

Abstract
Background
Modelling and simulation are being increasingly utilized to support the discovery and development of new anti-malarial drugs. These approaches require reliable in vitro data for physicochemical properties, permeability, binding, intrinsic clearance and cytochrome P450 inhibition. This work was conducted to generate an in vitro data toolbox using standardized methods for a set of 45 anti-malarial drugs and to assess changes in physicochemical properties in relation to changing target product and candidate profiles.
Methods
Ionization constants were determined by potentiometric titration and partition coefficients were measured using a shake-flask method. Solubility was assessed in biorelevant media and permeability coefficients and efflux ratios were determined using Caco-2 cell monolayers. Binding to plasma and media proteins was measured using either ultracentrifugation or rapid equilibrium dialysis. Metabolic stability and cytochrome P450 inhibition were assessed using human liver microsomes. Sample analysis was conducted by LC–MS/MS.
Results
Both solubility and fraction unbound decreased, and permeability and unbound intrinsic clearance increased, with increasing Log D7.4. In general, development compounds were somewhat more lipophilic than legacy drugs. For many compounds, permeability and protein binding were challenging to assess and both required the use of experimental conditions that minimized the impact of non-specific binding. Intrinsic clearance in human liver microsomes was varied across the data set and several compounds exhibited no measurable substrate loss under the conditions used. Inhibition of cytochrome P450 enzymes was minimal for most compounds.
Conclusions
This is the first data set to describe in vitro properties for 45 legacy and development anti-malarial drugs. The studies identified several practical methodological issues common to many of the more lipophilic compounds and highlighted areas which require more work to customize experimental conditions for compounds being designed to meet the new target product profiles. The dataset will be a valuable tool for malaria researchers aiming to develop PBPK models for the prediction of human PK properties and/or drug–drug interactions. Furthermore, generation of this comprehensive data set within a single laboratory allows direct comparison of properties across a large dataset and evaluation of changing property trends that have occurred over time with changing target product and candidate profiles.
Keywords: Physiologically-based pharmacokinetic modelling, Anti-malarial drugs, Ionization constant, Partition coefficient, Biorelevant solubility, Protein binding, Blood to plasma partitioning, Microsomal stability, CYP inhibition
Background
The number of deaths due to malaria has dropped substantially in recent years, from more than 800,000 in 2000 [1] to approximately 435,000 in 2017 [2]. This reduction has been attributed in large part to the widespread use of artemisinin-based combination therapy (ACT) and insecticide-treated bed nets as well as improved vector control. However, the most recent estimates from the World Health Organization (WHO) suggest that the malaria incidence rate per 1000 population at risk has been steady at 59 for the past 3 years suggesting that progress in reducing infection has reached a standstill [2]. The factors contributing to these trends are many, including parasite resistance to existing drugs, mosquito resistance to insecticides, lack of sustained and predictable financing for malaria eradication programmes in disease endemic countries, poor performance of regional health systems and various regional conflicts [3].
Since 2000, there has been a considerable increase in anti-malarial drug discovery leading to a relatively healthy pipeline of promising new drug candidates in preclinical and clinical development [4]. Over this same time period, new drug approvals have included new artemisinin-based combinations, new combinations of other existing drugs, and new and improved formulations, each of which has contributed significantly to the anti-malarial arsenal. However there have been only two new drug approvals containing new chemical entities (Synriam, a combination of the novel ozonide arterolane or OZ277 and piperaquine, and Krintafel/Kozenis containing tafenoquine) over this same period, and of these, only tafenoquine has undergone stringent regulatory approval by International Conference on Harmonization (ICH) members or observers. This scenario reflects the relatively limited emphasis on anti-malarial drug discovery prior to about 2000, and the inevitable timeframe required to progress new compounds through discovery, translational and clinical development. The situation is further exacerbated by the need for combination therapies, preferably delivered in a single dose, to treat all parasitic forms and reduce the development of drug resistance, and the associated complexity of obtaining efficacy, safety, and pharmacokinetic data for individual agents before they are combined.
Given this landscape, it is essential that improved methods to accelerate the discovery and development of malaria drugs are implemented so that new and more convenient medicines can be made available to patients in a shorter period of time. Modelling and simulation tools have received considerable attention in recent years, are well established in the industry [5–10] and are being increasingly recognized by regulatory authorities [11–15]. These approaches are now also being applied in the discovery and development of anti-malarial drugs as recently reviewed by Andrews et al. [16]. The availability of improved preclinical models for assessing efficacy against human parasitic infections [17], as well as the establishment of volunteer infection studies (VIS) [18–21], has reduced the time required to establish preclinical and clinical proof of concept and provides a rich supply of data for the development of pharmacokinetic/pharmacodynamic models [22–25].
Fundamental to many of these modelling initiatives is the use of physiologically-based pharmacokinetic (PBPK) modelling. This predictive tool is a mechanistic whole-body distribution model that incorporates compound specific data (e.g. physicochemical, permeability, binding and clearance) along with physiological (e.g. tissue composition, volume and organ blood flow) and population specific data to simulate absorption, distribution and elimination profiles. As highlighted in a recent white paper, these methods are being increasingly recognized by the FDA for first-in-human dose selection and to predict clinical drug–drug interactions [6, 14, 26–28]. Previous reports have highlighted the need for reliable compound specific data to improve the predictability of PBPK models [6]. While there are numerous in silico methods available for predicting physicochemical properties, there are still inherent flaws in being able to accurately predict certain parameters that impact the outcome of PBPK predictions.
The current work was undertaken as part of a broad collaboration between the Medicines for Malaria Venture, the Bill & Melinda Gates Foundation and Simcyp (Certara UK Limited) to demonstrate the utility of PBPK modelling and simulation to accelerate the discovery and development of fixed-dose combinations for new anti-malarial drugs. The first stage of the project, which is the subject of this manuscript, was to generate in vitro data to support PBPK modelling, including physicochemical, permeability and binding properties, intrinsic clearance, and cytochrome P450 inhibition constants for a set of legacy anti-malarial drugs and drug candidates in preclinical and clinical development using standardized conditions. The second stage, which will be published separately, was to use the data set to build PBPK models for legacy compounds and make them available to malaria researchers. These models will be used for different applications such as simulations of drug–drug interactions of new combinations containing legacy compounds. The final stage, which is still on-going, is to implement the PBPK methodology into candidate selection and clinical development of new anti-malarial drug combinations. This manuscript reports the in vitro data set generated for a total of 45 compounds of which 23 are legacy drugs, 2 are active metabolites, and 20 are preclinical and clinical development compounds (including 2 recently introduced new drugs) with details of the methodology used to obtain these data.
Methods
Materials
All compounds were obtained from the Medicines for Malaria Venture, Geneva, Switzerland. Structures, salt forms and current development status for all compounds in the data set are shown in Additional file 1: Table S1. The data set includes 20 development compounds that are either in preclinical or clinical development or have been recently approved (OZ277 or Arterolane and tafenoquine), 23 legacy compounds that are currently used clinically or have been used in the past, and two active metabolites (desethylamodiaquine and cycloguanil). The launched drug list was obtained from the public database ChEMBL (https://www.ebi.ac.uk/chembl/) and included 274 oral drugs launched between 2000 and 2017 excluding enzymes, oligopeptides, polymers, buffering agents, and amino acids, and drugs that have been withdrawn or discontinued.
Molecular property descriptors
Molecular property descriptors were calculated using ChemAxon JChem for Excel version 18.5.0.196 (ChemAxon, Budapest, Hungary). For the ChEMBL oral drug set, SMILES strings were used to calculate the molecular property descriptors using ChemAxon.
Instrumentation and sample analysis
Sample analysis was conducted by LC–MS/MS using a Waters Acquity UPLC system (Waters Corporation, Milford, MA) coupled to either a triple quadrupole mass spectrometer (Waters Micromass Quattro Premier, Waters Micromass Quattro Ultima PT, Waters Xevo TQ, or Waters Xevo TQD) for quantitative analysis or a time of flight mass spectrometer (Waters Xevo G2 QToF) for the assessment of metabolism. For samples where concentrations were high (e.g. some of the partitioning and solubility samples), detection was conducted by UV absorption rather than MS/MS. Details of the sample preparation procedures are provided within each of the individual method sections. In all cases, quantitation was conducted by comparison of the sample response (peak area ratio using diazepam as an internal standard) to the response for a set of calibration standards prepared in the same matrix, bracketing the expected concentration range and analysed at the same time as the study samples. Representative analytical conditions are shown in Additional file 1: Table S2 with typical validation data shown in Additional file 1: Table S3.
Ionization constants
Ionization constants were calculated using in silico methods and measured experimentally. In silico methods included the ADMET Predictor module embedded within the PBPK software package, GastroPlus, ver. 9.6 (Simulations Plus, Inc, Lancaster, CA) and ChemAxon JChem for Excel. Calculated values from the public database ChEMBL (https://www.ebi.ac.uk/chembl/, ACD Labs ver. 12.01) were included for compounds available within the ChEMBL database.
Ionization constants were measured by potentiometric titration using a Metrohm 809 Titrando autotitrator (Metrohm AG, Switzerland) equipped with an 800 Dosino burette (2 mL), an 800 stirring unit and a jacketed reaction vessel capable of titrating volumes between 2 and 10 mL. The autotitrator was controlled by Tiamo software (Version 1.3). pH measurements were conducted using a Metrohm LL Micro glass electrode which was calibrated on the day of use with calibration standards at pH 2, 4, 7 and 10. All reagents were standardized (directly or indirectly) against potassium hydrogen phthalate (Sigma-Aldrich, A.C.S. Acidimetric Standard). Titrant solutions were protected from carbon dioxide absorption by flushing with nitrogen before sealing or by the incorporation of a drying tube filled with self-indicating soda lime into the titration reaction vessel set-up.
A stock solution of each compound was prepared in DMSO typically at a concentration of 5 mM. Aliquots were introduced directly into the titration vessel and diluted 1:10 with water (typical final compound concentration of 0.5 mM). Titrations were performed in triplicate with standardized hydrochloric acid or potassium hydroxide (10 mM) and titrant volume increments of 1 µL, resulting in a minimum of 100 data points for each titration. pKa values were obtained by fitting the data to the Henderson–Hasselbalch equation [29] and averaging the results about the 0.5 equivalent point (first pKa) and the 1.5 equivalent point (second pKa where present) of the titration.
Partition coefficients
Partition coefficients were calculated using in silico methods and measured experimentally and. In silico methods included ADMET Predictor and ChemAxon. Calculated values from the public database ChEMBL (https://www.ebi.ac.uk/chembl/, ACD Labs ver. 12.01) were included for compounds available in the ChEMBL database.
Partition coefficients between octanol and pH 7.4 buffer were measured using a shake flask method. A stock solution of test compound in octanol was prepared at a concentration between 3 and 30 mg/mL based on the expected partition coefficient value. This stock solution was then diluted 3- and 10-fold with octanol and used to prepare the octanol phase for the partitioning experiments. Two different dilutions were used to confirm that there were no saturation effects. Phosphate buffered saline was prepared by combining 67 mM disodium hydrogen orthophosphate and sodium dihydrogen orthophosphate (both prepared in 43 mM NaCl) to a final pH of 7.4.
Partitioning experiments were conducted by mixing equal volumes of the octanol (containing test compound) and aqueous phases and placing on a vibrating plate mixer in an incubator at 37 °C. At 24 and 48 h, the samples were centrifuged (10,000 rpm × 3 min) and duplicate aliquots of the octanol phase removed and diluted first with isopropanol (1:9) and then with 50–80% aqueous methanol depending on the compound properties. An aliquot of the aqueous phase was carefully removed and centrifuged again to ensure no contamination from the octanol phase before sampling in duplicate and diluting with aqueous methanol for analysis. Diluted samples were analysed by LC–MS along with calibration standards (Additional file 1: Table S2) and partition coefficients were calculated from the ratio of the mean octanol to buffer concentration after accounting for the dilution factors. The partitioning results for the two time points were used to confirm that the partitioning experiment had reached equilibrium.
