

Abstract
The advances made in the treatment of HIV-1 infection represent a major success of modern biomedical research, prolonging healthy life and reducing virus transmission. There remain, however, many challenges relating primarily to side effects of long-term therapy and the ever-present danger of the emergence of drug-resistant strains. To counter these threats, there is a continuing need for new and better drugs, ideally targeting multiple independent steps in the HIV-1 replication cycle. The most successful current drugs target the viral enzymes: protease (PR), reverse transcriptase (RT), and integrase (IN). In this review, we outline the advances made in targeting the Gag protein and its mature products, particularly capsid and nucleocapsid, and highlight possible targets for future pharmacological intervention.
Graphical Abstract
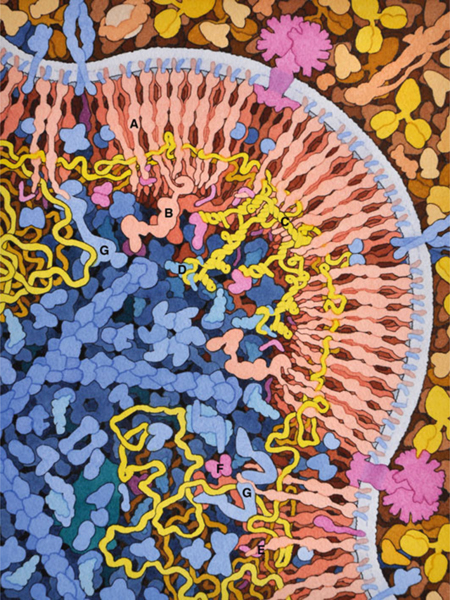
HIV budding. HIV Gag protein (A) and Gag-pol (B) form arrays on the cell surface, capturing two copies of HIV genome (in yellow), which dimerize through a specific sequence (C) and bind to a cellular transfer RNA (D) that will act as a primer for reverse transcription. Viral proteins Vpr (E) and Vif (F) are also incorporated. Several cellular proteins of the ESCRT system (G) are involved in the process of budding
1. Introduction
1.1. The AIDS Epidemic
Human immunodeficiency virus (HIV), as the causative agent of acquired immunodeficiency syndrome (AIDS), is one of the most serious threats to human health throughout the world; in the absence of a protective vaccine, it is likely to remain a major human pathogen for the foreseeable future. There are two viruses responsible for AIDS in humans, HIV-1 and HIV-2, representing multiple independent zoonotic transmissions of simian immunodeficiency virus (SIV). HIV-1 passed into humans via a closely related chimpanzee virus (SIVcpz), while HIV-2 was transferred from sooty mangabeys (Sharp and Hahn 2011). HIV-1 is the main cause of the global HIV pandemic (34 million infected people as of 2011); HIV-2 accounts for 1–2 million infections, almost exclusively in Africa, and AIDS in these patients generally progresses more slowly (Gottlieb et al. 2008; Nyamweya et al. 2013; UNAIDS 2013). People infected with HIV-1, in the absence of treatment, can harbor the virus for a decade or more without showing obvious clinical manifestations. Eventually, viral replication depletes the body of circulating CD4+ T cells below 200 cells per cm3; this depletion of a critical immune cell leads to a wide range of AIDS-defining opportunistic infections (Morgan et al. 2002; Swanstrom and Coffin 2012). Following the onset of AIDS, life expectancy is limited to a few years in the absence of antiretroviral therapy (Morgan et al. 2002). While disease prevention through behavioral changes has played a significant role in controlling the spread of the pandemic, a massive international effort has been directed toward developing therapies to prevent viral replication and restore immune function in patients (Fonner et al. 2012; Arts and Hazuda 2012). The resulting drugs have significantly improved the prognosis for people infected with HIV-1, enhancing their life expectancy and reducing their capacity to transmit the virus to others (Arts and Hazuda 2012). While many of the available drugs show efficacy against HIV-2, HIV-1 will be the focus of this review.
1.2. The HIV-1 Replication Cycle
As described in a number of recent reviews (Flexner 2007; Freed and Martin 2013; Telenti and Johnson 2012), defining the HIV-1 replication cycle has revealed a variety of targets for intervention (Fig. 1). Briefly, the replication process begins when an infectious viral particle encounters a target cell, typically a CD4+ T cell. The surface envelope (Env) glycoprotein gp120 binds the primary receptor, CD4, and a co-receptor, either CCR5 or CXCR4, triggering conformational changes in the transmembrane Env glycoprotein gp41 that promote fusion of the viral and cellular membranes (Wilen et al. 2012). Fusion releases the viral core into the cytoplasm of the target cell where reverse transcription of the viral RNA to double- stranded DNA occurs (Hu and Hughes 2012). Reverse transcription appears to be coupled in some way to at least partial uncoating and transport of the incoming viral protein/nucleic acid complex to the nuclear pore (Hulme et al. 2011). The precise timing of these early events and formation and composition of the so-called pre-integration complex (PIC) remain under active investigation. The PIC interacts with various transportins and nucleoporins (e.g., transportin 3 (TNPO3), NUP153, and NUP358) to drive import of the PIC into the nucleus (Matreyek and Engelman 2013). Once inside the nucleus, the viral cDNA is integrated into the host genome by the viral integrase (IN) and the cellular cofactor, lens epithelium-derived growth factor (LEDGF/p75) (Craigie and Bushman 2012; Krishnan and Engelman 2012). The integrated DNA, referred to as the provirus, is transcribed by the host cell machinery to generate full-length and various spliced mRNAs encoding the viral proteins (Karn and Stoltzfus 2012). The full-length RNA serves as both the genome encapsidated into assembling particles and the mRNA for Gag and GagPol polyprotein precursors (Bell and Lever 2013).
Fig. 1.

Schematic of the HIV-1 replication cycle. Viral and cell components are labeled in italics, processes in plain text, and processes that can be inhibited by current anti-retrovirals are boxed. MA, red; CA, blue; NC, green; p6, orange; Env, purple; viral RNA, cyan; viral cDNA, brown
The Gag polyprotein precursor, Pr55Gag (often simply referred to as “Gag”), is the major driver in virus assembly and release (Sundquist and Krausslich 2012). Specific functions of individual domains within Gag are described below. GagPol is produced by a rare (approximately 5 % of translation events)-1 ribosomal frame-shift that takes place at a so-called slippery sequence located near the 5′ end of the p6-coding region of Gag (Bell and Lever 2013). Frameshifting thus eliminates most of the p6 and instead results in the translation of the coding regions for the viral enzyme protease (PR), reverse transcriptase (RT), and IN. Gag and GagPol are translated in the cytoplasm but are rapidly transported to cholesterol-rich (so-called lipid raft) microdomains in the PM (Ono and Freed 2005; Waheed and Freed 2010). Targeting is directed in part by the phospholipid phosphatidylinositol-(4,5)-bisphosphate (PI[4,5]P2) on the inner leaflet of the PM (Ono et al. 2004; Saad et al. 2006). Subsequent binding of nucleic acid, usually the genomic RNA, promotes Gag multimerization and assembly of the immature Gag lattice (Muriaux and Darlix 2010). Env is translated at the endoplasmic reticulum (ER) as a gp160 precursor and is co-translationally inserted into the ER membrane and glycosylated (Checkley et al. 2011). gp160 traffics to the PM through the Golgi apparatus where it is cleaved by a furin-like protease to generate the gp120 and gp41 subunits of the Env complex. During assembly, the Env glycoproteins are incorporated into the immature Gag lattice as non-covalently associated heterotrimers (three molecules each of gp120 and gp41) (Checkley et al. 2011; Johnson 2011). The immature particle undergoes budding and release from the PM; the membrane scission event required for virus release is mediated through the recruitment by Gag of the endosomal sorting complexes required for transport (ESCRTs), a cellular budding and membrane scission apparatus that normally functions in multivesicular body (MVB) formation and cytokinesis (Votteler and Sundquist 2013; Weiss and Gottlinger 2011). During or shortly after release, the viral particle undergoes maturation, triggered by PR-mediated cleavage of Gag and GagPol polyprotein precursors to liberate the mature Gag and Pol proteins and permit the conversion of the immature Gag lattice into the mature conical core (Sundquist and Krausslich 2012). The maturation process also increases the fusogenicity of the Env glycoproteins (Murakami et al. 2004; Wyma et al. 2004). The mature, infectious particle is at this point competent to carry out a new round of infection.
In addition to the Gag, Pol, and Env proteins encoded by all replication-com-petent orthoretroviruses, HIV-1 also encodes regulatory proteins that greatly increase viral gene transcription (Tat) and nuclear–cytoplasmic export of unspliced viral RNAs (Rev) and several “accessory” proteins (Vpu, Vif, Nef, and Vpr) that to a large extent function in counteracting the innate antiviral response (Freed and Martin 2013). These regulatory and accessory proteins are described in a number of recent reviews and will not be discussed further here (Landi et al. 2011; Le Tortorec et al. 2011; Malim and Emerman 2008; Romani and Cohen 2012; Sharkey 2013).
1.3. The Diverse Roles of Gag During HIV-1 Replication
The Gag precursor is comprised of four major domains, matrix (MA), capsid (CA), nucleocapsid (NC) and p6, and two spacer peptides located between CA and NC (SP1) and between NC and p6 (SP2) (Fig. 2) (Bell and Lever 2013). The MA domain of Gag bears a bipartite membrane-binding domain, consisting of an N- terminal, covalently attached myristic acid and a highly basic patch of residues (Chukkapalli and Ono 2011). MA binds directly to the PM-specific phospholipid PI (4,5)P2, thereby ensuring that Gag is localized to the PM rather than to more abundant intracellular membranes (Chukkapalli et al. 2008; Ono et al. 2004; Saad et al. 2006). Solution nuclear magnetic resonance (NMR) spectroscopy data suggest a model whereby binding of MA to PI(4,5)P2 triggers exposure of the myristate moiety, allowing the latter to insert into the lipid bilayer and anchor Gag at the membrane (Saad et al. 2006). The MA domain also facilitates the incorporation of the viral Env glycoprotein complex into nascent virions (Freed and Martin 1995; Johnson 2011). The CA domain promotes Gag multimerization by engaging in multiple intermolecular interactions during assembly (Sundquist and Krausslich 2012). As a mature product, CA is the principle viral protein responsible for the assembly of the capsid core during virus maturation (Briggs and Krausslich 2011). Recent evidence also suggests that CA, as part of the incoming reverse transcription complex (RTC), engages cellular transportins and nuclear pore components to regulate nuclear import of the PIC (Matreyek and Engelman 2013). The full-length viral genomic RNA is recruited into particles via direct interactions with the NC domain of Gag. RNA binding by NC is driven by its overall positive charge, with specificity for viral genomic RNA imparted by two zinc finger motifs that interact with the RNA packaging signal (Muriaux and Darlix 2010; Lu et al. 2011). By binding nucleic acids, NC also promotes Gag multimerization during assembly and functions as a nucleic acid chaperone at a number of steps in the replication cycle (Rein 2010). The p6 domain of Gag contains the so-called late domains necessary for recruiting the ESCRT machinery to promote virus budding and release (Votteler and Sundquist 2013; Weiss and Gottlinger 2011). The spacer peptides regulate the kinetics of Gag processing, and SP1 forms part of a sequence contiguous with the C-terminus of CA that is necessary for Gag–Gag interactions during assembly (Datta et al. 2011; de Marco et al. 2012; Lee et al. 2012).
