

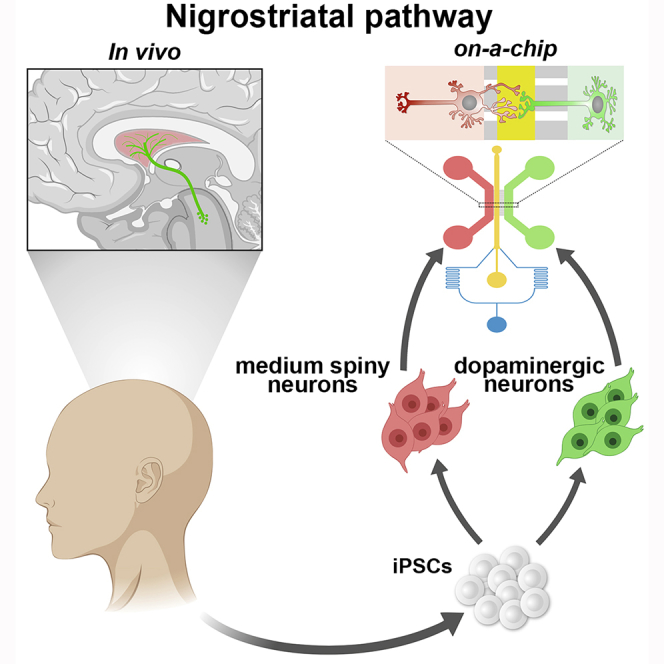
Summary
Stem cell-derived neurons are generally obtained in mass cultures that lack both spatial organization and any meaningful connectivity. We implement a microfluidic system for long-term culture of human neurons with patterned projections and synaptic terminals. Co-culture of human midbrain dopaminergic and striatal medium spiny neurons on the microchip establishes an orchestrated nigro-striatal circuitry with functional dopaminergic synapses. We use this platform to dissect the mitochondrial dysfunctions associated with a genetic form of Parkinson’s disease (PD) with OPA1 mutations. Remarkably, we find that axons of OPA1 mutant dopaminergic neurons exhibit a significant reduction of mitochondrial mass. This defect causes a significant loss of dopaminergic synapses, which worsens in long-term cultures. Therefore, PD-associated depletion of mitochondria at synapses might precede loss of neuronal connectivity and neurodegeneration. In vitro reconstitution of human circuitries by microfluidic technology offers a powerful system to study brain networks by establishing ordered neuronal compartments and correct synapse identity.
Graphical Abstract
Highlights
-
•
Long-term stable reconstitution of the human nigro-striatal neuronal circuit on-a-chip
-
•
Stable synaptic connectivity of the iPSC-derived nigro-striatal neuronal connections
-
•
Dopaminergic-specific synaptic identity of the iPSC-derived nigro-striatal pathway
-
•
PD-OPA1 DA axons show a severe loss and impairment of mitochondria
Iannielli et al. implement a microfluidic system for long-term and stable culture of iPSC-derived neurons with patterned organization of their projections and synaptic terminals. Culture of the iPSC-derived medium spiny and dopaminergic neurons on-a-chip establishes a well-organized nigro-striatal circuit with functional dopaminergic synapses.
Introduction
The introduction of induced pluripotent stem cell (iPSC) technology has provided a powerful system for the in vitro generation of somatic cells affected in human diseases and the mechanistic understanding of the underlying pathological processes (Grandy et al., 2019, Shi et al., 2017). By recapitulating the milestones of embryonic cell lineage differentiation, iPSCs can be efficiently induced in vitro in specific neuronal derivatives providing the exact subtypes that are specifically affected in a variety of neurological disorders (Brennand et al., 2015, Kirwan et al., 2015, Tao and Zhang, 2016). These advances in the differentiation protocols in terms of cell specificity and efficiency have enormously accelerated the successful in vitro modeling of numerous neurological disorders (Fujimori et al., 2018, Tiscornia et al., 2011, Wen et al., 2016). However, the majority of protocols generate cultures of neurons that lack both spatial organization and defined synaptic connectivity. In addition, conventional neuronal cultures fail to recapitulate meaningful circuitries between different neuronal cell types as they are established within the brain. These limitations pose relevant barriers for establishing iPSC-derived neuronal models informative for those diseases that arise from dysfunctions in only selected brain circuitries (Brennand, 2013, Guo et al., 2017, Mertens et al., 2018, Saha and Jaenisch, 2009). This is indeed the case for the majority of the neurodegenerative diseases, such as Parkinson’s disease (PD), amyotrophic lateral sclerosis, dementia, ataxia, and neuropathies, in which only selective networks in the nervous system result particularly susceptible. In fact, in several genetic forms of these diseases, neurodegeneration occurs in selected neuronal networks, although the responsible mutated genes are widely expressed. Thus, it remains poorly understood why some neuronal connections are vulnerable whereas others are resistant to exactly the same pathogenetic insult. To gain insight into this crucial question, it would be extremely advantageous to establish a system whereby different iPSC-derived neuronal subtypes can be cultured together and organized to form a patterned network as normally occurring in vivo.
Microfluidic platforms obtained with microfabrication technologies have initially made it possible to develop two-compartment miniaturized devices with which neuronal soma and axons can be spatially and fluidically isolated for investigating processes selectively in one of these two districts (Neto et al., 2016, Park et al., 2006, Taylor et al., 2005). This microfluid system has been used largely by neuroscientists, and this initial design has been developed in commercialized chips available worldwide. Subsequently, the introduction of a central channel connected with microgrooves with two lateral chambers has constituted an organized space in which neuronal compartments and synapses can be studied separately (Taylor et al., 2010). These chips have been used with cultures of mouse primary neurons to study synaptic biology, axon-specific processes, and disease-relevant mechanisms (Coquinco et al., 2014, Taylor et al., 2010, Virlogeux et al., 2018). Recently, microdevice platforms were successfully used to maintain iPSC-derived neuronal networks (Gribaudo et al., 2019). However, it remains unclear if the microfluidic environment is permissive for long-term culture of human neurons and whether it preserves their subtype identities and synaptic maturation. In addition, to the best of our knowledge a brain-on-chip system with which a defined neuronal circuit endowed with specific synaptic connectivity can be successfully established is lacking. Important applications of this platform would be of relevance for research in PD, a disease that leads to the early degeneration of the nigro-striatal pathway responsible for the onset of the movement deficits.
We have recently modeled using iPSC technology a genetic form of PD caused by dominant heterozygous mutations in the dynamin-related GTPase optic atrophy type 1 (OPA1) gene (Iannielli et al., 2018). OPA1-PD iPSC-derived neurons showed a fragmented mitochondrial network, impaired oxidative metabolism, reduced ATP intracellular levels, and heightened oxidative stress (Iannielli et al., 2018). Importantly, mutant neurons exhibited a reduced survival upon extensive in vitro culture. These data suggested that OPA1 mutant patients with a complex phenotype could present higher risk for developing parkinsonism during disease progression in elderly patients (Chevrollier et al., 2008, Iannielli et al., 2018, Zanna et al., 2008). However, the chain of mechanistic events that connects mitochondrial impairment with neuronal cell death is not yet fully understood. This is of particular relevance because the largest group of PD-causative genes encodes for crucial modulators of mitochondrial quality control and homeostasis (Giannoccaro et al., 2017). Herein, we sought to determine how mitochondria dysfunctions might impair the maintenance of the nigro-striatal network in a human system. To this end, we adapted a microfluidic platform to organize a patterned circuitry between human iPSC-derived midbrain dopaminergic neurons (DANs) and striatal medium spiny neurons (MSNs) for in vitro modeling of the nigro-striatal connection. This platform provided us with the opportunity to study pathologically relevant processes in long-term cultures of iPSC neuronal derivatives with spatially orchestrated functional connectivity mirroring the brain nigro-striatal pathway affected in PD.