Solubility in biorelevant media
Solubility of each active pharmaceutical ingredient was evaluated at 37 °C in pH 7.4 phosphate buffered saline (prepared as described for the partitioning experiments), fasted (FaSSIF-V2) and fed (FeSSIF-V2) state simulated intestinal fluids and fasted state simulated gastric fluid (FaSSGF) as described by Jantratid et al. [30]. Compounds were accurately weighed into individual screw cap polypropylene tubes and media added to give a nominal target compound concentration of 2 mg/mL (maximum concentration tested for most compounds). Samples were vortexed and placed in a 37 °C incubator on an orbital mixer (IKA® VXR basic Vibrax® orbital mixer) set at 600 rpm. Sampling times were 1 h for FaSSGF or 5–6 h for FaSSIF-V2, FeSSIF-V2, and PBS. These times were used to reflect the maximal likely residence times within the stomach and small intestine, respectively. Sampling was carried out by centrifuging each sample at 10,000 rpm for 3 min, transferring 300 µL aliquots into fresh Eppendorf tubes and centrifuging these tubes again at 10,000 rpm for 3 min. Triplicate aliquots of the supernatant were then removed and diluted 1:2 in 50% aqueous methanol and then again in 50% aqueous acetonitrile to be within the analytical concentration range. Samples were analysed by LC–MS along with calibration standards (Additional file 1: Table S2).
Permeability
Bidirectional permeability was assessed across Caco-2 cell monolayers as described previously [31]. Briefly, permeability experiments were performed using either aqueous transport buffer (pH 7.4 Hanks balanced salt solution containing 20 mM HEPES) or human plasma (Australian Red Cross Blood Service) in both the apical and basolateral chambers. Donor solutions were prepared by spiking stock solutions into transport media to give a final compound concentration in the range of 10–20 µM (using buffer as the transport medium) or 10–50 µM (using plasma as the transport medium; note that the unbound donor concentration will vary depending on the fraction unbound). The final DMSO concentration in the donor solution was 0.1% v/v. Donor solutions were equilibrated at 37 °C for up to 4 h before centrifuging at 4000 rpm for 5 min to remove any compound that may have precipitated.
Compound flux was assessed over a maximum period of 90–180 min, with samples taken periodically from the acceptor chamber. Samples from the donor chamber were taken at the start and end of the experiment. Donor and acceptor samples for lucifer yellow and rhodamine 123 were analysed by fluorescence (FLUOstar OPTIMA plate reader; BMG Lab Technologies, Offenburg, Germany) with the excitation/emission wavelengths set at 430/535 nm for lucifer yellow and 500/525 nm for rhodamine 123. Donor and acceptor samples were stored frozen at − 80 °C until analysis by LC–MS (Additional file 1: Table S2) with sample preparation as described previously [31]. The mass balance and apparent permeability coefficient (Papp) were calculated as previously described [31].
Where human plasma was used as the transport medium, Papp values were calculated as shown above, with correction for the fraction unbound (fu) in the donor solution with fu determined at a similar concentration to that used in the transport experiment. The apparent flux of lucifer yellow was based on an endpoint measurement assuming no lag time. The efflux ratio was calculated as the ratio of the mean B–A to A–B Papp values.
Solubility limited absorbable dose calculations
The solubility limited absorbable dose (SLAD) was calculated as previously described [32] using Eq. (1):
| 1 |
where Ssi is the estimated solubility in the small intestine (based on the measured FaSSIF solubility), V is the fluid volume (500 mL), Mp is the permeability multiplier (equivalent to the absorption number (An = Peff × tres/R, where Peff is the predicted effective human jejunal permeability, tres is the mean residence time in the small intestine (3.32 h [32]), and R is the radius of the small intestine (1 cm) [33])) with a minimum value of 1 for poorly permeable compounds. Predicted Peff values were obtained from a calibration plot of literature Peff values [34, 35] vs measured Caco-2 Papp [31] using either buffer or plasma as the transport medium (see Results section). The maximum value for Caco-2 Papp was conservatively taken to be 3 × 10−4 cm/s giving a maximum value for Peff of ~ 1 × 10−3 cm/s which is consistent with previous reports [33, 35].
In vitro protein binding
Media sources
Pooled human plasma (n = 3–4 donors) was obtained by centrifugation of blood (collected by the Australian Red Cross Blood Service, Melbourne, Australia or the Volunteer Blood Donor Registry, Clinical Translation Centre, Walter & Eliza Hall Institute of Medical Research, Parkville, Australia), or sourcing pooled plasma directly from commercial sources (Innovative Research Inc, MI) and stored frozen at − 80 °C. On the day of the experiment, frozen plasma was thawed and either neat or diluted plasma aliquots were spiked with a compound stock solution (prepared in 20/40/40 (v/v) DMSO/acetonitrile/water) to give a final nominal concentration of 1000–2000 ng/mL and maximum final DMSO and acetonitrile concentrations of 0.2% (v/v) and 0.4% (v/v), respectively.
A suspension of human liver microsomes (HLM, XenoTech LLC, Lenexa, KS, USA) was prepared in 0.1 M phosphate buffer (pH 7.4) at a protein concentration of 0.4 mg/mL immediately prior to the experiment. An aliquot of the HLM matrix was spiked with compound stock solution as described above to give a final concentration of 0.5–1 µM with final DMSO and acetonitrile concentrations of 0.004% (v/v) and 0.1% (v/v), respectively.
Albumax medium was prepared as per the manufacturer’s instructions and contained Albumax II (lipid rich bovine serum albumin; 5.0 g/L, Gibco, Thermo Fisher Scientific), RPMI 1640 powder (Gibco; 1 sachet or 10.4 g/L; contains l-glutamine 0.3 g/L and sodium bicarbonate 2.1 g/L), HEPES (5.94 g/L) and neomycin (100 mg/L). An aliquot of Albumax medium was spiked with a compound stock solution as described above to give a final concentration of 500 ng/mL with final DMSO and acetonitrile concentrations of 0.2% (v/v) and 0.4% (v/v), respectively.
Dulbecco’s Modified Eagle’s Medium (DMEM) containing GlutaMAX-I was purchased from Invitrogen (Thermo Fisher Scientific) and stored at 4 °C. Medium was prepared by adding heat inactivated foetal calf serum (FCS, final 10% v/v), penicillin (final 100 U/mL), streptomycin (final 100 µg/mL) d-glucose (final 4.0 mg/mL) and sodium pyruvate (final 0.1 mg/mL). Aliquots of medium were spiked with compound stock solutions as described above to give a final concentration of 1000 ng/mL and maximum final DMSO and acetonitrile concentrations of 0.2% (v/v) and 0.4% (v/v), respectively.
Protein binding via ultracentrifugation (UC)
An ultracentrifugation method adapted from a previous publication [36] was initially used to assess plasma protein binding and binding in the other media. Spiked plasma, Albumax or DMEM/FCS medium was vortex mixed briefly and aliquots (n = 3–4) transferred to ultracentrifuge tubes which were allowed to equilibrate for 30–45 min at 37 °C in an atmosphere of 5% (for plasma or Albumax) or 10% (for DMEM/FCS) CO2 before being transferred to a rotor (Beckman Rotor type 42.2 Ti; 223,000×g). The rotor was maintained for a further 15 min under the same CO2 atmosphere and the pH was confirmed to be within pH 7.4 ± 0.1 before the rotor was sealed and subjected to ultracentrifugation at 37 °C for 4.2 h. For microsomes, samples were equilibrated for 30–45 min at 37 °C under ambient atmosphere since microsomes are suspended in phosphate buffer and, therefore, not subject to the same pH shifts as for the other bicarbonate buffered media and plasma. Additional ultracentrifuge tubes containing spiked matrix were maintained at 37 °C, 5% or 10% CO2 or normal atmosphere conditions, with aliquots being taken within 0.5 h of the start and at the end of ultracentrifugation to serve as controls for the assessment of stability and to obtain a measure of the total concentration (Ctotal). Following ultracentrifugation, the pH was checked and an aliquot of protein-free supernate was taken from each ultracentrifuge tube for determination of the unbound concentration (Cunbound).
Total matrix and protein free samples were analysed using a matrix matching approach [37] whereby each sample was mixed in a 1:1 ratio with the opposite blank medium (i.e. blank total matrix or blank protein free buffer). For example, plasma samples were mixed with blank pH 7.4 buffer whereas plasma supernatant samples were mixed with blank plasma. Each of the sample sets were then assayed against a common calibration curve prepared in a 1:1 mixture of total matrix and protein free pH 7.4 buffer. All samples were stored at − 80 °C until analysis by LC–MS (Additional file 1: Table S2). The unbound fraction in plasma or medium was calculated using the average values for Ctotal and Cunbound (n = 3–4 for each). The standard deviation for fu was calculated using the propagation of errors approach as described previously [38]. The potential for compound degradation was assessed by comparing the average value for Ctotal at the start and end of the experiment.
Protein binding via rapid equilibrium dialysis (RED)
For compounds that were found to have lower fu values (nominally fu < 0.1) by ultracentrifugation, binding was further assessed by RED using diluted plasma. Plasma was diluted 1:10 with pH 7.4 phosphate buffer (prepared by mixing 0.1 M sodium dihydrogen phosphate and 0.1 M disodium hydrogen phosphate (both containing 0.04 M NaCl) to pH 7.4) and spiked with compound to achieve a total measured concentration of ~ 1000–3000 ng/mL. Diluted plasma was vortex mixed briefly and aliquots (n = 3–4) were transferred to RED (Thermo Fisher Scientific, Waltham, MA) units that were placed at 37 °C under ambient atmosphere on a plate shaker. The pH of the diluted plasma was confirmed to be within pH 7.4 ± 0.1, and dialysis was conducted for a period of 6 or 24 h (see further details for the 24 h conditions below). At the end of the dialysis period, samples were removed from both the donor and dialysate chambers of the RED units. Validation experiments confirmed that the pH of 10% plasma and dialysate at the end of the experiment were each within 7.4 ± 0.1. Samples were matrix matched as described above and stored at − 80 °C until analysis by LC–MS (Additional file 1: Table S2). Stability was confirmed as for the UC assay. Fraction unbound values were calculated for each individual RED unit and the mean and SD calculated for n = 3–4 replicates.
For compounds that were very highly bound (fu < 0.01) in plasma and highly lipophilic (Log D ≥ 3.5) with the potential for loss due to adsorption to the dialysis units and slow equilibration, additional measures were incorporated to ensure that the system was at steady state [39, 40]. These measures included (i) incorporating a presaturation period to saturate non-specific binding sites on the RED chamber and dialysis membrane prior to dialysis, (ii) adding unbound compound to the dialysate chamber at the start of the dialysis period to accelerate the attainment of steady state, and (iii) using a 24 h dialysis period. Briefly, the RED device was exposed for two 30 min periods and one overnight period to fresh solutions of compound prepared in pH 7.4 buffer at approximately 10% of the total dialysis concentration. Following the preincubations, solutions were removed from the RED device and discarded. To initiate the dialysis, spiked diluted plasma was added to the donor chamber and pH 7.4 phosphate buffer spiked with compound (at 1–2% of the total diluted plasma concentration) was added to dialysate chamber and dialysis allowed to proceed for 24 h at 37 °C under ambient atmosphere on a plate shaker. Samples were removed and analysed as described above.
For binding assessments using 10% plasma, the unbound fraction (fu) in neat plasma was calculated using the average values for Ctotal and Cunbound and Eq. (2), where D is the dilution factor [41]:
| 2 |
Blood to plasma partitioning
Human whole blood was collected and supplied by the Volunteer Blood Donor Registry (Clinical Translation Centre, Walter & Eliza Hall Institute of Medical Research, Parkville, Australia) and used on the day of collection. The haematocrit (Hct) was determined by centrifugation (13,000×g for 3 min using Clemets® Microhaematocrit centrifuge and Safecap® Plain Self-sealing Mylar wrapped capillary tubes) to ensure it was between 0.40 and 0.48. An aliquot was centrifuged (Heraeus, Multifuge 3 S-R; 4500×g) for 10 min to obtain plasma required for matrix matching purposes as described below.
Aliquots of whole blood were spiked with compound stock solutions (prepared in 20/40/40 (v/v) DMSO/acetonitrile/water) to give a final nominal concentration of 1000 ng/mL with final DMSO and acetonitrile concentrations of 0.2% (v/v) and 0.4% (v/v), respectively. Two aliquots of the spiked whole blood were transferred to fresh microcentrifuge tubes and maintained at 37 °C/5% CO2 in a humidified incubator. The pH was confirmed to be 7.4 ± 0.1 at the start and end of the incubation. At each time point (30 min and 4 h), one whole blood tube was removed from the incubator and mixed by gentle inversion, after which four replicate blood samples were taken and matrix matched with an equal volume of blank plasma. The remainder of the blood sample was centrifuged (Eppendorf, Mini Spin plus; 6700×g) for 2 min for the collection of 4 replicate plasma samples which were similarly matrix matched with an equal volume of blank whole blood. The 1:1 mixtures of blood/plasma were mixed, snap frozen in dry ice and stored at − 80 °C until analysis by LC–MS (Additional file 1: Table S2) against calibration standards prepared in the same mixed matrix. Any further distribution of compound into RBCs at this stage was irrelevant as the cells were lysed during the sample preparation and the total concentration in the mixed matrix was measured for both the calibration standards and samples.
