 | 91.51 ± 0.13 | 14.1 ± 3.2 | 93.08 ± 0.14 |
| 8eb,m | 7.7 ± 1.1 | 89.50 ± 0.14 | 9.9 ± 1.9 | 90.50 ± 0.15 |
| 8fb,o | 9.9 ± 1.5 | 90.50 ± 0.14 | 13.9 ± 4.2 | 92.00 ± 0.18 |
| 8gb,m | 3.9 ± 0.3 | 81.28 ± 0.15 | 14.5 ± 4.2 | 91.56 ± 0.23 |
| 8hb,o | 3.5 ± 0.2 | 79.92 ± 0.14 | 10.9 ± 1.5 | 93.04 ± 0.09 |
| 13ab,e | 13.2 ± 2.2 | 92.85 ± 0.10 | 2.3 ± 0.1 | 74.77 ± 0.09 |
| 13bb,e | 9.9 ± 1.1 | 92.58 ± 0.09 | 10.7 ± 1.2 | 92.39 ± 0.09 |
| 13cb,e | 12.5 ± 3.1 | 92.30 ± 0.10 | 9.2 ± 0.9 | 91.38 ± 0.09 |
| 13db,e | 9.8 ± 2.8 | 90.00 ± 0.32 | 14.5 ± 4.8 | 92.50 ± 0.23 |
| 13ib,f | 10.6 ± 2.7 | 91.00 ± 0.20 | 12.0 ± 4.0 | 93.00 ± 0.30 |
| 8im | 3.4 ± 0.2 | 81.44 ± 0.16 | 1.5 ± 0.1 | 59.83 ± 0.17 |
| 8jc,m | 10.8 ± 1.0 | 93.00 ± 0.09 | 2.8 ± 0.1 | 76.59 ± 0.08 |
| 8kc,p | 2.3 ± 0.1 | 72.50 ± 0.11 | 1.9 ± 0.1 | 69.20 ± 0.11 |
| 8lc,p | 10.7 ± 1.5 | 92.20 ± 0.15 | 4.4 ± 0.3 | 85.54 ± 0.05 |
| 8mc,o | 16.5 ± 1.8 | 94.80 ± 0.07 | 16.6 ± 2.0 | 94.91 ± 0.07 |
| 13ec,e | 8.5 ± 0.5 | 91.60 ± 0.10 | 4.4 ± 0.2 | 85.62 ± 0.08 |
| 13fc,e | 12.1 ± 1.9 | 93.00 ± 0.12 | 4.2 ± 0.2 | 85.11 ± 0.07 |
| 8np | 1.7 ± 0.1 | 66.86 ± 0.13 | 1.9 ± 0.1 | 68.96 ± 0.12 |
| 8od,p | 3.4 ± 0.1 | 81.50 ± 0.11 | 2.3 ± 0.1 | 73.85 ± 0.11 |
| 8pd,m | 1.8 ± 0.1 | 63.50 ± 0.32 | 3.6 ± 0.3 | 79.50 ± 0.20 |
| 8qd,o | 14.2 ± 3.0 | 92.85 ± 0.11 | 1.5 ± 0.3 | 61.31 ± 0.15 |
| 13gd,e | 12.1 ± 1.4 | 93.40 ± 0.11 | 11.4 ± 1.4 | 93.13 ± 0.09 |
| 13hd,e | 38.6 ± 7.7 | 97.80 ± 0.06 | 70.8 ± 25.8 | 98.39 ± 0.04 |
Compounds were incubated with HLM or MLM for 60 minutes and quantified by LC-MS/MS.
8a related analogs in red.
8i related analogs in blue.
8n related analogs in green.
D-ring N-hetero analogs.
A-ring N-hetero analogs.
D-ring “ortho”.
D-ring “meta”.
D-ring “para”.
Interestingly, the position of the methoxy substituent on the “D-ring” also seemed to have an impact on the metabolic stability of the PQs, and the effects observed differed in HLM and MLM, as indicated by a comparison of ortho-OCD3 ligands with their para-OCD3 analogs (8e vs 8d; 8h vs 8b; 8m vs 8l; 8q vs 8o). A similar mixed half-life improvement in the presence of HLM and MLM was observed when meta-OCD3-isomers were compared with the para-OCD3 series (8c vs 8d; 8g vs 8b; 8j vs 8l; 8p vs 8o). The introduction of ortho-OCD3 substituents appeared to have the largest effect on metabolic stability in the 8i and 8n series (8m and 8q, respectively), but notably this effect was not the case in the 8a series (8f and 8h). Meta-OCD3 substituted ligands generally had the least improvement on metabolic stability in all three series (8e, 8g, 8j, and 8p). Clearly, the observed mixed effect on metabolic stability governed by the position of the OCD3 group on the “D-ring” was in line with the activity of CYP450 enzymes in HLM versus MLM. Therefore, it is not possible, at present, to predict trends in the metabolic stability between the para- versus ortho- versus meta-methoxy substituted analogs as the HLM and MLM enzymes appeared non-discriminate in regards to the position of the methoxy group on the “D-ring” during the microsomal studies.
The N-heterocyclic ligands also exhibited better metabolic stability than the parent compounds. The N-hetero ligands are more polar (see cLogP data in Table 4) than their respective parent ligands and therefore should not undergo metabolism as quickly as the more lipophilic parent ligands, since the CYP450 enzymes primarily consist of lipophilic membrane-associated proteins.46 Here, the N-hetero ligand 13a when compared to the parent ligand 8a exhibited a four-fold increase in half-life in the presence of HLM, yet a slightly decreased half-life was observed in the presence of MLM. Interestingly, the “A-ring” N-hetero ligand 13i displayed a four-fold increase in the presence of both HLM and MLM. Similar improvement was seen when 13e was compared to its parent compound 8k where a nearly four-fold increased half-life (HLM) and a two-fold increased half-life (MLM) was observed. In the case of 8n, the N-hetero ligand 13g exhibited a seven-fold increase in half-life in both HLM and MLM which is significant.
Table 4:
Solubility and cLogP of Compounds Indicating the Effect of Adding the Nitrogen Atom to the A- or D-Ringa
| |||
|---|---|---|---|
| Compound | cLogPe | Solubility (μM) in DI Water, pH = 7f |
Solubility (μM) in 0.01M HCl, pH = 2f |
| 8a | 2.76 | 24.3 ± 3.8 | 27.8 ± 0.9 |
| 13ab | 2.17 | 67.2 ± 7.9 | 115.4 ± 14.9 |
| 13ib | 2.28 | 103.7 ± 28.5 | 135.9 ± 2.9 |
| 8i | 3.56 | 6.8 ± 0.3 | 2.6 ± 0.2 |
| 8kc | 3.56 | 6.4 ± 0.5 | 8.5 ± 1.9 |
| 13ec | 2.93 | 0.8 ± 0.1 | 0.9 ± 0.2 |
| 8n | 3.71 | 8.3 ± 0.2 | 3.3 ± 0.1 |
| 13gd | 3.08 | 7.3 ± 0.2 | 7.2 ± 0.4 |
Solubility experiments performed in water at pH = 7 and pH =2 to mimic physiological and gastrointestinal conditions and to examine the impact of N-hetero substitution on hydrogen chloride salt formation.
8a related analogs in red.
8i related analogs in blue.
8n related analogs in green.
cLogP values calculated using ChemBioDraw® Ultra (ver.13.0.0.3015).
Calculated using an LCMS. n = 3
Notably, the effect of incorporation of both the “D-ring” N-hetero (pyridine-like) substituents into and the deuterated methoxy groups onto the lead ligands, exhibited even further improvement in the metabolic stability over either case individually. Here, the OCD3 substituted N-hetero ligand 13f when compared to the OCH3 substituted N-hetero ligand 13e exhibited a 40% increased half-life (HLM) and no change in respect to half-life in the presence of MLM. Additionally, the OCD3 substituted N-hetero ligand 13h exhibited a three-fold increased stability (HLM) and six-fold increased stability (MLM) with respect to their half-lives when compared to the OCH3 substituted N-hetero ligand 13g. Of special note, the OCD3 substituted N-heterocyclic compound 13h displayed the best metabolic stability of all the analogs in the series with half-lives of 38.6 hours (HLM) and 70.8 hours (MLM), respectively.
The effect of deuteration of the “A-ring” and / or “D-ring” methoxy groups was also examined in the case of the 13a related analogs (13b, 13c and 13d). A definitive metabolic rate difference between the “A-ring” methoxy group and the “D-ring” methoxy group (see 13c, “A-ring” OCD3 group and 13b, “D-ring” OCD3 group) as compared to 13a was not evident; the effect was much more subtle than the metabolic stability observed in the α6 selective 8a series above and again was different in the presence of HLM or MLM. In the case of di-OCD3 analog 13d, deuteration of both methoxy groups did not have an additive effect on the metabolic stability in comparison to the mono-OCD3 analogs 13c and 13b. Obviously, the concurrent effect on the metabolic stability between N-hetero and OCD3 substitution is too complex to draw clear conclusions on the impact of deuteration of the “A-ring” versus “D-ring” methoxy groups in the case of 13a related analogs. It is very possible that retarding the rate of the O-demethylation reaction in both the “A-ring” and “D-ring” has directed the metabolism rapidly towards another metabolic pathway.
Metabolite Study.
Analysis of the metabolites from the HLM and MLM study confirmed that the primary metabolic function of CYP450 enzymes34 with respect to methoxyarylpyrazoloquinolinones proceeded by O-demethylation of the methoxy groups (OCH3) in either the “A-ring” or “D-ring” to form hydroxyarylpyrazoloquinolinones. This is in agreement with Nelson et al.35 Furthermore, this study confirmed the kinetic DIE33 and illustrated that kH > kD for compounds with both a OCD3 and a OCH3 group in either the “A-ring” or “D-ring” (Scheme 4) was in operation. Qualitative assessment by mass spectroscopy confirmed for “D-ring” OCD3 substituted ligand 8b (Scheme 4A), that primary metabolite I (in bold) existed at a much higher concentration than metabolite II and the di-desmethoxy metabolite III in both (HLM and MLM) metabolism studies after sixty minutes. Similarly, for “A-ring” OCD3 substituted ligand 8c (Scheme 4B), primary metabolite IV (in bold) was present at a higher concentration than either metabolite V or VI. Examples A and B in Scheme 4, clearly indicated that the kinetic DIE predominated in the metabolic pathway (kH > kD ) of the O-demethylation (OCD3 versus OCH3) of these ligands over which ring (“A-ring” or “D-ring”) the deuterated versus non-deuterated methoxy groups was placed upon.
Scheme 4:

Metabolite Study. O-Demethylation of OCH3 Versus OCD3 by CYP450 Enzymes in HLM and MLMa
aMetabolites found confirmed kH > kD in both HLM and MLM using an LCMS. kH = O-demethylation of OCH3, kD = O-demethylation of OCD3, CYP450 = metabolism by Cytochrome P450 enzymes. Primary metabolites (I, IV, VII and X) are highlighted in examples A and B respectively.
For the case of N-hetero substituted ligand 13b (Scheme 4A), primary metabolite VII (in bold) existed at a much higher concentration than metabolite VIII and the di-desmethoxy metabolite IX in both the HLM and MLM metabolism study after 60 minutes. Similarly, for “A-ring” OCD3 substituted ligand 13c (Scheme 4B), primary metabolite X (in bold) was present at a higher concentration than either metabolite XI or XII. Examination of examples A and B in Scheme 4 not only indicated that substitution of OCD3 for OCH3 predominated during metabolism over the effect of which ring (“A-ring” versus “D-ring)” the deuterated methoxy group was placed on, but also showed that the kinetic DIE prevailed over the effect of N-hetero substitution onto the “D-ring” of the ligands on the rate of metabolism.
In the cases of 8o and 8j, it was also confirmed (see Scheme 4C and 4D) that the primary metabolic pathway occurred via O-demethylation of the methoxy groups since primary metabolite XIII was found for 8o and primary metabolite XIV was found for 8j. In summary, this metabolite study confirmed the importance of deuteration of the methoxy groups to positively impact the metabolic stability of this series of GABAAR α6 subtype selective ligands. In addition, the metabolic rate of these ligands was governed predominately by the kinetic DIE over the position of the deuterated versus non-deuterated methoxy group on the “A-ring” versus “D-ring” or the impact of a “D-ring” N-hetero substituent, as mentioned above.
Study of GABAAR α6β3γ2 Subtype Efficacy and Selectivity.
The three lead ligands (8a, 8n and 8i) all displayed a pronounced functional selectivity for the GABAAR α6β3γ2 subtype,10, 29 and modulated the EC3-GABA induced currents strongly with efficacies at 30 μM ranging from ~400% (8i) to ~ 900% (8a and 8n) potentiation (Figure 4). Electrophysiological experiments confirmed that within experimental error, replacement of OCH3 by OCD3 groups did not significantly change the efficacy of five different deuterated ligands for modulating GABA-induced currents at the α6β3γ2 GABAAR subtype (Figure 5), as compared to the three lead compounds. It thus could be assumed that this also would hold true for the efficacy of these ligands for modulating GABA-induced currents at the other receptor subtypes that was zero or close to zero for the three lead compounds up to a concentration of 1 μM (Figure 4), and that these compounds would exhibit a comparable α6β3γ2 GABAAR selectivity. Specifically, the deuterated analogs of 8a (8b, 8c, 8d) and 8n (8o) exhibited the best efficacy and selectivity.
Figure 4:

GABAAR α6β3γ2 subtype selectivity of the lead ligands: 8a, 8n and 8ia
aConcentration response curves of the change of GABA EC3 currents (modulation, referenced to 100% for the EC3 current) by increasing compound concentrations. 8a (A), 8n (B) and 8i (C) preferentially modulate GABA currents of α6β3γ2 receptors.
Figure 5:

The effect of substituting OCD3 for OCH3 groups on the lead ligands on GABAAR α6β3γ2 subtype activitya
aElectrophysiological experiments confirmed that OCD3 groups do not change the activity (within experimental error) of the ligands for the α6β3γ2 subtype. 8a related ligands (A). 8n related ligands (B). 8i related ligands (C).
In a different series of compounds based on PZ-II-028 (8k)25 which exhibited higher efficacy but poor α6 selectivity, as compared with 8a,10 the methoxy group on the “D-ring” was moved from para to meta (8i) and to ortho (8m) positions. These modifications dramatically changed the properties of the compounds. As a result the meta-OCH3 substituted compound, LAU 159 (8i), in contrast to its non-selective parent compound 8k was a selective modulator of the α6β3γ2 subtype,28 while the ortho-OCD3 analogue 8m was completely inactive (Figure 6). The same turned out to be true for the non-deuterated analogue of 8m (LAU 165).28 Since LAU 165 was not able to inhibit the effects of 8i,28 it might not be able to bind to the α6+β3- interface. Since deuteration does not change the activity of compounds at the receptor, this conclusion also holds true for 8m. Consequently, 8m can be further investigated in in vivo studies as a negative control compound from the same chemotype to establish that an observed biological effect was connected with the modulation of the α6β3γ2 subtype.
Figure 6:

GABAAR α6β3γ2 subtype activity of “D-ring” ortho-OCD3 isomer 8ma
aTwo-point electrophysiological experiments displayed that ligand 8m with a “D-ring” ortho-OCD3 substituent and “A-ring” 8-chloro substituent had no activity at the GABAAR α6β3γ2 subtype.
In an additional study, ligands with heteroatoms in the core scaffold with substituents at positions analogous to the parent ligands (8a, 8n and 8k) were also investigated. Incorporation of the N-hetero atoms into the “A-ring” or “D-ring” of the PQ core was accompanied by a drop in efficacy at the α6β3γ2 subtype relative to parent compounds 8a and 8k, but not relative to the parent compound 8n, as revealed in Figure 7. Four compounds retained some efficacy (13b, 13c, 13h, and 13i), while one N-hetero ligand 13f displayed much poorer efficacy. The deuterated N-hetero analog 13c was then characterized in the whole panel of six receptors (Figure 8). While the maximum efficacy of 13c was weaker than that for the lead ligands, the α6 subtype selectivity was very good and if its bioavailability is as good or better, this compound may be a good alternative to 8a for use in in vivo studies.
Figure 7:

The effect of N-hetero substitution onto the “A-ring” or “D-ring” of the lead ligands on GABAAR α6β3γ2 subtype activitya
aElectrophysiological experiments exhibited that N-hetero substitution onto the “A-ring” or “D-ring” of the parent ligands decreased the activity of the ligands for the α6β3γ2 subtype. 8a related ligands (A). 8n related ligands (B). 8k related ligands (C).
Figure 8:

GABAAR α6β3γ2 subtype selectivity of the N-hetero ligand 13ca
aConcentration response curves of the change of GABA EC3 currents (modulation, referenced to 100% for the EC3 current) by increasing compound concentrations. Ligand 13c preferentially modulated GABA currents of α6β3γ2 receptors. In the α1-5β3γ2 receptors, lack of activity was confirmed in two-point experiments.
PDSP Receptor Study.