Results
PD-OPA1 iPSC-Derived DANs Show a Loss of Mitochondria along Neurites in Conventional Neuronal Cultures
To determine the impact of OPA1 mutations on post-mitotic neurons, control, mutant, and gene-reconstituted PD-OPA1 iPSCs were differentiated into mass neuronal cultures enriched in DANs. In a two-step differentiation protocol, iPSCs were induced into FOXA2/Nestin+ neural progenitors and then matured into neurons through exposure to BDNF, GDNF, DAPT, and ascorbic acid for 6 weeks, obtaining mixed neuronal cultures with a significant fraction of DANs (Figures 1A–1D) (Kirkeby et al., 2012, Kriks et al., 2011). Subsequently, neuronal cultures were stained for TOMM20 and tyrosine hydroxylase (TH) to visualize the mitochondrial network and its localization in DANs. Notably, the number of mitochondria along neurites was markedly reduced in PD-OPA1 compared with control TH+ neurons (Figures 1E–1M and 1T). In addition, the remaining PD-OPA1 mitochondria network appeared more fragmented (Figures 1E–1M and 1T). Notably, the reconstitution of the total OPA1 protein levels in mutant PD-OPA1 iPSCs by lentiviral-mediated gene transfer of the functional human gene (Iannielli et al., 2018) was sufficient to rescue the mitochondria deficits in neuronal derivatives (Figures 1N–1T). These data indicate that OPA1 deficiency is responsible for the altered mitochondrial morphology with concomitant loss of mitochondria in the neurites of PD-OPA1 patient neurons. However, this neuronal network cannot distinguish between axons and dendrites, and therefore the downstream alterations caused by this mitochondrial deficiency cannot be determined using this system.
Figure 1.

Mitochondrial Axonal Localization in Normal Donor (ND), Mutant, and Gene-Complemented PD-OPA1 Neurons
(A–D) Representative pictures (A–C) and quantitative analysis (D) of dopaminergic neurons (DANs) co-expressing TH and MAP2.
(E–S) Representative images of mitochondrial morphology stained with TOMM20 (green) in ND (E–G), mutant (H–M), and gene-complemented PD-OPA1 (N–S) neurons.
(T) Quantification of mitochondrial numbers and fragmented appearance along the neurites of mutant compared with control and gene-complemented PD-OPA1 neurons. TOMM20 puncta are normalized respect to TH staining area (n = 20 DANs for each experiments).
Data are mean ± SEM; n = 3 independent experiments. ∗∗p < 0.01 to ND neurons,°p < 0.01 to gene-complemented mutant OPA1 neurons. Statistical analysis was performed using one-way ANOVA, followed by Tukey post-test. Scale bars, 50 μm (A–C) and 100 μm (E–S).
In Vitro Reconstitution of the Brain Nigro-striatal Pathway with Human iPSC-Derived Neurons on Microfluidic Devices
Conventional neuronal cultures obtained by in vitro iPSC differentiation in the dish are a mix of neurons that lack spatial organization and do not reconstitute native circuitries. In these conditions it remains challenging to analyze specific sub-cellular compartments and validate any physiological connections between different neuronal cell types. This random ensemble of neurons is also inadequate to distinguish between axonal and dendritic compartments and their relative mitochondria content. In order to establish an ordered neuronal network and reconstitute the nigro-striatal connection in vitro, we sought to implement a microfluidic system suitable to promote the functional integration of human iPSC-derived neurons in culture (Figures 2A and 2B). We fabricated a polydimethylsiloxane (PDMS) microdevice previously used with mouse primary neurons consisting of two lateral compartments connected with a central channel through an array of microgrooves (Figure S1). Neurons are plated and cultured in the two lateral chambers, and with time they extend axons and dendrites into the microgrooves to reach the central (“synaptic”) channel, where synaptic contacts are established. The central channel is equipped with a perfusion system to maintain a separate culture environment when necessary. Moreover, this channel is placed at unequal distance from the lateral chambers where neurons are plated. On one side is a set of 500-μm-long microgrooves, which because of their length can be completely navigated only by DAN axons but not dendrites; on the other side is a set of 75-μm-long microgrooves that direct the growth of MSN neurites into the synaptic channel (Figure S1). However, to enrich for the growth of MSN dendrites within the microchannels, a gradient of laminin between the two compartments was applied while maintaining the same concentration of poly-d-lysine. With this design, MSNs and DANs, seeded separately in the two lateral compartments, project into the central channel where synaptic contacts are compartmentalized. Specifically, the asymmetric configuration of the microgroove system directs the growth of DAN axons and MSN dendrites into the synaptic channel, thus reconstituting the nigro-striatal connection.
Figure 2.

In Vitro Reconstitution of the Human Nigro-striatal Circuitry in a Microfluidic Device
(A) Illustration of the nigro-striatal pathway in the brain. Substantia nigra (SN) DANs project to the dorsal striatum (Str) to connect with the medium spiny neurons (MSNs).
(B) Schematic design of the three-compartment microfluidic device with the DA and striatal lateral chambers connected via microgrooves with the central synaptic channel (SC).
(C–H) Representative images of TH+ DANs seeded in the microfluidic device.
(I–K) Representative images of DARPP32+ MSNs seeded in the microfluidic device.
(L) Quantification of DANs and MSNs relative to respectively the GFP- or tdTomato-transduced cells, after 6 weeks of culture in the microfluidic platform.
Data are mean ± SEM; n = 3 independent experiments. Scale bars, 100 μm.
First, we optimized differentiation of control and PD-OPA1 iPSCs into either striatal MSNs or midbrain DANs in the dish. To generate the former neuronal cell type, iPSCs were initially treated with the BMP/SMAD inhibitors LDN, dorsomorphin, and SB431542, followed by 9 day exposure to activin (Arber et al., 2015). After this first induction phase, differentiating iPSCs were switched to the maturation medium supplemented with neurotrophins, DAPT, and ascorbic acid. Immunostaining on cultures differentiated for 5 weeks revealed a strong induction of the MSN markers GABA and CTIP2 (Figures S2A–S2H). Moreover, the definitive MSN marker DARPP32 was co-expressed in 60%–82% of MAP2+ neurons depending on each iPSC line (Figures S2A–S2H). To generate midbrain DANs, we applied previously established protocols (Kirkeby et al., 2012, Kriks et al., 2011) that combined BMP/SMAD inhibition, SHH/FGF8 exposure, and WNT activation at different time windows to induce iPSC differentiation into an intermediate step of proliferating FOXA2+ midbrain neural progenitors (Figures S2I and S2J). Subsequently, we found that the same maturation medium for MSNs was equally effective to mature midbrain DAN precursors into a culture of post-mitotic neurons with a fraction of TH+/MAP2+ between 68% and 84% depending on each iPSC line (Figures S2I–S2O).
To efficiently populate the microdevices, we sought to plate iPSC-derived striatal and DA neuronal precursors and let them to mature into the microfluidic chambers. Initially, we screened for different coating reagents to obtain an optimal adhesion of the neuronal precursors on the microfluidic material. Among others, the poly-l-lysine/laminin pre-treatment allows the most convenient procedure associated, with the highest cell adhesion efficiency peaking to ∼80% of the plated cells. To assess the survival rate of MSN and DAN progenitors after plating into the microfluidic devices, we performed live/dead cell double-fluorescence staining at both 3 and 7 days after initial seeding. For both neuronal populations, the survival rate was ∼75% at day 3, which did not change at day 7 (Figure S3). Thus, these results indicated that early MSN and DAN precursors can adapt well to the conditions imposed by the microfluidic environment with good survival efficiency.