Compound stability in whole blood was assessed by comparing the compound concentrations measured at 30 and 240 min. The apparent whole blood-to-plasma partitioning ratio (B/P) was calculated as the ratio of the average concentration in the blood sample to that in the plasma fraction of the same whole blood sample. A standard deviation (SD) for each B/P value was calculated using the propagation of errors approach as described previously [38].
In vitro metabolism in human liver microsomes
The metabolic stability assay was adapted from a previously published method [42]. Test compound spiking solutions (prepared in 5/95 DMSO/acetonitrile) were added to in duplicate to a suspension of human liver microsomes (0.4–0.5 mg/mL) prepared in 0.1 M phosphate buffer (pH 7.4) containing 1 U/mL glucose-6-phosphate dehydrogenase to give a final concentration of 1 µM for all compounds except JPC3210 and MMV052 which were run at 0.5 µM. Mixtures were equilibrated briefly (~ 5–10 min) at 37 °C. The metabolic reaction was initiated by the addition of an NADPH-regenerating system to give final concentrations of 1.3 mM NADP, 3.5 mM glucose-6-phosphate, and 3.3 mM MgCl2. Reactions were quenched at 2, 5, 15, 30 and 60 min by the addition of acetonitrile containing 150 ng/mL diazepam as internal standard. Control samples (containing no cofactor) were included (quenched at 2, 30 and 60 min) to monitor degradation in the absence of cofactor. Concentrations were determined by LC–MS (Additional file 1: Table S2) by comparison to the response for a single point calibration standard prepared in quenched microsomal matrix.
Test compound concentration versus time data were fit using an exponential decay function to determine the first-order rate constant for substrate depletion. Where deviation from first-order kinetics was evident, only the initial linear portion of the logarithmic profile was utilized to determine the initial degradation rate constant (k, min−1). Each substrate depletion rate constant was then used to calculate the in vitro intrinsic clearance value (CLint, in vitro, µL/min/mg protein) using Eq. (3).
| 3 |
The limit of sensitivity of this assay was considered to be 15% loss of substrate over the assay duration. For compounds showing < 15% loss over 60 min, intrinsic clearance is quoted as < 7 µL/min/mg protein. Unbound in vitro CLint values were obtained by dividing the measured CLint by the measured fu in microsomes.
Cytochrome P450 inhibition
The CYP inhibition assay was based on a previous publication with minor modifications [43]. The method uses human liver microsomes and a substrate-specific interaction approach which relies on the formation of a metabolite that is mediated by a specific CYP isoform. The specific CYP-mediated metabolic pathways, substrates, substrate Km values, positive control inhibitors and specific incubation conditions are shown in Additional file 1: Table S4. Multiple concentrations of each test compound (0.25 to 20 µM) or positive control inhibitor along with each substrate were added to a suspension of human liver microsomes in 0.1 M phosphate buffer (pH 7.4) containing 1 U/mL glucose-6-phosphate dehydrogenase at 37 °C. The final total organic solvent concentration (from the different spiking solutions) was 0.5% (v/v) for each sample. The reactions were initiated by the addition of an NADPH-regenerating system to give final concentrations of 1.4 mM NADP, 3.8 mM glucose-6-phosphate, and 3.5 mM MgCl2. Samples were quenched by the addition of ice-cold acetonitrile containing diazepam as the analytical internal standard. Concentrations of the substrate-specific metabolites in quenched samples were determined by LC–MS (Additional file 1: Table S5) relative to calibration standards prepared in quenched microsomal matrix. Control samples were included to confirm that the LC–MS assay of the specific metabolites was not affected by the presence of test compound (or potential test compound metabolites).
The inhibitory effect of each test compound and positive control inhibitor was based on the reduction in the formation of the specific CYP-mediated metabolite (represented as percent inhibition of enzyme activity) relative to metabolite formation in the absence of inhibitor (i.e. control for maximal enzyme activity). Where the inhibition of probe metabolite formation exceeded 50%, the inhibitor concentration resulting in 50% inhibition (IC50) was obtained by non-linear curve fitting of the percent inhibition vs inhibitor concentration using a 4-parameter sigmoidal function (GraphPad Prism, GraphPad Software, San Diego). Minimum and maximum inhibition values were constrained to 0 and 100%, respectively, unless reasonable model fitting could only be achieved without constraints. Where less than 50% inhibition was observed at the highest concentration tested (e.g. 20 µM in this assay), the IC50 value is reported as being > 20 µM. Where IC50 values could be measured, the inhibition constant (Ki) was then calculated by dividing the IC50 value by (1 + [S]/Km) where [S] is the substrate concentration and Km the Michaelis-Menten constant with an assumption of competitive inhibition. The Km was determined under the same incubation conditions by measuring the rate of metabolite formation (pmol/min/mg protein) as a function of substrate concentration (Additional file 1: Table S4).
Results
Molecular properties
A comparison of the key properties for the legacy and development compounds is shown graphically in Fig. 1 and tabulated values are shown in Table 1. Median values for legacy and development compounds were not significantly different and median parameters were also comparable to those for oral drugs launched between 2000 and 2017.
Fig. 1.

Molecular properties for the anti-malarial data sets and oral drugs launched between 2000 and 2017. Vertical bars represent the median and interquartile range
Table 1.
Calculated molecular properties (ChemAxon)
| Compound | Mass (Da) | cLog P | HBD/HBA | tPSA (Å2) | FRB | AROM | Fsp3 |
|---|---|---|---|---|---|---|---|
| Development compounds | |||||||
| AQ-13 | 291.82 | 3.00 | 1/3 | 30.6 | 7 | 2 | 0.44 |
| Artemisone | 401.52 | 2.21 | 0/7 | 74.3 | 1 | 0 | 1.00 |
| DSM265 | 415.33 | 5.68 | 1/4 | 55.1 | 4 | 3 | 0.21 |
| DSM421 | 358.28 | 3.34 | 1/5 | 68.0 | 4 | 3 | 0.29 |
| ELQ300 | 475.85 | 7.20 | 1/4 | 56.8 | 6 | 4 | 0.13 |
| Ferroquine | 433.76 | 3.54 | 1/3 | 30.6 | 7 | 2 | 0.26 |
| JPC3210 | 398.45 | 4.51 | 2/3 | 49.7 | 6 | 2 | 0.48 |
| KAF156 | 411.46 | 3.07 | 2/4 | 77.8 | 4 | 3 | 0.27 |
| KAE609 | 390.24 | 4.17 | 3/2 | 56.9 | 0 | 3 | 0.21 |
| M5717 | 462.57 | 3.53 | 1/5 | 58.9 | 7 | 3 | 0.41 |
| MMV048 | 393.38 | 2.70 | 1/5 | 85.9 | 4 | 3 | 0.11 |
| MMV052 | 525.73 | 5.91 | 2/6 | 73.8 | 4 | 1 | 0.81 |
| MMV253 | 465.58 | 4.23 | 2/8 | 88.2 | 6 | 3 | 0.50 |
| NPC1161B | 434.36 | 4.86 | 2/4 | 71.0 | 8 | 3 | 0.32 |
| OZ277 | 392.54 | 3.11 | 2/5 | 84.4 | 4 | 0 | 0.95 |
| OZ439 | 469.62 | 5.44 | 0/6 | 49.4 | 5 | 1 | 0.79 |
| P218 | 360.41 | − 0.04 | 3/8 | 138 | 10 | 2 | 0.39 |
| SJ733 | 468.41 | 3.64 | 1/4 | 86.1 | 5 | 3 | 0.17 |
| Tafenoquine | 463.50 | 4.97 | 2/5 | 80.3 | 10 | 3 | 0.38 |
| TDD-E209 | 501.64 | 6.06 | 0/6 | 50.6 | 5 | 1 | 0.79 |
| Legacy compounds | |||||||
| Amodiaquine | 355.87 | 3.80 | 2/4 | 49.6 | 6 | 3 | 0.25 |
| Desethylamodiaquine | 327.81 | 2.99 | 3/4 | 61.8 | 5 | 3 | 0.17 |
| Artemether | 298.38 | 3.48 | 0/5 | 46.2 | 1 | 0 | 1.00 |
| Artesunate | 384.43 | 3.10 | 1/7 | 103 | 5 | 0 | 0.89 |
| Atovaquone | 366.84 | 5.00 | 1/3 | 57.2 | 2 | 2 | 0.27 |
| Azithromycin | 749.00 | 2.44 | 5/13 | 183 | 7 | 0 | 0.97 |
| Chloroquine | 319.88 | 3.93 | 1/3 | 30.6 | 8 | 2 | 0.50 |
| Chlorproguanil | 288.18 | 2.99 | 5/5 | 87.3 | 2 | 1 | 0.27 |
| Clindamycin | 424.98 | 1.04 | 4/6 | 104 | 7 | 0 | 0.94 |
| Dapsone | 248.30 | 1.27 | 2/4 | 86.2 | 2 | 2 | 0.00 |
| Dihydroartemisinin | 284.35 | 2.84 | 1/5 | 57.2 | 0 | 0 | 1.00 |
| Doxycyclin | 444.44 | − 3.34 | 6/9 | 186 | 2 | 1 | 0.41 |
| Halofantrine | 500.43 | 8.06 | 1/2 | 24.7 | 11 | 3 | 0.46 |
| Lumefantrine | 528.94 | 9.19 | 1/2 | 24.7 | 10 | 3 | 0.33 |
| Mefloquine | 378.32 | 4.11 | 2/3 | 49.7 | 4 | 2 | 0.47 |
| Naphthoquine | 409.96 | 5.22 | 3/4 | 61.8 | 5 | 3 | 0.38 |
| Piperaquine | 535.52 | 5.27 | 0/6 | 41.2 | 6 | 4 | 0.38 |
| Primaquine | 259.35 | 1.64 | 2/4 | 61.8 | 6 | 2 | 0.40 |
| Proguanil | 253.73 | 2.38 | 5/5 | 87.3 | 2 | 1 | 0.27 |
| Cycloguanil | 251.72 | 1.70 | 2/5 | 81.6 | 1 | 1 | 0.27 |
| Pyrimethamine | 248.71 | 2.75 | 2/4 | 79.1 | 2 | 2 | 0.17 |
| Pyronaridine | 518.06 | 4.22 | 2/7 | 79.0 | 7 | 4 | 0.38 |
| Quinine | 324.42 | 2.51 | 1/4 | 46.8 | 4 | 2 | 0.45 |
| Sulfadoxine | 310.33 | 0.58 | 2/7 | 114 | 4 | 2 | 0.17 |
| Sulfamethoxazole | 253.28 | 0.79 | 2/4 | 95.4 | 2 | 2 | 0.10 |
Ionization and partitioning properties
Calculated (using ADMET Predictor) and measured pKa values are shown in Table 2 and Fig. 2a. Calculated pKa values obtained using ChemAxon and ACD Labs (where available) are shown in Additional file 1: Table S6 for comparison. Of the 45 compounds in the data set, 12 are neutral at physiological pH whereas 26 are positively or partially positively charged weak bases, 4 are negatively charged weak acids, and 2 exist as zwitterions at physiological pH. Of the compounds that are neutral at physiological pH, 9 have weakly basic pKa values below 7.4 and are therefore positively charged at low pH conditions present in stomach. Several compounds (OZ439, TDD-E209, atovaquone, halofantrine, lumefantrine, naphthoquine) were sufficiently insoluble that pKa values could not be determined experimentally with the methods used in this work. Others (azithromycin, dapsone, doxycyclin, piperaquine, pyronaridine) contain multiple overlapping pKa values that precluded accurate measurement. For some compounds, there were two or more predicted pKa values within the range of 2–12 however only one ionization could be measured (KAE609, MMV253, M5717, JPC3210, amodiaquine, N-desethylamodiaquine, sulfadoxine).
Table 2.