In addition to this GABAAR α6β3γ2 subtype selectivity, a screening of the ligands against a panel of 46 receptors, transporters and ion channels including the hERG channel confirmed selective binding of these ligands towards the Bz/GABAA receptors ((S) Table S1). The 8i related analogs only displaced the radiolabeled probe [3H]flunitrazepam from the benzodiazepine (BZP) rat brain site of GABAARs, except for 8l, that also weakly interacted with DOR ([3H]DADLE). No affinity of these analogs was observed for the other 45 receptors ((S) Table S1). The BZP rat brain site is a homogenized mixture of rat brain cells and membranes consisting of all GABAAR subtypes (α1-α6) where the binding affinity reflects affinity at the α+γ- site. The 8a related structures showed no activity in all assays except the BZP rat brain site of GABAARs, serotonin (5-HT7, [3H]LSD), and μ-opioid (MOR, [3H]DAMGO) receptors. However, individual ligands such as 8a, also showed activity at Dopamine D5 ([3H]SCH233930), or 13a at Muscarinic M4 ([3H]QNB) receptors. The 8n related structures again only showed activity at the BZP rat brain site of GABAARs, but individual compounds weakly interacted also with Serotonin 5-HT2B ([3H]LSD), 5-HT2C ([3H]Mesulergine) and 5-HT7 ([3H]LSD) receptors, or μ-opioid (MOR, [3H]DAMGO) receptors. Most of these individual additional interactions resulted in about 50% inhibition of binding of the respective ligand at a 10 μM drug concentration, suggesting an IC50 of about 10 μM. Secondary binding studies ((S) Table S2) were then conducted to determine the binding affinities for any ligand that displaced >50% radioligand in the primary (PDSP) assay, which demonstrated that off-target binding for most ligands was 1,000 times less potent compared to the primary target. The ortho isomers of 8b and 8o (8h and 8q, respectively) exhibited significantly less binding to the BZP rat brain site, while the ortho-OCD3 isomer of 8i (8m) still displayed a reasonable binding affinity to the BZP rat brain site, but less as compared to its para-OCD3 isomer 8l. Thus, the ortho isomer 8m was identified as a control compound because of its lack of efficacy exhibited at the GABAAR α6β3γ2 subtype, as described above. Overall, the library of potential α6 ligands do have high specificity for binding at the BZP rat brain site and importantly do not bind at the hERG channel nor any of the other 44 receptors, transporters and channels examined in a significant fashion. However, it cannot be ruled out from the PDSP assays that these compounds do not interact with binding sites at these receptors for which no radiolabeled ligands are available or at other receptors, transporters and channels that were not investigated in this study. Thus, although it is proposed that these α6 subtype selective ligands bind at both the α6+β3- (PQ) site as positive allosteric modulators and at the α6+γ2- (DI-Bz) site as null modulators of GABAARs,8, 10, 11 there is no way to assess their binding affinity at the α6+β3- (PQ) site at present. Deuterated and N-hetero analogs of the parent compounds did not display a meaningful change in binding behavior in the panel of radioligand binding assays explored (B. Roth et al., NIMH Psychoactive Drug Screening Program, UNC, available at http://pdsp.med.unc.edu) when compared to the parent OCH3 lead compounds.
Study of Solubility and cLogP.
The introduction of N-hetero atoms into either the “A-ring” (13i) or “D-ring” (13a) of α6 ligands such as 8a increased the water solubility, as compared to the parent ligands in both neutral and acidic media only significantly in the case of the lead ligand 8a (see Table 4). In the case of 8k, introduction of a pyridine-like nitrogen atom into the “D-ring” (13e) actually decreased the water solubility by almost 10-fold in both neutral and acidic aqueous media. Additionally, N-hetero introduction into the “D-ring” of 8n resulted in only a minimal impact on the neutral and acidic solubility in the case of 13g. Finally, 8i had similar solubility as 8k and 8n. Ultimately, the aqueous solubility for 8a was the best over the other non-N-hetero substituted ligands; however, N-hetero ligands 13i and 13a displayed significant improvement in water solubility. The acidic water solubility in 0.01 M HCl (pH = 2) of the ligands did not improve over the water solubility (pH = 7) substantially in any of the cases explored, presumably because the nitrogen of the N-hetero substituent lacks sufficient basicity to positively impact hydrogen chloride salt formation. Because the “A-ring” analog 13i was difficult to synthesize, only the deuterated analogs of 8a and 13a were carried forward for pharmacokinetic studies. Of note, the calculated cLogP (octanol-water partition coefficient37) values (Table 4) also dropped in all cases for the N-hetero analogs as expected; however, this did have an important positive effect on metabolic stability, as indicated by a comparison of 8a and 13a, 8k and 13e, or 8n and 13g. Interestingly, however, N-hetero substitution together with deuteration resulted in an 11-fold increase in metabolic stability in HLM, and a 30-fold increase in stability in MLM in the 8n series (compare 8o and 13h in Table 3).
Study of Potential Cytotoxicity in HEK293 (Kidney) cells and HEPG2 (Liver) cells.
An examination of the cell viability, indicated that these ligands are non-toxic even at 400 μM in the presence of HEK293 as well as HEPG2 cell lines (see (S) Table S3 and Figure S1 for details). In both cell lines, substitution of OCD3 for OCH3 in either the “A-ring” or “D-ring” of the parent ligands did not have a negative impact on cell viability. Additionally, variation of the substitution pattern of the methoxy groups on the “D-ring” to the meta position (8e and 8j) or ortho position (8f and 8m) had no impact on cytotoxicity. Finally, N-hetero atom insertion into the “D-ring” to provide 13c, 13e and 13g also had no impact of cell viability. All of the potential α6β3γ2 subtype selective ligands tested here were nontoxic.
Study of the Lack of Rotarod Motor Impairment.
Analysis of the data from the rotarod studies indicated that none of the GABAAR-α6 subtype selective PQ analogs were incapacitating in mice on the rotarod when dosed by oral gavage at 40mg/kg. Deuteration of the methoxy groups of the parent ligands ((S) Figure S2), and introduction of N-hetero substituents into the “D-ring”, as well as deuteration of the N-hetero analogs ((S) Figure S2) resulted in no further change of the physiological response in contrast to the effect on motor impairment of diazepam at 5mg/kg.
Pharmacokinetic Study.
In vivo plasma and brain kinetic profiles of six ligands (8a, 8b, 8c, 8d, 13c, and 8m) suspended in a standard vehicle47 and dosed by IP injection at 10 mg/kg in male Wistar rats, are illustrated in Figure 9. The time-concentration curves of all ligands tested indicated they were rapidly absorbed and distributed in the brain. The non-deuterated molecule 8a demonstrated a low brain-to-plasma partition coefficient (Kp = AUC0-t, brain/AUC0-t, plasma) of 0.08 ± 0.01. The three deuterated analogs 8b, 8c and 8d had increased Kp values, equal to 0.13 ± 0.01, 0.16 ± 0.02, and 0.17 ± 0.02, respectively, together with longer half-lives, both in plasma and brain. The brain AUC0-12 value of the N-hetero ligand 13c was only 45%, 36% and 55% of that obtained with 8b, 8c and 8d, respectively. The ortho-OCD3 substituted ligand 8m exhibited a reasonable Kp value of 0.15 ± 0.01 which additionally illustrated its usefulness in further in vivo studies as a control compound with a favorable pharmacokinetic profile.
Figure 9:

Pharmacokinetic Profiles of Select Ligandsa
aPlasma and brain concentration–time profiles of 8a (A), 8b (B), 8c (C), 8d (D), 13c (E), and 8m (F) after IP administration of a 10 mg kg−1 dose (n = 3 per time point). Cmax = maximum concentration in brain or plasma; tmax = time of maximum concentration in brain or plasma; t1/2 = terminal elimination half-life; AUC0–12 = area under the concentration versus time curve from zero to last measurable time point.
Because the three deuterated analogs of 8a had generally similar values of pharmacokinetic parameters, it was decided to pursue the study of 8b and 8d. The key ligands 8b and 8d were employed for calculation of the estimated free concentrations available for binding to receptors in brain tissue and free fractions were found to be 0.194 and 0.105, respectively. These values gave rise to more than two-fold higher AUC values of estimated free brain concentrations of 8b as compared to 8d (651.72 ± 217.70 ng h g−1 and 288.78 ± 61.07 ng h g−1, respectively). Having in mind the electrophysiological activity presented in Figure 5A, the free (unbound) brain concentrations of 8b in the range close to 100 nM were sufficiently high to mediate α6β3γ2 modulation in vivo. Hence, the “D-ring” substituted OCD3 ligand 8b was chosen to be tested in a battery of basic behavioral paradigms. The “A-ring” OCD3 ligand 8c was not pursued further because it requires more steps to synthesize than ligand 8b.
The concentration–time profiles of 8a and 8b in rat plasma and brain after oral (gavage) as well as after intravenous (IV) administration, with the calculated pharmacokinetic parameters, are presented in Figure 10. The values of absolute oral bioavailability of 8a and 8b from nanoemulsion formulations, calculated on the basis of the plasma AUC values obtained following two routes of administration, were 66.9% and 100.0%, respectively. These data reveal that deuteration of 8a resulted in optimization of pharmacokinetic behavior after oral administration, which was further supported by respective values of brain bioavailability of 8a and 8b (82.6% vs. 89.5%).
Figure 10:

Pharmacokinetic profiles in plasma and brain following oral gavage (PO) or IV administration of 8a (A) and 8b (B)a
aPlasma and brain absolute bioavailability profiles of 8a (A), and 8b (B) presented in respective graphs as Fsystemic and Fbrain is calculated as a ratio of respective AUC0–24 values obtained after IV and oral gavage (PO) administration of a 2 mg kg−1 dose (n = 3 per time point). Cmax = maximum concentration in brain or plasma; tmax = time of maximum concentration in brain or plasma; t1/2 = terminal elimination half-life; AUC0–24 = area under the concentration versus time curve from zero to last measurable time point.
In Vivo behavioral studies on 8b versus the control compound 8m.
Since the ligand 8b exhibited a longer duration of action, it was subjected to in vivo studies after IP administration at a 10 mg/kg dose. In addition, the control ligand 8m of the same chemotype, was also studied in parallel. Behavioral assays were run on these ligands to determine the occurrence of anxiolysis, assess muscle strength and basic sensorimotor activity on the elevated plus maze, grip strength and cued water-maze training, respectively (Table 5). The nanoemulsion formulation48 employed for the behavioral studies, which permits a more reliable dosing, did not grossly affect the pharmacokinetic profile of 8b obtained with the suspension formulation (data not shown). Moreover, the corresponding placebo (nanoemulsion alone) was not behaviorally active when compared to saline (data not shown). The changes in behavioral parameters analyzed above, which are generally observed with typical Bzs or Bz site ligands,47 were not elicited by either of the two ligands. In addition, motor coordination and balance on the rotarod were retained after IP doses as high as 55 mg/kg 8b and 60 mg/kg 8m. Taken together, these results indicate that selective in vivo potentiation of GABAARs containing the α6 subtype did not mimic any of the behavioral effects of classical Bzs supporting the role of the GABAAR α6 (DI) receptor in the following in vivo data.
Table 5:
The influence of IP 10 mg kg−1 doses of 8b and 8m on the selected parameters of the grip strength, elevated plus maze, and cued water-mazea
| Compound | Grip Strength (kg kg−1)b | Elevated Plus Maze | Cued Water Maze | ||||
|---|---|---|---|---|---|---|---|
| Total Distance (m) | Closed Arm Entries | % Open Arm Entries | Total Distance (m) | Average Swim Speed (m s−1) | % Peripheral Ring Distance | ||
| Placebo | 2.79 ± 0.16 | 6.71 ± 0.56 | 3.63 ± 0.42 | 23.07 ± 5.47 | 8.04 ± 1.16 | 0.21 ± 0.01 | 67.44 ± 4.60 |
| 8b | 2.67 ± 0.18 | 5.84 ± 0.50 | 2.63 ±0.42 | 22.36 ± 8.76 | 11.01 ± 1.41 | 0.20 ± 0.01 | 67.2 ± 3.83 |
| 8m | 2.66 ± 0.19 | 6.14 ± 0.73 | 3.50 ± 0.39 | 20.60 ± 6.51 | 9.93 ± 1.50 | 0.22 ± 0.01 | 69.34 ± 4.37 |
Three behavioral in vivo studies exhibited that selective GABAAR α6 potentiation had no classical DS effects.
The peak force of experimenter’s pull necessary to overcome the strength of the animal’s grip.
In Vivo hyperlocomotor response studies on 8b versus the control compound 8m.
In rats, 8b, but not 8m at an IP dose of 10 mg/kg induced a hyperlocomotor response (Figure 11A); in the repeated experiment, the effect of 10 mg/kg 8b did not reach statistical significance (Figure 11C), while in the third experiment it was present at both the dose of 3 and 10 mg/kg (Figure 11E). In mice, both 8b and 8a induced a hyperlocomotor action in a wide range of IP doses (3, 10 and 30 mg/kg, Figure 11B and 11D, respectively). The hyperlocomotor effect of 10 mg/kg 8b was present even in mice that received a sedative IP dose of diazepam (3 mg/kg) in combination (Figure 11F), suggesting that 8b is able to antagonize the effects of diazepam at Bz binding sites and additionally elicit locomotor stimulation presumably exerted through GABAARs containing the α6 subunit.
Figure 11:

The measurement of influences on locomotor activity of rats (left panel: A,C,E) or mice (right panels: B,D,F)a
aThe locomotor activity tests were performed in rats (A,C,E) and mice (B,D,F) in six experiments in total. The ligands used were 8a, 8b, 8m and diazepam (DZP), while control groups were placebo nanoemulsion (PLA) and saline (SAL) via IP administration. The number of animals per group is given within columns. The results of ANOVA are as follow: (A) F (3,28)=5.335, p=0.005; (B) F (2,21)=2.542, p=0.103; (C) F (4,23)=3.685, p=0.018; (D) F (4,35)=9.932, p<0.001; (E) F (4,35)=5.745, p<0.001; (F) F (3,16)=10.345, p<0.001. Post hoc significant differences are as follows: *,** and ***, p<0.05, p<0.01, and p<0.001 vs. PLA; ++ and +++, p<0.01 and p<0.001 vs. SAL; #, p<0.05 vs. 8b, 30 mg/kg; ¥¥, p<0.01 vs. 8m, 10 mg/kg; §§ and §§§, p<0.01 and p<0.001 vs. PLA + DZP 3 mg/mg.
Discussion and Conclusions
GABAARs containing the α6 subunit are primarily located in the granule cells of cerebellum,5, 12 but are also expressed in other brain regions.13, 14 So far, not much is known about the function of these receptors in the brain, but recent evidence indicates that they might play a role in a variety of diseases. These include disorders of the trigeminal system,18 neuropsychiatric disorders with sensori-motor gating deficits (such as tic disorders, certain symptoms of schizophrenia, obsessive compulsive disorder and attention deficit disorders),15, 16 and depression,14 as well as migraine.17 Recently, some PQs were identified as the very first compounds that are able to modulate α6-GABAARs with high selectivity.10, 11, 28, 29 Based on three highly α6-GABAAR-selective PQ lead compounds (8a, 8n and 8i), a total of 42 PQs were synthesized in the present study with the aim to increase their limited water solubility,31 as well as their metabolic stability. Exchanging deuterium for hydrogen has been shown previously to positively affect the metabolic stability of drugs while retaining the pharmacologic profile of active compounds.32 Despite these successes, deuteration sometimes has also been observed to have no effect on metabolic stability.49,50 In the present study, the substitution of OCD3 groups for OCH3 groups in the lead ligands (8a, 8n and 8i) resulted in the deuterated α6β3γ2 GABAAR subtype selective ligands (8b, 8c, 8d, 8j and 8o) with an improved metabolic stability in both HLM and MLM assays (Table 3) and improved pharmacokinetic profile (Figure 9). However, the half-life of compounds not only increased but also decreased on deuteration depending on the structure of the compound investigated and the position of the incorporated deuterium and was different in HLM and MLM (Table 3). The changes in half-life observed on deuteration were thus not predictable. The same was true for the N-heterocyclic compounds (13a, 13e, 13g and 13i) and their deuterated derivatives (13b, 13c, 13d, 13f and 13h) (Table 3). The N-heterocyclic compounds were synthesized to increase the water solubility of the lead compounds. But, although the water solubility was increased in some of the N-heterocyclic compounds as compared to the lead compounds (compare 8a with 13a and 13i), in other cases the water solubility was unchanged (compare 8n with 13g), or even decreased (compare 8k with 13e) in the N-heterocyclic compounds (Table 4).
As expected, the deuterated derivatives exhibited an efficacy comparable to the respective lead compound for α6β3γ2 receptors (Figure 5). In contrast, incorporation of N-atoms into the “A-ring” or “D-ring” of the PQ core was accompanied by a drop in efficacy at the α6β3γ2 subtype relative to parent compounds 8a and 8k, but not relative to the parent compound 8n, as revealed in Figure 7.