Maturation and Subtype Neuronal Identity of iPSC-Derived Neurons in Microfluidic Settings
Long-term culture in a microfluidic platform might potentially alter the maturation of the iPSC-derived neurons or even alter their specific identity. To assess this possibility, MSNs and DANs were plated in the two lateral chambers and were transduced after 2 days with lentiviruses expressing either GFP or tdTomato. Neurons were then examined after 6 weeks of culture in the microfluidics system in the presence of maturation medium. Double GFP/tdTomato staining showed virtually separate staining between GFP+ DANs and tdTomato+ MSNs, indicating that viral infections remained selectively confined within each of the two lateral chambers (Figures 2C–2E). Importantly, the ratio of TH+ DA7Ns relative to either GFP+ cells or DAPI nuclei was approximately 60%–85% in both cases and comparable with that obtained when iPSCs were differentiated into conventional culture dishes (Figures 2F–2H and 2L). Likewise, 60%–80% of tdTomato+ cells expressed the mature striatal marker DARPP32 (Figures 2I–2L) with comparable efficiency with that reached in standard differentiating conditions. After 6 weeks of culture in the microdevices, a large fraction of DANs expressed both vesicular monoamine transporter 2 (VMAT2; NCBI: SLC18A2) and DOPA decarboxylase (DDC), two key players of the DA enzymatic machinery, with virtual complete overlapping between the TH+ and VMAT2+ neuronal populations (Figure S4).
These data suggest that the manipulation and microenvironment generated in the microfluidic system is permissive to both the survival and maturation of iPSC-derived neurons, maintaining a specific subtype neuronal identity even after considerable time in culture.
Functional Connectivity of the Human Nigro-striatal Pathway with Active DA Synapses
Next, we evaluated whether DANs and MSNs could extend projections into the central chamber and form synapses. Initially, to evaluate the nature of the neuronal projections within the microchannels, we used antibodies recognizing intrinsic components of the axonal (SMI-312) and dendritic (MAP2) compartments. Of note, TH/SMI-312/βIII-tubulin triple immunofluorescence showed that ∼88% of the axonal projections in the synaptic chamber have a DA identity (Figures S5A–S5I). In addition, SMI-312/MAP2/βIII-tubulin triple staining in the MSN lateral chamber revealed that ∼68% of the βIII-tubulin-positive projections were also decorated with MAP2, suggesting that a large fraction of the MSN neurites entering in the microchannels connected with the synaptic chamber are dendrites (Figures S5J–S5R). Next, to assess the generation of synaptic terminals, we performed double immunofluorescence for Synapsin (SYN) and Homer, two resident proteins of the pre- and post-synaptic terminal, respectively. The presence of multiple SYN/Homer abutting dots indicated the formation of synaptic contacts between DANs and MSNs within the central chamber (Figures 3A–3F). To ascertain the stability of these contacts and the effective establishment of connectivity between DANs and MSNs, we adapted the rabies virus (RV) transneuronal tracing system to the microfluidic settings. EnvA-pseudotyped (G)-deleted RV (dG-RV) has been a transformative tool for retrograde transsynaptic tracing between two interconnected neuronal populations in the nervous system (Callaway and Luo, 2015, Luo et al., 2018). Following G protein trans-complementation in the starter cells, dG-RV can produce functional viral particles that can spread retrogradely across synapses and infect pre-synaptic neurons lacking G protein expression, which are therefore unable to support further viral spreading (Kim et al., 2016). To visualize neuronal connectivity into the microdevices, the MSN containing microwell was exposed to a multicistronic lentivirus co-expressing TVA, G protein, and a nuclear GFP. One week later, the same neurons were transduced with the mCherry expressing dG-RVs (Figure 3G). With this strategy, the mCherry fluorescence acted as a reporter for the assembling and spreading of dG-RVs from the MSNs. Indeed, a few days after dG-RV transduction, the majority of the MSNs expressed mCherry indicating productive infectivity and effective spreading within this neuronal population (Figures 3H–3J). In addition, several DANs in the separate lateral chamber of the device exhibited mCherry staining (Figures 3K–3M). Importantly, all the mCherry+ DANs were negative for nuclear GFP staining, confirming that the fluorescence was not caused by direct dG-RV transduction but rather was a result of retrograde viral spreading from infected MSNs crossing a monosynaptic terminal connecting the two neuronal pairs.
Figure 3.

The Human Nigro-striatal Pathway Establishes a Stable Synaptic Connectivity
(A–F) Representative pictures of single and double staining for Synapsin and Homer to visualize pre- and post-synaptic terminals, respectively (arrowheads in D, E, and F).
(G) Illustration of the lentiviral and rabies viral vectors used in this experiment. The tracing vector transduces the cells with a nuclear GFP, the TVA receptor, and G replication factor necessary for rabies infection. Neurons infected with the tracing virus are termed starter MSNs. After rabies viral infection, these cells express mCherry, and because of the presence of GP, the rabies viruses spread retrogradely to the traced neurons.
(H–M) Representative images of connectivity between MSNs and DANs. Starter MSNs are positive for both GFP and mCherry (H–J) indicating rabies viral productive infectivity.
(K–M) DANs express mCherry, but not GFP, indicating active spreading of the rabies viruses from MSNs and DANs and stable connectivity between the two neuronal networks.
Scale bars, 100 μm.
Because synaptic activity is a strong prerequisite to validate this platform for modeling synaptic dysfunctions in neurological disorders, we sought to determine the identity and functionality of the synaptic terminals within the central channel. Moreover, the previous description of synaptic contacts does not necessarily prove their identity, because a small contaminant of non-DANs/MSNs is also present within both populations. Initially, to prove the presence of active monoaminergic synapses, we used the fluorescent probe FFN206, which is a specific and sensitive substrate of VMAT2 (Hu et al., 2013). FFN206 is taken up by VMAT2-expressing neurons and associates with the transporter within the acidic synaptic vesicles, maintaining a robust and sustained fluorescence intensity (Hu et al., 2013). FFN206 was mixed with artificial cerebrospinal fluid (ACSF) buffer and perfused within the central channel 5 weeks after plating of the neurons in the microdevices (Figure 4A). To unambiguously identify the axonal terminals within the central chamber, the DAN-containing microwell was exposed to a Synapsin-GFP-expressing lentivirus for 24 h. Lentiviral infection in the microdevice transduced a large fraction of neurons and remained confined into the microwell, without any evident diffusion between the microgrooves and the other surrounding chambers. KCl-containing medium (56 mM) was perfused in the central channel and the lateral chamber containing the DANs for 10 min before starting the imaging sessions. Live confocal imaging detected numerous FFN206+ puncta, most of which, but not all, were co-localized with GFP+ fibers in the central chamber (Figures 4B–4D). Likely during live imaging experiments, not all the GFP+ fibers could be readily detectable for a low fluorescence signal, thus plausibly explaining why not all FFN206+ dots could be mapped on GFP+ axonal branches. These data indicate that numerous monoaminergic synapses are established in the medial channel and show active recycling of VMAT2+ vesicles in DAT terminals. To determine whether dopamine release from the dopaminergic pre-synaptic terminals is sufficient to trigger a post-synaptic response in connected MSNs, we used the dLight1 system. dLight1 is an engineered dopamine receptor containing a circularly permuted GFP whose intensity is directly related to dopamine signaling (Patriarchi et al., 2018). Thus, MSNs were transduced with two independent lentiviruses expressing either the tdTomato or dLight1 cassette and subjected to video recordings after enhancing DAN activity through KCl perfusion (Figure 4E). Remarkably, we were able to score GFP signal transients along tdTomato+ MSN dendrites within the central synaptic channel (Figures 4F–4I). These results suggest that DA pre-synaptic terminals are functionally connected with the post-synaptic compartments of striatal MSNs and able to elicit functional activation of G protein-coupled DA receptors.