Calculated (ADMET predictor) and measured pKa and Log D7.4
| Compound | Calculated pKa | Measured pKaa | Calculated Log D7.4 |
Measured Log Db7.4 |
|---|---|---|---|---|
| Ionized or partially ionized bases at physiological pH | ||||
| Cycloguanil | 10.5 (B) | 11.4 ± 0.3 (B) | − 1.80 | − 1.10 |
| Doxycyclin | 9.13 (B), 3.35 (A), 9.98 (A) | CNDe | − 0.75 | − 0.20 |
| Pyronaridine | 7.65 (B), 6.39 (B), 5.20 (B), 10.1 (A) | CNDe | 5.61 | 0.23 |
| Proguanil | 10.0 (B), 6.64 (B) | CNDc | 0.21 | 0.27 |
| Primaquine | 9.92 (B), 3.88 (B) | 10.2 ± 0.12 (B), 3.3 ± 0.02 (B) | 0.40 | 0.54 |
| Chloroquine | 9.86 (B), 7.25 (B) | 9.9 ± 0.1 (B), 8.4 ± 0.1 (B) | 2.42 | 0.93 |
| Chlorproguanil | 9.79 (B), 6.29 (B) | CNDc | 0.84 | 1.10 |
| Azithromycin | 8.72 (B), 7.63 (B) | CNDe | 1.64 | 1.10 |
| AQ-13 | 9.63 (B), 7.28 (B) | 7.6 ± 0.2 (B) | 1.89 | 1.30 |
| Desethylamodiaquine | 10.3 (B), 6.21 (B), 8.14 (A) | 8.5 ± 0.05 (B), 7.1 ± 0.01 (B) | 3.51 | 1.30 |
| Quinine | 7.95 (B), 3.87 (B) | 8.5 ± 0.05 (B), 4.2 ± 0.03 (B) | 1.99 | 1.80 |
| Clindamycin | 7.44 (B) | 7.1 ± 0.07 (B) | 1.62 | 1.90 |
| KAF156 | 8.08 (B), 3.83 (B) | 8.4 ± 0.06 (B), 4.5 ± 0.02 (B) | 1.80 | 2.06 |
| M5717 | 8.67 (B), 6.23 (B), 2.54 (B), 10.9 (A) | 8.7 ± 0.11 (B), 6.8 ± 0.04 (B)g | 2.49 | 2.50 g |
| OZ277 | 9.38 (B) | 8.9 ± 0.16 (B) | 1.28 | 2.60 |
| Mefloquine | 8.52 (B) | 8.5 ± 0.04 (B) | 2.66 | 2.70 |
| Amodiaquine | 7.95 (B), 6.25 (B), 10.3 (A) | 7.0 ± 0.02 (B) | 4.27 | 2.95 |
| NPC1161B | 9.94 (B), 3.61 (B) | 9.3 ± 0.03 (B), 6.0 ± 0.01 (B) | 3.24 | CNDf |
| Ferroquine | 8.08 (B), 6.74 (B) | 8.4 ± 0.06 (B), 7.5 ± 0.02 (B) | 5.41 | 3.39 |
| Naphthoquine | 8.48 (B), 6.48 (B), 10.7 (A) | CNDd | 5.36 | 4.18 |
| Tafenoquine | 10.0 (B), 4.00 (B) | 8.7 ± 0.09 (B), 6.0 ± 0.10 (B) | 2.61 | 4.24 |
| MMV253 | 8.03 (B), 4.63 (B), 3.03 (B), 2.52 (B) | 8.0 ± 0.03 (B), 5.5 ± 0.03 (B) | 3.76 | 4.42 |
| MMV052 | 8.75 (B) | 8.3 ± 0.06 (B) | 4.58 | 5.40 |
| Piperaquine | 7.60 (B), 5.93 (B), 5.15 (B), 4.36 (B) | CNDe | 5.59 | CNDf |
| Halofantrine | 9.20 (B) | CNDd | 5.78 | CNDf |
| TDD-E209 | 7.40 (B) | CNDd | 5.84 | CNDf |
| Lumefantrine | 8.66 (B) | CNDd | 7.34 | CNDf |
| Ionized acids at physiological pH | ||||
| Sulfadoxine | 2.01 (B), 6.40 (A) | 6.2 ± 0.01 (A) | − 0.28 | − 0.780 |
| Sulfamethoxazole | 6.15 (A) | 6.1 ± 0.01 (A) | − 0.25 | − 0.780 |
| Artesunate | 4.51 (A) | 4.7 ± 0.04 (A) | − 0.38 | − 0.120 |
| Atovaquone | 4.28 (A) | CNDd | 2.58 | 5.30 |
| Zwitterionic or partially zwitterionic at physiological pH | ||||
| P218 | 7.22 (B), 4.26 (A) | 7.3 ± 0.003 (B), 4.9 ± 0.002 (A) | 0.49 | 0.080 |
| JPC3210 | 8.20 (B), 10.5 (A) | 5.3 ± 0.04 (A) | 5.42 | 5.30 |
| Neutral at physiological pH | ||||
| Dapsone | 3.06 (B), 2.18 (B) | CNDe | 0.97 | 0.86 |
| Dihydroartemisinin | NA | NA | 2.16 | 2.30 |
| DSM421 | 3.04 (B) | CNDc | 3.51 | 2.36 g |
| Pyrimethamine | 6.57 (B) | 6.9 ± 0.10 (B) | 2.47 | 2.41 |
| MMV048 | 4.18 (B) | 4.0 ± 0.07 (B) | 2.81 | 2.50 |
| Artemisone | 5.22 (B) | CNDc | 1.36 | 2.82 |
| Artemether | NA | NA | 2.80 | 3.70 |
| SJ733 | 3.16 (B), 10.7 (A) | 4.1 ± 0.03 (B) | 3.34 | 3.90 |
| DSM265 | 3.23 (B) | CNDc | 4.59 | 4.03 |
| KAE609 | 3.95 (B), 10.7 (A), 10.1 (A) | 5.1 ± 0.02 (B) | 4.36 | CNDf |
| OZ439 | 6.38 (B) | CNDd | 5.03 | CNDf |
| ELQ300 | NA | NA | 5.28 | CNDf |
A acidic pKa, B basic pKa, CND could not determine, NA not applicable
aValues for pKa represent the mean ± SD for n = 3 titrations
bValues for Log D represent the average ratio for n = 2–3 replicate measurements of each partitioning phase (i.e. buffer or octanol); replicate measurements for each phase differed by less than 10%
cNo ionization detected
dSolubility-limited
eMultiple overlapping pKa values
fAqueous phase concentrations below the analytical LLQ
Fig. 2.

Relationship between calculated (using ADMET Predictor) and measured a pKa and b Log D7.4 values for development (green) and legacy (blue) compounds. Solid black lines represent the lines of best fit and labelled points are those that differed the most between the measured and calculated values
Overall, there was good agreement between the measured and calculated (ADMET Predictor) values for the majority of compounds, with the slope (1.03 ± 0.08) not differing significantly (p = 0.73) from unity (Fig. 2a). For a few compounds (artemisone, DSM265, DSM421), no ionizations could be detected in spite of the calculated pKa values being within a measurable range (i.e. 2–12) suggesting that ADMET Predictor overestimated the basicity of the nitrogens in these structures. This is supported by the solubility results for these three compounds (see below) which showed no major increase in solubility under low pH conditions (FaSSGF, pH 1.6) compared to more neutral pH (FaSSIF, pH 6.5). For these three compounds, the pKa calculations using ChemAxon (Additional file 1: Table S6) were more in line with the experimental results. Poor calculated predictions were obtained for JPC3210 (both acidic and basic groups), tafenoquine (less basic group), and NPC1161B (less basic group). For JPC3210 and tafenoquine, the ChemAxon (Additional file 1: Table S6) calculated values still differed considerably from the measured whereas the ChemAxon values for NPC1161B were somewhat more consistent with the measured values. As highlighted previously, it is unlikely that a single software package will be accurate for all compounds [44], however a rigorous assessment of the different calculation packages was outside the scope of this work.
Calculated (ADMET Predictor) and measured Log D7.4 values are shown in Table 2 and Fig. 2b. Calculated values using ChemAxon and ACD Labs (where available) are shown in Additional file 1: Table S6 for comparison. In general, the development compounds were somewhat more lipophilic than the legacy compounds, with 10 of 20 having calculated Log D7.4 values above 3.5 compared to only 6 out of 23 for the legacy compounds. Measured values were obtained for all compounds with the exception of halofantrine, lumefantrine, OZ439, ELQ300, KAE609 and piperaquine where concentrations in the aqueous phase were below the analytical limit of quantitation. Even though there was more scatter than for the pKa data, the slope (0.84 ± 0.092) of the calculated (ADMET Predictor) vs measured relationship (Fig. 2b) was not significantly different from unity (p = 0.074) suggesting that the calculated Log D7.4 values still provide a reasonable estimate of the true Log D7.4. Exceptions to this included pyronaridine, ferroquine and atovaquone and for each of these the ChemAxon or ACD calculated values (Additional file 1: Table S6) were somewhat more consistent with the measured values.
Solubility
Measured solubility values determined in fasted state simulated gastric fluid (FaSSGF), fasted and fed state simulated intestinal fluids (FaSSIF and FeSSIF, both version 2 [30]) and phosphate buffered saline (pH 7.4) are shown in Table 3. Given the high prevalence of weak bases in the data set, it is not surprising that the majority of compounds had high solubility in simulated gastric fluid with most exceeding the maximum tested concentration of 2 mg/mL (2.6–8.1 mM). The notable exceptions to this were the neutral compounds or those showing minimal or no ionization (artemisone, DSM265, ELQ300, artemether), the weak acid (atovaquone) and the highly lipophilic weak bases (OZ439, MMV052, TDD-E209, halofantrine, lumefantrine). In general, solubility was considerably lower in FaSSIF and decreased with increasing Log D7.4 (Fig. 3a) but improved for most compounds in FeSSIF reflecting an increase in solubilization in the presence of bile salts and mixed micellar phases. Compounds that exhibited high solubility (> 2 mg/mL) in all media tested included the charged compounds (M5717, chloroquine, chlorproguanil, clindamycin, primaquine, and proguanil) and the very polar compound doxycycline. Development compounds KAF156 and P218 also had very good solubility (> 400–500 µg/mL) in all media tested.
Table 3.