The synthesis of 42 novel ligands also expanded the SAR library of potential α6β3γ2 subtype selective ligands (Table 1, Table 2, and (S) Table S4). 8k had previously been identified as a ligand with three times higher α6β3γ2 efficacy at 10 μM than 8a and 8n, but the selectivity amongst α1-6β3γ2 GABAARs was much poorer for 8k.10 Hence, the 8-chloro substituent in “Ring A” of 8k appeared to play a role in the increased potentiation of GABA-currents at several GABAAR subtypes (α1-6β3γ2), whereas a 7-methoxy substituent in “Ring A” of 8a appeared to be the key for α6β3γ2 subtype selectivity. The example of 8n (Figure 4B) provided evidence that the 7-position can also be substituted with an electron withdrawing group such as bromine, and still retain a similar α6β3γ2 efficacy and selectivity as 8a.
The “D-ring” substituent seems to play a role in the binding affinity at the Bz site23–25 as well as in the α6β3γ2 efficacy mediated via the PQ site.10, 28, 29 Thus, substitution of ortho-methoxy groups onto the “D-ring” of 7-methoxy (8h) and 7-bromo (8q) compounds resulted in a significantly reduced affinity at the BZP rat brain site as shown in (S) Table S1 and Table S2. The affinity at the BZP rat brain site was measured by a compound-induced displacement of [3H]flunitrazepam binding to brain membranes. It thus represents the affinity at the DS-Bz binding site of all GABAA receptors present in brain membranes and no conclusions can be made on a possible interaction of these compounds with the DI-Bz site at α6β3γ2 receptors. Interestingly, for the 8-chloro ligand 8m, that also contains an ortho-methoxy group in the “D-ring”, the binding affinity at the BZP rat brain site was retained, although it was about 50-100-fold reduced compared to that of other compounds investigated ((S) Table S2). However, 8m displayed almost no efficacy via the PQ site of the α6β3γ2 subtype (Figure 6) rendering this ligand an ideal “control compound” for future in vivo experiments.
N-heterocyclic fragments have often been used in drug design to alter bioavailability via manipulation of the aqueous solubility, lipophilicity and polarity of molecules.51
As detailed in this study, introduction of an N-atom into the “D-ring” of the parent ligand 8a resulted in 13a with an about 3-fold increased water solubility (Table 4) and a 3.6-fold increased half-life in HLM (Table 3). The 7-OCD3 derivative of 13a, compound 13c, exhibited a similarly increased half-life as 13a in HLM (Table 3) while retaining α6β3γ2 subtype selective efficacy (Figure 8). The “D-ring” N-hetero substituted analogs of 8i and 8n (13e and 13g, respectively) did not exhibit improved aqueous solubility and were not pursued further in this study.
Compounds 8b, 8c, 8d and 13c, the deuterated derivatives of 8a, all exhibited a similarly enhanced metabolic stability (within experimental error) over the parent 8a with half-lives of 8.3 – 12.5 hours (HLM) and 9.2 – 14.2 hours (MLM) (Table 3). Pharmacokinetic studies, however, displayed an almost two-fold increase in the brain concentrations (Cmax) for the deuterated analogs (8b, 8c and 8d), whereas the Cmax for deuterated N-hetero analog 13c was considerably less than that of the parent 8a (Figure 9). Metabolic stability alone thus does not necessarily result in higher brain concentrations. Many other factors mediate brain exposure such as: lipid solubility, charge, tertiary structure and degree of protein binding.52 Despite the increased solubility of 13c (see OCH3 analog, 13a in Table 4), the decreased lipophilicity (cLogP = 2.17) may have retarded the ability to cross the BBB by the introduction of the “D-ring” N-hetero substituent.53
In summary, the deuterated para analogs (8b, 8c, 8d and 8o) of 8a and 8n displayed the ideal combination of α6β3γ2 GABAAR subtype selectivity and efficacy (Figure 4, Figure 5). The deuterated meta analog 8j was not pursued further in this study due to its poorer efficacy compared to 8b, 8c, 8d and 8o. The 8n related ligands (8o and 13h) exhibited efficacies comparable to 8n (Figure 5 and Figure 7, respectively), but increased metabolic stability (Table 3) and will be explored in the future with pharmacokinetic studies. The deuterated, “D-ring” N-hetero substituted 13h was the metabolically most stable ligand of the series developed, to date (Table 3). In the future, a number of other ligands including the additional compounds reported in this work ((S) Table S4) with a variety of substituents in the “A-ring” and “D-ring” of the PQ scaffold will have to be investigated for their α6β3γ2 GABAAR-selectivity to obtain a more accurate SAR profile.
Due to its ease of synthesis, the functionally α6β3γ2 GABAAR-selective ligand 8b was ultimately chosen as the lead ligand from this work for future in vivo studies although analogs 8c and 8d, exhibited a similarly increased brain concentration (Cmax) as 8b over the parent ligand 8a (Figure 9). Deuteration of the methoxy group in the “D-ring” (8b) was achieved in four synthetic steps (Scheme 1), whereas, deuteration of the “A-ring” methoxy group in 8c required six synthetic steps. The di-OCD3 substituted ligand 8d required nine convergent steps but did not result in a beneficial increase in bioavailability over the mono-OCD3 substituted ligand 8b to warrant the extra synthetic steps required to obtain it.
As indicated in the rotarod experiments ((S) Table S2) as well as in the elevated plus maze, grip strength and cued water maze experiments (Table 5), 8b was devoid of many effects characteristic of classical Bzs (sedation, motor incoordination, anxiolytic action). While the predominant lack of significant influences on behavioral parameters are expected to improve the safety and tolerability profile of this ligand, the unexpected mild hyperlocomotor activity (Figure 11), induced in rodents by 8b, but not 8m, requires further examination in order to identify the underlying biological substrate. Such activity may reflect a subtle increase in motivational drive and might be useful in depressive conditions;54 this speculative interpretation needs to be evaluated in future studies.
Cellular viability assays on HEK293 cells and HEP2G cells indicated that the ligands described herein are non-toxic even at 400 μM ((S) Table S3). In addition, a screen of these compounds with a panel (PDSP) of a wide range of 46 membrane receptors, channels and transporters including the hERG channel indicated that there were no off-target effects (no hERG inhibition at > 10 μM) ((S) Table S1 and Table S2). This result coupled with the lack of adverse Bz-like effects in basic behavioral tests, suggest that 8b is a potential agonist-like clinical agent targeted for α6β2/3γ2 GABAergic receptors. Further work in this area will define its application in those diseases that are of practical relevance.
Experimental Section
General Procedures.
All reactions were performed in oven-dried round-bottom flasks with magnetic stir bars or overhead mechanical stirrers under an argon atmosphere unless the reaction conditions were supposed to contain water. Organic solvents were purified when necessary by standard methods55 or purchased from Sigma-Aldrich.™ Chemicals were purchased from either Sigma Aldrich™, Oakwood Chemical, Alfa Aesar, Matrix Scientific, or Acros Organic. The progress of the reactions was monitored by TLC on a silica gel plate (25% EtOAc in hexanes or 10% MeOH in DCM). The 1H and 13C NMR data were obtained on Bruker Spectrospin 300 MHz and GE 500 MHz instruments with the chemical shifts in δ (ppm) reported relative to TMS. The HRMS spectral data was obtained on a LCMS-IT-TOF by Shimadzu Scientific. Purity of all final compounds was 98% or higher and was determined by HPLC on a LC-MS with Shimadzu LCMS 2020, (Shimadzu Scientific Instruments, Columbia, MD) using a PDA detector at 254 nm. The column was a Shimadzu C18 3 μm 50 × 4.6mm reversed phase LC column. LC mobile phase: 90% acetonitrile (w. 0.1% TFA) and 10% H2O (w/ 0.1% TFA) with a flow rate of 1 mL min−1, column temperature: 25 °C, injection size: 1.0μL.
Chemistry
General Procedure for the Synthesis of N-(Hydroxyphenyl)acetamides (2a-c).
N-(4-Hydroxyphenyl)acetamide (2a).
To a mixture of 4-aminophenol 1a (50.0 g, 458.2 mmol) and THF (200 mL), acetic anhydride (49.1 g, 481.1 mmol) was added dropwise over 30 min while keeping the temperature below 50 °C. The reaction mixture was then allowed to stir for 30 min at 50 °C and then allowed to cool to rt. The reaction mixture was then diluted with hexanes (200 mL) to precipitate 2a. After stirring for 1 h, the solid was filtered and washed with hexanes (50 mL x 2). The solid was dried under vacuum at 40 °C to afford 2a as a white crystalline solid (62.7 g, 90.0%): mp 170-171 °C; 1H NMR (300 MHz, DMSO) δ 9.64 (s, 1H), 9.13 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 1.98 (s, 3H); 13C NMR (75 MHz, DMSO) δ 167.95, 153.57, 131.49, 121.26, 115.44, 24.19; HRMS (ESI) m/z calculated for C8H10NO2 (M + H)+ 152.0706; found 152.0715. The 1H NMR and 13C NMR spectroscopic properties of 2a were identical to those reported in the literature.56
N-(3-Hydroxyphenyl)acetamide (2b).
The title compound was prepared by a similar procedure as that used for 2a using 3-aminophenol 1b (25.0 g, 229.1 mmol) and THF (100 mL), acetic anhydride (24.5 g, 240.5 mmol) to afford 2b as a white crystalline solid (33.2 g, 96.0%): mp 145-148 °C; 1H NMR (300 MHz, DMSO) δ 9.77 (s, 1H), 9.32 (s, 1H), 7.18 (s, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.42 (dd, J = 7.9, 2.1 Hz, 1H), 2.01 (s, 3H); 13C NMR (75 MHz, DMSO) δ 168.60, 158.01, 140.81, 129.72, 110.55, 110.18, 106.60, 24.50; HRMS (ESI) m/z calculated for C8H10NO2 (M + H)+ 152.0706; found 152.0715. The 1H NMR and 13C NMR spectroscopic properties of 2b were identical to those reported in the literature.56
N-(2-Hydroxyphenyl)acetamide (2c).
The title compound was prepared by a similar procedure as that used for 2a using 2-aminophenol 1c (25.0 g, 229.1 mmol) and THF (100 mL), acetic anhydride (24.5 g, 240.5, mmol) to afford 2c as a light brown solid (33.1 g, 95.7%): mp 211-213 °C; 1H NMR (300 MHz, DMSO) δ 9.75 (s, 1H), 9.31 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 6.85 (ddd, J = 33.6, 14.6, 7.2 Hz, 3H), 2.10 (s, 3H); 13C NMR (75 MHz, DMSO) δ 169.44, 148.34, 126.88, 125.08, 122.80, 119.40, 116.38, 24.05; HRMS (ESI) m/z calculated for C8H10NO2 (M + H)+ 152.0706; found 152.0716. The 1H NMR and 13C NMR spectroscopic properties of 2c were identical to those reported in the literature.56
General Procedure for the Synthesis of N-(Methoxy-d3-phenyl)acetamides (3a-c).
N-(4-Methoxy-d3-phenyl)acetamide (3a).
To a mixture of N-(4-hydroxyphenyl)acetamide 2a (62.0 g, 410.1 mmol), potassium carbonate (113.4 g, 615.2 mmol), and acetone (230 mL), iodomethane-d3 (100 g, 689.8 mmol) was added dropwise over 30 min. The reaction mixture was then allowed to stir for 24 h at 20-25 °C in a RB flask equipped with a dry ice condenser. The reaction mixture was diluted with EtOAc (300 mL) and H2O (300 mL). The biphasic mixture, which resulted, was allowed to stand for 15 min and the layers were separated. The aq layer was extracted with EtOAc (200 mL) and the combined organic layers were washed with 10% aq potassium carbonate solution (200 mL). The organic layer was dried (MgSO4). The solvents were removed under reduced pressure and the residue was slurried with hexanes (200 mL). The solid was filtered and washed with hexanes (50 mL x 2). The solid was dried under vacuum at 40 °C to afford 3a as an off-white solid (71.7 g, 99%): mp 125-126 °C; 1H NMR (300 MHz, DMSO) δ 9.77 (s, 1H), 7.48 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 3.38 (s, 3H), 2.00 (s, 3H); 13C NMR (75 MHz, DMSO) δ 168.20, 155.48, 132.94, 121.01, 114.21, 24.23; HRMS (ESI) m/z calculated for C9H9D3NO2 (M + H)+ 169.1051; found 169.1071.
N-(3-Methoxy-d3-phenyl)acetamide (3b).
The title compound was prepared by a similar procedure as that used for 3a using N-(3-hydroxyphenyl)acetamide 2b (35.0 g, 231.5 mmol), potassium carbonate (64.0 g, 463.1 mmol), acetone (140 mL), and iodomethane-d3 (50.3 g, 347.3 mmol) to afford 3b as an off-white solid (38.9 g, 99%): mp 89-91 °C; 1H NMR (300 MHz, DMSO) δ 9.89 (s, 1H), 7.27 (s, 1H), 7.18 (t, J = 8.1 Hz, 1H), 7.10 (d, J = 8.2 Hz, 1H), 6.60 (dd, J = 7.8, 2.0 Hz, 1H), 2.03 (s, 3H); 13C NMR (75 MHz, DMSO) δ 168.76, 159.93, 140.96, 129.89, 111.71, 108.76, 105.30, 24.53; HRMS (ESI) m/z calculated for C9H9D3NO2 (M + H)+ 169.1051; found 169.1062.
N-(2-Methoxy-d3-phenyl)acetamide (3c).
The title compound was prepared by a similar procedure as that used for 3a using N-(2-hydroxyphenyl)acetamide 2c (30.0 g, 198.5 mmol), potassium carbonate (54.9 g, 396.9 mmol), acetone (140 mL), and iodomethane-d3 (50.3 g, 347.3 mmol) to afford 3c as an off-white solid (31.9 g, 99%): mp 82-83 °C; 1H NMR (300 MHz, DMSO) δ 9.10 (s, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.22 – 6.97 (m, 2H), 6.97 – 6.79 (m, 1H), 2.08 (s, 3H); 13C NMR (75 MHz, DMSO) δ 168.87, 149.98, 127.87, 124.63, 122.45, 120.57, 111.50, 24.30; HRMS (ESI) m/z calculated for C9H9D3NO2 (M + H)+ 169.1051; found 169.1060.
General Procedure for the Synthesis of Methoxy-d3-phenylhydrazines (4a-c).
4-Methoxy-d3-phenylhydrazine (4a).
A mixture of N-(4-methoxy-d3-phenyl)acetamide 3a (30 g, 178.4 mmol), concentrated hydrochloric acid (72 mL), and H2O (72 mL) was heated to 90 °C and held at 90 °C for 2 h to hydrolyze the amide functionality. The reaction mixture was then cooled to 0 to 5°C and a solution of sodium nitrite (12.9 g, 187.7 mmol) in H2O (25 mL) was slowly added drop-wise to the reaction mixture. Upon completion of the addition, the reaction mixture was allowed to stir for an additional 15 min at 0 to 5 °C. The reaction mixture was then cooled to −25 to −20°C and a solution of tin (II) chloride (74.4 g, 392.4 mmol) and concentrated hydrochloric acid (150 mL) was added drop-wise to the reaction mixture over 30 min. Upon completion of the addition, the reaction mixture was allowed to stir for an additional 4 h at −25 to −20 °C. The reaction mixture was then diluted with diethyl ether (300 mL) and the solids were filtered off and washed with diethyl ether (100 mL x 3). The tin adduct of the product was then dissolved in a mixture of sodium hydroxide (60 g), H2O (250 mL) and DCM (250 mL). After stirring for 2 h at 0 to 5 °C, the solids completely dissolved. The layers were separated and the aq layer was extracted with DCM (100mL x 3). The combined organic layers were dried (MgSO4). The solvents were removed under reduced pressure and the residue was slurried with hexanes (50 mL). The solid was then filtered and washed with hexanes (50 mL x 2). The solid was dried under vacuum at rt to afford 4a as a pale orange crystalline solid (16.6 g, 66%): 1H NMR (300 MHz, MeOD) δ 6.91 – 6.85 (m, 2H), 6.85 – 6.78 (m, 2H), 4.88 (s, 3H); 13C NMR (75 MHz, DMSO) δ 152.05, 147.27, 114.60, 113.38; HRMS (ESI) m/z calculated for C7H8D3N2O (M+H)+ 142.1054; found 142.1063.
3-Methoxy-d3-phenylhydrazine (4b).
The title compound was prepared by a similar procedure as that used for 4a using N-(3-methoxy-d3-phenyl)acetamide 3b (25 g, 148.6 mmol), concentrated hydrochloric acid (60 mL), and H2O (60 mL), then, sodium nitrite (10.8 g, 156.1 mmol) in H2O (21 mL), and finally, tin (II) chloride (62.0 g, 327.0 mmol) and concentrated hydrochloric acid (125 mL) to afford 4b as an orange-red oil (5.4 g, 26%): 1H NMR (300 MHz, DMSO) δ 6.98 (t, J = 8.0 Hz, 1H), 6.65 (s, 1H), 6.51 – 6.27 (m, 2H), 6.16 (d, J = 7.9 Hz, 1H), 3.91 (s, 2H); 13C NMR (75 MHz, DMSO) δ 160.68, 154.54, 129.69, 104.96, 102.72, 97.52; HRMS (ESI) m/z calculated for C7H8D3N2O (M+H)+ 142.1054; found 142.1060. This material was employed in a later experiment with no further purification.