Figure 4.

DA-Specific Synaptic Identity and Functionality in the Human Nigro-striatal Pathway
(A) Illustration of the protocol used for the FFN206 dye staining. DANs are infected with lentiviruses (LVs) expressing GFP under the control of the Synapsin promoter. FFN206 dye is added directly into the synaptic chamber.
(B–D) Representative images of FFN206-positive puncta localized on DA axonal branches.
(E) Schematics of the experimental setup to visualize dLight1 transients in MSN dendrites in the synaptic channel.
(F) GFP immunofluorescence to highlight dLight1-expressing MSNs.
(G–I) Representative image of dLight1 transients (arrows in H) captured along MSN dendrites (G and H) and relative quantification of the signal intensity over time (I); n = 3 independent experiments.
Scale bars, 100 μm.
PD-OPA1 DA Axonal Projections Display a Strong Loss of Mitochondria with Altered Morphology and Reduced Mobility
The integration of the 500-μm-long microchannel array on the pre-synaptic side makes it possible to stabilize only the axonal projections, which can extend to this distance to reach the central chamber and establish synaptic contacts. Thus, we took advantage of this design to better define the consequences of OPA1 mutations on the mitochondrial network along the axons. To this aim, the A495V mutant, gene-complemented PD-OPA1 and control iPSCs were differentiated in either DANs or MSNs, plated in the microdevices, and matured for 4 weeks. Next, we evaluated the morphology and dynamics of the mitochondrial network specifically in DA axonal compartments, focusing on the distal part of the projections close to the synaptic chamber. Triple TOMM20/TH/βIII-tubulin immunofluorescence highlighted the mitochondrial content along the TH+ axons, showing a marked loss of mitochondria in this compartment of the mutant patient compared with gene-corrected and control neurons (Figures 5A and 5D). A similar reduction was obtained visualizing the fraction of active mitochondria with a functional membrane potential highlighted with the MitoTracker orange live staining (Figure 5B). In a closer inspection, we noted the occasional accumulation and aberrant clustering of mitochondria exclusively in the mutant axons (Figures 5C and 5D). To assess the mitochondrial dynamics along axons, we established a time-lapse video recording on axonal projections of neurons transduced with lentiviral particles expressing Mito-dsRed in the pre-synaptic chamber (Figure 5E). Although the PDMS material raised some intrinsic fluorescence background, this did not prevent a detailed imaging of mitochondria over time. Interestingly, video recording analysis showed that movements of mutant mitochondria were significantly reduced with an increasing number of stalled Mito-dsRed+ organelles along the axons (Figures 5F and 5G; Videos S1 and S2). Median speed and traveled distance were particularly affected in the anterograde direction indicating an impairment of mutant mitochondria to move effectively toward the synapses. In summary, these results demonstrate that this microfluidic device offers a convenient setting for performing extended video imaging of sub-cellular organelles focusing on selected neuronal compartments. This setting enabled us to reveal that in OPA1-mutant DA neuronal axons, the mitochondria are severely reduced in number and exhibit poor motility in particular along the anterograde direction to reach the synaptic terminals.
Figure 5.

PD-OPA1 DA Axons Show Loss of Mitochondria with Impaired Dynamics
(A) Representative images of mitochondrial morphology stained with TOMM20 (red) in control, mutant, and gene-complemented PD-OPA1 TH+/βIII-tubulin (βIII-Tub)-positive DA axons.
(B–D) Visualization and quantification of MitoTracker orange live signal and HSP60 immunofluorescence confirming the reduction and aberrant clustering of mitochondria in PD-OPA1 axons (arrows in C).
(E) Illustration of the microdevice infected with the Mito-dsRed expressing lentivirus to visualize mitochondria in DA axons.
(F and G) Representative kymographs (F) and kinetics analysis (G) show an impairment of PD-OPA1 mutant mitochondria trafficking along the DA axonal projections.
Data are mean ± SEM; n = 3 or 4 independent experiments. ∗p < 0.05 and ∗∗∗p < 0.001. Statistical analysis was performed using one-way ANOVA, followed by Tukey post-test or Student’s t test. Scale bars, 10 μm.
A significant fraction of PD-OPA1 mutant mitochondria are stalled or show less mobility along the DA axonal terminals.
PD-OPA1 DA Neurons Exhibit a Reduced Number of Active Synapses in the Nigro-striatal Circuitry
Given the evident loss of mitochondria in the OPA1-mutant DA neuronal axons, we asked whether this defect could impair the formation and maintenance of functional synapses. This question is extremely difficult to answer using mass neuronal cultures because it remains hard to specifically trace and quantify the DA pre-synaptic terminals in such a condition. In contrast, this microdevice system conveys all the DA axons within the central chamber, which acts as a unique container for all the synapses, allowing a precise quantitative assessment. Thus, PD-OPA1 and control DANs and MSNs were cultured in microdevices and accurate quantification of synapses was obtained by counting the number of SYN or Homer1 positive dots at 3 and 10 weeks after initial plating. These measurements were performed only in areas of the central chamber in which all the associated microchannel contained a neurite on both the DAN and MSN side. At the earliest time point, PD-OPA1 neurons exhibited a 30% reduction in either type of synaptic puncta compared with control cultures (Figures 6A–6M). This loss of synaptic staining was not found in the gene-complemented PD-OPA1 neurons at the same time point, indicating OPA1 loss as the direct cause for the synapse damage. These results suggest that early establishment of neuronal connectivity is already affected by reduced OPA1 levels. Then, we assessed the long-term maintenance of synaptic contacts at 10 weeks in PD-OPA1 and control DANs and MSNs. Remarkably, comparing this late with the early time point, we scored an increased loss of synaptic puncta within the PD-OPA1 mutant neurons that could be reverted by reintroduction of OPA1 (Figures 6A–6M). These data strongly suggest that PD-OPA1 neuronal cultures suffered for a progressive loss of synaptic staining that persisted over a long period of time in culture. To further corroborate these findings, we performed FFN206 staining in neuronal cultures maintained for 10 weeks in the microdevices. Notably, the total number of FFN206-positive dots in the synaptic chamber was reduced by 40% in PD-OPA1 compared with control and gene-corrected neuronal cultures (Figures 6N–6W). Altogether, these results showed that PD-OPA1 neuronal cultures on-chip exhibited an early impairment in synapse formation that was further worsened by the progressive loss of synaptic contacts occurring over a long period of culture in vitro.
Figure 6.

PD-OPA1 DANs Show an Increasing Loss of Synaptic Terminals over Time in Culture
(A–L) Representative images of Synapsin and Homer immunostaining for visualizing synaptic puncta in microdevices seeded with control (A–C), mutant (D–I), and gene-complemented (J–L) PD-OPA1 neurons.
(M) Quantification of the Synapsin and Homer staining show a time-dependent reduction of synaptic terminals in PD-OPA1 mutant neurons.
(N–W) Representative pictures (N–V) and quantification (W) of FFN206+ signal confirming a significant reduction of DA active pre-synaptic terminals.