Measured solubility in fasted and fed state simulated intestinal fluids (FaSSIF and FeSSIF), fasted state simulated gastric fluid (FaSSGF), and pH 7.4 phosphate buffered saline
| Compound | Solubility (µg/mL)/(µM)a | |||
|---|---|---|---|---|
| FaSSGF (1 h) | FaSSIF-V2 (5–6 h) | FeSSIF-V2 (5–6 h) | PBS7.4 (5–6 h) | |
| Ionized or partially ionized bases at physiological pH | ||||
| Cycloguanil | > 2000/> 7950 | > 2000/> 7950 | 533/2120 | > 2000/> 7950 |
| Doxycyclin | > 2000/> 4500 | > 2000/> 4500 | > 2000/> 4500 | > 2000/> 4500 |
| Pyronaridine | > 2000/> 3800 | > 2000/> 3860 | > 2000/> 3800 | 357/689 |
| Proguanil | > 2000/> 7880 | > 2000/> 7880 | > 2000/> 7880 | > 2000/> 7880 |
| Primaquine | > 2000/> 7700 | > 2000/> 7700 | > 2000/> 7700 | > 2000/> 7700 |
| Chloroquine | > 2000/> 6250 | > 2000/> 6250 | > 2000/> 6250 | > 2000/> 6250 |
| Chlorproguanil | > 2000/> 6940 | 1100/3810 | 1670/5780 | 1420/4930 |
| Azithromycin | 1180/1570 | 1440/1930 | > 2000/> 2670 | > 2000/> 2670 |
| AQ-13 | > 2000/> 6850 | > 2000/> 6850 | > 2000/> 6850 | 1480/5070 |
| Quinine | 1925/5930 | 1960/6050 | 1930/5930 | 806/2480 |
| Clindamycin | > 2000/> 4700 | > 2000/> 4700 | > 2000/> 4700 | > 2000/> 4700 |
| KAF156 | > 2000/> 4850 | 1600/3880 | > 2000/> 4850 | 560/1360 |
| M5717 | > 3000/> 6490b | > 3000/> 6490b | > 3000/> 6486b | > 3000/> 6490b |
| OZ277 | > 2000/> 5000 | > 2000/> 5100 | > 2000/> 5000 | 198/504 |
| Mefloquine | 740/1960 | 584/1540 | > 2000/> 5200 | 290/767 |
| Amodiaquine | > 2000/> 5620 | 1120/3150 | > 2000/> 5620 | 26.3/73.9 |
| NPC1161B | 1520/3500 | 19.9/45.8 | > 2000/> 4600 | 5.44/12.5 |
| Ferroquine | > 2000/> 4600 | 320/738 | > 2000/> 4600 | 4.9/11 |
| Naphthoquine | > 2000/> 4880 | 410/1000 | 941/2300 | 28.5/69.5 |
| Tafenoquine | > 2000/> 2800 | 15.8/34.1 | 1310/2820 | 18.3/39.5 |
| MMV253 | > 2000/> 4300 | 569/1220 | > 2000/> 4300 | 19.0/40.8 |
| MMV052 | 13.0/24.7 | 1140/2170 | > 2000/> 3800 | 49.6/94.3 |
| Piperaquine | > 2000/> 3700 | 103/192 | 60.7/113 | < 0.5/< 0.9 |
| Halofantrine | 12.7/25.4 | 69.9/140 | 739/1480 | < 0.05/< 0.1 |
| TDD-E209 | 211/421 | 87.2/174 | > 2000/> 4000 | 0.056/0.112 |
| Lumefantrine | 12.6/23.8 | 0.063/0.119 | 14.4/27.2 | < 0.05/< 0.1 |
| Ionized acids at physiological pH | ||||
| Sulfadoxine | 324/1040 | 858/2770 | 474/1530 | > 1900/> 6400 |
| Sulfamethoxazole | 758/2990 | > 2000/> 7900 | > 2000/> 7900 | > 2000/> 7900 |
| Artesunate | 321/835 | 1690/4390 | 1720/4490 | 1680/4370 |
| Atovaquone | 0.26/0.71 | 1.83/4.99 | 6.41/17.5 | < 0.05/< 0.1 |
| Zwitterionic or partially zwitterionic at physiological pH | ||||
| P218 | > 2000/> 5550 | 408/1130 | 525/1460 | 1070/2960 |
| JPC3210 | 213/535 | 7.60/19.1 | 131/329 | 0.800/2.01 |
| Neutral at physiological pH | ||||
| Dapsone | 946/3810 | 323/1300 | 423/1700 | 227/914 |
| Dihydroartemisinin | 140/492 | 159/559 | 246/865 | 130/457 |
| DSM421 | 116/324b | 92.7/259b | 119/332b | 85.5/239 |
| Pyrimethamine | > 2000/> 8000 | 111/446 | 120/483 | 43/175 |
| MMV048 | 1200/3040 | 6.73/17.1 | 7.47/19.0 | 4.52/11.5 |
| Artemisone | 89.1/222 | 115/286 | 278/692 | 86.1/214 |
| Artemether | 95/320 | 166/556 | 1040/3480 | 109/365 |
| SJ733 | > 2000/> 4270 | 101/216 | 119/254 | 115/246 |
| DSM265 | 6.84/16.5b | 5.12/12.3b | 27.6/66.5b | 2.04/4.9 |
| KAE609 | 1540/3940 | 111/284 | 1240/3180 | 30.4/77.9 |
| OZ439 | 79.0/168 | 40.6/87 | 526/1120 | 0.085/0.181 |
| ELQ300 | 0.929/1.95 | 0.524/1.10 | 0.065/0.137 | 0.102/0.214 |
Fig. 3.

Relationship between a FaSSIF solubility (grey bar represents the maximum solubility range assessed), b Caco-2 permeability (circled symbols represent compounds with high efflux ratios), c fraction unbound in plasma, and d unbound intrinsic clearance (open symbols represent “less than” values) and measured Log D7.4. Where measured Log D values were not available, calculated (ADMET Predictor) values were used. Symbols represent data for development (green) and legacy (blue) anti-malarial compounds. Caco-2 Papp data for chloroquine, quinine, amodiaquine, naphthoquine, mefloquine, piperaquine, atovaquone and halofantrine are from Katneni et al. [31]
Permeability
The measurement of flux across Caco-2 monolayers was used as a means to determine the apparent permeability coefficient (Papp) which was then converted to a predicted effective human jejunal permeability (Peff) using a calibration plot of reported human Peff values [34, 35] and measured Caco-2 Papp for a series of control compounds [31]. The general performance of the Caco-2 test system was assessed on the basis of the permeability data for the minimally permeable marker, lucifer yellow, the high permeability marker, propranolol, and the efflux ratio for a P-gp efflux marker, rhodamine 123 (Additional file 1: Table S7). The wide range of physicochemical properties across the data set necessitated the use of two different transport buffers consisting of either aqueous pH 7.4 buffer or human plasma as recently described [31]. Results for mass balance and Papp are shown in Table 4 and data for control compounds under the two conditions can be found in Katneni et al. [31].
Table 4.
Caco-2 mass balance and bidirectional permeability coefficients
| Compound | Matrix | A–B mass bal (%) | A–B Paapp (10−6 cm/s) | B–A mass bal (%) | B–A Paapp (10−6 cm/s) | Efflux ratio |
|---|---|---|---|---|---|---|
| Ionized or partially ionized bases at physiological pH | ||||||
| Cycloguanil | Buffer | 100 | 1.2 ± 0.070 | 100 | 0.77 ± 0.05 | 0.7 |
| Doxycyclin | Buffer | 99 | 3.4 ± 0.23 | 110 | 4.2 ± 0.29 | 1.2 |
| Pyronaridine | Plasma | 63 | 16 ± 2.2 | 87 | 41 ± 3.0 | 2.6 |
| Proguanil | Buffer | 82 | 3.5 ± 0.75 | 95 | 7.9 ± 0.51 | 2.3 |
| Primaquine | Buffer | 66 | 29 ± 4.5 | 99 | 34 ± 1.5 | 1.2 |
| Chloroquineb | Plasma | 61 | 16 ± 2.5 | 110 | 22 ± 4.6 | 1.4 |
| Chlorproguanil | Plasma | 79 | 19 ± 3.0 | 100 | 61 ± 7.4 | 3.2 |
| Azithromycin | Buffer | 98 | 0.37, 0.38 | 87 | 13 ± 3.9 | 33 |
| AQ-13 | Plasma | 54 | 25 ± 2.3 | 92 | 32 ± 0.8 | 1.2 |
| Quinineb | Buffer | 70 | 39 ± 5.0 | 92 | 40 ± 4.5 | 1.0 |
| Clindamycin | Buffer | 75 | 17 ± 1.1 | 92 | 48 ± 9.1 | 2.8 |
| KAF156 | Plasma | 86 | 47 ± 7.6 | 96 | 230 ± 17 | 4.9 |
| M5717 | Buffer | 60 | 34 ± 2.6 | 81 | 39 ± 7.6 | 1.1 |
| OZ277 | Plasma | 67 | 42 ± 2.8 | 100 | 70 ± 0.38 | 1.7 |
| Mefloquineb | Plasma | 70 | 180 ± 20 | 86 | 150 ± 10 | 0.83 |
| Amodiaquineb | Plasma | 81 | 90 ± 8.3 | 88 | 63 ± 9.6 | 0.7 |
| NPC1161B | Plasma | 76 | > 300 | 97 | > 300 | – |
| Ferroquine | Plasma | 66 | 32 ± 6.8 | 110 | 24 ± 4.1 | 0.8 |
| Naphthoquineb | Plasma | 72 | 230 ± 32 | 94 | 180 ± 10 | 0.78 |
| Tafenoquine | Plasma | 79 | > 300 | 110 | v300 | – |
| MMV253 | Plasma | 84 | 150 ± 29 | 94 | 150 ± 23 | 1.0 |
| MMV052 | Plasma | 77 | > 300 | 110 | > 300 | – |
| Piperaquineb | Plasma | 100 | > 300 | 100 | > 300 | – |
| Halofantrineb | Plasma | 83 | > 300 | 87 | > 300 | – |
| TDD-E209 | Plasma | 93 | > 300 | 100 | > 300 | – |
| Lumefantrine | Plasma | 110 | CND | 110 | CND | CND |
| Ionized acids at physiological pH | ||||||
| Sulfadoxine | Buffer | 93 | 15 ± 2.5 | 110 | 23 ± 2.8 | 1.6 |
| Sulfamethoxazole | Buffer | 96 | 4.5 ± 0.29 | 100 | 5.9 ± 0.26 | 1.3 |
| Artesunate | Buffer | 81 | 3.4, 2.9 | 84 | 2.8 ± 0.48 | 0.9 |
| Atovaquone b | Plasma | 99 | > 300 | 97 | > 300 | – |
| Zwitterionic or partially zwitterionic at physiological pH | ||||||
| P218 | Buffer | 96 | 0.71 ± 0.04 | 100 | 15 ± 1.7 | 21 |
| JPC3210 | Plasma | 97 | > 300 | 100 | > 300 | – |
| Neutral at physiological pH | ||||||
| Dapsone | Buffer | 86 | 45 ± 7.3 | 98 | 56 ± 1.3 | 1.2 |
| Dihydroartemisinin | Buffer | 85 | 50 ± 2.3 | 91 | 49 ± 1.2 | 1.0 |
| DSM421c | Buffer | 70 | 33 ± 2.5 | 98 | 46 | 1.4 |
| Pyrimethamine | Buffer | 75 | 60 ± 5.6 | 100 | 59 ± 2.9 | 1.0 |
| MMV048 | Buffer | 82 | 41 ± 2.1 | 98 | 53 ± 1.9 | 1.3 |
| Artemisone | Buffer | 98 | 58.8, 64.5 | 99 | 47 ± 1.7 | 0.8 |
| Artemether | Buffer | 73 | 39.1, 46.3 | 95 | 42 ± 5.6 | 1.0 |
| SJ733 | Buffer | 93 | 14.0, 13.4 | 99 | 50 ± 2.0 | 3.6 |
| DSM265 | Buffer | 83 | 52.4, 69.7 | 93 | 49 ± 5.1 | 0.9 |
| KAE609 | Plasma | 93 | > 300 | 98 | > 300 | – |
| OZ439 | Plasma | 91 | > 300 | 110 | > 300 | – |
| ELQ300 | Plasma | 99 | > 300 | 110 | > 300 | – |
The use of plasma as the transport medium for the more lipophilic compounds significantly improved the mass balance as shown in Fig. 4 allowing permeability values to be measured even for the more lipophilic compounds. As expected, Papp increased with increasing Log D7.4 (Fig. 3b) with only a few of the more polar compounds showing low A-B Papp values (i.e. < 5 × 10−6 cm/s) including P218, azithromycin, cycloguanil, doxycycline, proguanil, and sulfamethoxazole. Several of the highly lipophilic compounds had Papp values in excess of 300 × 10−6 cm/s and, other than P218 and azithromycin (efflux ratios of 21 and 33, respectively), no compounds exhibited high efflux ratios (i.e. efflux ratios were generally less than 4).
Fig. 4.
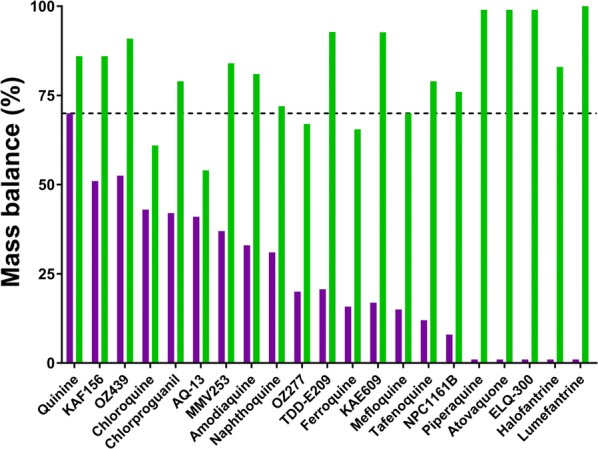
Mass balance for Caco-2 permeability experiments conducted using either pH 7.4 buffer (purple bars) or human plasma (green bars) as the transport matrix. Data for chloroquine, quinine, amodiaquine, naphthoquine, mefloquine, piperaquine, atovaquone and halofantrine are from Katneni et al. [31]
Figure 5 illustrates the relationship between human jejunal Peff values from the literature [34, 35] and the measured Caco-2 A-B Papp for control compounds using either pH 7.4 buffer or plasma as the transport medium [31]. Also shown is the relationship from Sun et al. [45] for passively permeating compounds using pH 7.4 buffer as the Caco-2 transport medium showing the similarity in the relationships across the different studies. As shown previously, Papp values are typically higher using plasma as the transport medium compared to buffer due to improved sink conditions [31] and as a result the relationship using plasma is shifted marginally to the right.
Fig. 5.