2-Methoxy-d3-phenylhydrazine (4c).
The title compound was prepared by a similar procedure as that used for 4a using N-(2-methoxy-d3-phenyl)acetamide 3c (25 g, 148.6 mmol), concentrated hydrochloric acid (60 mL), and H2O (60 mL), then, sodium nitrite (10.7 g, 156.1 mmol) in H2O (21 mL), and finally, tin (II) chloride (62.0 g, 327.0 mmol) and concentrated hydrochloric acid (125 mL) to afford 4c as a pale pink solid (12.5 g, 60%): 1H NMR (300 MHz, DMSO) δ 7.01 (dd, J = 7.8, 1.3 Hz, 1H), 6.92 – 6.71 (m, 2H), 6.61 (td, J = 7.7, 1.4 Hz, 1H), 5.92 (s, 1H), 3.92 (s, 2H); 13C NMR (75 MHz, DMSO) δ 146.33, 141.94, 121.26, 117.23, 111.20, 110.07; HRMS (ESI) m/z calculated for C7H8D3N2O (M+H)+ 142.1054; found 142.1050.
General Procedure for the Synthesis of Methoxy-d3-anilines (5a-b).
4-Methoxy-d3-aniline (5a).
A mixture of N-(4-methoxy-d3-phenyl)acetamide 3a (20.0 g, 118.9 mmol), 12 M hydrochloric acid (20 mL, 240 mmol), and H2O (60 mL) was heated at 90-95 °C for 2 h. The reaction mixture was then allowed to cool to 20-25 °C and the pH was adjusted to 14 with a solution of sodium hydroxide (20g, 500 mmol) and H2O (20 mL). The product was then extracted from the aq layer with DCM (50 mL x 4). The combined organic layers were dried (MgSO4). Removal of the solvents under reduced pressure afforded 5a as a dark orange oil (14.4 g, 96%): 1H NMR (300 MHz, DMSO) δ 5.75 – 5.62 (m, 2H), 5.62 – 5.47 (m, 2H), 3.50 (s, 2H); 13C NMR (75 MHz, DMSO) δ 150.34, 141.51, 114.62, 113.89; HRMS (ESI) m/z calculated for C7H7D3NO (M + H)+ 127.0945; found 127.0948. This material was employed in a later experiment with no further purification.
3-Methoxy-d3-aniline (5b).
The title compound was prepared by a similar procedure as that used for 5a using N-(3-methoxy-d3-phenyl)acetamide 3b (20.0 g, 118.9 mmol), 12 M hydrochloric acid (20 mL, 240 mmol), and H2O (60 mL) to afford 5b as a golden yellow oil (13.5 g, 90%): 1H NMR (300 MHz, CDCl3) δ 7.10 (t, J = 8.0 Hz, 1H), 6.35 (dddd, J = 11.9, 11.2, 3.4, 2.0 Hz, 3H), 4.00 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 160.78, 147.54, 130.16, 108.14, 104.22, 101.28; HRMS (ESI) m/z calculated for C7H7D3NO (M + H)+ 127.0945; found 127.0947. This material was employed in a later experiment with no further purification.
General Procedure for the Synthesis of Ethyl-4-hydroxy-quinoline-3-carboxylates (6a-f).
Ethyl-4-hydroxy-6-methoxy-d3-quinoline-3-carboxylate (6a).
A mixture of 4-methoxy-d3-aniline 5a (10 g, 81.2 mmol), diethyl ethoxymethylenemalonate (21.1 g, 97.4 mmol) and diphenyl ether (100 mL) was slowly heated to 230 °C. The EtOH, which evolved, was collected in a Dean-Stark trap. Once the EtOH formation had ceased, the reaction mixture was heated for an additional 30 min at 230 °C. The reaction mixture was then cooled to 80 °C and diluted with hexanes (100 mL). Upon cooling to 20-25 °C the solid, which formed, was collected by filtration and washed with hexanes (50 mL x 2). The solid was dried under vacuum at 40 °C to afford 6a as a light brown solid (9.9 g, 49%). 1H NMR (300 MHz, TFA) δ 11.66 (s, 1H), 9.15 (s, 1H), 8.05 (d, J = 9.2 Hz, 1H), 7.97 – 7.74 (m, 2H), 4.67 (q, J = 7.1 Hz, 2H), 1.67 – 1.39 (m, 3H); 13C NMR (75 MHz, TFA) δ 171.68, 167.54, 160.89, 141.86, 134.62, 129.91, 121.73, 121.28, 104.58, 102.17, 64.41, 11.97; HRMS (ESI) m/z calculated for C13H11D3NO4 (M+H)+ 251.1106; found 251.1115.
Ethyl-4-hydroxy-7-methoxy-d3-quinoline-3-carboxylate (6b).
The title compound was prepared by a similar procedure as that used for 6a using 3-methoxy-d3-aniline 5b (10 g, 81.2 mmol), diethyl ethoxymethylenemalonate (21.1 g, 97.4 mmol) and diphenyl ether (100 mL) to afford 6b as a brown solid (13.0 g, 64%): 1H NMR (300 MHz, TFA) δ 11.64 (s, 1H), 9.23 (s, 1H), 8.57 (d, J = 9.3 Hz, 1H), 7.59 (dd, J = 9.4, 2.3 Hz, 1H), 7.48 (d, J = 2.2 Hz, 1H), 4.71 (q, J = 7.2 Hz, 2H), 1.58 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, TFA) δ 171.89, 167.92, 167.64, 144.50, 142.44, 126.28, 121.93, 114.05, 103.81, 99.25, 64.29, 12.01; HRMS (ESI) m/z calculated for C13H11D3NO4 (M+H)+ 251.1106; found 251.1081.
Ethyl-6-chloro-4-hydroxyquinoline-3-carboxylate (6c).
The title compound was prepared by a similar procedure as that used for 6a using 4-chloroaniline 5c (45.5 g, 356.7 mmol), diethyl ethoxymethylenemalonate (80.9 g, 374.1 mmol) and diphenyl ether (200 mL) to afford 6c as an off-white crystalline solid (85.1 g, 95%): 1H NMR (300 MHz, TFA) δ 11.66 (s, 1H), 9.32 (d, J = 4.5 Hz, 1H), 8.62 (d, J = 2.5 Hz, 1H), 8.12 (d, J = 13.0 Hz, 2H), 4.82 – 4.55 (m, 2H), 1.53 (dd, J = 11.8, 7.0 Hz, 3H); 13C NMR (75 MHz, TFA) δ 172.51, 167.19, 144.95, 138.35, 137.62, 137.58, 123.58, 121.35, 120.82, 105.30, 64.70, 11.96; HRMS (ESI) m/z calculated for C12H11ClNO3 (M+H)+ 252.0422; found 252.0411.
Ethyl-7-bromo-4-hydroxyquinoline-3-carboxylate (6d).
The title compound was prepared by a similar procedure as that used for 6a using 3-bromoaniline 5d (8.7 g, 58.1 mmol), diethyl ethoxymethylenemalonate (10.9 g, 58.1 mmol) and diphenyl ether (40 mL) to afford 6d as a light brown solid (11.5 g, 77%): 1H NMR (300 MHz, TFA) δ 11.64 (s, 1H), 9.38 (s, 1H), 8.57 (d, J = 8.9 Hz, 1H), 8.43 (s, 1H), 8.15 (d, J = 8.9 Hz, 1H), 4.75 (q, J = 7.1 Hz, 2H), 1.60 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, TFA) δ 173.48, 167.33, 145.75, 139.71, 134.19, 134.06, 125.60, 122.70, 118.57, 105.08, 64.74, 12.01; HRMS (ESI) m/z calculated for C12H10BrNO3 (M+H)+ 295.9917; found 295.9894.
Ethyl-4-hydroxy-7-methoxyquinoline-3-carboxylate (6e).
The title compound was prepared by a similar procedure as that used for 6a using 3-methoxyaniline 5e (50.0 g, 406.0 mmol), diethyl ethoxymethylenemalonate (87.8 g, 406.0 mmol) and diphenyl ether (200 mL) to afford 6e as a light brown solid (37.1 g, 37%): 1H NMR (300 MHz, TFA) δ 11.63 (s, 1H), 9.22 (d, J = 6.3 Hz, 1H), 8.56 (dd, J = 9.1, 6.7 Hz, 1H), 7.66 – 7.54 (m, 1H), 7.47 (d, J = 4.2 Hz, 1H), 4.69 (dd, J = 13.8, 6.9 Hz, 2H), 4.13 (d, J = 6.4 Hz, 3H), 1.57 (q, J = 6.8 Hz, 3H); 13C NMR (75 MHz, TFA) δ 171.88, 167.91, 167.62, 144.49, 142.43, 126.28, 121.92, 114.05, 103.81, 99.24, 64.28, 55.45, 12.00; HRMS (ESI) m/z calculated for C13H14NO4 (M+H)+ 248.0917; found 248.0908.
Ethyl-4-hydroxy-7-methoxy-1,6-naphthyridine-3-carboxylate (6f).
The title compound was prepared by a similar procedure as that used for 6a using 4-amino-2-methoxypyridine 11c (10.0 g, 80.6 mmol), diethyl ethoxymethylenemalonate (17.4 g, 80.6 mmol) and diphenyl ether (40 mL) to afford 6f as a light brown solid (3.1 g, 15%): 1H NMR (500 MHz, TFA) δ 11.63 (s, 1H), 9.40 (s, 1H), 8.18 (d, J = 7.7 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 4.74 (q, J = 7.0 Hz, 2H), 3.99 (s, 3H), 1.63 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, TFA) δ 175.82, 164.29, 162.82, 149.50, 148.69, 145.32, 111.96, 108.86, 100.58, 64.48, 36.92, 12.07; HRMS (ESI) m/z calculated for C12H13N2O4 (M+H)+ 249.0870; found 249.0880.
General Procedure for the Synthesis of Ethyl-4-chloro-quinoline-3-carboxylates (7a-f).
Ethyl-4-chloro-6-methoxy-d3-quinoline-3-carboxylate (7a).
A mixture of ethyl-4-hydroxy-6-methoxy-d3-quinoline-3-carboxylate 6a (10.0 g, 40.0 mmol), N,N-dimethylformamide (0.1 mL, 1.5 mmol), and DCM (75 mL) was heated to 35-40°C. Oxalyl chloride (5.6 g, 44.0 mmol) was added dropwise to the reaction mixture over 30 min. The reaction mixture was heated for 6 h at reflux (38-40 °C). The pale yellow solution, which resulted, was then allowed to cool to 20-25 °C. The reaction mixture was brought to pH = 10 (pH paper) by slowly adding a 25% solution of aq potassium carbonate (10 g) in H2O (40 mL). The layers were then separated and the aq layer was extracted with DCM (40 mL). The combined organic layers were then washed with a 25% solution of aq potassium carbonate (5 g) in H2O (20 mL). The combined organic layers were dried (MgSO4). The solvents were then removed under reduced pressure and the residue was slurried with hexanes (20 mL). The solid, which formed, was filtered and washed with hexanes (5 mL x 2). The solid was dried under vacuum at 40 °C to afford 7a as an off-white solid (8.5 g, 79%): 1H NMR (300 MHz, DMSO) δ 8.95 (s, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.67 – 7.45 (m, 2H), 4.42 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO) δ 164.49, 159.57, 147.25, 145.36, 139.99, 131.67, 126.91, 125.20, 123.87, 102.96, 62.36, 14.47; HRMS (ESI) m/z calculated for C13H10D3ClNO3 (M+H)+ 269.0767; found 269.0762.
Ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate (7b).
The title compound was prepared by a similar procedure as that used for 7a using ethyl-4-hydroxy-7-methoxy-d3-quinoline-3-carboxylate 6b (13.0 g, 51.9 mmol), N,N-dimethylformamide (0.1 mL, 1.5 mmol), DCM (100 mL), and oxalyl chloride (7.2 g, 57.1 mmol) to afford 7b as an off-white solid (11.5 g, 82%): 1H NMR (300 MHz, DMSO) δ 9.07 (s, 1H), 8.23 (d, J = 9.2 Hz, 1H), 7.54 – 7.38 (m, 2H), 4.40 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO) δ 164.29, 162.85, 151.53, 150.75, 142.41, 126.84, 122.11, 121.07, 120.56, 108.27, 62.15, 14.49; HRMS (ESI) m/z calculated for C13H10D3ClNO3 (M+H)+ 269.0767; found 269.0774.
Ethyl-4,6-dichloroquinoline-3-carboxylate (7c).
The title compound was prepared by a similar procedure as that used for 7a using ethyl-6-chloro-4-hydroxyquinoline-3-carboxylate 6c (85.1 g, 338.1 mmol), N,N-dimethylformamide (1.0 mL, 12.9 mmol), DCM (640 mL), and oxalyl chloride (47.2 g, 371.9 mmol) to afford 7c as an off-white solid (81.9 g, 90%): 1H NMR (300 MHz, DMSO) δ 9.13 (s, 1H), 8.30 (d, J = 2.2 Hz, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.97 (dd, J = 9.0, 2.3 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO) δ 164.01, 150.53, 147.73, 141.04, 134.30, 133.34, 132.20, 126.53, 124.37, 124.08, 62.59; HRMS (ESI) m/z calculated for C12H10Cl2NO2 (M+H)+ 270.0083; found 270.0089. The spectroscopic properties of 7c were identical to those reported in the literature.40
Ethyl-7-bromo-4-chloroquinoline-3-carboxylate (7d).
The title compound was prepared by a similar procedure as that used for 7a using ethyl-7-bromo-4-hydroxyquinoline-3-carboxylate 6d (11.0 g, 37.1 mmol), N,N-dimethylformamide (0.1 mL, 1.3 mmol), DCM (50 mL), and oxalyl chloride (5.2 g, 40.9 mmol) to afford 7d as an off-white solid (7.2 g, 61%): 1H NMR (300 MHz, DMSO) δ 9.15 (s, 1H), 8.36 (d, J = 1.9 Hz, 1H), 8.28 (d, J = 9.0 Hz, 1H), 7.98 (dd, J = 9.0, 1.9 Hz, 1H), 4.44 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO) δ 164.04, 151.40, 149.74, 142.34, 132.67, 131.88, 127.50, 126.64, 124.70, 123.96, 62.55, 14.46; HRMS (ESI) m/z calculated for C12H10BrClNO2(M+H)+ 313.9578; found 313.9568.
Ethyl-4-chloro-7-methoxyquinoline-3-carboxylate (7e).
The title compound was prepared by a similar procedure as that used for 7a using ethyl-4-hydroxy-7-methoxyquinoline-3-carboxylate 6e (37.1 g, 150.0 mmol), N,N-dimethylformamide (0.5 mL, 6.5 mmol), DCM (150 mL), and oxalyl chloride (20.9 g, 165.0 mmol) to afford 7e as an off-white solid (36.3 g, 91%): 1H NMR (300 MHz, DMSO) δ 9.08 (s, 1H), 8.25 (d, J = 9.2 Hz, 1H), 7.57 – 7.37 (m, 2H), 4.41 (q, J = 7.1 Hz, 2H), 3.98 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO) δ 164.32, 162.83, 151.62, 150.81, 142.02, 126.85, 122.12, 121.11, 120.56, 108.36, 62.15, 56.45, 14.49; HRMS (ESI) m/z calculated for C13H13ClNO3 (M+H)+ 266.0578; found 266.0578. The 1H NMR spectroscopic properties of 7e were identical to those reported in the literature.41
Ethyl-4-chloro-7-methoxy-1,6-napththyridine-3-carboxylate (7f).
The title compound was prepared by a similar procedure as that used for 7a using ethyl-4-hydroxy-7-methoxy-1,6-naphthyridine-3-carboxylate 6f (3.0 g, 120.9 mmol), N,N-dimethylformamide (0.1 mL, 1.3 mmol), DCM (40 mL), and oxalyl chloride (1.7 g, 13.3 mmol) to afford 7f as an off-white solid (1.3 g, 40.4%): 1H NMR (500 MHz, CDCl3) δ 8.99 (s, 1H), 7.44 (d, J = 7.4 Hz, 1H), 6.71 (d, J = 7.5 Hz, 1H), 4.47 (q, J = 7.0 Hz, 2H), 3.60 (s, 3H), 1.44 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 164.43, 160.49, 157.34, 153.76, 145.84, 138.50, 126.66, 118.46, 106.93, 62.27, 37.76, 14.18; HRMS (ESI) m/z calculated for C12H12ClN2O3 (M+H)+ 267.0531; found 267.0531.
General Procedure for the Synthesis of PQs (8a-t, 13a-i).
7-Methoxy-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8a).