Data are mean ± SEM; n = 3 independent experiments. ∗p < 0.05 and ∗∗p < 0.01. Statistical analysis was performed using one-way ANOVA, followed by Tukey post-test. Scale bars, 100 μm.
Discussion
The brain is a highly organized organ in which defined neuronal circuitries represent the elementary functional units that control sensory information and drive behavior. However, physiological connectivity is not retained in conventional mass neuronal cultures normally derived from iPSC in vitro differentiation, thus resulting in a disordered network of neurons. This represents a barrier for a better understanding of disease-specific dysfunctions restricted to specific neuronal systems. This is the case for PD in which the selective vulnerability and degeneration of the nigro-striatal pathway are responsible for the motor behavior abnormalities. The lack of a faithful in vitro model of this neuronal network has certainly hindered the comprehension of the PD-associated pathophysiological processes. Herein, we have developed a robust system combining microfluidics, microfabrication, and iPSC differentiation procedures in which the nigro-striatal pathway can be established and maintained both with healthy and diseased human neurons. We defined the conditions to plate, differentiate, and maintain human neurons in long-term cultures within a microfluidic environment. In addition, we established a procedure to culture two independent neuronal populations on the same chip and promote the formation of lasting and functional connections. iPSC-derived DANs and MSNs in long-term cultures on the chip maintained healthy and stereotyped morphology and expressed multiple key markers of their neuronal subtype identity. However, future single-cell gene expression analyses are needed to define if other minor neuronal populations can be differentiated from iPSCs with these protocols and establish confounding connectivities on the chip. In addition, transcriptome studies will be also helpful to determine whether the microfluidic environment can alter particular molecular pathways or limit specific metabolic processes in these neuronal cultures.
Nonetheless, we showed that iPSC-derived DANs and MSNs can form stable synapses with active pre-synaptic terminals that can activate post-synaptic DA receptors on MSNs. The establishment of this neuronal circuit uncovers multiple opportunities for basic biology and disease-relevant studies. In particular, the spatial and fluidic isolation of the synaptic compartment obtained with this system offers an advantageous setting to evaluate the effects of toxins, inflammatory signals, and other chemicals on human synaptic function. Additionally, the detrimental effects of aggregated α-synuclein and β-amyloid toxic species on synaptic activity described in murine models can be further addressed in this human culture system (Brahic et al., 2016, Cavaliere et al., 2017).
We herein exploited this platform for assessing the early effects on human neurons caused by OPA1 mutations associated with a genetic form of PD. Interestingly, axons of PD-OPA1 DANs exhibited a robust loss of mitochondria, with the few remaining displaying an abnormal morphology and reduced mobility. These defects are likely responsible for a marked impoverishment of mitochondria on pre-synaptic terminals, which are crucial for generating local energy for promoting synaptic vesicle trafficking. In line with this effect, quantification of either synaptic puncta or FFN206-positive axonal terminals confirmed a reduced number of synapses that further diminished in long-term cultures. These data cannot be influenced by the relative survival of the neuron and their soma in the lateral chambers, as quantifications were performed exclusively in areas of the synaptic channel in which all connected microgrooves contained neuronal projections on both the DAN and MSN side of the microdevice. These results provide strong evidence that synaptic degeneration is an early event triggered by OPA1-dependent mitochondrial dysfunctions (Figure 7) and corroborate recent studies in PD animal models and patients. In fact, mouse mutants for genes critically involved in mitochondrial dynamics or oxidative phosphorylation might develop neurodegeneration which starts with the loss of synapses and axons before it progresses to degeneration of the cell bodies (Berthet et al., 2014, Kayser et al., 2016). More broadly, loss of synaptic terminals might be an early pathophysiological process shared between sporadic cases of PD. In fact, postmortem studies in human PD brains have shown that the degree of neurodegenerative changes of the DA axonal terminals is more severe than in the soma residing in the substantia nigra (Kordower et al., 2013). In addition, high-resolution positron emission tomographic (PET) imaging for the DA transporter (DAT) capable of visualizing and quantifying DAT in the whole nigro-striatal pathway indicated that in the early stage of PD, DAT loss is markedly increased in the striatal terminals rather than in cell bodies in the midbrain (Fazio et al., 2018). Altogether these findings support the view that damage of synaptic terminals in the caudo-putamen is an early event in PD and likely challenges the subsequent survival of neurons during disease progression. Future studies with this microfluidic system will be focused to evaluate whether neurons derived by iPSCs from idiopathic PD patients will exhibit similar synaptic instability and dissect the underlying causes.
Figure 7.

OPA1 Mutations Associated with PD Cause an Early Loss of DA Synapses Mediated by Mitochondria Dysfunctions
Illustration depicting mitochondrial morphology and dynamics in healthy and OPA1-mutant DANs and their axonal projections. PD-OPA1-mutant DANs show a significant loss of mitochondria along axonal projections with altered morphology and impaired dynamics. These deficits cause a significant loss of mitochondria on the DA pre-synaptic terminals, which causes a lack of local bioenergetics, leading to progressive synaptic degeneration.
In the present work, we showed that the VMAT2 ligand FFN206 can enable fast and precise visualization of monoaminergic synapses, providing a straightforward approach to quantify synapses in the central chamber of the microdevices. This approach is suitable for pharmacological testing and small-molecule screening campaigns to identify compounds capable of protecting synaptic function either in PD neurons or following exposure to toxins.
In summary, we established long-term cultures of human neurons in a microfluidic platform with a design that enables the reconstitution of the nigro-striatal pathway and the formation of functional DA synapses. The generation of functional in vitro circuitries with defined post- and pre-synaptic neuronal elements provides an accurate and sophisticated system to investigate both physiological and pathological processes. The high versatility of the different microfabrication modalities has prompted the generation of microdevices with a large variety of shapes and complexities. We anticipate that further refinement of these technologies will lead to more elaborated designs to accommodate multiple neuronal populations for faithful and systematic modeling of complex brain networks.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TH | Immunological Sciences | AB-10312 |
| Mouse monoclonal anti-MAP2 | Immunological Sciences | MAB-10334 |
| Rabbit polyclonal anti-TOMM20 | Novus | NBP1-81556 |
| Chicken polyclonal anti-GFP | ThermoFisher | A10262 |
| Rabbit polyclonal anti-DARP-32 | Millipore | AB10518 |
| Rabbit polyclonal anti-Homer1 | Synaptic Systems | 160-003 |
| Mouse monoclonal anti-Synapsin1 | Synaptic Systems | 106001 |
| Rabbit polyclonal anti-bIII-Tub | Covance | PRB-435P |
| Rabbit polyclonal anti-GABA | Sigma-Aldrich | A2052 |
| Rat monoclonal anti-CTIP2 | Abcam | AB18465 |
| Rabbit polyclonal anti-FOXP1 | Abcam | AB16645 |
| Mouse monoclonal anti-NESTIN | Millipore | MAB5326 |
| Rabbit recombinant monoclonal anti-FOXA2 | Abcam | AB108422 |
| Rabbit polyclonal anti-VMAT2 | Abcam | AB70808 |
| Rabbit polyclonal anti-DDC | Novus | NBP1-56918 |
| Mouse monoclonal anti-SMI-312 | BioLegend | 837904 |
| Bacterial and Virus Strains | ||
| Rabies virus | Viral Core facility of the Charitè Berlin University | N/A |
| Critical Commercial Assays | ||
| MitoTracker Orange CMTMRos | ThermoFisher | M7510 |
| FFN206, fluorescent VMAT2 substrate | Abcam | AB144554 |
| LIVE/DEAD Viability/Cytotoxicity Kit, for mammalian cells | ThermoFisher | L3224 |
| Experimental Models: Cell Lines | ||
| ND iPSCs | Iannielli et al., 2018 | N/A |
| A495V OPA1 iPSCs | Iannielli et al., 2018 | N/A |
| G488R OPA1 iPSCs | Iannielli et al., 2018 | N/A |
| A495V+OPA1 iPSCs | Iannielli et al., 2018 | N/A |
| G488R+OPA1 iPSCs | Iannielli et al., 2018 | N/A |
| Recombinant DNA | ||
| pFUGW-tdtomato | Addgene | 22478 |
| LV-GFP | Laboratory of Luigi Naldini | N/A |
| pCMV-dLight1.1 | Addgene | 111053 |
| LV-Syn-GFP | Laboratory of Flavia Valtorta | N/A |
| pLV-mitoDsRed | Addgene | 44386 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 6 | Prism | https://www.graphpad.com/scientific-software/prism/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vania Broccoli (broccoli.vania@hsr.it). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Cell cultures
OPA1 mutant and control iPSC lines were generated as previously described (Iannielli et al., 2018). iPSC lines were maintained in feeder-free conditions in mTeSR1 (Stem Cell Technologies) and seeded in HESC qualified Matrigel (Corning)-coated 6-well plates.