Fig. 2.

Schematic of HIV-1 Gag indicating major functional motifs. The myristic acid and highly basic region of MA mediate membrane interactions of Gag. Residues in MA that have been shown to affect Env incorporation are indicated with dashed vertical lines. CA is divided into N-terminal and C-terminal domains, NTD and CTD, respectively. The NTD promotes pentamer formation, while the CTD, which also contains the MHR, is required for CA dimerization and multimerization. NC contributes to Gag assembly by binding nucleic acid, typically the viral genome, via its zinc finger motifs, leading to long-range Gag multimerization. p6 contains the late domains PTAP and YPXL, which bind TSG101 and ALIX, respectively, thereby recruiting the ESCRT machinery to facilitate virus budding from the cell membrane. MA, red; CA, blue; NC, green; p6, orange. Spacer peptides SP1 and SP2 are indicated, as is the approximate length of the Gag precursor (500 amino acids)
1.4. The Current State of Antiretroviral Drug Therapy
There are currently more than two dozen drugs approved for use against HIV-1, targeting several critical steps in the virus replication cycle (Fig. 1) (Arts and Hazuda 2012; Ballantyne and Perry 2013). The first drugs used to treat HIV-1 were nucleoside analogs, which inhibit reverse transcription by binding to the RT enzyme in place of authentic deoxynucleoside triphosphates (Arts and Hazuda 2012). These nucleoside RT inhibitors (NRTIs) are incorporated into the growing DNA chain, but cannot be extended, resulting in chain termination (Arts and Hazuda 2012). NRTIs showed marked efficacy in the short term, but resistance rapidly developed, and in many cases, resistance to one compound conferred resistance to several members of the class. This resistance drove research into new drug targets, the most successful of which have been the protease inhibitors (PIs), which prevent maturation of released HIV-1 virions, and the non-nucleoside RT inhibitors (NNRTIs), which bind RT allosterically to prevent enzymatic function. More recently, integrase strand-transfer inhibitors (INSTIs) have been developed and are being used with increasing frequency (Arts and Hazuda 2012). Drugs blocking gp120 binding to the co-receptor CCR5 or that block viral fusion with target cells by interacting with gp41 have been licensed, but are less widely used (Haqqani and Tilton 2013).
To counter the emergence of drug resistance, patients are typically treated with cocktails of three drugs simultaneously, with at least two independent mechanisms of action. Common regimens will combine two NRTIs with a PI, an NNRTI or an INSTI (Arts and Hazuda 2012). Such regimens have proved highly effective, extending life expectancy and slowing spread of the AIDS epidemic where treatment is available. Patients on combination antiretroviral therapy (cART) often show little or no clinical evidence of viral infection; however, the treatment is not a cure. Although in most treated patients virus can be detected only with highly sensitive methods, viral loads will rapidly rebound if treatment is stopped (Palmer 2013). If the dosing regimen is not followed diligently, suboptimal inhibition of replication can lead to emergence of viral resistance, compromising the efficacy of the therapy (Arts and Hazuda 2012); even when the regimen is followed rigorously, side effects from long-term therapy reduce the life expectancy of HIV-1-infected patients relative to age-matched uninfected individuals (Nakagawa et al. 2012). Consequently, there continues to be a need for new drugs with superior characteristics (e.g., reduced toxicity and more convenient dosing regimens) and novel targets in the viral replication cycle.
Importantly for the purposes of this review, at present, there are no licensed drugs directly targeting the Gag protein. The therapeutic potential of Gag as an antiviral target is illustrated by the efficacy of the PIs, which prevent PR from cleaving Gag into its mature products (Fig. 2), thereby blocking maturation. Mat- uration is required for HIV-1 infectivity and can influence the function of other virion components. For example, Env is less fusogenic in immature than in mature particles (Murakami et al. 2004; Wyma et al. 2004).
In this review, we summarize recent progress in developing compounds that target the HIV-1 Gag protein and speculate on possible future avenues for therapeutic development. We will focus on the inhibitors of maturation, CA and NC, as these have yielded the most promising results. Compounds that act by disrupting the CA lattice have demonstrated excellent efficacy in tissue culture-based experiments, providing proof of concept for the viability of these inhibitors. The best NC inhibitors go a step further and can suppress viral infection in animal models. Finally, the first-generation HIV-1 maturation inhibitor was used successfully in human subjects, while research continues to improve efficacy and reduce viral resistance in the second generation of these compounds. Following discussion of these topics, we will also address targets such as MA and p6 that, while currently undeveloped, could in the future provide viable approaches to novel antiviral therapy.
2. Maturation
Given the potency of PIs, it is perhaps not surprising that one of the most successful attempts to target Gag so far involves blocking maturation of the viral core. A class of compounds known as the maturation inhibitors prevents the PR-mediated cleavage between CA and SP1, leading to the accumulation of CA–SP1 precursor and loss of viral infectivity. Because incomplete processing at the CA–SP1 junction strongly interferes with virion maturation and particle infectivity (Checkley et al. 2010; Muller et al. 2009), even a partial block to cleavage at this site can elicit a potent antiviral effect. The first compound reported to block CA–SP1 processing was a betulinic acid derivative, 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid (DSB), also known as YK-FH312, PA-457, or Bevirimat (BVM). BVM was originally described as a compound capable of inhibiting processing at the CA–SP1 junction, causing aberrant virion morphology, and inhibiting the replication of HIV-1 in culture with an IC50 of ~10 nM (Li et al. 2003). This work, and subsequent studies, identified the CA–SP1 junction as the likely site of inhibitor binding, and a panel of resistant mutants were identified with amino acid substitutions in either the carboxy (C)-terminus of CA or the amino (N)-terminus of SP1 (Adamson et al. 2006; Li et al. 2003, 2006; Nguyen et al. 2011; Zhou et al. 2006). An additional feature of maturation inhibitors that may contribute to their antiviral activity is their ability to stabilize the immature Gag lattice (Keller et al. 2011, 2013).
The in vitro results described above were followed by successful phase I and phase II clinical trials, demonstrating safety and efficacy in a small number of patients (Martin et al. 2007a, b; Smith et al. 2007; Yebra and Holguin 2008). The outcomes of larger phase IIb trials, however, were mixed. While efficacy was seen in nine out of twenty patients, eleven displayed no response (Van Baelen et al. 2009). Patients who did not respond to BVM therapy were infected with viral strains containing polymorphisms in the CA–SP1 region that rendered them less susceptible to the compound (Adamson et al. 2010; Van Baelen et al. 2009); mutations in SP1 residue 6–8, in particular replacing valine at position 7 with either alanine or methionine, were associated with high resistance to BVM. The V7A polymorphism is found in the consensus sequences for subtypes C, D, F, and G; subtype C is the dominant strain in large parts of Africa and Asia and accounts for approximately 50 % of HIV-1 infections globally (Hemelaar et al. 2011) (http://www.hiv.lanl.gov/). The prevalence of resistance-conferring polymorphisms led to the discontinuation of BVM as a potential therapeutic agent (Margot et al. 2009; Seclen et al. 2010; Verheyen et al. 2010).
In 2009, a second, structurally unrelated molecule, PF-46396, was shown to inhibit CA–SP1 processing with an IC50 against laboratory isolates between 17 and 5000 nM (Blair et al. 2009). Clinical isolates likewise displayed variable sensitivity (Blair et al. 2009). PF-46396 exhibited lower potency in vitro than BVM, but appeared to function by a similar mechanism, as virus passage experiments selected for a variety of resistance mutations, again clustering in the CA–SP1 junction region (Blair et al. 2009; Waki et al. 2012). In addition, some of the mutations conferring resistance to PF-46396 were far upstream of the CA–SP1 junction; these included several in the major homology region (MHR) of CA (Waki et al. 2012). The MHR is a highly conserved motif in retroviral CA domains that plays an important but still incompletely understood role in Gag assembly [for review, see Freed (1998), Mateu (2009)]. These MHR mutations were associated with loss of fitness and drug dependency—they could replicate only in the presence of PF-46396 (Waki et al. 2012).
BVM and PF-46396 provide proof of concept that inhibitors of CA–SP1 processing can potently inhibit HIV-1 replication and, in the case of BVM, can reduce viral loads in infected patients. In addition, these compounds are not only active against PI-resistant viruses, but PI-resistant mutants have a reduced capacity to acquire mutations conferring BVM resistance. This is presumably due to a cumulative fitness cost, implying potential synergy between these drug classes (Adamson et al. 2009). Another study highlighted the complex relationship between mutations in PR and susceptibility to BVM, finding a more diverse range of BVM resistance mutations when selection was performed in a PI-resistant background (Fun et al. 2011). New maturation inhibitors are being produced based on the BVM scaffold, and it now appears likely that compounds can be identified that surpass BVM in terms of efficacy against a broad range of viral isolates, including those containing polymorphisms in the CA–SP1 boundary region (Coric et al. 2013; Dang et al. 2012, 2013; Qian et al. 2012). Extensive analysis of resistance mutations (Adamson et al. 2006; Waki et al. 2012) and structural analyses using NMR (Coric et al. 2013) have provided information that begins to delineate the boundaries of the maturation inhibitor-binding pocket; this information, coupled with direct structural analysis, may enable the rational design of additional classes of compounds that, like BVM and PF-46396, target CA–SP1 cleavage.
Although currently available maturation inhibitors block CA–SP1 processing, it is theoretically possible that small molecules could be developed that target other cleavage sites within Gag. Indeed, several studies have demonstrated that Gag- processing intermediates can exhibit potent trans-acting inhibitory activity (Checkley et al. 2010; Lee et al. 2009; Muller et al. 2009), highlighting the potential utility of this approach.