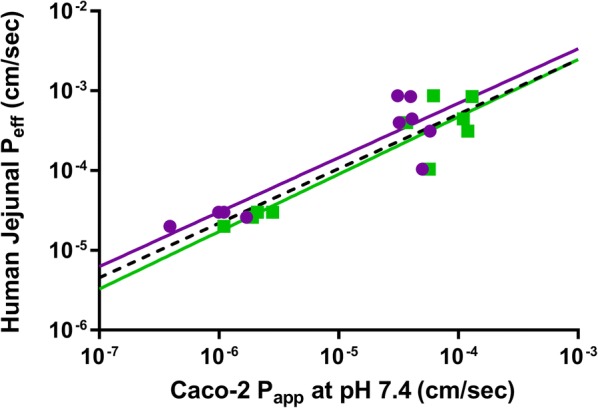
Relationship between human jejunal permeability (Peff [34, 35]) and Caco-2 A-B apparent permeability (Papp [31]) at pH 7.4 measured using either pH 7.4 buffer (purple) or human plasma (green) as the transport medium. Solid lines represent the lines of best fit to the Log transformed data (for buffer: slope = 0.6989, y-int = − 0.3941; for plasma: slope = 0.7255, y-int = − 0.4239). The reported relationship from Sun et al. [45] is shown by the dashed black line (slope = 0.6836, y-int = − 0.5579)
Solubility limited absorbable dose
Measured solubility (FaSSIF) and predicted human permeability (Peff based on Caco-2 Papp values and the calibration data shown in Fig. 5) were used to estimate the solubility limited absorbable dose (SLAD, Table 5) as described previously [32]. SLAD values ranged from less than 10 mg for the compounds showing very low FaSSIF solubility (in spite of good permeability for several of these) to greater than 2 g for compounds demonstrating both high solubility and high permeability.
Table 5.
Estimated solubility limited absorbable dose (SLAD)
| Compound | Permeability | Solubility/dose | |||
|---|---|---|---|---|---|
| Predicted human Peff (10−4 cm/s) | Permeability classificationa | An | Ssi (µg/mL) | SLAD (mg) | |
| Ionized or partially ionized bases at physiological pH | |||||
| Doxycyclin | 0.70 | Intermediate | 1.0 | > 2000 | > 1000 |
| Pyronaridine | 1.4 | High | 1.6 | > 2000 | > 1610 |
| Proguanil | 0.71 | Intermediate | 1.0 | > 2000 | > 1000 |
| Primaquine | 3.0 | High | 3.6 | > 2000 | > 3560 |
| Chloroquine | 1.4 | High | 1.6 | > 2000 | > 1620 |
| Chlorproguanil | 1.5 | High | 1.8 | 1100 | 998 |
| Azithromycin | 0.16 | Low | 1.0 | 1440 | 720 |
| AQ-13 | 1.9 | High | 2.2 | > 2000 | > 2250 |
| Quinine | 3.7 | High | 4.4 | 1960 | 4280 |
| Clindamycin | 2.1 | High | 2.5 | > 2000 | > 2530 |
| KAF156 | 2.9 | High | 3.5 | 1600 | 2780 |
| M5717 | 3.4 | High | 4.0 | > 3000 | > 6050 |
| OZ277 | 2.7 | High | 3.2 | > 2000 | > 3240 |
| Mefloquine | 7.7 | High | 9.2 | 584 | 2690 |
| Amodiaquine | 4.7 | High | 5.6 | 1120 | 3130 |
| Ferroquine | 2.2 | High | 2.7 | 320 | 426 |
| Naphthoquine | 9.2 | High | 11 | 410 | 2250 |
| Tafenoquine | 11 | High | 13 | 15.8 | 105 |
| MMV253 | 6.8 | High | 8.1 | 569 | 2300 |
| MMV052 | 11 | High | 13 | 1140 | 7580 |
| Piperaquine | 11 | High | 13 | 103 | 685 |
| Halofantrine | 11 | High | 13 | 69.9 | 466 |
| TDD-E209 | 11 | High | 13 | 87.0 | 579 |
| Lumefantrine | CND | CND | CND | CND | CND |
| Ionized acids at physiological pH | |||||
| Sulfadoxine | 1.9 | High | 2.2 | 858 | 959 |
| Sulfamethoxazole | 0.85 | Intermediate | 1.0 | > 2000 | > 1000 |
| Artesunate | 0.66 | Intermediate | 1.0 | 1690 | 845 |
| Atovaquone | 11 | High | 13 | 1.83 | 12 |
| Zwitterionic or partially zwitterionic at physiological pH | |||||
| P218 | 0.24 | Low | 1.0 | 408 | 204 |
| JPC3210 | 11 | High | 13 | 7.60 | 51 |
| Neutral at physiological pH | |||||
| Dapsone | 4.1 | High | 4.9 | 323 | 786 |
| Dihydroartemisinin | 4.4 | High | 5.2 | 159 | 413 |
| DSM421 | 3.3 | High | 3.9 | 93.0 | 182 |
| Pyrimethamine | 4.9 | High | 5.9 | 111 | 326 |
| MMV048 | 3.8 | High | 4.5 | 6.70 | 15 |
| Artemisone | 5.0 | High | 6.0 | 115 | 345 |
| Artemether | 3.9 | High | 4.7 | 166 | 388 |
| SJ733 | 1.8 | High | 2.2 | 101 | 109 |
| DSM265 | 4.8 | High | 5.7 | 5.10 | 14 |
| KAE609 | 11 | High | 13 | 111 | 738 |
| OZ439 | 11 | High | 13 | 40.6 | 270 |
| ELQ300 | 11 | High | 13 | 0.520 | 3 |
Binding and whole blood partitioning
Binding properties were assessed in human plasma, Albumax medium (used for the majority of in vitro P. falciparum activity assays), 10% FCS in DMEM (used in several standard parasite in vitro assays), and human liver microsomes (Table 6). For Albumax, DMEM/FCS and HLMs, an ultracentrifugation method was used to separate bound and free fractions.
Table 6.
Binding to plasma and media proteins
| Compound | Fraction unbound ± SD | |||
|---|---|---|---|---|
| Human Plasmaa | Albumaxa | 10% FCS in DMEMa | HLMa | |
| Ionized or partially ionized bases at physiological pH | ||||
| Cycloguanil | 0.75 ± 0.040 | 0.88 ± 0.057 | 0.94 ± 0.064 | 0.72 ± 0.083 |
| Doxycyclin | 0.23 ± 0.090b | 0.41 ± 0.023 | 0.44 ± 0.070 | 0.93 ± 0.045 |
| Pyronaridine | 0.090 ± 0.012b | 0.69 ± 0.023 | Not assessed | 0.29 ± 0.018 |
| Proguanil | 0.34 ± 0.010 | 0.80 ± 0.021 | Not assessed | 0.33 ± 0.014 |
| Primaquine | 0.26 ± 0.023 | 0.96 ± 0.056 | 0.78 ± 0.025 | 0.56 ± 0.050 |
| Chloroquine | 0.54 ± 0.048 | 0.97 ± 0.069 | Not assessed | 0.50 ± 0.032 |
| Chlorproguanil | 0.10 ± 0.0070 | 0.50 ± 0.020 | Not assessed | 0.052 ± 0.004 |
| Azithromycin | 0.34, 0.35b | 0.76 ± 0.065 | Not assessed | 0.70 ± 0.060 |
| AQ-13 | 0.43 ± 0.022 | 0.85 ± 0.028 | Not assessed | 0.66 ± 0.048 |
| Desethylamodiaquine | 0.17 ± 0.0020 | 0.75 ± 0.074 | Not assessed | not assessed |
| Quinine | 0.37 ± 0.040b | 0.69 ± 0.090 | Not assessed | 0.57 ± 0.036 |
| Clindamycin | 0.19 ± 0.011 | 0.65 ± 0.066 | Not assessed | 0.67 ± 0.084 |
| KAF156 | 0.069 ± 0.013b | 0.38 ± 0.017 | 0.477 ± 0.032 | 0.14 ± 0.0060 |
| M5717 | 0.24 ± 0.0020b | 0.55 ± 0.041 | 0.638 ± 0.065 | 0.50 ± 0.041 |
| OZ277 | 0.086 ± 0.0080 | 0.29 ± 0.021 | Not assessed | 0.044 ± 0.0050 |
| Mefloquine | 0.015 ± 0.0010b | 0.27 ± 0.019 | Not assessed | 0.050 ± 0.0060 |
| Amodiaquine | 0.089 ± 0.0080 | 0.67 ± 0.033 | Not assessed | 0.91 ± 0.094 |
| NPC1161B | 0.00074 ± 0.00016c | 0.035 ± 0.0020 | 0.043 ± 0.004 | CNDe |
| Ferroquine | 0.041 ± 0.0010c | 0.52 ± 0.031 | 0.364 ± 0.026 | 0.27 ± 0.032 |
| Naphthoquine | 0.018 ± 0.0010c | 0.27 ± 0.035 | Not assessed | 0.21 ± 0.015 |
| Tafenoquine | 0.00070 ± 0.00017c | CNDe | 0.038 ± 0.001 | 0.0020 ± 0.00030 |
| MMV253 | 0.017 ± 0.00020c | 0.48 ± 0.026 | Not assessed | 0.11 ± 0.017 |
| MMV052 | 0.00040 ± 0.00010c | 0.014 ± 0.0010 | Not assessed | 0.0010 ± 0.00010 |
| Piperaquine | 0.0003 ± 0.0001c | 0.019 ± 0.0040 | 0.092 ± 0.0030 | 0.013 ± 0.0020 |
| Halofantrine | < 0.0001c | 0.0080 ± 0.0010 | Not assessed | 0.0020 ± 0.00020 |
| TDD-E209 | < 0.0001c | 0.0070 ± 0.0010 | Not assessed | 0.00030 ± 0.000010 |
| Lumefantrine | < 0.0001c | CNDe | Not assessed | CNDe |
| Ionized acids at physiological pH | ||||
| Sulfadoxine | 0.036 ± 0.0010 | 0.50 ± 0.030 | 0.86 ± 0.044 | 0.93 ± 0.086 |
| Sulfamethoxazole | 0.32 ± 0.024 | 0.67 ± 0.120 | 0.87 ± 0.058 | 1.00 ± 0.033 |
| Artesunate | 0.27 ± 0.0080d | 0.14 ± 0.0070d | 0.72 ± 0.026 | CNDd |
| Atovaquone | < 0.0001c | CNDe | CNDe | ~ 0.0020 |
| Zwitterionic or partially zwitterionic at physiological pH | ||||
| P218 | 0.13 ± 0.013 | 0.41 ± 0.022 | 0.684 ± 0.017 | 0.88 ± 0.041 |
| JPC3210 | 0.0028 ± 0.00020c | 0.21 ± 0.012 | 0.227 ± 0.012 | 0.029 ± 0.0030 |
| Neutral at physiological pH | ||||
| Dapsone | 0.27 ± 0.019 | 0.63 ± 0.11 | Not assessed | 0.86 ± 0.059 |
| Dihydroartemisinin | 0.21 ± 0.010d | 0.34 ± 0.018d | Not assessed | 0.87 ± 0.10d |
| DSM421 | 0.039 ± 0.007 | 0.43 ± 0.020 | 0.615 ± 0.043 | 0.77 ± 0.065f |
| Pyrimethamine | 0.095 ± 0.0010b | 0.46 ± 0.090 | 0.48 ± 0.034 | 0.54 ± 0.097 |
| MMV048 | 0.11 ± 0.0070b | 0.46 ± 0.022 | 0.653 ± 0.060 | 0.61 ± 0.014 |
| Artemisone | 0.12 ± 0.0060 | 0.46 ± 0.013 | Not assessed | 0.44 ± 0.024 |
| Artemether | 0.038 ± 0.0090c | 0.46 ± 0.039 | Not assessed | 0.50 ± 0.052 |
| SJ733 | 0.085 ± 0.012c | 0.40 ± 0.040 | Not assessed | 0.58 ± 0.069 |
| DSM265 | 0.0018 ± 0.00010c | 0.18 ± 0.0050f | 0.366 ± 0.041 | 0.28 ± 0.0070f |
| KAE609 | 0.0016 ± 0.00010c | 0.044 ± 0.0040 | Not assessed | 0.019 ± 0.0010 |
| OZ439 | < 0.0001c | 0.010 ± 0.0010 | Not assessed | 0.0010 ± 0.00010 |
| ELQ300 | 0.00010 ± 0.000010c | CNDe | 0.001 ± 0.001 | 0.002 ± 0.00010 |
CND could not determine
aMean ± SD, n = 3–4 replicates; ultracentrifugation used unless indicated otherwise
bRED device with diluted plasma correcting for the dilution factor with 6 h dialysis
cRED device with diluted plasma correcting for the dilution factor; presaturation of device and 24 h dialysis
dInstability evident; where values are given they represent an estimate only
eCompound not detected in free fraction
Since the physicochemical and protein binding properties varied considerably across the data set, it was necessary to use a range of methods to minimize experimental artefacts for plasma (e.g. compound adsorption to the dialysis chamber or membrane, or slow equilibration) and obtain measurable unbound concentrations. Preliminary plasma fu values for control and anti-malarial compounds using the different methods are shown in Additional file 1: Tables S8 and S9, respectively, and the final fu values for the anti-malarial compounds obtained under the optimized conditions are shown in Table 6. Fraction unbound values ranged from < 0.0001 for the most lipophilic compounds to > 0.4 for some of the more polar compounds and generally correlated with Log D7.4 as shown in Fig. 3c.