Method Details
Neuronal differentiation
Medium spiny neurons (MSNs) were generated as previously described with appropriated optimization (Arber et al., 2015). For differentiation, iPSCs were plated on HESC qualified Matrigel (Corning)-coated 6-well plates in mTeSR1. When cells reached > 80% confluence, differentiation was then initiated by switching to DMEM-F12/Neurobasal media (2:1) supplemented with N2 (1:100, ThermoFischer) and retinol-free B27 (1:50, Life Technologies). For the first 9 days, cultures were supplemented with SB431542 (10 μM, Sigma-Aldrich), LDN-193189 (100 nM, Stemgent), and Dorsomorphin (200 nM, Sigma-Aldrich). During this period medium was replaced every 2-3 days. After 9 days, cells were dissociated with Accutase (Sigma-Aldrich) and plated on Matrigel-coated 6-weel plates in to DMEM-F12/Neurobasal media (2:1) supplemented with N2 and retinol-free B27 supplemented with Y27632 (10 μM, Milteny Biotec) and activin A (25 ng/ml Sigma-Aldrich). The medium was changed the following day to remove Y27632. During this period medium was replaced every 2-3 days. After 9 days, cells were dissociated with Accutase and plated on poly-L-lysine/laminin-coated 24-well plates or on microfluidic device. Human BDNF (10 ng/ml, PeproTech), human GDNF (10 ng/ml, PeproTech), DAPT (10 μM, Sigma-Aldrich) and ascorbic acid (10 μM, Sigma-Aldrich) were added from day 20 to promote neuronal maturation and survival.
Dopaminergic neurons were generated as previously described with small modifications (Kirkeby et al., 2012, Kriks et al., 2011). iPSCs were dissociated with Accutase and plated on matrigel-coated 6-well plates in mTeSR1 medium. When cells reached > 80% confluence, the medium was replaced by differentiation medium containing DMEM-F12/Neurobasal media (2:1) supplemented with N2 (Life Technologies) and retinol-free B27 (Life Technologies). For the first 9 days, cultures were supplemented with SB431542 (10 μM), Noggin (200 ng/ml), Sonic Hedgehog (50 ng/ml) and CHIR99021 (0,8 μM, Stemgent). Half medium was changed every 2-3 days. After 9 days, cells were dissociated with Accutase and plated on Matrigel-coated 6-weel plates in to DMEM-F12/Neurobasal media (2:1) supplemented with N2 and retinol-free B27 supplemented with FGF8b (100ng/ml, Sigma Aldrich). During this period medium was replaced every 2-3 days. After 9 days, cells were dissociated with Accutase and plated on poly-L-lysine/laminin-coated 24-well plates or on microfluidic device for the final maturation. BDNF (10 ng/ml), GDNF (10 ng/ml), DAPT (10 μM, Sigma-Aldrich) and ascorbic acid (10 μM, Sigma-Aldrich) were added from day 20 to promote neuronal maturation and survival.
Microfluidic device fabrication
Microfluidic devices were fabricated through soft-lithography of PDMS on master molds. The design (Figure S1) features two chambers for cultures of neuronal cell bodies (1.5mm width, 50μm height, 7mm length) connected by an array of approximately 130 microchannels for axon guidance (3μm width, 6μm height, 625μm length). A central synaptic channel (50μm width, 50μm height) is interposed asymmetrically between the culture chambers to monitor and fluidically manipulate synapses only, at a distance of 75μm and 500μm from MSNs and DANs culture channels, respectively. The design of the lateral chambers and synaptic channel was printed on a high-resolution photomask (64.000 DPI) for subsequent master mold fabrication steps. Master molds were fabricated through standard two-layer photolithography of SU-8 (Microchem, Germany) on silicon wafers. In particular, a first layer of SU8-2005 with a thickness of 6μm was exposed on a mask-less laser writer (MLA100, Heidelberg, Germany) to obtain the microgroove array alone. Subsequently, a second layer of SU-8 2050 was deposited to a thickness of 50μm and exposed through a standard mask aligner and photomask to obtain the lateral chambers and the synaptic channel. To obtain microfluidic layers, liquid PDMS was poured on silicon wafers at a pre-polymer to curing agent mixing ratio of 10:1 (w/w) and cured at 65°C for 3h. Cast PDMS was peeled-off the molds, trimmed and through-holes were punched to obtain culture channel access ports (8mm diameter) and synaptic chamber access ports (3mm diameter). Finally, PDMS microfluidic layers were plasma bonded (Harrick Plasma, USA) to glass coverslides and stored until use.
Neuronal cultures in microfluidic devices
The microfluidic devices were autoclaved and coated with a mixture of poly-D-lysin (0.1 mg/ml) in the MSN and synaptic chambers, and with a mix of poly-D-lysin (0.1 mg/ml) + laminin (10 μg/ml) in the DA chamber overnight at 4°C. The following day microfluidic devices were washed 3 times with maturation medium (DMEM-F12/Neurobasal media (2:1) supplemented with N2, retinol-free B27, BDNF, GDNF, DAPT and ascorbic acid) and placed at 37°C before neurons were plated. Dissociated MSNs and DANs were resuspended with maturation medium and 80.000 cells were plated in each of the corresponding chamber.
Constructs, plasmid, and lentiviruses
Lentiviral replication-incompetent, VSV-G-coated lentiviral particles were packaged in 293T cells. For the lentiviruses expressing tdTomato, the constuct was purchased by Addgene (#22478), while the construct expressing GFP is a kind gift of L. Naldini. For the LV-Syn-GFP, GFP coding sequence was cloned under the control of Synapsin promoter in a lentiviral vector. The dLight1.1 construct was purchased by Addgene (#111053) and cloned into a lentiviral vector downstream to the EF-1α promoter. The construct for the RV infection is a kind gift of M. Parmar (Grealish et al., 2014). Rabies viral particles were generated by the Viral Core facility of the Charitè Berlin University.
MitoTracker Orange
Neurons seeded in microfluidic device were incubated with 50 nM of MitoTracker Orange (Molecular Probes) for 30 min at 37°C, washed with PBS and acquired by confocal microscope (Leica TCS SP5, Germany). For the analysis, the images were collected using a X63/1.4 oil objective and analyzed using Mito-Morphology macro in ImageJ.