3. CA
CA is the domain primarily responsible for the structure of both the immature Gag lattice and the mature viral core. Compounds targeting CA could thus block or perturb the assembly of either of these critical structures. CA folds into two domains, an N-terminal domain (NTD) and a C-terminal domain (CTD). The CTD appears to be the major determinant in CA dimerization and multimerization (Franke et al. 1994; Gitti et al. 1996). The CTD also contains the MHR, which, as mentioned above, plays an essential role in Gag assembly. The NTD bears a Pro-rich loop that binds cyclophilins, most notably cyclophilin A (CypA) (Luban et al. 1993). In the immature particle, the Gag precursor, Pr55Gag, assembles into a hexagonal lattice. The lattice contains gaps that allow it to form a spherical structure underlying the viral membrane (Briggs et al. 2009). After Gag processing by PR, the mature CA protein reassembles to form the conical capsidcore in which CA again adopts a curved hexagonal arrangement. The hexagonal CA lattice is closed off at both ends by a defined number of CA pentamers: seven at the wide end and five at the narrow end. In the mature core, the characteristic CA hexamers and pentamers are formed by NTD–NTD and intermolecular NTD–CTD interactions, while CTD–CTD interac- tions connect hexamers into an extended lattice (Ganser-Pornillos et al. 2007, 2009). Although both the immature and mature Gag lattices are predominantly hexameric, inter-subunit contacts differ significantly in these two structures (Bharat et al. 2012). Consistent with this myriad of interactions, scanning mutagenesis revealed that most residues in CA are essential for efficient virus replication (Rihn et al. 2013). Mutations in CA that either stabilize or destabilize the capsid core disrupt virus infectivity (Forshey et al. 2002), indicating that core stability is fine-tuned to allow ordered disassembly during the early phase of the virus replication cycle. One could thus envision CA-based inhibitors that act by either stabilizing or destabilizing the core. Indeed, as described below, compounds with both types of activities have been described.
Growing evidence suggests that the capsid core does not fully disassemble postentry but rather remains intact, to an undetermined extent, until the core docks with the nuclear pore. Structures that appear to be conical capsid cores have been visualized by scanning electron microscopy at nuclear pores (Arhel et al. 2007), and more recently, several factors that restrict HIV-1 infection as part of the cellular innate immune response have been shown to interact with CA. The species-specific retroviral restriction factor TRIM5α (Stremlau et al. 2004) and the related protein TRIMCyp (Sayah et al. 2004) block infection by binding to CA on the incoming core. Binding to CA accelerates viral uncoating, potentially as a consequence of TRIM5α assembling on top of the hexameric CA lattice (Ganser-Pornillos et al. 2011). The antiviral activity of the newly discovered HIV-1 restriction factor Mx2 (also known as MxB), which is associated with a defect in viral nuclear import, is also determined by CA; mutations in the Pro-rich loop in CA that binds CypA allow HIV-1 to evade Mx2 restriction (Goujon et al. 2013; Kane et al. 2013; Liu et al. 2013). It is conceivable that the antiviral activity of these restriction factors could be harnessed therapeutically, perhaps by upregulating their expression or by interfer- ing with the ability of CA mutations to evade their inhibitory potential.
CA molecules on the incoming core interact not only with restriction factors but also with transportins and nuclear pore components that promote HIV-1 infection by facilitating nuclear entry of the viral PIC. In dividing cells, the nuclear mem- brane breaks down during mitosis, allowing unfettered access of the incoming PIC to the host cell DNA. However, this dissolution of the nuclear Env does not take place in non-dividing cells, requiring active transport of the PIC through nuclear pores for the viral DNA to gain access to the host chromosomes. CA was shown to be the major viral determinant responsible for the ability of lentiviruses to infect non-dividing cells (Yamashita et al. 2007). Host factors responsible for HIV-1 nuclear import, for example, the karyopherin TNPO3 and the nuclear pore proteins Nup153 and Nup358, were initially identified in genomewide RNAi screens as host factors essential for HIV-1 infection (Brass et al. 2008; Konig et al. 2008). Follow- up studies provided evidence for direct interaction between some of these host factors and CA. TNPO3 binds to the viral core (Valle-Casuso et al. 2012), and the sensitivity to TNPO3 depletion can be altered by mutations in CA (De Iaco and Luban 2011; Krishnan et al. 2010). Although the precise mechanism by which TNPO3 promotes HIV-1 nuclear import remains to be defined, a recent study (De Iaco et al. 2013) suggests that TNPO3 regulates the nuclear import of cleavage and polyadenylation specificity factor subunit 6 (CPSF6), a factor that also binds CA (Lee et al. 2010). In TNPO3-depleted cells, CPSF6 accumulates in the cytosol, where it binds to and stabilizes the viral core, thereby preventing nuclear import (De Iaco et al. 2013). Nup358 contains a cyclophilin domain that binds the Pro-rich loop of CA originally identified as the CypA binding site (Bichel et al. 2013; Luban et al. 1993). It has been suggested that Nup358 and CypA binding to the incoming capsid core serves to protect HIV-1 from being recognized by the cellular innate immune response (Rasaiyaah et al. 2013).
An increasing number of small molecules and peptide-based compounds have been shown to inhibit various aspects of CA function (Table 1; Fig. 3). Antiviral activity is mediated by disrupting CA–CA interactions in the immature Gag lattice, the mature CA core, or both. The first CA-targeting compounds to be developed were CAP-1 and capsid assembly inhibitor (CAI) (Sticht et al. 2005; Tang et al. 2003). These were effective at blocking CA assembly in vitro and, in the case of CAP-1, inhibited virus infection of cells. However, the IC50s were relatively high, and CAI, a peptide inhibitor, was unable to cross cell membranes. CAI was obtained through a phage display screen for CA-binding peptides (Sticht et al. 2005) and was shown by X-ray crystallography to bind a hydrophobic cavity in the CA–CTD, thereby destabilizing the CTD–dimer interface (Ternois et al. 2005) (Fig. 3b). The CAI peptide was modified by intramolecular hydrocarbon “stapling” to stabilize its conformation and enhance its membrane permeability (Zhang et al. 2008). The resulting molecule, NYAD-1, was subsequently modified to improve solubility in water, generating NYAD-13 (Bhattacharya et al. 2008). NYAD-1 and NYAD-13 targeted the same site in CA as CAI, but displayed enhanced binding affinity and membrane permeability and demonstrated efficacy against numerous laboratory and clinical isolates of HIV-1 (Bhattacharya et al. 2008; Zhang et al. 2008). Modification of the sites of NYAD-1 stapling resulted in several peptides that displayed dual activity; they not only bound CA but also blocked virus entry in a V3-loop- dependent manner (Zhang et al. 2013). The stapled peptide NYAD-201 was designed to mimic the dimerization domain of CA (Zhang et al. 2011). Like NYAD-1, this peptide can cross the cell membrane and inhibit virus production (Zhang et al. 2011). Several peptides were designed to mimic helical domains of the CA–CTD that are involved in inter-subunit interactions. These peptides, CAC1 and derivatives, inhibited CA assembly in vitro and displayed increased potency when used in combination (Bocanegra et al. 2011; Garzon et al. 2004). Thus far, these peptides remain comparatively weak binders and none is able to enter cells, although some cell uptake can be achieved when the CAC1 peptides are provided together with a cell-penetrating peptide (Bocanegra et al. 2011).
Table 1.
Capsid-targeting compounds with demonstrated activity against HIV-1
| Compound | Membrane permeable? | EC50 (μM) | Phenotype | Binding site on CA |
|---|---|---|---|---|
| CAP-1 | Yes | ≈70 (infection) | Blocks infectivity | CA NTD, helices 1 and 2, and residues 59–63 |
| CAI | No | Not done | Blocks assembly | CA CTD, residues 169–191 |
| NYAD-1 | Yes | 4–22 (culture) | Blocks assembly | CA CTD, residues 169–191 |
| CAC1M | No | >170 (transfected into cells) | Blocks assembly | CA CTD, residues 150–220 |
| BD 1 | Yes | 0.07 (culture) | Blocks assembly | CA NTD, helices 1, 2, 3 and 7 |
| BM 1 | Yes | 0.062 (culture) | Blocks maturation | CA NTD, helices 2, 3 and 7 |
| PF-3450074 | Yes | 0.3–0.6 (infection) | Destabilizes the core post-entry | CA NTD, helices 3, 4, 5 and 7 |
| BMMP | Yes | 25–50 (infection) | Destabilizes the core post-entry | Unknown |
| BI-1/2 | Yes | 1.4–7.5 (culture) | Stabilizes the core | CA NTD, helices 3, 4, 5 and 7 |
Fig. 3.

Crystal structures of the NTD (a) and CTD (b) of CA. Binding sites of CA inhibitors are indicated, where known, as transparent overlays. a The binding site of BM, BD, and CAP-1 is known as “site 1”; the binding site of PF-3450074 and BI-1/2 is known as “site 2.” b The binding site of CAI, NYAD-1, and CAC1 at the CTD dimer interface is indicated. Helix (h) numbers are indicated. Structures for NTD and CTD were generated in Pymol based on Protein Data Bank (PDB) coordinates 1GWP and 1A80, respectively (Gamble et al. 1997; Tang et al. 2002)
In addition to the CA-targeted peptides described above, a number of small molecules have been reported that disrupt CA assembly both in vitro and in cell-based assays. An early example was CAP-1, which was identified through computational screening of compound libraries for molecules that bind pockets in CA. Binding to CA was verified by NMR titration analysis (Tang et al. 2003). CAP-1 was shown to disrupt particle assembly in cell-based assays and to alter virion morphology at a high (100 μM) concentration (Tang et al. 2003). By combining NMR and X-ray crystallography approaches, CAP-1 was shown to bind via an induced-fit mechanism into a pocket (referred to as “site 1” in Fig. 3a) at the base of the CA–NTD normally occupied by the aromatic ring of Phe-32 (Kelly et al. 2007). Binding at this site and displacement of Phe-32 likely disrupt intermolecular NTD– CTD interactions within the hexamer (Kelly et al. 2007).
Several studies over the past few years have used high-throughput in vitro CA assembly assays to screen compound libraries for inhibitors of CA–CA interactions. The most recent additions to the growing family of CA-binding inhibitors are the benzodiazepine (BD) and benzimidazol (BM) compounds (Fader et al. 2011; Lemke et al. 2012). The best of this series display antiviral activity in culture with IC50s under 100 nM. These compounds bind to the above-mentioned CAP-1 pocket (“site 1”; Fig. 3a) at the base of the CA–NTD. Despite binding the same site in CA, the BD and BM compounds differ in their mechanism of action: The BD family inhibits assembly of the immature Gag lattice, preventing virus particle production. By contrast, the BM family only weakly inhibits virus assembly, but efficiently disrupts virus maturation and therefore infectivity.