A mixture of ethyl-4-chloro-7-methoxyquinoline-3-carboxylate 7e (4 g, 15.1 mmol), 4-methoxyphenylhydrazine hydrochloride 4d (3.15 g, 18.1 mmol), triethylamine (3.66g, 36.1 mmol) and xylenes (32 mL) was heated to reflux (138 °C) and held at reflux for 2 h. The yellow-orange slurry, which resulted, was cooled to 100 °C and diluted with EtOH (32 mL). The reaction mixture was heated at reflux at 80°C for 30 min and then allowed to cool to 20-25 °C. The solids, which remained, were collected by filtration and washed with a 1:1 mixture of EtOH (2.5 mL x 2) and hexanes (2.5 mL x 2) and then washed with hexanes (5 mL x 2). The solid was dried under vacuum at 40 °C to afford 8a as a yellow powder (2.0 g, 41%): 1H NMR (300 MHz, DMSO) δ 12.59 (s, 1H), 8.65 (s, 1H), 8.10 (t, J = 8.7 Hz, 3H), 7.34 – 7.12 (m, 2H), 7.01 (d, J = 9.1 Hz, 2H), 3.87 (s, 3H), 3.78 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.45, 160.85, 156.22, 143.11, 139.33, 137.42, 134.10, 124.05, 120.68, 115.77, 114.25, 112.68, 106.87, 102.26, 55.98, 55.68; HRMS (ESI) m/z calculated for C18H16N3O3 (M+H)+ 322.1186; found 322.1178; HPLC purity, 99.9%. The 1H NMR spectroscopic properties of 8a were identical to those reported in the literature.25
7-Methoxy-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8b).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxyquinoline-3-carboxylate 7e (2 g, 7.4 mmol), 4-methoxy-d3-phenylhydrazine 4a (1.25 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8b as a yellow powder (1.5 g, 62.5%): 1H NMR (300 MHz, DMSO) δ 12.60 (s, 1H), 8.66 (s, 1H), 8.10 (t, J = 9.7 Hz, 3H), 7.18 (s, 2H), 7.01 (d, J = 8.4 Hz, 2H), 3.88 (s, 3H); 13C NMR (75 MHz, DMSO) δ 160.73, 160.43, 156.39, 143.09, 139.34, 137.43, 134.08, 124.08, 120.68, 115.80, 114.24, 112.69, 106.87, 102.28, 56.00; HRMS (ESI) m/z calculated for C18H13D3N3O3 (M+H)+ 325.1374; found 325.1372; HPLC purity, 99.2%.
7-Methoxy-d3-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8c).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 4-methoxyphenylhydrazine hydrochloride 4d (1.56 g, 8.9 mmol), triethylamine (1.81g, 17.6 mmol) and xylenes (16 mL) to afford 8c as a yellow powder (1.0 g, 41%): 1H NMR (300 MHz, DMSO) δ 12.59 (s, 1H), 8.65 (s, 1H), 8.10 (t, J = 8.7 Hz, 3H), 7.17 (d, J = 2.0 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 3.78 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.45, 160.86, 156.22, 143.11, 139.32, 137.42, 134.11, 124.05, 120.68, 115.76, 114.25, 112.66, 106.87, 102.25, 55.68; HRMS (ESI) m/z calculated for C18H13D3N3O3 (M+H)+ 325.1374; found 325.1371; HPLC purity, 99.9%.
7-Methoxy-d3-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8d).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 4-methoxy-d3-phenylhydrazine 4a (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8d as a yellow powder (1.2 g, 50.0%): 1H NMR (300 MHz, DMSO) δ 12.57 (s, 1H), 8.65 (s, 1H), 8.10 (t, J = 9.0 Hz, 3H), 7.17 (d, J = 5.7 Hz, 2H), 7.01 (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, DMSO) δ 161.45, 160.85, 156.21, 143.12, 139.37, 137.48, 134.10, 124.05, 120.69, 115.76, 114.24, 112.68, 106.86, 102.30; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1549; HPLC purity, 98.5%.
7-Methoxy-d3-2-(3-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8e).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 3-methoxy-d3-phenylhydrazine 4b (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8e as a yellow powder (1.5 g, 62.0%): 1H NMR (300 MHz, DMSO) δ 12.62 (s, 1H), 8.66 (s, 1H), 8.13 (d, J = 9.4 Hz, 1H), 7.93 – 7.73 (m, 2H), 7.34 (t, J = 8.2 Hz, 1H), 7.18 (d, J = 6.7 Hz, 2H), 6.74 (d, J = 8.2 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.13, 161.02, 159.97, 143.50, 141.73, 139.59, 137.58, 129.94, 124.18, 115.84, 112.56, 111.30, 109.60, 106.83, 104.81, 102.32; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1545; HPLC purity, 99.9%.
7-Methoxy-d3-2-(2-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8f).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 2-methoxy-d3-phenylhydrazine 4c (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford (8f) as a yellow powder (1.8 g, 75.0%): 1H NMR (300 MHz, DMSO) δ 12.47 (s, 1H), 8.57 (s, 1H), 7.99 (d, J = 8.7 Hz, 1H), 7.51 – 7.24 (m, 2H), 7.22 – 6.93 (m, 4H); 13C NMR (75 MHz, DMSO) δ 162.19, 160.63, 155.66, 142.97, 139.04, 137.26, 129.88, 129.67, 128.51, 123.87, 120.65, 115.49, 112.96, 112.91, 105.83, 102.15; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1552; HPLC purity, 99.9%.
7-Methoxy-2-(3-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8g).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-quinoline-3-carboxylate 7e (2 g, 7.5 mmol), 3-methoxy-d3-phenylhydrazine 4b (1.28 g, 9.0 mmol), triethylamine (0.91g, 9.0 mmol) and xylenes (16 mL) to afford (8g) as a yellow powder (0.9 g, 37.0%): 1H NMR (300 MHz, DMSO) δ 12.61 (s, 1H), 8.66 (d, J = 2.9 Hz, 1H), 8.14 (dd, J = 9.4, 3.0 Hz, 1H), 7.97 – 7.69 (m, 2H), 7.34 (td, J = 8.2, 2.9 Hz, 1H), 7.18 (d, J = 1.3 Hz, 2H), 6.74 (d, J = 8.0 Hz, 1H), 3.89 (s, 3H); 13C NMR (75 MHz, DMSO) δ 162.13, 161.03, 159.99, 143.51, 141.74, 139.58, 137.59, 129.94, 124.19, 115.84, 112.60, 111.31, 109.61, 106.84, 104.83, 102.36, 56.02; HRMS (ESI) m/z calculated for C18H13D3N3O3 (M+H)+ 325.1374; found 325.1365; HPLC purity, 98.8%.
7-Methoxy-2-(2-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8h).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-quinoline-3-carboxylate 7e (2 g, 7.5 mmol), 2-methoxy-d3-phenylhydrazine 4c (1.28 g, 9.0 mmol), triethylamine (0.91g, 9.0 mmol) and xylenes (16 mL) to afford 8h as a yellow powder (1.6 g, 65.6%): 1H NMR (300 MHz, DMSO) δ 12.46 (d, J = 4.9 Hz, 1H), 8.57 (d, J = 5.8 Hz, 1H), 7.99 (d, J = 8.7 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 7.15 (dd, J = 9.7, 6.0 Hz, 3H), 7.03 (t, J = 7.5 Hz, 1H), 3.87 (s, 3H); 13C NMR (75 MHz, DMSO) δ 184.22, 162.19, 160.62, 155.66, 142.95, 139.04, 137.26, 129.88, 129.66, 128.51, 123.88, 120.65, 115.49, 112.97, 112.94, 105.83, 102.15, 55.94; HRMS (ESI) m/z calculated for C18H13D3N3O3 (M+H)+ 325.1374; found 325.1367; HPLC purity, 99.9%.
8-Chloro-2-(3-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8i).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4,6-dichloro-quinoline-3-carboxylate 7c (2 g, 7.4 mmol), 3-methoxyphenylhydrazine hydrochloride 4e (1.55 g, 8.9 mmol), triethylamine (1.80g, 17.8 mmol) and xylenes (16 mL) to afford 8i as a yellow powder (0.7 g, 30.0%): 1H NMR (300 MHz, DMSO) δ 12.85 (s, 1H), 8.69 (s, 1H), 8.15 (d, J = 1.9 Hz, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.70 (dt, J = 9.0, 5.4 Hz, 2H), 7.34 (t, J = 8.1 Hz, 1H), 6.83 – 6.65 (m, 1H), 3.81 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.99, 159.98, 142.44, 141.52, 140.02, 134.81, 131.11, 130.62, 129.97, 122.17, 121.62, 120.42, 111.47, 110.04, 106.80, 104.96, 55.59; HRMS (ESI) m/z calculated for C17H13ClN3O2 (M+H)+ 326.0691; found 326.0693; HPLC purity, 99.5%. The spectroscopic properties of 8i were identical to those reported in the literature.28
8-Chloro-2-(3-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8j).
The title compound was prepared by a similar procedure as that used for 8a using of ethyl-4,6-dichloro-quinoline-3-carboxylate 7c (2 g, 7.4 mmol), 3-methoxy-d3-phenylhydrazine hydrochloride 4b (1.45 g, 8.1 mmol), triethylamine (1.87g, 18.5 mmol) and xylenes (16 mL) to afford 8j as a yellow powder (2.0 g, 87.0%): 1H NMR (300 MHz, DMSO) δ 12.85 (s, 1H), 8.71 (s, 1H), 8.17 (s, 1H), 8.00 – 7.49 (m, 4H), 7.35 (t, J = 7.7 Hz, 1H), 6.77 (d, J = 7.4 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.01, 160.01, 142.48, 141.54, 140.10, 134.76, 131.15, 130.72, 130.01, 122.14, 121.68, 120.45, 111.42, 110.04, 106.87, 104.95; HRMS (ESI) m/z calculated for C17H10D3ClN3O2 (M+H)+ 329.0879; found 329.0854; HPLC purity, 98.9%.
8-Chloro-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8k).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4,6-dichloro-quinoline-3-carboxylate 7c (2 g, 7.4 mmol), 4-methoxyphenylhydrazine hydrochloride 4d (1.55 g, 8.9 mmol), triethylamine (1.80g, 17.8 mmol) and xylenes (16 mL) to afford 8k as a yellow powder (1.7 g, 71.0%): 1H NMR (300 MHz, DMSO) δ 12.95 (d, J = 5.6 Hz, 1H), 8.72 (d, J = 6.2 Hz, 1H), 8.14 (d, J = 2.1 Hz, 1H), 8.08 (d, J = 9.0 Hz, 2H), 7.70 (dt, J = 8.9, 5.5 Hz, 2H), 7.02 (d, J = 9.1 Hz, 2H), 3.79 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.37, 156.48, 141.97, 139.79, 134.59, 133.87, 131.04, 130.46, 122.08, 121.50, 120.89, 120.48, 114.29, 106.86, 55.71; HRMS (ESI) m/z calculated for C17H13ClN3O2 (M+H)+ 326.0691; found 326.0682; HPLC purity, 98.6%. The 1H NMR spectroscopic properties of 8k were identical to those reported in the literature.25
8-Chloro-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8l).
The title compound was prepared by a similar procedure as that used for 8a using of ethyl-4,6-dichloro-quinoline-3-carboxylate 7c (2 g, 7.4 mmol), 4-methoxy-d3-phenylhydrazine 4a (1.25 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8i as a yellow powder (1.3 g, 53.6%): 1H NMR (300 MHz, DMSO) δ 12.89 (s, 1H), 8.74 (s, 1H), 8.24 – 7.89 (m, 3H), 7.86 – 7.56 (m, 2H), 7.02 (d, J = 8.9 Hz, 2H); 13C NMR (75 MHz, DMSO) δ 161.38, 156.49, 141.98, 139.86, 134.59, 133.84, 131.05, 130.49, 122.09, 121.52, 120.91, 120.49, 114.29, 106.88; HRMS (ESI) m/z calculated for C17H10D3ClN3O2 (M+H)+ 329.0879; found 329.0855; HPLC purity, 99.5%.
8-Chloro-2-(2-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8m).
The title compound was prepared by a similar procedure as that used for 8a using of ethyl-4,6-dichloro-quinoline-3-carboxylate 7c (2 g, 7.4 mmol), 2-methoxy-d3-phenylhydrazine 4c (1.25 g, 8.9 mmol), triethylamine (0.9g, 8.9 mmol) and xylenes (16 mL) to afford 8m as a yellow powder (1.0 g, 41.0%): 1H NMR (300 MHz, DMSO) δ 12.74 (s, 1H), 8.66 (s, 1H), 8.03 (s, 1H), 7.69 (p, J = 9.0 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.15, 155.64, 141.87, 139.59, 134.48, 130.83, 130.23, 129.91, 129.85, 128.22, 121.91, 121.40, 120.76, 120.68, 113.00, 105.81; HRMS (ESI) m/z calculated for C17H10D3ClN3O2 (M+H)+ 329.0879; found 329.0858; HPLC purity, 99.9%.
7-Bromo-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8n).
The title compound was prepared by a similar procedure as that used for 8a using of ethyl-7-bromo-4-chloro-quinoline-3-carboxylate 7d (2 g, 6.3 mmol), 4-methoxyphenylhydrazine hydrochloride 4d (1.33 g, 7.6 mmol), triethylamine (1.54g, 15.3 mmol) and xylenes (16 mL) to afford 8n as a yellow powder (1.4 g, 60.0%): 1H NMR (300 MHz, DMSO) δ 12.75 (s, 1H), 8.74 (s, 1H), 8.09 (dd, J = 17.7, 8.8 Hz, 3H), 7.89 (d, J = 1.6 Hz, 1H), 7.68 (dd, J = 8.6, 1.6 Hz, 1H), 7.02 (d, J = 9.1 Hz, 2H), 3.79 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.37, 156.47, 142.38, 140.08, 136.98, 133.85, 129.65, 124.51, 122.95, 122.22, 120.87, 118.22, 114.31, 107.21, 55.71; HRMS (ESI) m/z calculated for C17H13BrN3O2 (M+H)+ 370.0186; found 370.0176; HPLC purity, 99.0%.
7-Bromo-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8o).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-7-bromo-4-chloro-quinoline-3-carboxylate 7d (2 g, 6.3 mmol), 4-methoxy-d3-phenylhydrazine 4a (1.08 g, 7.6 mmol), triethylamine (0.77g, 7.6 mmol) and xylenes (16 mL) to afford 8o as a yellow powder (1.0 g, 42.0%): 1H NMR (300 MHz, DMSO) δ 12.75 (s, 1H), 8.74 (d, J = 4.9 Hz, 1H), 8.09 (dd, J = 17.8, 8.7 Hz, 3H), 7.88 (s, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 8.8 Hz, 2H); 13C NMR (75 MHz, DMSO) δ 161.35, 156.49, 141.88, 140.06, 136.95, 133.83, 129.65, 124.51, 122.93, 122.18, 120.86, 118.22, 114.24, 107.22; HRMS (ESI) m/z calculated for C17H10D3BrN3O2 (M+H)+ 373.0374; found 373.0359; HPLC purity, 99.8%.
7-Bromo-2-(3-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8p).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-7-bromo-4-chloro-quinoline-3-carboxylate 7d (1.5 g, 4.8 mmol), 3-methoxy- d3-phenylhydrazine 4b (0.81 g, 5.7 mmol), triethylamine (0.58g, 5.7 mmol) and xylenes (12 mL) to afford 8p as a yellow powder (0.7 g, 40.0%): 1H NMR (300 MHz, DMSO) δ 12.78 (s, 1H), 8.76 (s, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.95 – 7.76 (m, 3H), 7.71 (d, J = 8.6 Hz, 1H), 7.35 (t, J = 8.2 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.00, 160.00, 142.81, 141.51, 140.33, 137.11, 130.03, 129.75, 124.65, 123.22, 122.26, 118.16, 111.39, 109.98, 107.20, 104.92; HRMS (ESI) m/z calculated for C17H10D3BrN3O2 (M+H)+ 373.0374; found 373.0366; HPLC purity, 99.7%.
7-Bromo-2-(2-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8q).
The title compound was prepared by a similar procedure as that used for 8a using of ethyl-7-bromo-4-chloro-quinoline-3-carboxylate 7d (2 g, 6.3 mmol), 2-methoxy-d3-phenylhydrazine 4c (1.08 g, 7.6 mmol), triethylamine (0.77g, 7.6 mmol) and xylenes (16 mL) to afford 8q as a yellow powder (1.2 g, 50.6%): 1H NMR (300 MHz, DMSO) δ 12.60 (s, 1H), 8.64 (d, J = 16.3 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.86 (s, 1H), 7.64 (d, J = 8.5 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 7.7 Hz, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.14, 158.75, 155.64, 142.23, 139.79, 136.89, 129.84, 129.45, 128.23, 124.36, 122.64, 122.04, 120.69, 118.50, 112.99, 106.18; HRMS (ESI) m/z calculated for C17H10D3BrN3O2 (M+H)+ 373.0374; found 373.0386; HPLC purity, 99.9%.
8-Methoxy-d3-2-(4-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8r).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-6-methoxy-d3-quinoline-3-carboxylate 7a (2 g, 7.4 mmol), 4-methoxy-d3-phenylhydrazine 4a (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8r as a yellow powder (0.9 g, 37.0%): 1H NMR (300 MHz, DMSO) δ 12.77 (s, 1H), 8.64 (s, 1H), 8.11 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 9.1 Hz, 1H), 7.57 (s, 1H), 7.28 (d, J = 9.1 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H); 13C NMR (75 MHz, DMSO) δ 161.59, 157.98, 156.34, 143.00, 138.11, 134.10, 130.13, 121.70, 120.92, 120.50, 119.95, 114.23, 105.70, 102.96; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1562; HPLC purity, 99.9%.