Table 7 shows values for the whole blood to plasma partitioning ratio across the data set. While many of the values are close to 1, some of the compounds showing very high plasma protein binding (e.g. DSM265, ELQ-300, JPC3210, atovaquone, lumefantrine) have restricted distribution into red blood cells (B/P value 0.5–0.6) while others (4-aminoquinolines, proguanil and chlorproguanil) appear to concentrate in red blood cells (B/P > 3).
Table 7.
Whole blood to plasma partitioning
| Compound | B/Pa | Haematocrit (Gender) |
|---|---|---|
| Ionized or partially ionized bases at physiological pH | ||
| Cycloguanil | 0.71 ± 0.07 | 0.48 (M) |
| Doxycyclin | 0.78 ± 0.06 | 0.43 (M) |
| Pyronaridine | 9.0 ± 0.83 | 0.48 (M) |
| Proguanil | 3.30 ± 0.14 | 0.46 (M) |
| Primaquine | 0.82 ± 0.07 | 0.44 (M) |
| Chloroquine | 3.5 ± 0.09 | 0.42 (F) |
| Chlorproguanil | 3.3 ± 0.38 | 0.43 (M) |
| Azithromycin | 1.6 ± 0.19 | 0.48 (M) |
| AQ-13 | 3.15 ± 0.20 | 0.42 (M) |
| Desethylamodiaquine | 3.26 ± 0.35 | 0.50 (M) |
| Quinine | 0.67 ± 0.02 | 0.44 (M) |
| Clindamycin | 0.61 ± 0.04 | 0.42 (M) |
| KAF156 | 1.3 ± 0.02 | 0.42 (F) |
| M5717 | 1.3 ± 0.02 | 0.42 (F) |
| OZ277 | 1.10 ± 0.10 | 0.46 (M) |
| Mefloquine | 1.1 ± 0.07 | ~ 0.43 (M) |
| Amodiaquine | 1.1 ± 0.18 | ~ 0.43 (M) |
| NPC1161B | 1.09 ± 0.07 | 0.44 (M) |
| Ferroquine | 1.6 ± 0.15 | ~ 0.43 (M) |
| Naphthoquine | 1.14 ± 0.10 | ~ 0.43 (M) |
| Tafenoquine | 1.3 ± 0.04 | ~ 0.43 (M) |
| MMV253 | 1.0 ± 0.05 | ~ 0.43 (M) |
| MMV052 | 1.6 ± 0.14 | ~ 0.43 (M) |
| Piperaquine | 0.57 ± 0.05 | ~ 0.43 (M) |
| Halofantrine | 0.68 ± 0.06 | 0.43 (M) |
| TDD-E209 | 0.51 ± 0.08 | 0.44 (M) |
| Lumefantrine | 0.48 ± 0.09 | 0.48 (M) |
| Ionized acids at physiological pH | ||
| Sulfadoxine | 0.57 ± 0.03 | 0.46 (M) |
| Sulfamethoxazole | 0.65 ± 0.07 | 0.43 (M) |
| Artesunate | CNDb | 0.44 (M) |
| Atovaquone | 0.52 ± 0.03 | 0.44 (M) |
| Zwitterionic or partially zwitterionic at physiological pH | ||
| P218 | 0.56 ± 0.03 | ~ 0.43 (M) |
| JPC3210 | 0.54 ± 0.04 | ~ 0.43 (M) |
| Neutral at physiological pH | ||
| Dapsone | 1.1 ± 0.06 | 0.43 (M) |
| Dihydroartemisinin | CNDb | 0.42 (M) |
| DSM421 | 0.53 ± 0.03 | 0.44 (M) |
| Pyrimethamine | 0.84 ± 0.03 | 0.43 (M) |
| MMV048 | 0.77 ± 0.03 | 0.42 (F) |
| Artemisone | CNDb | 0.44 (M) |
| Artemether | CNDb | 0.42 (M) |
| SJ733 | 0.72 ± 0.02 | 0.42 (F) |
| DSM265 | 0.54 ± 0.02 | 0.44 (M) |
| KAE609 | 0.66 ± 0.04 | 0.42 (F) |
| OZ439 | 0.78 ± 0.05 | 0.44 (M) |
| ELQ300 | 0.53 ± 0.05 | 0.44 (M) |
aMean ± SD, n = 3–4 measurements
bCND = could not determine, unstable in blood and plasma
In vitro metabolism
Intrinsic clearance (CLint) was assessed in human liver microsomes and values for total and unbound CLint are shown in Table 8. Out of 45 compounds in the dataset, 22 showed less than 15% degradation over the 60 min incubation precluding the determination of CLint. Of the compounds where degradation was detected, intrinsic clearance was relatively low (< 20 µL/min/mg) for most compounds, however unbound CLint values varied considerably reflecting the high degree of binding for many compounds in the data set. Fraction unbound values could not be measured for NPC1161B, artesunate, or lumefantrine precluding estimation of unbound CLint values. Data for control compounds included in each assay are shown in Additional file 1: Table S10. Figure 3d illustrates that unbound in vitro CLint values were highly correlated with Log D7.4.
Table 8.
In vitro metabolism in human liver microsomes
| Compound | HLM CLint (µL/min/mg)a | Unbound CLint (µL/min/mg) |
|---|---|---|
| Ionized or partially ionized bases at physiological pH | ||
| Cycloguanil | < 7b | < 9.72c |
| Doxycyclin | < 7b | < 7.53c |
| Pyronaridine | 8.6 | 29.7 |
| Proguanil | < 7b | < 21.2c |
| Primaquine | 7.70 | 13.8 |
| Chloroquine | < 7b | < 14c |
| Chlorproguanil | < 7b | < 135c |
| Azithromycin | < 7b | < 10c |
| AQ-13 | < 7b | < 10.6c |
| Desethylamodiaquine | 13.3 | CNDd |
| Quinine | 16.5 | 28.9 |
| Clindamycin | 32.0 | 47.8 |
| KAF156 | 14.7 | 105 |
| M5717 | 11.1 | 22.2 |
| OZ277 | < 7b | < 159 |
| Mefloquine | < 7b | < 140c |
| Amodiaquine | 308 | 339 |
| NPC1161B | < 7b | CND |
| Ferroquine | 29.2 | 108 |
| Naphthoquine | 14.4 | 68.6 |
| Tafenoquine | < 7b | < 3500c |
| MMV253 | 13.2 | 120 |
| MMV052 | 13.0 | 13,000 |
| Piperaquine | 20.1 | 1540 |
| Halofantrine | 26.9 | 13,500 |
| TDD-E209 | 25.5e | 85,000 |
| Lumefantrine | < 7b | CNDd |
| Ionized acids at physiological pH | ||
| Sulfadoxine | < 7b | < 7.53 |
| Sulfamethoxazole | < 7b | < 7.00 |
| Artesunate | 146 | CNDd |
| Atovaquone | < 7b | < 3500c |
| Zwitterionic or partially zwitterionic at physiological pH | ||
| P218 | < 7b | < 7.95c |
| JPC3210 | 9.50 | 328 |
| Neutral at physiological pH | ||
| Dapsone | < 7b | < 8.14c |
| Dihydroartemisinin | 16.6 | 19.1 |
| DSM421 | < 7b | < 9.09c |
| Pyrimethamine | < 7b | < 13c |
| MMV048 | < 7b | < 11.5c |
| Artemisone | 97.7 | 222 |
| Artemether | 163 | 326 |
| SJ733 | 142 | 245 |
| DSM265 | < 7b,e | < 25c |
| KAE609 | 24.1 | 1270 |
| OZ439 | 72.1 | 72,100 |
| ELQ300 | < 7b | < 3500c |
CYP inhibition
The ability of compounds to inhibit the five major CYP isoforms was assessed in human liver microsomes. Data for the anti-malarial compounds are presented in Table 9 and positive control inhibitors are shown in Additional file 1: Table S11. The majority of compounds showed no inhibition up to the highest concentration tested (20 µM). The most frequently inhibited isoform was CYP2D6 where 17 compounds had IC50 values below 10 µM with 10 below 3 µM. Surprisingly, only 5 compounds showed evidence of inhibiting CYP3A4 with IC50 values in the range of 3–13 µM.
Table 9.
CYP inhibition in human liver microsomes
| Compound | IC50 (µM) (% inhibition at max conc)/Ki (µM) | |||||
|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 (test) | CYP3A4 (midaz) | |
| Ionized or partially ionized bases at physiological pH | ||||||
| Cycloguanil | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 6.0/3.7 | > 20 (nmi) | > 20 (nmi) |
| Doxycyclin | CNDa | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (22%) | > 20 (nmi) |
| Pyronaridine | > 20 (30%) | > 20 (nmi) | > 20 (nmi) | 1.2/0.75 | > 20 (nmi) | > 20 (28%) |
| Proguanil | > 20 (nmi) | > 20 (nmi) | > 20 (27%) | 3.5/2.2 | > 20 (24%) | > 20 (nmi) |
| Primaquine | < 0.25/< 0.15 | > 20 (20%) | > 20 (34%) | > 20 (33%) | > 20 (nmi) | > 20 (31%) |
| Chloroquine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 6.1/3.8 | > 20 (nmi) | > 20 (24%) |
| Chlorproguanil | > 20 (nmi) | > 20 (nmi) | > 20 (39%) | 1.4/0.87 | > 20 (41%) | > 20 (nmi) |
| Azithromycin | > 20 (nmi) | > 20 (nmi) | > 20 (29%) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| AQ-13 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 13/8.3 | > 20 (nmi) | > 20 (nmi) |
| Desethylamodiaquine | > 18 (nmi) | > 18 (nmi) | > 18 (nmi) | 2.6/1.6 | CNDa | > 18 (nmi) |
| Quinine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 6.0/3.7 | > 20 (nmi) | > 20 (nmi) |
| Clindamycin | > 20 (nmi) | > 20 (nmi) | > 20 (32%) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| KAF156 | > 20 (nmi) | 19/14 | > 20 (40%) | 1.2/0.75 | 2.7/1.6 | 2.5/1.8 |
| M5717 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| OZ277 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (46%) | > 20 (26%) |
| Mefloquine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 16/10 | > 20 (29%) | > 20 (nmi) |
| Amodiaquine | > 20 (27%) | > 20 (nmi) | > 20 (nmi) | 0.88/0.55 | > 20 (nmi) | > 20 (nmi) |
| NPC1161B | 6.5/3.8 | > 20 (37%) | 9.6/5.7 | > 20 (39%) | 13.6/8.2 | > 20 (37%) |
| Ferroquine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 0.83/0.52 | > 20 (42%) | 7.3/5.2 |
| Naphthoquine | > 20 (33%) | > 20 (nmi) | > 20 (nmi) | 1.1/0.68 | > 20 (38%) | > 20 (41%) |
| Tafenoquine | > 20 (20%) | 14/10 | > 20 (42%) | > 20 (35%) | 3.8/2.3 | > 20 (40%) |
| MMV253 | > 20 (nmi) | > 20 (nmi) | CNDa | > 20 (35%) | > 20 (37%) | CNDa |
| MMV052 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| Piperaquine | > 20 (nmi) | CNDa | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 4.5/3.2 |
| Halofantrine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 0.27/0.19 | > 20 (19%) | > 20 (24%) |
| TDD-E209b | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (43%) | > 20 (33%) |
| Lumefantrine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 2.9/1.8 | > 20 (nmi) | > 20 (nmi) |
| Ionized acids at physiological pH | ||||||
| Sulfadoxine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| Sulfamethoxazole | CNDa | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| Artesunate | 20/12 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 20 (nmi) | > 20 (nmi) |
| Atovaquone | > 20 (nmi) | > 20 (26%) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| Zwitterionic or partially zwitterionic at physiological pH | ||||||
| P218 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| JPC3210 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 0.70/0.43 | > 20 (nmi) | > 20 (nmi) |
| Neutral at physiological pH | ||||||
| Dapsone | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| Dihydroartemisinin | 11/6.2 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) |
| DSM421b | > 20 (nmi) | > 20 (16%) | > 20 (nmi) | > 20 (33%) | > 20 (nmi) | CNDa |
| Pyrimethamine | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (36%) | > 20 (nmi) | > 20 (nmi) |
| MMV048 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (16%) | > 20 (nmi) | CNDa |
| Artemisone | NA | NA | NA | NA | NA | NA |
| Artemether | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (37%) | > 20 (39%) |
| SJ733 | > 20 (nmi) | > 20 (33%) | > 20 (38%) | 16/9.6 | > 20 (37%) | > 20 (33%) |
| DSM265b | > 20 (nmi) | > 20 (25%) | > 20 (19%) | 7.1/4.4 | > 20 (34%) | CND |
| KAE609 | 4.5/2.7 | 5.5/4.0 | < 0.25/< 0.15 | 5.9/3.7 | > 20 (30%) | > 20 (nmi) |
| OZ439 | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | > 20 (nmi) | 5.1/3.1 | 12/8.6 |
| ELQ300 | > 20 (nmi) | 7.5/5.4 | > 20 (41%) | 8.0/5.0 | > 20 (nmi) | CNDa |
Discussion
The objective of this work was to collect in vitro ADME data using standardized conditions for a set of legacy and development anti-malarial compounds to facilitate discovery and development activities, and more specifically, to enhance modelling and simulation approaches being applied to predictions of human dose, pharmacokinetic profiles and drug–drug interactions. The parameters evaluated included those that represent input parameters for PBPK modelling, namely pKa, Log D7.4, solubility in biorelevant media, effective human intestinal permeability, plasma protein binding, blood to plasma partitioning, unbound intrinsic clearance and inhibition of the major CYP isoforms. In addition, binding values were obtained with media used for in vitro activity assessment for the major parasite assay formats such that intrinsic unbound activity can be compared across platforms and incorporated into pharmacokinetic/pharmacodynamic (PKPD) modelling. Included in the data set were 23 legacy drugs (including three that have been withdrawn due to toxicity issues), two active metabolites (desethylamodiaquine and cycloguanil), and 20 compounds in preclinical (a few which have since been discontinued) or clinical development (including recently introduced compounds, tafenoquine and OZ277).