A second class of CA-binding inhibitors acts early during infection, destabilizing the incoming viral core and inhibiting reverse transcription and possibly nuclear import. The best-described member of this class is the Pfizer compound, PF-3450074 (Blair et al. 2010; Shi et al. 2011). A second compound possessing what appears to be a similar mechanism of action has been described, 2-(benzothiazol-2-ylmethylthio)-4-methylpyrimidine (BMMP) (Shi et al. 2011; Urano et al. 2011). Both of these compounds disrupt CA assembly in vitro, but in cells the inhibition seems to be imposed predominantly post-entry but before integration. PF-3450074 binds the CA–NTD at a site distinct from the CAP-1 pocket (depicted as “site 2” in Fig. 3a). Two additional compounds were recently described by Boehringer Ingelheim, BI-1 and BI-2 (Lamorte et al. 2013). These pyrrolopyrazolones share the binding site of PF-3450074 (site 2; Fig. 3a), but surprisingly appear to stabilize rather than destabilize the CA lattice. Intriguingly, the binding pocket of PF-3450074 and BI-1/2 (site 2) is also the interaction site for the host proteins CPSF6 and the nucleoporin NUP153 (Lee et al. 2010; Matreyek et al. 2013; Price et al. 2012) [for review, see Matreyek and Engelman (2013)]. These observations raise the possibility that BI-1 and BI-2 may disrupt viral nuclear import by competitively interfering with the binding between host factors and the incoming capsid. Cyclosporin A (CsA) and its non-immuno- suppressive analogs prevent the binding of cyclophilins to CA; these compounds have long been known to impair HIV-1 replication (Luban et al. 1993; Thali et al. 1994), but an understanding of their mechanism of action has remained elusive. It has recently been suggested that by blocking the binding of CypA or Nup358 to CA, cyclosporins “unmask” the viral core, allowing it to be recognized by restriction factors (De Iaco and Luban 2014) or other components of the host innate immune response (Rasaiyaah et al. 2013).
A small-scale study of cyclosporin treatment in HIV-1/hepatitis C virus (HCV)-co-infected patients demonstrated a strong anti-HCV effect, but no significant inhibition of HIV-1 (Flisiak et al. 2008). Similarly, trials of cyclosporin as an addition to standard ART did not reveal an advantage compared to ART alone (Lederman et al. 2006; Markowitz et al. 2010). It is possible that therapeutically effective concentrations of these compounds may be difficult to achieve in patients. In addition, HIV-1 isolates have been identified that replicate independently of cyclophilin A and are insensitive to, or even dependent upon, cyclophilin inhibitors (Aberham et al. 1996; Ptak et al. 2008). Residue 87 of CA is typically histidine; glutamine or proline at this position confers resistance to cyclosporine. A survey of known HIV-1 isolates in the Los Alamos database (http://www.hiv.lanl.gov/) suggests that these resistant polymorphisms are present in greater than 20 % of sequences, and examples can be found in most subtypes (Gallay et al. 2013). Inhibition of viral nuclear import by targeting the interaction between CA and karyopherins and nuclear pore components may represent a feasible approach to inhibiting HIV-1 replication; however, the ability of the virus to exploit multiple nuclear import pathways (Lee et al. 2010) makes this approach challenging.
As has been the case with many antiretroviral compounds (Adamson and Freed 2008), identification of resistant mutants arising during in vitro propagation of CA-binding inhibitors has provided insights into the compounds’ target and mechanism of action. For example, mutations that confer resistance to BI-2 cluster around CA–NTD site 2 and prevent the enhancement of capsid stabilization conferred by this compound (Lamorte et al. 2013). While some of the mutations conferring resistance to the BM inhibitor prevent compound binding to CA, other resistance mutations in CA are located outside the inhibitor-binding site and stabilize CA assembly, suggesting an indirect mechanism of resistance that offsets the destabilizing activity of the BM compounds (Lemke et al. 2012).
A third region of CA–NTD has been identified as the binding site for a family of benzimidazole CA assembly inhibitors. These compounds bind between helix 6 and the cyclophilin-binding loop but do not prevent cyclophilin binding (Goudreau et al. 2013b).
Progress continues to be made studying the structure of CA. Models of the immature CA lattice and the conformational shifts that occur during retroviral maturation have been examined by cryo-electron tomography and cryo-EM of Mason–Pfizer monkey virus (M-PMV) Gag (Bharat et al. 2012). A pseudoatomic model of the immature HIV-1 CA lattice was developed by fitting the HIV-1 CA crystal coordinates onto the M-PMV cryo-EM map (Bharat et al. 2012). A cryo-EM approach coupled with a large-scale molecular dynamics simulation has generated an all-atom model for the HIV-1 capsid core (Zhao et al. 2013). These high-resolution models of both immature and mature structures should aid in continued exploration of the CA protein as a potential therapeutic target. While it is not clear that any of the CA-targeted inhibitors reported thus far represent viable leads for clinical development, ongoing progress from both the drug discovery and structural biology perspectives suggests that further efforts in this direction are warranted.
4. NC
NC is a small (7 kDa), basic, nucleic acid-binding protein containing two zinc fingers (Fig. 4). The presence of one or two zinc fingers in NC is one of the most conserved structural elements in orthoretroviral Gag proteins. As part of the Gag precursor, the basic residues in the NC domain are critical for the nucleic acid-binding function that, during assembly, promotes Gag multimerization. The zinc fingers confer specificity to the nucleic-acid-binding properties of the NC domain, allowing Gag to recruit the viral genomic RNA into the virus particle by binding the packaging signal in the genomic RNA. In addition to its roles in Gag assembly and genomic RNA encapsidation, as a mature protein, NC promotes post-entry events including reverse transcription. These activities are attributed to the ability of NC to act as a nucleic acid chaperone (Levin et al. 2010).
Fig. 4.

Structure of HIV-1 NC. Residues 12–53 of NC are shown in green. The zinc coordinating residues are shown, with side chains, in red around the two zinc ions (gray spheres). Structure generated using Pymol, based on PDB coordinates 1ESK
Several classes of inhibitors have been developed that disrupt NC activity [reviewed by (de Rocquigny et al. 2008)]. These include zinc-ejecting compounds, zinc-finger-binding non-zinc ejectors, peptidomimetics, and RNA aptamers. Zinc ejectors block NC function by displacing the zinc ions from the zinc fingers. Early zinc-ejecting compounds displayed activity in vitro and in culture, but toxicity precluded clinical development (Morcock et al. 2005; Rice et al. 1993, 1995). Zincejecting compounds include 2,2′-dithiobis[benzamides] (DIBAs) and pyrimidino-alkanoyl thioesters (PATEs), molecules with improved specificity compared to earlier ejectors (Goel et al. 2002). The most recently developed class of zinc-ejecting compounds, the S-acyl-2-mercaptobenzamide thioesters (SAMTs), shows much improved efficacy and specificity relative to earlier compounds (Miller Jenkins et al. 2010). These compounds enter the cells as pro-drugs before being acylated intracellularly. They covalently modify the C-terminal zinc finger of NC, irreversibly preventing zinc binding. Members of this family have been developed that exhibit low toxicity (CC50s >100 μM) and EC50s in the low-micromolar range, e.g., SAMT-247 EC50 0.6–5.7 μM (Miller Jenkins et al. 2010).
Several groups have performed screens to identify small molecules that disrupt NC/nucleic acid interactions. Breuer et al. reported two compounds capable of binding NC in vitro and inhibiting HIV-1NL4–3 in single-cycle infectivity assays with EC50s of 0.32 and 3.5 μM (Breuer et al. 2012). Activity against replicating virus in primary T cells was also apparent (Breuer et al. 2012). Another family of NC inhibitors was described by Boerhinger Ingelheim that binds simultaneously to both zinc fingers of NC, thereby blocking NC interactions with RNA. These compounds inhibit HIV-1 replication in culture with low-μM EC50s but display a relatively low therapeutic index (Goudreau et al. 2013a). As yet, these molecules have not been tested in clinical trials or screened for the development of resistance; they do in some cases, however, show encouraging signs of activity with lower toxicity to host cells, and the SAMT molecules have been evaluated in rhesus macaques infected with SIV/HIV (SHIV) chimeras (Wallace et al. 2009).
NC inhibitors have also been identified by using an assay specific for NC binding to cTAR DNA, in an attempt to target the nucleic acid chaperone activity of NC (Shvadchak et al. 2009). The assay was able to uncover several hits with low-micromolar IC50s in vitro; however, these compounds have not thus far been tested in cells.
Although 25 years of research has yet to identify a clinically effective HIV-1 inhibitor that targets NC, the importance of NC in multiple steps of the virus replication cycle provides a strong argument that these efforts should continue.
5. MA
The MA domain of Gag plays two primary roles during the virus replication cycle: It directs Gag to the plasma membrane early in the assembly process, and it promotes the incorporation of the viral Env glycoproteins into nascent virions. The N-terminus of MA is covalently modified with a myristic acid moiety; this N-terminal myristylation is essential for membrane binding. The myristate moiety has been shown to be oriented in two distinct conformations: A folded-back conformation in which it is sequestered in a hydrophobic groove in the globular core of MA, and an exposed conformation (Tang et al. 2004) (Fig. 5a, b). The specificity of membrane association is conferred by sequences downstream in MA, in particular a highly basic patch of residues located between residues 17 and 31. Mutation of these residues induces the mistargeting of Gag to a late endosomal or MVB compartment within the cell (Freed et al. 1994; Ono and Freed 2004; Ono et al. 2000). Early structural data suggested that the basic patch would juxtapose the membrane (Massiah et al. 1994), leading to the proposal that these positively charged residues might interact with negatively charged phospholipids on the inner leaflet of the lipid bilayer (Hermida-Matsumoto and Resh 1999; Zhou and Resh 1996). The most significant phospholipid in this regard is PI(4,5)P2; depletion of this phosphoinositide causes Gag to be mislocalized in the cell in much the same way as is induced by mutation of the basic patch (Ono et al. 2004). A direct interaction between MA and PI(4,5)P2 was demonstrated by NMR (Fig. 5a, b); interestingly, not only were electrostatic interactions between MA and PI(4,5)P2 evident in this structure but also PI(4,5)P2 binding to MA led to increased exposure of the N-terminal myristic acid (Saad et al. 2006) (Fig. 5b). One of the acyl chains of PI(4,5)P2 was observed to pack into a hydrophobic groove in MA distinct from the above-mentioned myristate groove (Saad et al. 2006). Whether such packing occurs in the context of membrane-bound PI(4,5)P2 remains to be established; in such a scenario, the acyl chain would have to be extruded from the lipid bilayer to be available for packing interactions with MA.
Fig. 5.