8-Methoxy-d3-2-(3-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8s).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-6-methoxy-d3-quinoline-3-carboxylate 7a (2 g, 7.4 mmol), 3-methoxy-d3-phenylhydrazine 4b (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8s as a yellow powder (1.8 g, 74.0%): 1H NMR (300 MHz, DMSO) δ 12.80 (s, 1H), 8.65 (s, 1H), 7.99 – 7.80 (m, 2H), 7.67 (d, J = 9.1 Hz, 1H), 7.59 (s, 1H), 7.41 – 7.21 (m, 2H), 6.76 (d, J = 8.2 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.25, 159.98, 158.04, 143.42, 141.76, 138.36, 130.26, 129.94, 121.73, 120.45, 120.10, 111.53, 109.64, 105.70, 105.11, 103.16; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1550; HPLC purity, 99.9%.
8-Methoxy-d3-2-(2-methoxy-d3-phenyl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (8t).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-6-methoxy-d3-quinoline-3-carboxylate 7a (2 g, 7.4 mmol), 2-methoxy-d3-phenylhydrazine 4c (1.26 g, 8.9 mmol), triethylamine (0.90g, 8.9 mmol) and xylenes (16 mL) to afford 8t as a yellow powder (0.5 g, 20.0%): 1H NMR (300 MHz, DMSO) δ 12.65 (s, 1H), 8.57 (s, 1H), 7.65 (d, J = 9.1 Hz, 1H), 7.54 – 7.28 (m, 3H), 7.28 – 7.21 (m, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.05 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 162.38, 157.83, 155.76, 142.92, 137.98, 130.05, 129.94, 129.83, 128.54, 121.50, 120.75, 120.65, 119.64, 112.91, 104.61, 102.88; HRMS (ESI) m/z calculated for C18H10D6N3O3 (M+H)+ 328.1563; found 328.1546; HPLC purity, 99.9%.
2-Methoxy-d3-5-nitropyridine (10).
To a mixture of potassium t-butoxide (13.3 g, 111.8 mmol) and methanol-d4 (50 mL) was slowly added 2-chloro-5-nitropyridine 9 (15.0 g, 94.6 mmol). The exothermic reaction warmed up to 50 °C and then was heated at reflux at 65°C for 2 h to complete the reaction. The reaction mixture was then cooled to 20-25 °C and poured into H2O (750 mL). After stirring the mixture for 1 h the solid, which formed, was filtered and washed with H2O (25 mL x 2). The solid was dried under vacuum at 40 °C to afford 10 as a light yellow powder (13.0g, 87%). 1H NMR (300 MHz, DMSO) δ 9.08 (s, 1H), 8.47 (d, J = 9.1 Hz, 1H), 7.03 (d, J = 9.1 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 167.45, 145.02, 139.99, 135.01, 111.67; HRMS (ESI) m/z calculated for C6H4D3N2O3 (M+H)+ 158.0639; found 158.0634.
5-Amino-2-methoxy-d3-pyridine (11a).
A mixture of 2-methoxy-d3-5-nitropyridine 10 (13.0 g, 82.7 mmol), iron powder (15.9 g, 284.7 mmol), H2O (5 mL) and EtOH (50 mL) was heated to reflux (78 °C). Once at reflux, concentrated hydrochloric acid (1 mL, 83.3 mmol) was added dropwise. The reaction mixture was heated at reflux for 4 h to complete the reaction. Upon cooling to 20-25 °C, the mixture was filtered to remove the iron and the solids were washed 3 times with EtOH (25 mL x 3). Sodium bicarbonate (5.0 g) was added to the filtrate and the EtOH was removed under reduced pressure. Then, H2O (50 mL) and DCM (50 mL) were added to dissolve the residue. The layers were separated and the aq layer was extracted with DCM (50 mL x 2). The combined organic layers were dried (MgSO4). The solvents were then removed under reduced pressure to afford 11a as a clear orange-red oil (10.0 g, 95.1%): 1H NMR (300 MHz, DMSO) δ 7.57 (d, J = 2.7 Hz, 1H), 7.05 (dd, J = 8.6, 2.9 Hz, 1H), 6.56 (d, J = 8.7 Hz, 1H), 4.74 (s, 2H); 13C NMR (75 MHz, DMSO) δ 156.29, 139.77, 131.63, 126.85, 110.42; HRMS (ESI) m/z calculated for C6H6D3N2O (M+H)+ 128.0898; found 128.0910. This material was employed in the next experiment with no further purification.
General Procedure for the Synthesis of 5-Hydrazinyl-2-methoxypyridines (12a-b).
5-Hydrazinyl-2-methoxy-d3-pyridine (12a).
A mixture of 5-amino-2-methoxy-d3-pyridine 11a (10 g, 78.6 mmol), concentrated hydrochloric acid (24 mL), and H2O (24 mL) was cooled to 0 to 5°C and a solution of sodium nitrite (5.7 g, 82.5 mmol) and H2O (12 mL) was slowly added drop-wise to the reaction mixture. Upon completion of the addition, the reaction mixture was allowed to stir for an additional 15 min at 0 to 5 °C. The reaction mixture was then cooled to −25 to −20 °C and a solution of tin (II) chloride (32.8 g, 173.0 mmol) and concentrated hydrochloric acid (70 mL) was added drop-wise to the reaction mixture over 30 min. Upon completion of the addition, the reaction mixture was allowed to stir for an additional 2 h at −25 to −20 °C. The reaction mixture was then diluted with DCM (100 mL). A solution of potassium hydroxide (100 g) in H2O (200 mL) was added dropwise to the reaction mixture at 0 to 5 °C over 30 min. After stirring for 1 h at 0 to 5 °C, the solids completely dissolved. The layers were separated and the aq layer was extracted with DCM (50mL x 4). The combined organic layers were dried (MgSO4). The solvents were then removed under reduced pressure and the residue was slurried with hexanes (20 mL). The slurry was placed in a freezer at −20 °C for 24 h to fully precipitate the product. The solid was then filtered off and washed with hexanes (10 mL x 2). The solid was dried under vacuum at rt to afford 12a as a pale yellow-brown solid (6.4 g, 57%): 1H NMR (300 MHz, DMSO) δ 7.70 (d, J = 2.8 Hz, 1H), 7.31 – 7.19 (m, 1H), 6.61 (d, J = 8.8 Hz, 1H), 6.42 (s, 1H), 4.00 (s, 2H); 13C NMR (75 MHz, DMSO) δ 157.01, 144.07, 129.87, 125.27, 110.22; HRMS (ESI) m/z calculated for C6H7D3N3O (M+H)+ 143.1007; found 143.1005.
5-Hydrazinyl-2-methoxypyridine (12b).
The title compound was prepared by a similar procedure as that used for 12a using 5-amino-2-methoxypyridine 11b (10 g, 80.6 mmol), concentrated hydrochloric acid (24 mL) in H2O (24 mL), sodium nitrite (5.8 g, 84.6 mmol) in H2O (12 mL), and tin (II) chloride (33.6 g, 177.2 mmol) in concentrated hydrochloric acid (70 mL) to afford 12b as a pale yellow-brown solid (7.8 g, 70%): 1H NMR (300 MHz, DMSO) δ 7.70 (s, 1H), 7.20 (d, J = 8.7 Hz, 1H), 6.61 (d, J = 8.8 Hz, 1H), 6.41 (s, 1H), 3.97 (s, 2H), 3.73 (s, 3H); 13C NMR (75 MHz, DMSO) δ 156.98, 144.09, 129.85, 125.25, 110.22, 53.19; HRMS (ESI) m/z calculated for C6H10N3O (M+H)+ 140.0818; found 140.0814. The 1H NMR and 13C NMR spectroscopic properties of 12b were identical to those reported in the literature.45
7-Methoxy-2-(6-methoxypyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13a).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-quinoline-3-carboxylate 7e (2 g, 7.5 mmol), 5-hydrazinyl-2-methoxypyridine 12b (1.26 g, 9.0 mmol), triethylamine (0.91g, 9.0 mmol) and xylenes (16 mL) to afford 13a as a yellow powder (1.2 g, 49.0%): 1H NMR (300 MHz, DMSO) δ 12.65 (s, 1H), 8.92 (d, J = 2.4 Hz, 1H), 8.68 (s, 1H), 8.43 (dd, J = 9.0, 2.6 Hz, 1H), 8.24 – 7.91 (m, 1H), 7.29 – 7.02 (m, 2H), 6.92 (d, J = 9.0 Hz, 1H), 3.88 (s, 6H); 13C NMR (75 MHz, DMSO) δ 161.74, 160.98, 160.43, 143.86, 139.71, 137.45, 137.37, 131.88, 130.77, 124.13, 115.94, 112.59, 110.56, 106.22, 102.30, 56.00, 53.71; HRMS (ESI) m/z calculated for C17H15N4O3 (M+H)+ 323.1139; found 323.1133; HPLC purity, 99.8%.
7-Methoxy-2-(6-methoxy-d3-pyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13b).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-quinoline-3-carboxylate 7e (2 g, 7.5 mmol), 5-hydrazinyl-2-methoxy-d3-pyridine 12a (1.28 g, 9.0 mmol), triethylamine (0.91 g, 9.0 mmol) and xylenes (16 mL) to afford 13b as a yellow powder (1.2 g, 49.0%): 1H NMR (300 MHz, DMSO) δ 12.68 (s, 1H), 8.91 (d, J = 2.1 Hz, 1H), 8.68 (s, 1H), 8.42 (dd, J = 9.0, 2.4 Hz, 1H), 8.16 – 8.03 (m, 1H), 7.18 (d, J = 5.9 Hz, 2H), 6.92 (d, J = 9.0 Hz, 1H), 3.87 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.74, 160.98, 160.44, 143.85, 139.69, 137.44, 137.39, 131.86, 130.77, 124.13, 115.94, 112.58, 110.54, 106.22, 102.29, 56.00; HRMS (ESI) m/z calculated for C17H12D3N4O3 (M+H)+ 326.1327; found 326.1316; HPLC purity, 99.9%.
7-Methoxy-d3-2-(6-methoxypyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13c).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 5-hydrazinyl-2-methoxypyridine 12b (1.24 g, 8.9 mmol), triethylamine (0.90 g, 8.9 mmol) and xylenes (16 mL) to afford 13c as a yellow powder (1.0 g, 41.0%): 1H NMR (300 MHz, DMSO) δ 12.69 (s, 1H), 8.92 (s, 1H), 8.69 (s, 1H), 8.43 (d, J = 9.0 Hz, 1H), 8.12 (d, J = 9.4 Hz, 1H), 7.18 (s, 2H), 6.92 (d, J = 9.0 Hz, 1H), 3.88 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.75, 161.01, 160.44, 143.88, 139.73, 137.46, 137.39, 131.88, 130.79, 124.15, 115.96, 112.57, 110.58, 106.22, 102.31, 53.72; HRMS (ESI) m/z calculated for C17H12D3N4O3 (M+H)+ 326.1327; found 326.1324; HPLC purity, 99.9%.
7-Methoxy-d3-2-(6-methoxy-d3-pyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one 13d).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-d3-quinoline-3-carboxylate 7b (2 g, 7.4 mmol), 5-hydrazinyl-2-methoxy-d3-pyridine 12a (1.27 g, 8.9 mmol), triethylamine (0.90 g, 8.9 mmol) and xylenes (16 mL) to afford 13d as a yellow powder (1.7 g, 69.7%): 1H NMR (500 MHz, DMSO) δ 12.70 (s, 1H), 8.92 (d, J = 2.3 Hz, 1H), 8.69 (s, 1H), 8.43 (dd, J = 8.9, 2.4 Hz, 1H), 8.12 (d, J = 9.4 Hz, 1H), 7.19 (d, J = 7.4 Hz, 2H), 6.93 (d, J = 8.9 Hz, 1H).; 13C NMR (126 MHz, DMSO) δ 161.75, 160.99, 160.45, 143.87, 139.71, 137.45, 137.39, 131.87, 130.79, 124.15, 115.96, 112.57, 110.57, 106.23, 102.28.; HRMS (ESI) m/z calculated for C17H9D6N4O3 (M+H)+ 329.1515; found 329.1507; HPLC purity, 99.9%.
8-Chloro-2-(6-methoxypyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13e).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4,6-dichloroquinoline-3-carboxylate 7c (2 g, 7.4 mmol), 5-hydrazinyl-2-methoxypyridine 12b (1.24 g, 8.9 mmol), triethylamine (0.90 g, 8.9 mmol) and xylenes (16 mL) to afford 13e as a yellow powder (1.0 g, 41.0%): 1H NMR (300 MHz, DMSO) δ 12.96 (s, 1H), 8.92 (d, J = 2.6 Hz, 1H), 8.77 (s, 1H), 8.42 (dd, J = 8.9, 2.6 Hz, 1H), 8.14 (s, 1H), 7.89 – 7.60 (m, 2H), 6.93 (d, J = 9.0 Hz, 1H), 3.89 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.62, 160.64, 142.70, 140.12, 137.57, 134.58, 131.66, 131.15, 130.92, 130.64, 122.11, 121.58, 120.38, 110.59, 106.25, 53.74; HRMS (ESI) m/z calculated for C16H12ClN4O2 (M+H)+ 327.0643; found 327.0623; HPLC purity, 99.8%.
8-Chloro-2-(6-methoxy-d3-pyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13f).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4,6-dichloroquinoline-3-carboxylate 7c (2 g, 7.4 mmol), 5-hydrazinyl-2-methoxy-d3-pyridine 12a (1.26 g, 8.9 mmol), triethylamine (0.90 g, 8.9 mmol) and xylenes (16 mL) to afford 13f as a yellow powder (1.4 g, 57.0%): 1H NMR (300 MHz, DMSO) δ 12.92 (s, 1H), 8.90 (d, J = 1.7 Hz, 1H), 8.74 (d, J = 9.1 Hz, 1H), 8.40 (dd, J = 8.9, 2.4 Hz, 1H), 8.09 (s, 1H), 7.78 – 7.61 (m, 2H), 6.90 (d, J = 8.9 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 161.60, 160.63, 142.67, 140.06, 137.55, 134.55, 131.64, 131.13, 130.87, 130.60, 122.07, 121.56, 120.37, 110.55, 106.26; HRMS (ESI) m/z calculated for C16H9D3ClN4O2 (M+H)+ 330.0832; found 330.0811; HPLC purity, 99.7%.
7-Bromo-2-(6-methoxypyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13g).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-7-bromo-4-chloroquinoline-3-carboxylate 7d (2 g, 6.3 mmol), 5-hydrazinyl-2-methoxypyridine 12b (1.06 g, 7.6 mmol), triethylamine (0.77 g, 7.6 mmol) and xylenes (16 mL) to afford 13g as a yellow powder (1.6 g, 67.0%): 1H NMR (300 MHz, DMSO) δ 12.81 (s, 1H), 10.29 – 10.27 (m, 1H), 8.89 (s, 1H), 8.75 (s, 1H), 8.40 (dd, J = 8.9, 2.2 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.86 (s, 1H), 7.68 (d, J = 8.1 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 3.88 (s, 3H); 13C NMR (75 MHz, DMSO) δ 161.63, 160.61, 143.11, 140.38, 137.53, 136.95, 131.65, 130.88, 129.74, 124.54, 123.15, 122.26, 118.12, 110.61, 106.58, 53.74; HRMS (ESI) m/z calculated for C16H12BrN4O2 (M+H)+ 371.0138; found 371.0119; HPLC purity, 99.7%.
7-Bromo-2-(6-methoxy-d3-pyridin-3-yl)-2,5-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (13h).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-7-bromo-4-chloroquinoline-3-carboxylate 7d (1.20 g, 3.8 mmol), 5-hydrazinyl-2-methoxy-d3-pyridine 12a (0.65 g, 4.6 mmol), triethylamine (0.46 g, 4.6 mmol) and xylenes (16 mL) to afford 13h as a yellow powder (0.5 g, 35.0%): 1H NMR (300 MHz, DMSO) δ 12.77 (s, 1H), 8.88 (d, J = 2.3 Hz, 1H), 8.75 (d, J = 8.4 Hz, 1H), 8.39 (dd, J = 8.9, 2.5 Hz, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.83 (s, 1H), 7.65 (d, J = 8.6 Hz, 1H), 6.90 (d, J = 8.9 Hz, 1H); 13C NMR (75 MHz, DMSO) δ 161.60, 160.60, 143.08, 140.33, 137.51, 136.93, 131.63, 130.83, 129.70, 124.50, 123.11, 122.24, 118.10, 110.56, 106.58; HRMS (ESI) m/z calculated for C16H9D3BrN4O2 (M+H)+ 374.0326; found 374.0306; HPLC purity, 99.8%.
7-Methoxy-2-(4-methoxyphenyl)-2,5-dihydro-3H-pyrazolo[4,3-c][1,6]naphthyridin-3-one (13i).