Mitochondrial morphology
Mitochondrial morphology was assessed by TOMM20 immunostaining. Cellular fluorescence images were acquired with a Nikon Eclipse Ni microscope. Images were collected using a X63/1.4 oil objective and analyzed using Mito-Morphology macro in ImageJ.
FFN206 and dLight1 stainings
Neurons plated in microfluidic device were incubated with 5 μM of FFN206 in ACSF buffer containing 140 mM NaCl, 56mM KCl, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, 40mM NaOH and 10 mM HEPES for 15 min at 37°C. Then neurons were washed using ACSF buffer with 4 mM KCl and acquired by confocal microscope (Leica TCS SP8, Germany). For the analysis, the images were collected using a X63/1.4 oil objective and analyzed using ImageJ. For dLight1 experiment, MSNs plated in microfluidic device were infected with LV-tdTomato to visualize the neurons and with LV-dLight1 one week before registration. Neurons were incubated in ACSF buffer containing 140 mM NaCl, 56 mM KCl, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, 40 mM NaOH and 10 mM HEPES during imaging. Movies were acquired by an inverted fluorescence microscope (Nikon Eclipse Ni) using a X63/1.4 oil objective and analyzed using ImageJ. We calculated spatial movies and images of ΔF/F in response to KCl stimulation as [F(t)-F’baseline]/F’baseline, with F(t) the pixel-wise fluorescence value at each time, t, and mean fluorescence in time points prior to KCl application, Fbaseline.
Live/Dead labeling
Cells plated in microfluidic device were incubated with 5 μM of Live/Dead Assay Kit (ThermoFischer) for 10 min at 37°C. Then, cells were washed with PBS and acquired by inverted fluorescence microscope (Nikon, Eclipse Ti). For the analysis, the images were collected using a X20/0.45 objective and analyzed using ImageJ.
Video recording of mitochondrial dynamics
For mitochondrial movement in microfluidic device, dopaminergic neurons were infected with Mito-dsRed one week before registration. Live-cell recordings were performed using a fully automatized inverted Nikon-Ti microscope for HCA and super-resolution (Nikon Instruments), equipped with Crest Optics Spinning Disk module (Crest-Crisel Instruments) and Andor DU-888 EM-CCD camera for fast recordings and NIS-elements v.5 for acquisition (Nikon/Lim Instrumentation). A 6-line LED excitation device (Spectra, Lumencore) was employed for bleaching and photo-toxicity minimization. Specifically, dsRed was excited with a 555nm excitation line and detected through a TxRed BP filter (Chroma) placed within the spinning disk module. Video-recording were performed over 5-minute duration with 1 s timelapse, using either 60x oil (NA 1.40) or 100x oil (NA 1.46) objectives (Nikon Instruments) and matched pinholes (70 μm) in spinning disk. A total of three biological independent experiments were acquired for each sample condition, accounting for at least n = 29 independent recordings for each analyzed condition. Following acquisition, each recording was processed for intensity equalization over time, background suppression using ROI in image dark-filed and Richardson-Lucy type of deconvolution with n = 20 iterations (all through NIS-Elements v.5).
Kymograph analysis
Kymograph of individual movie (5 minutes recording) has been generated with ImageJ software. After image deconvolution, manual trace along the midline of a single axon has been processed using Reslice {/} method for the dsRed channel. Data were collected as XY coordinates and converted into time spent (seconds) to cover a fixed distance (micrometers) by the particle.
Immunostaining
Neurons were seeded on matrigel-coated glass coverslips and they were fixed for 20 min in ice in 4% paraformaldehyde (PFA, Sigma), solution in phosphate-buffered saline (PBS, Euroclone). Then, cells were permeabilized for 30 min in blocking solution, containing 0.5% Triton X-100 (Sigma-Aldrich) and 10% donkey serum (Sigma-Aldrich), and incubated overnight at 4°C with the primary antibodies in blocking solution. Then, cells were washed with PBS and incubated for 24 h at room temperature with Hoechst and with secondary antibodies.
Neurons in the microchambers were fixed for 20 min in ice in 4% paraformaldehyde (PFA, Sigma), solution in phosphate-buffered saline (PBS, Euroclone). The fixation buffer was rinsed three times with PBS and neurons were incubated for 1h at RT with a blocking solution containing 0.5% Triton X-100 (Sigma-Aldrich) and 10% donkey serum (Sigma-Aldrich). The compartment of interest was then incubated with primary antibodies overnight at 4°C and appropriate fluorescent secondary antibodies were incubated overnight at 4°C. The immunofluorescence was maintained in PBS for a maximum of one week in the dark at 4°C.
The following antibodies were used: anti-TH (1:200, Immunological Sciences), anti-MAP2 (1:500, Immunological Sciences), anti-TOMM20 (1:300, Novus), anti-GFP (1:500, Thermo), anti-DARP-32 (1:300, Millipore), anti-Homer1 (1:500, Synaptic Systems), anti-Synapsin1 (1:500, Synaptic Systems), anti-bIII-TUBULIN (1:500, Covance), anti-GABA (1:300, Sigma-Aldrich), anti-CTIP2 (1:500, Abcam), anti-FOXP1, anti-NESTIN (1:300, Millipore), anti-FOXA2 (1:300, Abcam), anti-SMI-312 (1:500, BioLegend), anti-VMAT2 (1:500, Abcam), anti-DDC (1:500, Novus). All the secondary antibodies used for the immunofluorescence staining are Alexa Fluor™.
Quantification and Statistical Analysis
All values are expressed as mean ± SEM. Differences between means were analyzed using the Student t test or one-way analysis of variance (ANOVA) depending on the number of groups and variables in each experiment. Data were then submitted to Tukey post hoc test using GraphPad Prism software. The null hypothesis was rejected when P value was < 0.05.
Data and Code Availability
The published article includes the datasets generated and analyzed during this study. No new code was generated during this study.
Acknowledgments
We are thankful to E. Cattaneo, A. Menegon, M. Parmar, and V. Tiranti for sharing of reagents. We acknowledge D. Bonanomi and all members of the Broccoli lab for helpful discussion and critical reading of the manuscript. This work was supported by the European Research Council (AdERC 340527 and ERC-PoC 842423).
Author Contributions
A.I. performed the experiments and analyzed the data. T.C. maintained iPSC cultures. A.R. established differentiation protocols for generating DANs and MSNs. G.S.U. fabricated and validated microdevices. C.C. performed and elaborated time-lapse video microscopy. M.V. performed confocal imaging acquisition and analysis. S.B. analyzed video imaging data. M.R. designed the microdevices and contributed to elaborating the experiments. V.B. supervised, coordinated, and supported the project and wrote the paper with A.I. and M.R.
Declaration of Interests
The authors declare no competing interests.
Published: December 24, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.111.