The MA myristyl switch. HIV-1 MA exists in two conformations: a In the cytoplasm, the hydrophobic myristic acid moiety is sequestered into a groove on the surface of the protein. b At the plasma membrane, MA binds to PI(4,5)P2 (di-C4-PI[4,5]P2 in this structure), causing a conformational shift and exposure of myristic acid. a and b were generated using Pymol and PDB coordinates 2H3I and 2H3Q, respectively (Saad et al. 2006). MA, red; myristic acid, green; myristate-binding groove, blue; PI(4,5)P2 binding residues, orange; di-C4-PI(4,5)P2, cyan
The second major function of MA involves the incorporation of the viral Env glycoproteins into virions [for review, see (Checkley et al. 2011)]. Single-amino-acid mutations in MA block the incorporation of full-length HIV-1 Env without affecting any other viral function (e.g., assembly or infectivity). The block to Env incorporation imposed by these MA mutations is relieved by truncating the HIV-1 gp41 cytoplasmic tail or by providing a foreign short-tailed Env glycoprotein in trans (pseudotyping). Interestingly, the incorporation of C-terminally truncated HIV-1 Env occurs efficiently in only a small subset of laboratory cell lines; in physiologically relevant cell types, such as primary CD4+ T cells and monocyte-derived macrophages, gp41 cytoplasmic tail truncations block Env incorporation (Akari et al. 2000; Murakami and Freed 2000). These findings lend support to the hypothesis that differentially expressed host factors contribute to the trafficking and/ or incorporation of HIV-1 Env and that such putative host factors bind the gp41 cytoplasmic tail. Tail-interacting protein of 47 kDa (TIP47) was suggested as a candidate for this function (Lopez-Verges et al. 2006), but these findings were not independently confirmed (Checkley et al. 2013). More recently, Rab11-FIP1c was reported to be a host factor that regulates Env incorporation (Qi et al. 2013); further study will clarify its role in HIV-1 replication.
Compounds have been described that target the PI(4,5)P2-binding groove, presumably preventing the targeting of Gag to the PM and myristate exposure during assembly (Saad et al. 2006; Zentner et al. 2013a, b). The most active compound inhibited HIV-1 production in cell culture with an IC50 in the 5–20 μM range (Zentner et al. 2013a). Although selection of resistant mutants was not performed, mutations engineered into the putative PI(4,5)P2-binding cleft (e.g., L21A and T81A) were found to abolish sensitivity to the compound.
Another potential target in MA is the myristate-binding groove. As mentioned above, the myristic acid moiety is in equilibrium between a folded-back conformation in which it packs into a hydrophobic groove in MA and an exposed conformation. Compounds that displace the myristate from its folded-back (“sequestered”) conformation should alter Gag interaction with the PM. It is not clear whether such compounds would decrease or increase Gag–membrane binding. However, mutations that disrupt myristate exposure impose defects in membrane association and virus assembly (Freed et al. 1994; Ono and Freed 1999; Saad et al. 2006). Conversely, mutations that increase membrane binding, potentially by triggering myristate exposure, cause replication defects at a post-entry stage (Kiernan et al. 1998). It therefore seems likely that perturbation of myristic acid exposure would be detrimental to virus replication.
Early structural studies indicated that both HIV-1 and SIV MA form a trimeric lattice upon crystallization (Hill et al. 1996; Rao et al. 1995). More recent work demonstrated that, when assembled on a two-dimensional membrane, MA or MA–CA fusions form hexamers of trimers, with the MA-induced trimers orienting themselves on top of the underlying hexameric lattice formed by CA (Alfadhli et al. 2009). Point mutations in MA that disrupt Env incorporation encircle a hole, or gap, in the hexamer-of-trimers lattice, suggesting that MA trimer formation may play an important role in Env incorporation. However, direct evidence for MA trimers in the context of HIV-1 particles was lacking. Recently, it was observed that mutations at the trimer interface could rescue a wide range of Env-incorporation-deficient MA mutants, leading to the proposal that MA trimers do indeed exist in virions and play an important role in Env incorporation (Tedbury et al. 2013). It therefore appears likely that compounds that bind the MA trimer interface, thereby altering trimer formation, would be disruptive to Env incorporation. Because of the critical requirement for Env during virion binding and entry, such compounds would likely display antiviral activity.
6. p6
The p6 region of HIV-1 Gag bears the so-called late domains that recruit cellular machinery required for virus release. At the core of this machinery is the ESCRT apparatus, composed of four multiprotein complexes (ESCRT-0, I, II, and III) and a variety of factors that interface directly or indirectly with these complexes. The hijacking of cellular ESCRT machinery by HIV-1 and other retroviruses (and, more broadly, non-retroviral enveloped viruses) has been reviewed elsewhere (McCul- lough et al. 2013; Votteler and Sundquist 2013) and will not be described in detail here. In brief, however, p6 contains two late domains: a Pro-Thr-Ala-Pro (PTAP) motif that binds directly to the ESCRT-I component Tsg101, and a Tyr-Pro-Xn-Leu sequence (YPXnL, where X is any residue, and n = 1–4 amino acids) that binds to the ESCRT-associated factor Alix. Although the dominant late domain for HIV-1 is the Tsg101-binding PTAP motif (Demirov et al. 2002; Gottlinger et al. 1991; Huang et al. 1995), the Alix-binding YPXnL motif is also required for efficient HIV-1 replication in relevant cell types (Fujii et al. 2009). The PTAP–Tsg101 and YPXnL–Alix interaction interfaces, for which structures are available (Fisher et al. 2007; Im et al. 2010; Lee et al. 2007; Pornillos et al. 2002; Zhai et al. 2008) (Fig. 6), could in theory be amenable to high-throughput screening for small-molecule inhibitors. It is not clear from a therapeutic perspective whether both interactions would need to be targeted simultaneously to achieve a high degree of antiviral potency or whether disrupting PTAP–Tsg101 binding would be sufficient.
Fig. 6.

Late-domain peptide binding to ESCRT proteins. a TSG101 ubiquitin E2 variant (UEV) domain bound to PTAP peptide. b View of UEV–PTAP interaction, facing into the binding groove. c ALIX V domain bound to YPLTSL peptide. d Close-up view of the ALIX-YPLTSL binding site. Host proteins are shown in gray, with binding sites in blue. Late-domain peptides are shown in orange, with interacting residues in red. Structures of late-domain interactions with TSG101 and ALIX are generated using Pymol with PDB coordinates 1M4P and 2RO2, respectively (Pornillos et al. 2002; Zhai et al. 2008)
p6 also contains a binding site for the HIV-1 accessory protein Vpr, thereby enabling Gag to recruit Vpr into virions. A number of functions for Vpr in culture have been described, and this protein appears to contribute to viral replication and pathogenesis in vivo [for reviews see (Kogan and Rappaport 2011; Planelles and Barker 2010)]. Whether potent inhibitors of p6–Vpr binding could be obtained remains to be determined.
7. Conclusions
The advances made in suppressing HIV-1 replication in infected patients represent one of the greatest success stories of modern medicine, and infected patients receiving treatment can now expect to live many decades longer than they would without therapy. This success has not been complete, however. Although manageable, HIV-1 infection remains typically incurable and the drugs used to control it cause harmful side effects in many patients. The evolution of drug resistance by HIV-1 continues to be an ongoing challenge. In this background, it is clear that improved drugs and drugs with novel targets are essential to maintain the efficacy of ART in the long term. As the protein primarily responsible for driving HIV-1 assembly, Gag is an attractive target for such efforts and the compounds and strategies discussed above exploit a range of distinct targets within the Gag protein, acting early and late in the replication cycle (Fig. 7). Compounds that inhibit maturation by blocking CA–SP1 cleavage or by binding CA possess encouraging properties and disrupt HIV-1 replication at reasonably low concentrations. Although much work remains to produce compounds suitable for use in humans, these studies demonstrate the potential value of compounds targeting Gag and suggest that in the future these inhibitors may join the armamentarium of drugs available to combat the ongoing AIDS pandemic. In addition to clinical benefits for HIV-infected patients, drug discovery efforts will also continue to provide novel and fundamental insights into the molecular mechanisms regulating HIV-1 replication.
Fig. 7.