The title compound was prepared by a similar procedure as that used for 8a using ethyl-4-chloro-7-methoxy-1,6-naphthyridine-3-carboxylate 7f (1.2 g, 4.5 mmol), 4-methoxyphenylhydrazine hydrochloride 4d (0.94 g, 5.4 mmol), triethylamine (1.09 g, 10.8 mmol) and xylenes (15 mL) to afford 13i as a yellow powder (1.0 g, 69.0%): 1H NMR (500 MHz, DMSO) δ 12.61 (s, 1H), 8.60 (s, 1H), 8.08 (d, J = 7.9 Hz, 2H), 7.83 (d, J = 7.3 Hz, 1H), 7.02 (d, J = 8.3 Hz, 2H), 6.45 (d, J = 7.1 Hz, 1H), 3.79 (s, 3H), 3.51 (s, 3H); 13C NMR (126 MHz, DMSO) δ 161.75, 160.99, 160.45, 143.87, 139.71, 137.45, 137.39, 131.87, 130.79, 124.15, 115.96, 112.57, 110.57, 106.23, 102.28.; HRMS (ESI) m/z calculated for C17H15N4O3 (M+H)+ 323.1139; found 323.1139; HPLC purity, 98.6%.
Biology
Microsomal stability assay.
The 4 μL of 1 mM test compound at a final concentration of 10μM dissolved in either DMSO, ACN, MeOH, or EtOH (depending on compound solubility) was preincubated at 37°C for 5 minutes on a digital heating shaking dry bath (Fischer scientific, Pittsburgh, PA) in a mixture containing 282 μL of H2O, 80 μL of phosphate buffer (0.5 M, pH 7.4), 20 μL of NADPH Regenerating System Solution A (BD Bioscience, San Jose, CA) and 4 μL of NADPH Regenerating System Solution B (BD Bioscience, San Jose, CA) in a total volume of 391.2 μL. Following preincubation, the reaction was initiated by addition of 8.8 μL of either HLM (BD Gentest, San Jose, CA), or MLM (Life technologies, Rockford, IL), at a protein concentration of 0.5 mg/mL. Aliquots of 50 μL were taken at time intervals of 0 (without microsomes), 10, 20, 30, 40, 50 and 60 minutes. Each aliquot was added to 100 μL of cold acetonitrile solution containing 1 μM/2 μM internal standard. This was followed by sonication for 10 seconds and centrifugation at 10,000 rpm for 5 minutes. The 100 μL of the supernatant was transferred into Spin-X HPLC filter tubes (Corning Incorporated, NY) and centrifuged at 13,000 rpm for 5 minutes. The filtrate was diluted 100 fold and subsequently analyzed by LC-MS/MS with Shimadzu LCMS 8040, (Shimadzu Scientific Instruments, Columbia, MD). The ratio of the peak areas of the internal standard and test compound was calculated for every time point and the natural log of the ratio was plotted against time to determine the linear slope (k). The metabolic rate (k*C0/C), half-life (0.693/k), and internal clearance (V*k) were calculated, where k is the slope, C0 is the initial concentration of test compound, C is the concentration of microsomes, and V is the volume of incubation in μL per microsomal protein in mg. All experiments (Table 3) were repeated three times in duplicate and followed the experimental procedures reported in Forkuo et al.57,58 and Jahan et al.59
Metabolite Assay.
Metabolite analysis by LCMS confirmed OCH3 rate > OCD3 rate and that the primary metabolites for the PQs was generated via O-demethylation of the methoxy groups to form hydroxypyrazoloquinolinones. The 60 min samples from the metabolism study above were injected on a LC-MS with Shimadzu LCMS 2020, (Shimadzu Scientific Instruments, Columbia, MD) using a SIM mode (Dual Ion Source (DUIS) ionization, simultaneous ESI and APCI) method calibrated to the respective retention times and mass units of each component and the respective peak intensities were measured. Compound peaks were resolved using a Shimadzu C18 3 μm 50 × 4.6mm reversed phase LC column. LC mobile phase: 90% acetonitrile (w. 0.1% TFA) and 10% H2O (w/ 0.1% TFA). Results shown in Scheme 4.
Electrophysiological Assay.
The details of electrophysiological experiments with Xenopus oocytes have been reported.21, 60 Mature female Xenopus laevis (Nasco, Fort Atkinson, WI, USA) were anaesthetized in a bath of ice-cold 0.17% Tricain (Ethyl-m-aminobenzoat, Sigma-Aldrich, St. Louis, MO, USA) before decapitation and transfer of the frog’s ovary to ND96 medium (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES; pH 7.5). Following incubation in 1mg/ml collagenase (Sigma-Aldrich, St. Louis, MO, USA) for 30 min, stage 5 to 6 oocytes were singled out of the ovary and defolliculated using a platinum wire loop. Oocytes were stored and incubated at 18°C in NDE medium (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, 1.8 mM CaCl2; pH 7.5) that was supplemented with 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and 2.5 mM pyruvate. Oocytes were injected with an aq solution of mRNA. A total of 2.5 ng of mRNA per oocyte was injected. Subunit ratio was 1:1:5 for αxβγ2 (x = 1,2,3,5) and 3:1:5 for α4/6β3γ2 and αβδ receptors. Injected oocytes were incubated for at least 36 h before electrophysiological recordings. Oocytes were placed on a nylon-grid in a bath of NDE medium. For current measurements, the oocytes were impaled with two microelectrodes (2–3MΩ), which were filled with 2M KCl. The oocytes were constantly washed by a flow of 6 mL·min−1 NDE that could be switched to NDE containing GABA and/or drugs. Drugs were diluted into NDE from DMSO solutions, which resulted, in a final concentration of 0.1% DMSO. Maximum currents measured in mRNA injected oocytes were in the microampere range for all receptor subtypes. To test for modulation of GABA induced currents by compounds, a GABA concentration that was titrated to trigger 3-5% of the respective maximum GABA-elicited current of the individual oocyte (EC3-5) was applied to the cell together with various concentrations of test compounds. All recordings were performed at room temperature at a holding potential of −60 mV using a Warner OC-725C TEV (Warner Instrument, Hamden, CT, USA) or a Dagan CA-1B Oocyte Clamp or a Dagan TEV-200A TEV (Dagan Corporation, Mineapolis, MN, USA). Data were digitized using a Digidata 1322A or 1550 data acquisition system (Axon Instruments, Union City, CA, USA), recorded using Clampex 10.5 software (Molecular Devices, Sunnyvale, CA, USA), and analyzed using Clampfit 10.5 and GraphPad Prism 6.0 (La Jolla, CA, USA) software. Concentration-response data were fitted using the Hill equation. Data are given as mean±SEM from at least three oocytes of two batches.
PDSP Compound-induced radioligand displacement assays for 46 receptors.
Binding affinity (Ki) determinations, receptor binding profiles, agonist and/or antagonist functional data, HERG data, MDR1 data, etc. as appropriate was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C(NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. See (S) Table S1 for the respective primary inhibition studies and (S) Table S2 for secondary binding affinity studies.
Solubility Assay and cLogP.
Solubility method: 10mg of each respective compound was dissolved in 2mL of DMAC. The water solubility (pH = 7) samples were prepared by adding 10 μL of the resulting DMAC solution to 2mL of DI H2O and sonicated at 37°C for 2 hours. The acidic solubility (pH = 2) samples were prepared by adding 10 μL of the resulting DMAC solution to 2mL of 0.01 M aq HCl and held at 37°C for 2 hours. The mixtures were then filtered using a 0.45 micron syringe filter. The resultant solutions were injected on a LC-MS with Shimadzu LCMS 2020, (Shimadzu Scientific Instruments, Columbia, MD) using a SIM mode (Dual Ion Source (DUIS) ionization, simultaneous ESI and APCI) method calibrated to the respective retention times and mass units of each compound. Compound peaks were resolved using a Shimadzu C18 3 μm 50 × 4.6mm reversed phase LC column. LC mobile phase: 90% acetonitrile (w. 0.1% TFA) and 10% H2O (w/ 0.1% TFA). Calibration curves using standard solutions (n = 5) of each compound dissolved in DMAC were used to calculate the unknown aq concentrations of each compound, n=3. The cLogP data was calculated using ChemBioDraw® Ultra (ver. 13.0.0.3015).37, 61 Data shown in Table 4.
Cytotoxicity Assay.
Human liver hepatocellular carcinoma (HEPG2) and human embryonic kidney 293T (HEK293T) cell lines were purchased (ATCC) and cultured in 75 cm2 flasks (CellStar). Cells were grown in DMEM/High Glucose (Hyclone, #SH3024301) media to which non-essential amino acids (Hyclone, #SH30238.01), 10 mM HEPES (Hyclone, #SH302237.01), 5 × 106 units of penicillin and streptomycin (Hyclone, #SV30010), and 10% of heat inactivated fetal bovine serum (Gibco, #10082147) were added. Cells were harvested using 0.05% Trypsin (Hyclone, #SH3023601), washed with PBS, and dispensed into sterile white, optical bottom 384-well plates (NUNC, #142762). After three hours, small molecule solutions were transferred with a Tecan Freedom EVO liquid handling system equipped with a 100 nL pin tool (V&P Scientific). The controls were 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (25 mM in DMSO, positive control) and DMSO (negative control). The cells were incubated for 48 hours followed by the addition of CellTiter-Glo™, a luminescence-based cell viability assay (Promega, Madison, WI). All luminescence readings were performed on a Tecan Infinite M1000 plate reader. The assay was carried out in quadruplet with three independent runs. The data was normalized to the controls and analyzed by nonlinear regression (GraphPad Prism), see (S) Table S3 and Figure S1.
Rotorod Motor Impairment Study.
Possible adverse CNS sensorimotor effects were evaluated using a rotarod apparatus ((S) Figure S2). Female Swiss Webster mice were dosed via oral gavage with 40 mg/kg of the indicated compound. The sensorimotor rotarod test was carried out after 10, 30 and 60 minutes. The diazepam positive control (5mg/kg), showed the greatest impairment of sensorimotor steadiness at 10 minutes, followed by 30 and 60 minutes. Several of the compounds were not soluble in the standard vehicle (50% PBS, 40% propylene glycol, 10% DMSO) and were subsequently administered via oral gavage at 40mg/kg in a vehicle of 2% polyethylene glycol and 2.5% hydroxymethyl cellulose solution. None of the compounds displayed motor impairment.
Pharmacokinetic Assay.
Pharmacokinetic study of 8a, 8b, 8c, 8d and 13c in male Wistar rats weighing 250-350 g was performed after an acute treatment with the 5 mg/ml suspension formulated in the vehicle consisting of 85% distilled H2O, 14% propylene glycol and 1% Tween 80. Single 10 mg/kg doses of each of the ligands were administered i.p. to three rats per time point (5, 20, 60, 180 and 720 min after dosing), at which blood and brain samples of the rats were collected after sacrificing. In addition, the absolute oral bioavailability of 8a and 8b was measured after respective single 2 mg/kg oral gavage (PO) or IV administration to three rats per post-dosing time 5, 30, 120, 480 and 1440 min (24 h). The ligands were extracted from the respective samples by solid phase extraction and their concentrations were determined by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). In a separate in vitro experiment, the rapid equilibrium dialysis assay was used to determine free fraction of 8b and 8d in rat plasma and brain tissue. See Figure 9 and Figure 10.
Battery of Behavioral Tests in Rats.
Behavioral studies (Table 5) in male Wistar rats weighing 200-300 g was performed with the biocompatible nanoemulsions of 8b and 8m formulated at 2 mg /mL−1. Nanoemulsions, with and without a ligand, were prepared by a procedure slightly modified from Djordjevic et al.,48 and injected IP in a 5 mL kg−1 volume (10 mg kg−1). They were composed of medium chain triglycerides, castor oil, soybean lecithin, sodium oleate, polyoxyethylensorbitan monooleate, butylhydroxytoluene, DMSO and ultra-pure H2O. Forty-five minutes after treatment, the rats (eight animals per group) were consecutively subjected to a battery of behavioral tests, consisting of the elevated plus maze, grip strength and cued water maze.
The elevated plus maze apparatus, constructed of black Plexiglas, was elevated to a height of 50 cm and consisted of two open (50×10 cm2) and two enclosed arms (50×10×40 cm3), connected by a junction area measured 10×10 cm2. Single rats were placed in the center of the maze, facing one of the enclosed arms, and their behavior was recorded for 5 min. An entry into an arm was scored when 90% of the animal crossed the virtual line separating the central square of the maze from an arm, whereas an exit occurred when more than 90% of the animal left the respective arm. Dependent variables chosen for analysis were total distance traveled, closed arm entries and percent of open arm entries.
The rat pulled by the tail grasps the trapeze connected to a force transducer of the grip strength meter (Ugo Basile, Milan, Italy, model 47105), and the apparatus measured the peak force of experimenter’s pull (in grams) necessary to overcome the strength of the animal’s forelimbs grip. Each animal was given three consecutive trials, and the median value of three measurements, normalized against body weight, was used for further statistics.
The cued water-maze experiment was performed in a 2 m diameter circular pool filled to a height of 30 cm with water at 22 ± 1 °C. The fixed escape platform (15 cm × 10 cm) submerged 2 cm below the water surface was flagged with an object protruding 15 cm above water level. Rats were given one swimming block, consisting of four trials. For each trial the rat was placed in the water at the same starting position, most distant to the platform. Once the rat has found and mounted the escape platform it was permitted to remain on the platform for 15 s. The rat was guided to the platform by the experimenter if it failed to locate it within 120 s. Dependent variables chosen for tracking were: total distance traveled, mean speed and % of distance swam in the peripheral annulus.
As a separate pilot experiment, the influence of 8b and 8m on motor performance of rats was assessed using an automated rotarod (Ugo Basile, Italy). Before testing, rats were trained for three days until they could remain for 180 s on the rod revolving at 15 rpm. On the fourth day, selection was made and rats fulfilling the given criteria were treated with the suspension formulations of 8b and 8m in the range 10-55 or 60 mg/kg (2-3 animals per group). Latency to fall from the rotarod was recorded 45 min later.
Spontaneous Locomotor Activity Assay in Rats and Mice.
Spontaneous locomotor activity (SLA) assay was performed in adult male Wistar rats or C57BL/6 mice in the apparatus consisting of four white and opaque Plexiglas chambers (40 × 25 × 35 cm) under dim red light (20 lux). Digital camera mounted above the apparatus recorded animal activity, which was tracked and analyzed using ANY-maze Video Tracking System software (Stoelting Co, Wood Dale, IL, US). Three experiments in both, rats and mice were performed; the treatments were administered IP and immediately afterwards, a single rat or mouse was placed in the center of the chamber with its activity being followed for a total of 60 min. Chambers were cleaned with 70% ethanol after every trial. See Figure 11.
Statistical analysis.
All in vivo data are expressed as mean ± SEM. Pharmacokinetic parameters were calculated using PK Functions for Microsoft Excel software (by Joel Usansky, Atul Desai, and Diane Tang-Liuwere). The animals were tracked by Any Maze software (Stoelting, Wood Dale, US) and behavioral parameters analyzed using one-way analysis of variance (ANOVA), with treatment as between-subject factor, with exception of the water-maze data, which were subjected to two-way repeated measures ANOVA (SigmaPlot 12.0, SPSS, Chicago, US).
Supplementary Material
Acknowledgments
We wish to thank the National Institutes of Health (NIH) – R01 NS076517 and R01 MH096463, the Austrian Science Fund I 2306-B27 and W1232, and the Ministry of Education, Science and Technological Development, R. Serbia – Grant No. 175076 for generous financial support. We also gratefully acknowledge the Shimadazu Laboratory for Advanced and Applied Analytical Chemistry and the Milwaukee Institute of Drug Discovery. The PDSP data was kindly provided by Dr. Bryan Roth (UNC, NIMH funded).