Supplemental Information
References
- Arber C., Precious S.V., Cambray S., Risner-Janiczek J.R., Kelly C., Noakes Z., Fjodorova M., Heuer A., Ungless M.A., Rodríguez T.A. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–1386. doi: 10.1242/dev.117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet A., Margolis E.B., Zhang J., Hsieh I., Zhang J., Hnasko T.S., Ahmad J., Edwards R.H., Sesaki H., Huang E.J., Nakamura K. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 2014;34:14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahic M., Bousset L., Bieri G., Melki R., Gitler A.D. Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol. 2016;131:539–548. doi: 10.1007/s00401-016-1538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K.J. Inducing cellular aging: enabling neurodegeneration-in-a-dish. Cell Stem Cell. 2013;13:635–636. doi: 10.1016/j.stem.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K.J., Marchetto M.C., Benvenisty N., Brüstle O., Ebert A., Izpisua Belmonte J.C., Kaykas A., Lancaster M.A., Livesey F.J., McConnell M.J. Creating patient-specific neural cells for the in vitro study of brain disorders. Stem Cell Reports. 2015;5:933–945. doi: 10.1016/j.stemcr.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E.M., Luo L. Monosynaptic circuit tracing with glycoprotein-deleted rabies viruses. J. Neurosci. 2015;35:8979–8985. doi: 10.1523/JNEUROSCI.0409-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaliere F., Cerf L., Dehay B., Ramos-Gonzalez P., De Giorgi F., Bourdenx M., Bessede A., Obeso J.A., Matute C., Ichas F., Bezard E. In vitro α-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017;103:101–112. doi: 10.1016/j.nbd.2017.04.011. [DOI] [PubMed] [Google Scholar]
- Chevrollier A., Guillet V., Loiseau D., Gueguen N., de Crescenzo M.A., Verny C., Ferre M., Dollfus H., Odent S., Milea D. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann. Neurol. 2008;63:794–798. doi: 10.1002/ana.21385. [DOI] [PubMed] [Google Scholar]
- Coquinco A., Kojic L., Wen W., Wang Y.T., Jeon N.L., Milnerwood A.J., Cynader M. A microfluidic based in vitro model of synaptic competition. Mol. Cell. Neurosci. 2014;60:43–52. doi: 10.1016/j.mcn.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Fazio P., Svenningsson P., Cselényi Z., Halldin C., Farde L., Varrone A. Nigrostriatal dopamine transporter availability in early Parkinson’s disease. Mov. Disord. 2018;33:592–599. doi: 10.1002/mds.27316. [DOI] [PubMed] [Google Scholar]
- Fujimori K., Ishikawa M., Otomo A., Atsuta N., Nakamura R., Akiyama T., Hadano S., Aoki M., Saya H., Sobue G., Okano H. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018;24:1579–1589. doi: 10.1038/s41591-018-0140-5. [DOI] [PubMed] [Google Scholar]
- Giannoccaro M.P., La Morgia C., Rizzo G., Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 2017;32:346–363. doi: 10.1002/mds.26966. [DOI] [PubMed] [Google Scholar]
- Grandy R., Tomaz R.A., Vallier L. Modeling disease with human inducible pluripotent stem cells. Annu. Rev. Pathol. 2019;14:449–468. doi: 10.1146/annurev-pathol-020117-043634. [DOI] [PubMed] [Google Scholar]
- Grealish S., Diguet E., Kirkeby A., Mattsson B., Heuer A., Bramoulle Y., Van Camp N., Perrier A.L., Hantraye P., Björklund A., Parmar M. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell. 2014;15:653–665. doi: 10.1016/j.stem.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaudo S., Tixador P., Bousset L., Fenyi A., Lino P., Melki R., Peyrin J.M., Perrier A.L. Propagation of α-synuclein strains within human reconstructed neuronal network. Stem Cell Reports. 2019;12:230–244. doi: 10.1016/j.stemcr.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Fumagalli L., Prior R., Van Den Bosch L. Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 2017;11:671. doi: 10.3389/fnins.2017.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Henke A., Karpowicz R.J., Jr., Sonders M.S., Farrimond F., Edwards R., Sulzer D., Sames D. New fluorescent substrate enables quantitative and high-throughput examination of vesicular monoamine transporter 2 (VMAT2) ACS Chem. Biol. 2013;8:1947–1954. doi: 10.1021/cb400259n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannielli A., Bido S., Folladori L., Segnali A., Cancellieri C., Maresca A., Massimino L., Rubio A., Morabito G., Caporali L. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018;22:2066–2079. doi: 10.1016/j.celrep.2018.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser E.-B., Sedensky M.M., Morgan P.G. Region-specific defects of respiratory capacities in the Ndufs4(KO) mouse brain. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0148219. e0148219–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.J., Jacobs M.W., Ito-Cole T., Callaway E.M. Improved monosynaptic neural circuit tracing using engineered rabies virus glycoproteins. Cell Rep. 2016;15:692–699. doi: 10.1016/j.celrep.2016.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkeby A., Grealish S., Wolf D.A., Nelander J., Wood J., Lundblad M., Lindvall O., Parmar M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1:703–714. doi: 10.1016/j.celrep.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Kirwan P., Turner-Bridger B., Peter M., Momoh A., Arambepola D., Robinson H.P.C., Livesey F.J. Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development. 2015;142:3178–3187. doi: 10.1242/dev.123851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower J.H., Olanow C.W., Dodiya H.B., Chu Y., Beach T.G., Adler C.H., Halliday G.M., Bartus R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S., Shim J.-W., Piao J., Ganat Y.M., Wakeman D.R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L., Callaway E.M., Svoboda K. Genetic dissection of neural circuits: a decade of progress. Neuron. 2018;98:865. doi: 10.1016/j.neuron.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J., Reid D., Lau S., Kim Y., Gage F.H. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 2018;52:271–293. doi: 10.1146/annurev-genet-120417-031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto E., Leitão L., Sousa D.M., Alves C.J., Alencastre I.S., Aguiar P., Lamghari M. Compartmentalized microfluidic platforms: the unrivaled breakthrough of in vitro tools for neurobiological research. J. Neurosci. 2016;36:11573–11584. doi: 10.1523/JNEUROSCI.1748-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.W., Vahidi B., Taylor A.M., Rhee S.W., Jeon N.L. Microfluidic culture platform for neuroscience research. Nat. Protoc. 2006;1:2128–2136. doi: 10.1038/nprot.2006.316. [DOI] [PubMed] [Google Scholar]
- Patriarchi T., Cho J.R., Merten K., Howe M.W., Marley A., Xiong W.-H., Folk R.W., Broussard G.J., Liang R., Jang M.J. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science. 2018;360:eaat4422. doi: 10.1126/science.aat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K., Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584–595. doi: 10.1016/j.stem.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Inoue H., Wu J.C., Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 2017;16:115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Zhang S.C. Neural subtype specification from human pluripotent stem cells. Cell Stem Cell. 2016;19:573–586. doi: 10.1016/j.stem.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M., Blurton-Jones M., Rhee S.W., Cribbs D.H., Cotman C.W., Jeon N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M., Dieterich D.C., Ito H.T., Kim S.A., Schuman E.M. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron. 2010;66:57–68. doi: 10.1016/j.neuron.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G., Vivas E.L., Izpisúa Belmonte J.C. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat. Med. 2011;17:1570–1576. doi: 10.1038/nm.2504. [DOI] [PubMed] [Google Scholar]
- Virlogeux A., Moutaux E., Christaller W., Genoux A., Bruyère J., Fino E., Charlot B., Cazorla M., Saudou F. Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington’s disease. Cell Rep. 2018;22:110–122. doi: 10.1016/j.celrep.2017.12.013. [DOI] [PubMed] [Google Scholar]
- Wen Z., Christian K.M., Song H., Ming G.-L. Modeling psychiatric disorders with patient-derived iPSCs. Curr. Opin. Neurobiol. 2016;36:118–127. doi: 10.1016/j.conb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanna C., Ghelli A., Porcelli A.M., Karbowski M., Youle R.J., Schimpf S., Wissinger B., Pinti M., Cossarizza A., Vidoni S. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131:352–367. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A significant fraction of PD-OPA1 mutant mitochondria are stalled or show less mobility along the DA axonal terminals.
Data Availability Statement
The published article includes the datasets generated and analyzed during this study. No new code was generated during this study.