Regions of the Gag molecule that may be targeted to inhibit HIV-1 replication. Domain colors as in Fig. 1. Text colors indicate the stage of the replication cycle that is primarily affected: assembly, red; maturation, blue; infection, green
Contributor Information
Philip R. Tedbury, Virus-Cell Interaction Section, HIV Drug Resistance Program, National Cancer Institute, Center for Cancer Research, Frederick, MD 21702-1201, USA
Eric O. Freed, Virus-Cell Interaction Section, HIV Drug Resistance Program, National Cancer Institute, Center for Cancer Research, Frederick, MD 21702-1201, USA
References
- Aberham C, Weber S, Phares W (1996) Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J Virol 70 (6):3536–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Ablan SD, Boeras I, Goila-Gaur R, Soheilian F, Nagashima K, Li F, Salzwedel K, Sakalian M, Wild CT, Freed EO (2006) In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat). J Virol 80(22):10957–10971. doi: 10.1128/JVI.01369-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Freed EO (2008) Recent progress in antiretrovirals–lessons from resistance. Drug Discov Today 13(9–10):424–432. doi: 10.1016/j.drudis.2008.02.003S1359-6446(08)00044-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Sakalian M, Salzwedel K, Freed EO (2010) Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV-1 maturation inhibitor bevirimat. Retrovirology 7:36. doi: 10.1186/1742-4690-7-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CS, Waki K, Ablan SD, Salzwedel K, Freed EO (2009) Impact of human immunodeficiency virus type 1 resistance to protease inhibitors on evolution of resistance to the maturation inhibitor bevirimat (PA-457). J Virol 83(10):4884–4894. doi: 10.1128/JVI.02659-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akari H, Fukumori T, Adachi A (2000) Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J Virol 74(10):4891–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A, Barklis RL, Barklis E (2009) HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387(2):466–472. doi: 10.1016/j.virol.2009.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P (2007) HIV-1 DNA Flap formation promotes uncoating of the preintegration complex at the nuclear pore. EMBO J 26(12):3025–3037. doi: 10.1038/sj.emboj.7601740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts EJ, Hazuda DJ (2012) HIV-1 antiretroviral drug therapy. Cold Spring Harbor Perspect Med 2 (4):a007161. doi: 10.1101/cshperspect.a007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne AD, Perry CM (2013) Dolutegravir: first global approval. Drugs 73(14):1627–1637. doi: 10.1007/s40265-013-0121-4 [DOI] [PubMed] [Google Scholar]
- Bell NM, Lever AM (2013) HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol 21(3):136–144. doi: 10.1016/j.tim.2012.11.006 [DOI] [PubMed] [Google Scholar]
- Bharat TA, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JA (2012) Structure of the immature retroviral capsid at 8 A resolution by cryo-electron microscopy. Nature 487(7407):385–389. doi: 10.1038/nature11169 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Zhang H, Debnath AK, Cowburn D (2008) Solution structure of a hydrocarbon stapled peptide inhibitor in complex with monomeric C-terminal domain of HIV-1 capsid. J Biol Chem 283(24):16274–16278. doi: 10.1074/jbc.C800048200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichel K, Price AJ, Schaller T, Towers GJ, Freund SM, James LC (2013) HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 10:81. doi: 10.1186/1742-4690-10-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair WS, Cao J, Fok-Seang J, Griffin P, Isaacson J, Jackson RL, Murray E, Patick AK, Peng Q, Perros M, Pickford C, Wu H, Butler SL (2009) New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob Agents Chemother 53 (12):5080–5087. doi: 10.1128/AAC.00759-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL (2010) HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6(12):e1001220. doi: 10.1371/journal.ppat.1001220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocanegra R, Nevot M, Domenech R, Lopez I, Abian O, Rodriguez-Huete A, Cavasotto CN, Velazquez-Campoy A, Gomez J, Martinez MA, Neira JL, Mateu MG (2011) Rationally designed interfacial peptides are efficient in vitro inhibitors of HIV-1 capsid assembly with antiviral activity. PloS ONE 6(9):e23877. doi: 10.1371/journal.pone.0023877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319(5865):921–926. doi: 10.1126/science.1152725 [DOI] [PubMed] [Google Scholar]
- Breuer S, Chang MW, Yuan J, Torbett BE (2012) Identification of HIV-1 inhibitors targeting the nucleocapsid protein. J Med Chem 55(11):4968–4977. doi: 10.1021/jm201442t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Krausslich HG (2011) The molecular architecture of HIV. J Mol Biol 410(4):491–500. doi: 10.1016/j.jmb.2011.04.021 [DOI] [PubMed] [Google Scholar]
- Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG (2009) Structure and assembly of immature HIV. Proc Nat Acad Sci USA 106(27):11090–11095. doi: 10.1073/pnas.0903535106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley MA, Luttge BG, Freed EO (2011) HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410(4):582–608. doi: 10.1016/j.jmb.2011.04.042S0022-2836(11)00471-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley MA, Luttge BG, Mercredi PY, Kyere SK, Donlan J, Murakami T, Summers MF, Cocklin S, Freed EO (2013) Reevaluation of the requirement for TIP47 in human immunodeficiency virus type 1 envelope glycoprotein incorporation. J Virol 87(6):3561–3570. doi: 10.1128/JVI.03299-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley MA, Luttge BG, Soheilian F, Nagashima K, Freed EO (2010) The capsid-spacer peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. Virology 400(1):137–144. doi: 10.1016/j.virol.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A (2008) Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J Virol 82(5):2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V, Ono A (2011) Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J Mol Biol 410(4):512–524. doi: 10.1016/j.jmb.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coric P, Turcaud S, Souquet F, Briant L, Gay B, Royer J, Chazal N, Bouaziz S (2013) Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur J Med Chem 62:453–465. doi: 10.1016/j.ejmech.2013.01.013 [DOI] [PubMed] [Google Scholar]
- Craigie R, Bushman FD (2012) HIV DNA integration. Cold Spring Harb Perspect Med 2(7): a006890. doi: 10.1101/cshperspect.a006890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Z, Ho P, Zhu L, Qian K, Lee KH, Huang L, Chen CH (2013) New betulinic acid derivatives for bevirimat-resistant human immunodeficiency virus type-1. J Med Chem 56(5):2029–2037. doi: 10.1021/jm3016969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Z, Qian K, Ho P, Zhu L, Lee KH, Huang L, Chen CH (2012) Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1 and bevirimat-resistant HIV-1 variants. Bioorg Med Chem Lett 22(16):5190–5194. doi: 10.1016/j.bmcl.2012.06.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A (2011) On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J Virol 85(9):4111–4121. doi: 10.1128/JVI.00006-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Luban J (2011) Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8:98. doi: 10.1186/1742-4690-8-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Luban J (2014) Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 11:11. doi: 10.1186/1742-4690-11-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J (2013) TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10:20. doi: 10.1186/1742-4690-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marco A, Heuser AM, Glass B, Krausslich HG, Muller B, Briggs JA (2012) Role of the SP2 domain and its proteolytic cleavage in HIV-1 structural maturation and infectivity. J Virol 86 (24):13708–13716. doi: 10.1128/JVI.01704-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rocquigny H, Shvadchak V, Avilov S, Dong CZ, Dietrich U, Darlix JL, Mely Y (2008) Targeting the viral nucleocapsid protein in anti-HIV-1 therapy. Mini Rev Med Chem 8(1):24–35 [DOI] [PubMed] [Google Scholar]
- Demirov DG, Orenstein JM, Freed EO (2002) The late domain of human immunodeficiency virus type 1 p6 promotes virus release in a cell type-dependent manner. J Virol 76(1):105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader LD, Bethell R, Bonneau P, Bos M, Bousquet Y, Cordingley MG, Coulombe R, Deroy P, Faucher AM, Gagnon A, Goudreau N, Grand-Maitre C, Guse I, Hucke O, Kawai SH, Lacoste JE, Landry S, Lemke CT, Malenfant E, Mason S, Morin S, O’Meara J, Simoneau B, Titolo S, Yoakim C (2011)Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg Med Chem Lett 21(1):398–404. doi: 10.1016/j.bmcl.2010.10.131S0960-894X(10)01592-1 [DOI] [PubMed] [Google Scholar]
- Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP (2007) Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128(5):841–852. doi: 10.1016/j.cell.2007.01.035 [DOI] [PubMed] [Google Scholar]
- Flexner C (2007) HIV drug development: the next 25 years. Nat Rev Drug Discov 6(12):959–966. doi: 10.1038/nrd2336 [DOI] [PubMed] [Google Scholar]
- Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, Siwak E, Cielniak I, Higersberger J, Kierkus J, Aeschlimann C, Grosgurin P, Nicolas-Metral V, Dumont JM, Porchet H, Crabbe R, Scalfaro P (2008) The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47(3):817–826. doi: 10.1002/hep.22131 [DOI] [PubMed] [Google Scholar]
- Fonner VA, Denison J, Kennedy CE, O’Reilly K, Sweat M (2012) Voluntary counseling and testing (VCT) for changing HIV-related risk behavior in developing countries. Cochrane Database Syst Rev 9:CD001224. doi: 10.1002/14651858.CD001224.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C (2002) Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76(11):5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke EK, Yuan HE, Luban J (1994) Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372(6504):359–362. doi: 10.1038/372359a0 [DOI] [PubMed] [Google Scholar]
- Freed EO (1998) HIV-1 gag proteins: diverse functions in the virus life cycle. Virology 251(1): 1–15 [DOI] [PubMed] [Google Scholar]
- Freed EO, Martin MA (1995) Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol 69(3):1984–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA (2013) HIVs and their replication In: Knipe DM, Howley PM (eds) Fields virology, 6th edn Lippincott Williams & Wilkins, Philadelphia, pp 1502–1560 [Google Scholar]
- Freed EO, Orenstein JM, Buckler-White AJ, Martin MA (1994) Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol 68(8):5311–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO (2009) Functional role of Alix in HIV-1 replication. Virology 391(2):284–292. doi: 10.1016/j.virol.2009.06.016S0042-6822(09)00363-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fun A, van Maarseveen NM, Pokorna J, Maas RE, Schipper PJ, Konvalinka J, Nijhuis M (2011) HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 8:70. doi: 10.1186/1742-4690-8-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallay PA, Ptak RG, Bobardt MD, Dumont JM, Vuagniaux G, Rosenwirth B (2013) Correlation of naturally occurring HIV-1 resistance to DEB025 with capsid amino acid polymorphisms. Viruses-Basel 5(3):981–997. doi: 10.3390/V5030981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP (1997) Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278(5339):849–853 [DOI] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M (2011) Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci USA 108 (2):534–539. doi: 10.1073/pnas.1013426108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Cheng A, Yeager M (2007) Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131(1):70–79 [DOI] [PubMed] [Google Scholar]
- Garzon MT, Lidon-Moya MC, Barrera FN, Prieto A, Gomez J, Mateu MG, Neira JL (2004) The dimerization domain of the HIV-1 capsid protein binds a capsid protein-derived peptide: a biophysical characterization. Protein Sci 13(6):1512–1523. doi: 10.1110/ps.03555304 (a publication of the Protein Society) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI (1996) Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273(5272):231–235 [DOI] [PubMed] [Google Scholar]
- Goel A, Mazur SJ, Fattah RJ, Hartman TL, Turpin JA, Huang M, Rice WG, Appella E, Inman JK (2002) Benzamide-based thiolcarbamates: a new class of HIV-1 NCp7 inhibitors. Bioorg Med Chem Lett 12(5):767–770 (S0960894X02000070) [DOI] [PubMed] [Google Scholar]