Abbreviations Used
- AUC
area under the curve
- BBB
blood-brain barrier
- Bz
benzodiazepine
- BZD
benzodiazepine
- Bz/GABAAR
benzodiazepine γ-aminobutyric acid type A receptor
- cLogP
octanol-water partition coefficient
- CNS
central nervous system
- CYP450
cytochrome P450
- DIE
deuterium isotope effect
- DI
diazepam insensitive
- DS
diazepam sensitive
- EC3
effective concentration eliciting 3% of maximal GABA current
- EC3-5
effective concentration eliciting 3-5% of maximal GABA current
- GABA
γ-aminobutyric acid
- GABAAR
γ-aminobutyric acid type A receptor
- hERG
human ether-à-go-go-related gene
- HLM
human liver microsomes
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- IC50
half maximal inhibitory concentration
- IP
intraperitoneal
- IV
intravenous
- Kp
brain-to-plasma partition coefficient
- LC
liquid chromatography
- LCMS
liquid chromatography-mass spectrometry
- LCMS-IT-TOF
liquid chromatography-mass spectrometry ion trap time-of flight
- MLM
mouse liver microsomes
- NMR
nuclear magnetic resonance
- PBS
phosphate-buffered saline
- PDA
photodiode array
- PDSP
Psychoactive Drug Screening Program
- PO
per os
- PQ
pyrazoloquinolinone
- SAR
structure-activity relationship
- TLC
thin-layer chromatography
References
- 1.Olsen RW; Sieghart W International Union of Pharmacology. LXX. Subtypes of Gamma-Aminobutyric Acid(A) Receptors: Classification on the Basis of Subunit Composition, Pharmacology, and Function. Update. Pharmacol. Rev 2008, 60, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olsen RW; Sieghart W GABA A Receptors: Subtypes Provide Diversity of Function and Pharmacology. Neuropharmacol. 2009, 56, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mortensen M; Patel B; Smart TG GABA Potency at GABA(A) Receptors Found in Synaptic and Extrasynaptic Zones. Front. Cell. Neurosci 2011, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisden W; Laurie DJ; Monyer H; Seeburg PH The Distribution of 13 GABAA Receptor Subunit mRNAs in the Rat Brain. I. Telencephalon, Diencephalon, Mesencephalon. J. Neurosci 1992, 12, 1040–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pirker S; Schwarzer C; Wieselthaler A; Sieghart W; Sperk G GABAA Receptors: Immunocytochemical Distribution of 13 Subunits in the Adult Rat Brain. Neurosci. 2000, 101, 815–850. [DOI] [PubMed] [Google Scholar]
- 6.Sieghart W Structure and Pharmacology of gamma-aminobutyric AcidA Receptor Subtypes. Pharmacol. Rev 1995, 47, 181–234. [PubMed] [Google Scholar]
- 7.Sieghart W; Ernst M Heterogeneity of GABAA Receptors: Revived Interest in the Development of Subtype-selective Drugs. Cur. Med. Chem 2005, 5, 217–242. [Google Scholar]
- 8.Ramerstorfer J; Furtmuller R; Sarto-Jackson I; Varagic Z; Sieghart W; Ernst M The GABAA Receptor alpha+beta- Interface: a Novel Target for Subtype Selective Drugs. J. Neurosci 2011, 31, 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieghart W; Ramerstorfer J; Sarto-Jackson I; Varagic Z; Ernst M A Novel GABA(A) Receptor Pharmacology: Drugs Interacting with the alpha(+) beta(−) Interface. Br. J. Pharmacol 2012, 166, 476–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varagic Z; Ramerstorfer J; Huang S; Rallapalli S; Sarto-Jackson I; Cook J; Sieghart W; Ernst M Subtype Selectivity of alpha+beta- Site Ligands of GABAA Receptors: Identification of the First Highly Specific Positive Modulators at alpha6beta2/3gamma2 Receptors. Br. J. Pharmacol 2013, 169, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varagic Z; Wimmer L; Schnurch M; Mihovilovic MD; Huang S; Rallapalli S; Cook JM; Mirheydari P; Ecker GF; Sieghart W; Ernst M Identification of Novel Positive Allosteric Modulators and Null Modulators at the GABAA Receptor alpha+beta- Interface. Br. J. Pharmacol 2013, 169, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nusser Z; Sieghart W; Somogyi P Segregation of Different GABAA Receptors to Synaptic and Extrasynaptic Membranes of Cerebellar Granule Cells. J. Neurosci 1998, 18, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutiérrez A; Khan ZU; De Blas AL Immunocytochemical Localization of the α6 Subunit of the γ-Aminobutyric acidA Receptor in the Rat Nervous System. J. Comp. Neuro 1996, 365, 504–510. [DOI] [PubMed] [Google Scholar]
- 14.Yang L; Xu T; Zhang K; Wei Z; Li X; Huang M; Rose GM; Cai X The Essential Role of Hippocampal alpha6 Subunit-Containing GABAA Receptors in Maternal Separation Stress-Induced Adolescent Depressive Behaviors. Behav. Brain Res 2016, 313, 135–143. [DOI] [PubMed] [Google Scholar]
- 15.Chiou LC; Wu CY; Lee HJ; Du JC; Hor CC; Ernst M; Sieghart W; Cook J The Cerebellar Alpha6-Containing GABA-A Receptor: A Novel Target for Neuropsychiatric Disorders In Neuroscience, San Diego, CA, USA, 2016. [Google Scholar]
- 16.Liao YH; Lee HJ; Huang WJ; Fan PC; Chiou LC Hispidulin Alleviated Methamphetamine-Induced Hyperlocomotion by Acting at alpha6 Subunit-Containing GABAA Receptors in the Cerebellum. Psychopharmacology. 2016, 233, 3187–3199. [DOI] [PubMed] [Google Scholar]
- 17.Fan PC; Huang M; Sieghart W; Ernst M; Knutson DE; Cook JM; Chiou LC The α6 Subunit-Containing GABAA Receptors: A Novel Target for Migraine Therapy. In The 18th Congress of International Headache Society (IHC2017), Vancouver, BC, Canada, 2017. [Google Scholar]
- 18.Kramer PR; Bellinger LL Reduced GABAA Receptor alpha6 Expression in the Trigeminal Ganglion Enhanced Myofascial Nociceptive Response. Neurosci. 2013, 245, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czernik AJ; Petrack B; Kalinsky HJ; Psychoyos S; Cash WD; Tsai C; Rinehart RK; Granat FR; Lovell RA; Brundish DE; Wade R CGS 8216: Receptor Binding Characteristics of a Potent Benzodiazepine Antagonist. Life Sci. 1982, 30, 363–372. [DOI] [PubMed] [Google Scholar]
- 20.Boast CA; Snowhill EW; Simke JP CGS 8216 and CGS 9896, Novel Pyrazoloquinoline Benzodiazepine Ligands with Benzodiazepine Agonist and Antagonist Properties. Pharmacol. Biochem. Behav 1985, 23, 639–644. [DOI] [PubMed] [Google Scholar]
- 21.Li X; Ma C; He X; Yu J; Han D; Zhang C; Atack JR; Cook JM Studies in Search of Diazepam-Insensitive Subtype Selective Agents for GABAA/Bz Receptors. Med. Chem. Res 2003, 11, 504–537. [Google Scholar]
- 22.Wong G; Gu ZQ; Fryer RI; Skolnick P Pyrazolo(4,3-c)quinolines with High Affinities for “Diazepam- Insensitive” (DI)GABAA/Benzodiazepine Receptors. Med. Chem. Res 1992, 2, 217–224. [Google Scholar]
- 23.Clayton T; Chen J; Ernst M; Richter L; Cromer B; Morton C; Ng H; Kaczorowski C; Helmstetter F; Furtmuller R; Ecker G; Parker M; Sieghart W; Cook J An Updated Unified Pharmacophore Model of the Benzodiazepine Binding Site on γ-Aminobutyric Acid Receptors: Correlation with Comparative Models. Curr. Med. Chem 2007, 14, 2755–2775. [DOI] [PubMed] [Google Scholar]
- 24.Clayton T; Poe MM; Rallapalli S; Biawat P; Savic MM; Rowlett JK; Gallos G; Emala CW; Kaczorowski CC; Stafford DC; Arnold LA; Cook JM A Review of the Updated Pharmacophore for the Alpha 5 GABA(A) Benzodiazepine Receptor Model. Int. J. Med. Chem 2015, 2015, 1–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P; Zhang W; Liu R; Harris B; Skolnick P; Cook JM Synthesis of Novel Imidazobenzodiazepines as Probes of the Pharmacophore for “Diazepam-Insensitive” GABAA Receptors. J. Med. Chem 1995, 38, 1679–1688. [DOI] [PubMed] [Google Scholar]
- 26.He X; Huang Q; Yu S; Ma C; McKernan R; Cook JM Studies of Molecular Pharmacophore/Receptor Models for GABAA/BzR Subtypes: Binding Affinities of Symmetrically Substituted Pyrazolo[4,3-c]quinolin-3-ones at Recombinant αxβ3γ2 Subtypes and Quantitative Structure-Activity Relationship Studies via a Comparative Molecular Field Analysis. Drug. Des. Discov 1999, 16, 77–91. [PubMed] [Google Scholar]
- 27.Huang Q; He X; Ma C; Liu R; Yu S; Dayer CA; Wenger GR; McKernan R; Cook JM Pharmacophore/Receptor Models for GABAA/BzR Subtypes (α1β3γ2, α5β3γ2, and α6β3γ2) via a Comprehensive Ligand-Mapping Approach. J. Med. Chem 2000, 43, 71–95. [DOI] [PubMed] [Google Scholar]
- 28.Treven M; Siebert DCB; Holzinger R; Bampali K; Fabjan J; Varagic Z; Wimmer L; Steudle F; Scholze P; Schnurch M; Mihovilovic MD; Ernst M Towards Functional Selectivity for alpha6beta3gamma2 GABAA Receptors: a Series of Novel Pyrazoloquinolinones. Br. J. Pharmacol 2018, 175, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiou L-C; Cook J; Ernst M; Fan P-C; Knutson D; Meirelles M; Mihovilovic M; Sieghart W; Varagic Z; Verma R; Wimmer L; Weizigmann C; Siebert DCB; Savic MM Preparation of Ligands Selective to Alpha 6 Subunit-Ccontaining GABAa Receptors and their Methods of Use. WO2016196961, 2016. [Google Scholar]
- 30.Mirheydari P; Ramerstorfer J; Varagic Z; Scholze P; Wimmer L; Mihovilovic MM; Sieghart W; Ernst M Unexpected Properties of delta-Containing GABAA Receptors in Response to Ligands Interacting with the alpha+ beta- Site. Neurochem. Res 2014, 39, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jedrychowski M; Nilsson E; Bieck P Metabolic Fate of CGS 8216, a Benzodiazepine Receptor Antagonist, in Rat and in Man. Psychopharmacology. 1986, 88, 529–530. [DOI] [PubMed] [Google Scholar]
- 32.Harbeson SL; Tung RD Deuterium Medicinal Chemistry: A New Approach to Drug Discovery and Development. Medchem News 2014, 1–14. [Google Scholar]
- 33.Wiberg KB The Deuterium Isotope Effect. Chem. Rev 1955, 55, 713–743. [Google Scholar]
- 34.Meunier B; de Visser SP; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- 35.Nelson SD; Trager WF The Use of Deuterium Isotope Effects to Probe the Active Site Properties, Mechanism of Cytochrome P450-Catalyzed Reactions, and Mechanisms of Metabolically Dependent Toxicity. Drug. Metab. Dispos 2003, 31, 1481–1498. [DOI] [PubMed] [Google Scholar]
- 36.Pajouhesh H; Lenz GR Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx. 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sangster J Octanol-Water Partition Coefficients of Simple Organic Compounds. J. Phys. Chem. Ref. Data. 1989, 18, 1111–1229. [Google Scholar]
- 38.Trudell MLI The Synthesis and Study of the Pharmacologic Activity of 7,12-Dihydropyrido[3,2-b:5,4-b’]diindoles A Novel Class of Rigid, Planar Benzodiaze-pine Receptor Ligands. II. The Total Synthesis of the Indole Alkaloid, (±)Suaveoline [Ph.D. Thesis]. University of Wisconsin-Milwaukee, Milwaukee, WI, 1989. [Google Scholar]
- 39.Frahn JL; Illman RJ Preparation of a 4-Methoxyphenylhydrazine and some other Arylhydrazines. Aust. J. Chem 1974, 27, 1361–1365. [Google Scholar]
- 40.Fryer RI; Zhang P; Rios R; Gu ZQ; Basile AS; Skolnick P Structure-Activity Relationship Studies at Benzodiazepine Receptor (BZR): a Comparison of the Substituent Effects of Pyrazoloquinolinone Analogs. J. Med. Chem 1993, 36, 1669–1673. [DOI] [PubMed] [Google Scholar]
- 41.Goncalves V; Brannigan JA; Whalley D; Ansell KH; Saxty B; Holder AA; Wilkinson AJ; Tate EW; Leatherbarrow RJ Discovery of Plasmodium Vivax N-Myristoyltransferase Inhibitors: Screening, Synthesis, and Structural Characterization of their Binding Mode. J. Med. Chem 2012, 55, 3578–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gould RG; Jacobs WA The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc 1939, 61, 2890–2895. [Google Scholar]
- 43.Salmon R Oxalyl Chloride-Dimethylformamide. In Encyclopedia of Regents for Organic Synthesis, J. Wiley: Chichester, West Sussex, 2001. [Google Scholar]
- 44.Yokoyama N; Ritter B; Neubert AD 2-Arylpyrazolo[4,3-c]quinolin-3-ones: a Novel Agonist, a Partial Agonist and an Antagonist of Benzodiazepines. J. Med. Chem 1982, 25, 337–339. [DOI] [PubMed] [Google Scholar]
- 45.Jeanty M; Blu J; Suzenet F; Guillaumet G Synthesis of 4- and 6-azaindoles via the Fischer Reaction. Org. Lett 2009, 11, 5142–5145. [DOI] [PubMed] [Google Scholar]
- 46.Berka K; Hendrychova T; Anzenbacher P; Otyepka M Membrane Position of Ibuprofen Agrees with Suggested Access Path Entrance to Cytochrome P450 2C9 Active Site. J. Phys. Chem. A. 2011, 115, 11248–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Savic MM; Majumder S; Huang S; Edwankar RV; Furtmuller R; Joksimovic S; Clayton T Sr.; Ramerstorfer J; Milinkovic MM; Roth BL; Sieghart W; Cook JM Novel Positive Allosteric Modulators of GABAA Receptors: Do Subtle Differences in Activity at alpha1 Plus alpha5 Versus alpha2 Plus alpha3 Subunits Account for Dissimilarities in Behavioral Effects in Rats? Prog. Neuropsychopharmacol. Biol. Psychiatry. 2010, 34, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dordevic SM; Cekic ND; Savic MM; Isailovic TM; Randelovic DV; Markovic BD; Savic SR; Timic Stamenic T; Daniels R; Savic SD Parenteral Nanoemulsions as Promising Carriers for Brain Delivery of Risperidone: Design, Characterization and in vivo Pharmacokinetic Evaluation. Int. J. Pharm 2015, 493, 40–54. [DOI] [PubMed] [Google Scholar]
- 49.Jenks WP Catalysis in Chemistry and Enzymology. Dover Publications Inc: Mineola, NY, 1987. [Google Scholar]
- 50.Northrop DB Steady-State Analysis of Kinetic Isotope Effects in Enzymic Reactions. Biochem. 1975, 14, 2644–2651. [DOI] [PubMed] [Google Scholar]
- 51.Gomtsyan A Heterocycles in Drugs and Drug Discovery. Chem. Heterocycl. Comp 2012, 48, 7–10. [Google Scholar]
- 52.Banks WA Characteristics of Compounds that Cross the Blood-Brain Barrier. BMC Neurol. 2009, 9 Suppl 1, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterhouse R Determination of Lipophilicity and Its Use as a Predictor of Blood–Brain Barrier Penetration of Molecular Imaging Agents. Mol. Imaging Biol 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]
- 54.Bewernick BH; Urbach AS; Broder A; Kayser S; Schlaepfer TE Walking Away from Depression-Motor Activity Increases Ratings of Mood and Incentive Drive in Patients with Major Depression. Psychiatry Res. 2017, 247, 68–72. [DOI] [PubMed] [Google Scholar]
- 55.Armarego WLF; Chai CLL Purification of Laboratory Chemicals, 7th Edition Butterworth-Heinemann: New York, NY, 2012. [Google Scholar]
- 56.Pouchert CJ; Behnke J The Aldrich Library of 13C and 1H FT NMR Spectra, 1st Edition Aldrich Chemical Co.: Milwaukee, WI, 1993; Vol. 2. [Google Scholar]
- 57.Forkuo GS; Guthrie ML; Yuan NY; Nieman AN; Kodali R; Jahan R; Stephen MR; Yocum GT; Treven M; Poe MM; Li G; Yu OB; Hartzler BD; Zahn NM; Ernst M; Emala CW; Stafford DC; Cook JM; Arnold LA Development of GABAA Receptor Subtype-Selective Imidazobenzodiazepines as Novel Asthma Treatments. Mol. Pharmacol 2016, 13, 2026–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forkuo GS; Nieman AN; Yuan NY; Kodali R; Yu OB; Zahn NM; Jahan R; Li G; Stephen MR; Guthrie ML; Poe MM; Hartzler BD; Harris TW; Yocum GT; Emala CW; Steeber DA; Stafford DC; Cook JM; Arnold LA Alleviation of Multiple Asthmatic Pathologic Features with Orally Available and Subtype Selective GABAA Receptor Modulators. Mol. Pharmacol 2017, 14, 2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jahan R; Stephen MR; Forkuo GS; Kodali R; Guthrie ML; Nieman AN; Yuan NY; Zahn NM; Poe MM; Li G; Yu OB; Yocum GT; Emala CW; Stafford DC; Cook JM; Arnold LA Optimization of Substituted Imidazobenzodiazepines as Novel Asthma Treatments. Eur. J. Med. Chem 2017, 126, 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramerstorfer J; Furtmuller R; Vogel E; Huck S; Sieghart W The Point Mutation Gamma 2F77I Changes the Potency and Efficacy of Benzodiazepine Site Ligands in Different GABAA Receptor Subtypes. Eur. J. Pharmacol 2010, 636, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khandelwal A; Bahadduri PM; Chang C; Polli JE; Swaan PW; Ekins S Computational Models to Assign Biopharmaceutics Drug Disposition Classification from Molecular Structure. Pharm. Res 2007, 24, 2249–2262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.