- Gottlieb GS, Eholie SP, Nkengasong JN, Jallow S, Rowland-Jones S, Whittle HC, Sow PS (2008) A call for randomized controlled trials of antiretroviral therapy for HIV-2 infection in West Africa. Aids 22(16):2069–2072. doi: 10.1097/QAD.0b013e32830edd44 (discussion 2073–2064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci USA 88 (8):3195–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreau N, Hucke O, Faucher AM, Grand-Maitre C, Lepage O, Bonneau PR, Mason SW, Titolo S (2013a) Discovery and structural characterization of a new inhibitor series of HIV-1 nucleocapsid function: NMR solution structure determination of a ternary complex involving a 2:1 inhibitor/NC stoichiometry. J Mol Biol 425(11):1982–1998. doi: 10.1016/j.jmb.2013.02.022 [DOI] [PubMed] [Google Scholar]
- Goudreau N, Lemke CT, Faucher AM, Grand-Maitre C, Goulet S, Lacoste JE, Rancourt J, Malenfant E, Mercier JF, Titolo S, Mason SW (2013b) Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem Biol 8 (5):1074–1082. doi: 10.1021/cb400075f [DOI] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH (2013) Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502(7472):559–562. doi: 10.1038/nature12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haqqani AA, Tilton JC (2013) Entry inhibitors and their use in the treatment of HIV-1 infection. Antiviral Res 98(2):158–170. doi: 10.1016/j.antiviral.2013.03.017 [DOI] [PubMed] [Google Scholar]
- Hemelaar J, Gouws E, Ghys PD, Osmanov S, Isolation W-UNfH, Characterisation (2011) Global trends in molecular epidemiology of HIV-1 during 2000–2007. Aids 25(5):679–689. doi: 10.1097/QAD.0b013e328342ff93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida-Matsumoto L, Resh MD (1999) Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55(gag) and p17MA. J Virol 73 (3):1902–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI (1996) Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci USA 93(7):3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WS, Hughes SH (2012) HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2 (10):37–58. doi: 10.1101/cshperspect.a006882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Orenstein JM, Martin MA, Freed EO (1995) p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol 69(11):6810–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme AE, Perez O, Hope TJ (2011) Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci USA 108(24):9975–9980. doi: 10.1073/pnas.1014522108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im YJ, Kuo L, Ren X, Burgos PV, Zhao XZ, Liu F, Burke TR Jr, Bonifacino JS, Freed EO, Hurley JH (2010) Crystallographic and functional analysis of the ESCRT-I /HIV-1 Gag PTAP interaction. Structure 18(11):1536–1547. doi: 10.1016/j.str.2010.08.010S0969-2126(10)00348-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MC (2011) Mechanisms for Env glycoprotein acquisition by retroviruses. AIDS Res Hum Retroviruses 27(3):239–247. doi: 10.1089/AID.2010.0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD (2013) MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502(7472):563–566. doi: 10.1038/nature12653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J, Stoltzfus CM (2012) Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2(2):a006916. doi: 10.1101/cshperspect.a006916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PW, Adamson CS, Heymann JB, Freed EO, Steven AC (2011) HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J Virol 85(4):1420–1428. doi: 10.1128/JVI.01926-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PW, Huang RK, England MR, Waki K, Cheng N, Heymann JB, Craven RC, Freed EO, Steven AC (2013) A two-pronged structural analysis of retroviral maturation indicates that core formation proceeds by a disassembly-reassembly pathway rather than a displacive transition. J Virol 87(24):13655–13664. doi: 10.1128/JVI.01408-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP (2007) Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol 373(2):355–366. doi: 10.1016/j.jmb.2007.07.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan RE, Ono A, Englund G, Freed EO (1998) Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J Virol 72(5):4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan M, Rappaport J (2011) HIV-1 accessory protein Vpr: relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 8:25. doi: 10.1186/1742-4690-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135(1):49–60. doi: 10.1016/j.cell.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L, Engelman A (2012) Retroviral integrase proteins and HIV-1 DNA integration. J Biol Chem 287(49):40858–40866. doi: 10.1074/jbc.R112.397760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A (2010) The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J Virol 84 (1):397–406. doi: 10.1128/JVI.01899-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW (2013) Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob AgentsChemother 57(10):4622–4631.doi: 10.1128/AAC.00985-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B (2011) One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr HIV Res 9(7):496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tortorec A, Willey S, Neil SJ (2011) Antiviral inhibition of enveloped virus release by tetherin/BST-2: action and counteraction. Viruses 3(5):520–540. doi: 10.3390/v3050520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman MM, Smeaton L, Smith KY, Rodriguez B, Pu M, Wang H, Sevin A, Tebas P, Sieg SF, Medvik K, Margolis DM, Pollard R, Ertl HC, Valdez H (2006) Cyclosporin A provides no sustained immunologic benefit to persons with chronic HIV-1 infection starting suppressive antiretroviral therapy: results of a randomized, controlled trial of the AIDS Clinical Trials Group A5138. J Infect Dis 194(12):1677–1685. doi: 10.1086/509261 [DOI] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN (2010) Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7(3):221–233. doi: 10.1016/j.chom.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH (2007) Structural basis for viral late-domain binding to Alix. Natl Struct Mol Biol 14(3):194–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Harris J, Swanstrom R (2009) A strongly transdominant mutation in the human immunodeficiency virus type 1 Gag gene defines an Achilles heel in the virus life cycle. J Virol. doi: 10.1128/JVI.00317-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Potempa M, Kolli M, Ozen A, Schiffer CA, Swanstrom R (2012) Context surrounding processing sites is crucial in determining cleavage rate of a subset of processing sites in HIV-1 Gag and Gag-Pro-Pol polyprotein precursors by viral protease. J Biol Chem 287(16):13279–13290. doi: 10.1074/jbc.M112.339374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier JF, Wardrop E, Faucher AM, Coulombe R, Banik SS, Fader L, Gagnon A, Kawai SH, Rancourt J, Tremblay M, Yoakim C, Simoneau B, Archambault J, Sundquist WI, Mason SW (2012) Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J Virol 86(12):6643–6655. doi: 10.1128/JVI.00493-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K (2010) Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol 7(6):754–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT (2003) PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci USA 100(23):13555–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zoumplis D, Matallana C, Kilgore NR, Reddick M, Yunus AS, Adamson CS, Salzwedel K, Martin DE, Allaway GP, Freed EO, Wild CT (2006) Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology 356(1–2):217–224. doi: 10.1016/j.virol.2006.07.023S0042-6822(06)00497-1 [DOI] [PubMed] [Google Scholar]
- Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C (2013) The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14(4):398–410. doi: 10.1016/j.chom.2013.08.015 [DOI] [PubMed] [Google Scholar]
- Lopez-Verges S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C (2006) Tail- interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci USA 103(40):14947–14952. doi: 10.1073/pnas.0602941103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Heng X, Summers MF (2011) Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol 410(4):609–633. doi: 10.1016/j.jmb.2011.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73(6):1067–1078 [DOI] [PubMed] [Google Scholar]
- Malim MH, Emerman M (2008) HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe 3(6):388–398. doi: 10.1016/j.chom.2008.04.008 [DOI] [PubMed] [Google Scholar]
- Margot NA, Gibbs CS, Miller MD (2009) Phenotyphic susceptibility to bevirimat among HIV-infected patient isolates without prior exposure to bevirimat. Paper presented at the 16th conference on retroviruses and opportunistic infections, Montreal, 8–11 Feb 2011 [Google Scholar]
- Markowitz M, Vaida F, Hare CB, Boden D, Mohri H, Hecht FM, Kalayjian RC, Conrad A, Mildvan D, Aberg J, Hogan C, Kilby JM, Balfour HH Jr, Schafer K, Richman D, Little S (2010) The virologic and immunologic effects of cyclosporine as an adjunct to antiretroviral therapy in patients treated during acute and early HIV-1 infection. J Infect Dis 201(9):1298–1302. doi: 10.1086/651664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Blum R, Doto J, Galbraith H, Ballow C (2007a) Multiple-dose pharmacokinetics and safety of bevirimat, a novel inhibitor of HIV maturation, in healthy volunteers. Clin Pharmacokinet 46(7):589–598. doi: 10.2165/00003088-200746070-00004 [DOI] [PubMed] [Google Scholar]
- Martin DE, Blum R, Wilton J, Doto J, Galbraith H, Burgess GL, Smith PC, Ballow C (2007b) Safety and pharmacokinetics of Bevirimat (PA-457), a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Antimicrob Agents Chemother 51(9):3063–3066. doi: 10.1128/AAC.01391-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI (1994) Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol 244(2):198–223. doi: 10.1006/jmbi.1994.1719S0022-2836(84)71719-0 [DOI] [PubMed] [Google Scholar]
- Mateu MG (2009) The capsid protein of human immunodeficiency virus: intersubunit interactions during virus assembly. FEBS J 276(21):6098–6109. doi: 10.1111/j.1742-4658.2009.07313.x [DOI] [PubMed] [Google Scholar]
- Matreyek KA, Engelman A (2013) Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses 5(10):2483–2511. doi: 10.3390/v5102483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Yucel SS, Li X, Engelman A (2013) Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 9(10):e1003693. doi: 10.1371/journal.ppat.1003693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Colf LA, Sundquist WI (2013) Membrane fission reactions of the mammalian ESCRT pathway. Ann Rev Biochem 82:663–692. doi: 10.1146/annurev-biochem-072909-101058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Jenkins LM, Ott DE, Hayashi R, Coren LV, Wang D, Xu Q, Schito ML, Inman JK, Appella DH, Appella E (2010) Small-molecule inactivation of HIV-1 NCp7 by repetitive intracellular acyl transfer. Natl Chem Biol 6(12):887–889. doi: 10.1038/nchembio.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcock DR, Thomas JA, Gagliardi TD, Gorelick RJ, Roser JD, Chertova EN, Bess JW Jr, Ott DE, Sattentau QJ, Frank I, Pope M, Lifson JD, Henderson LE, Crise BJ (2005) Elimination of retroviral infectivity by N-ethylmaleimide with preservation of functional envelope glycopro- teins. J Virol 79(3):1533–1542. doi: 10.1128/JVI.79.3.1533-1542.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Mahe C, Mayanja B, Okongo JM, Lubega R, Whitworth JA (2002) HIV-1 infection in rural Africa: is there a difference in median time to AIDS and survival compared with that in industrialized countries? Aids 16(4):597–603 [DOI] [PubMed] [Google Scholar]
- Muller B, Anders M, Akiyama H, Welsch S, Glass B, Nikovics K, Clavel F, Tervo HM, Keppler OT, Krausslich HG (2009) HIV-1 Gag processing intermediates trans-dominantly interfere with HIV-1 infectivity. J Biol Chem 284(43):29692–29703. doi: 10.1074/jbc.M109.027144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Ablan S, Freed EO, Tanaka Y (2004) Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78(2):1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Freed EO (2000) The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci USA 97(1):343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriaux D, Darlix JL (2010) Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol 7(6):744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa F, Lodwick RK, Smith CJ, Smith R, Cambiano V, Lundgren JD, Delpech V, Phillips AN (2012) Projected life expectancy of people with HIV according to timing of diagnosis. Aids 26(3):335–343. doi: 10.1097/QAD.0b013e32834dcec9 [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Feasley CL, Jackson KW, Nitz TJ, Salzwedel K, Air GM, Sakalian M (2011) The prototype HIV-1 maturation inhibitor, bevirimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology 8:101. doi: 10.1186/1742-4690-8-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyamweya S, Hegedus A, Jaye A, Rowland-Jones S, Flanagan KL, Macallan DC (2013) Comparing HIV-1 and HIV-2 infection: lessons for viral immunopathogenesis. Rev Med Virol 23(4):221–240. doi: 10.1002/rmv.1739 [DOI] [PubMed] [Google Scholar]
- Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO (2004) Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA 101(41):14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO (1999) Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol 73(5):4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO (2004) Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol 78(3):1552–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Freed EO (2005) Role of lipid rafts in virus replication. Adv Virus Res 64:311–358 [DOI] [PubMed] [Google Scholar]
- Ono A, Orenstein JM, Freed EO (2000) Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol 74(6):2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S (2013) Advances in detection and monitoring of plasma viremia in HIV-infected individuals receiving antiretroviral therapy. Curr Opin HIV AIDS 8(2):87–92. doi: 10.1097/COH.0b013e32835d80af [DOI] [PubMed] [Google Scholar]
- Planelles V, Barker E (2010) Roles of Vpr and Vpx in modulating the virus-host cell relationship. Mol Aspects Med 31(5):398–406. doi: 10.1016/j.mam.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Alam SL, Davis DR, Sundquist WI (2002) Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Natl Struct Biol 9(11):812–817. doi: 10.1038/nsb856 [DOI] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M (2009) X-ray structures of the hexameric building block of the HIV capsid. Cell 137(7):1282–1292. doi: 10.1016/j.cell.2009.04.063S0092-8674(09)00580-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC (2012) CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8(8):e1002896. doi: 10.1371/journal.ppat.1002896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Bethune MP, Neyts J, De Clercq E, Dumont JM, Scalfaro P, Besseghir K, Wenger RM, Rosenwirth B (2008) Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob Agents Chemother 52 (4):1302–1317. doi: 10.1128/AAC.01324-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Williams JA, Chu H, Chen X, Wang JJ, Ding L, Akhirome E, Wen X, Lapierre LA, Goldenring JR, Spearman P (2013) Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog 9(4): e1003278. doi: 10.1371/journal.ppat.1003278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian K, Bori ID, Chen CH, Huang L, Lee KH (2012) Anti-AIDS agents 90. novel C-28 modified bevirimat analogues as potent HIV maturation inhibitors. J Med Chem 55(18):8128–8136. doi: 10.1021/jm301040s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI (1995) Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 378(6558):743–747. doi: 10.1038/378743a0 [DOI] [PubMed] [Google Scholar]
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ (2013) HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503(7476):402–405. doi: 10.1038/nature12769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein A (2010) Nucleic acid chaperone activity of retroviral Gag proteins. RNA Biol 7(6):700–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WG, Schaeffer CA, Harten B, Villinger F, South TL, Summers MF, Henderson LE, Bess JW Jr, Arthur LO, McDougal JS et al. (1993) Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature 361(6411):473–475. doi: 10.1038/361473a0 [DOI] [PubMed] [Google Scholar]
- Rice WG, Supko JG, Malspeis L, Buckheit RW Jr, Clanton D, Bu M, Graham L, Schaeffer CA, Turpin JA, Domagala J, Gogliotti R, Bader JP, Halliday SM, Coren L, Sowder RC 2nd, Arthur LO, Henderson LE (1995) Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science 270(5239):1194–1197 [DOI] [PubMed] [Google Scholar]
- Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD (2013) Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 9(6):e1003461. doi: 10.1371/journal.ppat.1003461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani B, Cohen EA (2012) Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr Opin Virol 2(6):755–763. doi: 10.1016/j.coviro.2012.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF (2006) Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA 103 (30):11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, Luban J (2004) Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430(6999):569–573. doi: 10.1038/nature02777 [DOI] [PubMed] [Google Scholar]
- Seclen E, Mdel MG, Corral A, de Mendoza C, Soriano V, Poveda E (2010) High prevalence of natural polymorphisms in Gag (CA-SP1) associated with reduced response to bevirimat, an HIV-1 maturation inhibitor. Aids 24(3):467–469. doi: 10.1097/QAD.0b013e328335ce07 [DOI] [PubMed] [Google Scholar]
- Sharkey M (2013) Restriction of retroviral infection of macrophages. Curr Top Microbiol Immunol 371:105–122. doi: 10.1007/978-3-642-37765-5_4 [DOI] [PubMed] [Google Scholar]
- Sharp PM, Hahn BH (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1(1):a006841. doi: 10.1101/cshperspect.a006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhou J, Shah VB, Aiken C, Whitby K (2011) Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85(1):542–549. doi: 10.1128/JVI.01406-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvadchak V, Sanglier S, Rocle S, Villa P, Haiech J, Hibert M, Van Dorsselaer A, Mely Y, de Rocquigny H (2009) Identification by high throughput screening of small compounds inhibiting the nucleic acid destabilization activity of the HIV-1 nucleocapsid protein. Biochimie 91(7):916–923. doi: 10.1016/j.biochi.2009.04.014S0300-9084(09)00110-2 [DOI] [PubMed] [Google Scholar]
- Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE (2007) Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunode- ficiency virus infection. Antimicrob Agents Chemother 51(10):3574–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich HG (2005) A peptide inhibitor of HIV-1 assembly in vitro. Natl Struct Mol Biol 12(8):671–677. doi: 10.1038/nsmb964 [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427 (6977):848–853. doi: 10.1038/nature02343 [DOI] [PubMed] [Google Scholar]
- Sundquist WI, Krausslich HG (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2(7):a006924. doi: 10.1101/cshperspect.a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R, Coffin J (2012) HIV-1 pathogenesis: the virus. Cold Spring Harb Perspect Med 2 (12):a007443. doi: 10.1101/cshperspect.a007443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF (2003) Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol 327(5):1013–1020 [DOI] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF (2004) Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA 101 (2):517–522. doi: 10.1073/pnas.0305665101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Ndassa Y, Summers MF (2002) Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Natl Struct Biol 9(7):537–543. doi: 10.1038/nsb806 [DOI] [PubMed] [Google Scholar]
- Tedbury PR, Ablan SD, Freed EO (2013) Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog 9(11):e1003739. doi: 10.1371/journal.ppat.1003739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A, Johnson WE (2012) Host genes important to HIV replication and evolution. Cold Spring Harb Perspect Med 2(4):a007203. doi: 10.1101/cshperspect.a007203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternois F, Sticht J, Duquerroy S, Krausslich HG, Rey FA (2005) The HIV-1 capsid protein C- terminal domain in complex with a virus assembly inhibitor. Natl Struct Mol Biol 12(8):678–682. doi: 10.1038/nsmb967 [DOI] [PubMed] [Google Scholar]
- Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG (1994) Functional association of cyclophilin A with HIV-1 virions. Nature 372(6504):363–365. doi: 10.1038/372363a0 [DOI] [PubMed] [Google Scholar]
- UNAIDS (2013) 2013 global fact sheet. http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/20130923_FactSheet_Global_en.pdf
- Urano E, Kuramochi N, Ichikawa R, Murayama SY, Miyauchi K, Tomoda H, Takebe Y, Nermut M, Komano J, Morikawa Y (2011) Novel postentry inhibitor of human immunodeficiency virus type 1 replication screened by yeast membrane-associated two-hybrid system. Antimicrob Agents Chemother 55(9):4251–4260. doi: 10.1128/aac.00299-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-Casuso JC, Di Nunzio F, Yang Y, Reszka N, Lienlaf M, Arhel N, Perez P, Brass AL, Diaz- Griffero F (2012) TNPO3 is required for HIV-1 replication after nuclear import but prior to integration and binds the HIV-1 core. J Virol 86(10):5931–5936. doi: 10.1128/JVI.00451-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ (2009) HIV-1 susceptibility to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag SP1. Antimicrob Agents Chemother 53(5):2185–8. doi: 10.1128/AAC.01650-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen J, Verhofstede C, Knops E, Vandekerckhove L, Fun A, Brunen D, Dauwe K, Wensing AM, Pfister H, Kaiser R, Nijhuis M (2010) High prevalence of bevirimat resistance mutations in protease inhibitor-resistant HIV isolates. Aids 24(5):669–673. doi: 10.1097/QAD.0b013e32833160fa [DOI] [PubMed] [Google Scholar]
- Votteler J, Sundquist WI (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14 (3):232–241. doi: 10.1016/j.chom.2013.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed AA, Freed EO (2010) The role of lipids in retrovirus replication. Viruses 2(5):1146–1180. doi: 10.3390/v2051146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki K, Durell SR, Soheilian F, Nagashima K, Butler SL, Freed EO (2012) Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog 8(11): e1002997. doi: 10.1371/journal.ppat.1002997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace GS, Cheng-Mayer C, Schito ML, Fletcher P, Jenkins LMM, Hayashi R, Neurath AR, Appella E, Shattock RJ (2009) Human immunodeficiency virus type 1 nucleocapsid inhibitors impede trans infection in cellular and explant models and protect nonhuman primates from infection. J Virol 83(18):9175–9182. doi: 10.1128/JVI.00820-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss ER, Gottlinger H (2011) The role of cellular factors in promoting HIV budding. J Mol Biol 410(4):525–533. doi: 10.1016/j.jmb.2011.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen CB, Tilton JC, Doms RW (2012) HIV: cell binding and entry. Cold Spring Harb Perspect Med 2(8):23–36. doi: 10.1101/cshperspect.a006866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C (2004) Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78(7):3429–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Perez O, Hope TJ, Emerman M (2007) Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3(10):1502–1510. doi: 10.1371/journal.ppat.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yebra G, Holguin A (2008) The maturation inhibitor bevirimat (PA-457) can be active in patients carrying HIV type-1 non-B subtypes and recombinants. Antiviral Ther 13(8):1083–1085 [PubMed] [Google Scholar]
- Zentner I, Sierra LJ, Fraser AK, Maciunas L, Mankowski MK, Vinnik A, Fedichev P, Ptak RG, Martin-Garcia J, Cocklin S (2013a) Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. Chem Med Chem 8(3):426–432. doi: 10.1002/cmdc.201200577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner I, Sierra LJ, Maciunas L, Vinnik A, Fedichev P, Mankowski MK, Ptak RG, Martin-Garcia J, Cocklin S (2013b) Discovery of a small-molecule antiviral targeting the HIV-1 matrix protein. Bioorg Med Chem Lett 23(4):1132–1135. doi: 10.1016/j.bmcl.2012.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Q, Fisher RD, Chung HY, Myszka DG, Sundquist WI, Hill CP (2008) Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Natl Struct Mol Biol 15(1):43–49. doi: 10.1038/nsmb1319 [DOI] [PubMed] [Google Scholar]
- Zhang H, Curreli F, Waheed AA, Mercredi PY, Mehta M, Bhargava P, Scacalossi D, Tong X, Lee S, Cooper A, Summers MF, Freed EO, Debnath AK (2013) Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirology 10:136. doi: 10.1186/1742-4690-10-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Curreli F, Zhang X, Bhattacharya S, Waheed AA, Cooper A, Cowburn D, Freed EO, Debnath AK (2011) Antiviral activity of alpha-helical stapled peptides designed from the HIV-1 capsid dimerization domain. Retrovirology 8:28. doi: 10.1186/1742-4690-8-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhao Q, Bhattacharya S, Waheed AA, Tong X, Hong A, Heck S, Curreli F, Goger M, Cowburn D, Freed EO, Debnath AK (2008) A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol 378(3):565–580. doi: 10.1016/j.jmb.2008.02.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P (2013) Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497(7451):643–646. doi: 10.1038/nature12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chen CH, Aiken C (2006) Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J Virol 80 (24):12095–12101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Resh MD (1996) Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J Virol 70(12):8540–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]