Methodology considerations
Data for measured pKa and Log D7.4 suggested that the calculated values generated using the ADMET Predictor software provided a reasonable estimation for most compounds, however there were still cases where the calculated values differed significantly from the measured values. Given the importance of these two parameters as key determinants of tissue-to-plasma partitioning ratios in PBPK modelling, the results suggest that measured values for pKa and Log D7.4 should be generated and used whenever possible.
The two most challenging properties to measure were permeability and plasma protein binding due to the broad range of physicochemical properties across the data set and the fact that each of these assays is prone to artefacts for highly lipophilic compounds. For Caco-2 cell permeability, non-specific adsorption to the transport chambers and high retention in the cell monolayer due to the absence of effective sink conditions can result in very poor mass balance and an underestimation of the resulting permeability coefficient [31]. As shown in Fig. 4, many of the compounds in the data set had mass balance values well below 50% when a simple pH 7.4 aqueous buffer was used as the transport medium precluding the measurement of reliable Papp values. However, if plasma was used as the transport medium (with subsequent correction of Papp for the fraction unbound) [31], mass balance was improved to more than 70–80% for most compounds giving Papp values that are more in line with the expected permeation properties based on their molecular and physicochemical properties (as reviewed by [46]).
Given that commonly used in vitro permeability assays were not available at the time that most of the legacy anti-malarials were developed, there are few reports of measured apparent permeability values for these compounds in the literature. Even where values have been reported previously (Additional file 1: Table S12), interlaboratory variation in test conditions and measured Papp values makes it difficult to directly compare results [47]. Notwithstanding these issues, three compounds that have moderate to good solubility and good mass balance in the current studies (e.g. dihydroartemisinin, artemisone, and artesunate, Additional file 1: Table S12) showed similar Papp values compared to those reported previously. For several other compounds that exhibited poor mass balance using a standard aqueous buffer as the transport medium (e.g. naphthoquine, piperaquine, mefloquine, pyronaridine and amodiaquine, Fig. 4), measured Papp values using plasma as the transport buffer were considerably higher than those reported previously (Additional file 1: Table S12). For example, low to moderate Papp values have been reported for piperaquine, mefloquine, and amodiaquine [48, 49] whereas each of these was found to be highly permeable under the revised conditions.
To assess plasma protein binding, three different approaches were used depending on the matrix (e.g. media or plasma). Initially, ultracentrifugation was used for media and plasma based on a method adapted from that previously published by Nakai et al. [36]. Compared to equilibrium dialysis, this method has the advantage of not being plagued by non-specific compound adsorption to a dialysis membrane and is relatively straight forward and quick to conduct. For all media except plasma, the ultracentrifugation method was considered suitable given that the method was shown to remove > 99.9% of the total protein (assessed using the Bradford assay as described previously [50, 51]) and these media do not contain lipoproteins which have variable sedimentation rates [52]. The control of pH for the bicarbonate-buffered media (e.g. plasma, Albumax and DMEM/FCS) was necessary but could be readily achieved by equilibration of the samples and rotor in a suitable CO2 atmosphere (either 5 or 10% depending on the media) prior to sealing the rotor. For plasma, the ultracentrifugation method may potentially underestimate fu for very highly bound compounds or those that associate with lipoproteins due to residual protein in the supernatant fraction following ultracentrifugation [36]. Under the current conditions, supernatant protein concentrations following ultracentrifugation of neat plasma represented only about 0.2% of the total plasma protein concentration (assessed using the Bradford assay as described previously [50, 51]). However, total triglyceride levels (assessed using a colorimetric triglyceride assay kit, GPO-PAP, Roche Diagnostics) in the supernatant were approximately 17% of those in total plasma suggesting that the method does not satisfactorily remove the total lipoprotein pool. This is not likely an issue for many compounds but could be significant for highly lipophilic compounds that associate with the lipoprotein fraction, such as halofantrine [53]. Given these potential limitations, a conservative approach was applied and the UC binding results accepted only if the measured plasma fu values were equal to or greater than 0.1.
For compounds that were more highly bound in plasma (nominally those with fu < 0.1), a RED method was incorporated based on previous publications [39, 40, 54]. To increase the likelihood of being able to measure unbound concentrations in the dialysate, 10% human plasma diluted with pH 7.4 phosphate buffer was used with subsequent correction of the measured fu for the dilution factor [41]. It should be noted that the use of diluted plasma can lead to errors for compounds where the binding is very low as there will be minimal difference between the measured post-dialysis unbound and total concentrations. The use of diluted plasma is also prone to error if a compound is exclusively bound to α-1 acid glycoprotein due to the potential for saturation under the dilute conditions, although as highlighted previously, this situation is not common [40].
For the very highly bound compounds (nominally fu < 0.01), additional measures were taken to reduce the impact of non-specific adsorption to the dialysis membrane and accelerate the attainment of steady state equilibrium. These included (i) the use of a 24-h presaturation period exposing the dialysis unit and membrane to compound concentrations exceeding the expected unbound concentration, (ii) inclusion of a low concentration of compound (similar to the expected unbound concentration) in the dialysate at the start of the dialysis period, and (iii) a 24 h dialysis period [39, 40]. These conditions were considered the most stringent that could be practically incorporated under routine experimental conditions.
In several cases (e.g. for the control compounds propranolol, ketoprofen and warfarin and the anti-malarials ferroquine, KAF156, MMV048, DSM421, SJ733, amodiaquine, OZ277, pyrimethamine, sulfadoxine), values obtained using the RED method were comparable to those measured by UC even though the fu value was less than the conservative cut-off of 0.1 (Additional file 1: Tables S8 and S9). As shown in Additional file 1: Table S9, compounds showing low binding (i.e. fu > 0.1) generally had measured fu values that were in very good agreement with previously reported values (e.g. AQ-13, desethylamodiaquine, chloroquine, clindamycin, dapsone, doxycycline, M5717, primaquine, proguanil, quinine, and sulfamethoxazole). It should be noted that different batches of pooled plasma will introduce a degree of variability in the data even if the results for two methods are comparable.
For several of the more highly bound lipophilic compounds (e.g. DSM265, KAE609, tafenoquine, JPC3210, NPC1161B, MMV052, artemether, piperaquine), considerably lower fu values were obtained using the RED method (in either the 6 or 24 h dialysis format) compared to the UC method (Additional file 1: Table S9). Where literature reported values were obtained using equilibrium dialysis or erythrocyte partitioning methods, the current values using the RED method (6 or 24 h dialysis) were generally consistent with reported results (e.g. amodiaquine, artemether, mefloquine, pyrimethamine, sulfadoxine, Additional file 1: Table S9). In some cases (e.g. DSM265, KAE609, naphthoquine), the extra precautions taken to presaturate the dialysis unit and accelerate the attainment of steady state appeared to be unnecessary as the RED fu values were comparable for the 6 h and 24 h dialysis conditions. However, in other cases (e.g. tafenoquine, ELQ300, NPC1161B, MMV052, piperaquine) the additional measures allowed the measurement of unbound concentrations where they could not be measured without these more extreme conditions. Equally, for several of the highly bound compounds, the RED method incorporating presaturation and a long dialysis period gave considerably lower fu values than those previously reported using other methods (e.g. JPC3210, tafenoquine, lumefantrine, piperaquine, Additional file 1: Table S9).
As pointed out previously [39], it would be preferable to use multiple conditions to confirm convergence of the fu to a common value to provide confidence in the measured result. Ideally, one would also measure fraction unbound using multiple pooled plasma aliquots, however these additional precautions were not practical for the number of compounds examined here. Even with the more conservative presaturation RED method, fu values were still unable to be measured for halofantrine, lumefantrine, OZ439 and TDD-E209, and atovaquone. This could be due to extremely high binding, residual effects of non-specific adsorption, lack of steady state equilibrium under these experimental conditions, or a combination of these factors.
In this work, the binding measurements in microsome and Albumax media were conducted using the ultracentrifugation method, however the RED assay is equally applicable for these media. Issues related to non-specific adsorption and slow equilibration with more lipophilic compounds still need to be considered for these matrices in the same way as for plasma as described above. Out of the 45 compounds in the dataset, roughly half exhibited minimal degradation in hepatic microsomes under the standard conditions used here. Incubations were not extended past the 60 min time point given the risk of decreasing enzyme activity with time [55]. For a subset of compounds, the microsomal protein concentration was increased (to 2 mg/mL) in an attempt to obtain measurable levels of degradation (i.e. > 15%), but this approach was not successful. It is unknown at this stage whether the apparent stability results from inherently low unbound intrinsic clearance, or high microsomal binding, or a combination of the two, however it is noted that of the 22 compounds that showed minimal degradation, 8 were also highly bound to microsomal proteins (and of these 8, 5 had measured or calculated LogD7.4 values > 3), likely giving a false indication of their metabolic stability. These results emphasize the need for improved methods to assess intrinsic clearance for compounds that are highly metabolically stable and/or highly bound to microsomal proteins. Although not assessed as part of this work, additional studies should also be conducted using S9 fraction and hepatocytes to rule out the potential for non-CYP-mediated metabolic liabilities (e.g. due to metabolism by aldehyde or xanthine oxidases, or conjugative biotransformation).
Physicochemical property trends
Consistent with numerous reports in the literature regarding the links between lipophilicity and ADME properties [56–61], there was a notable correlation between several of the measured properties and Log D7.4. As shown in Fig. 3, both solubility and fraction unbound decreased, and permeability and unbound intrinsic clearance increased, with increasing Log D7.4 above a value of about 2. Except for one compound (P218), each of the development compounds had high permeability consistent with their relatively high Log D7.4, and accordingly, many had quite poor solubility in FaSSIF (≤ 100 µg/mL). The solubility-limited absorbable dose (SLAD) was calculated as described previously [32] taking into account both the predicted effective human permeability and the solubility properties. For the development compounds, 11 out of 20 had SLAD values below 400 mg (Table 5).
While the efficacious clinical dose for most of these compounds has not yet been finalized, the SLAD estimations highlight the likelihood that formulation approaches may be necessary to overcome solubility-limited absorption should these compounds continue to progress. However, as highlighted previously [32, 62], the solubility estimates based only on FaSSIF are likely conservative given that most of these compounds are weak bases and therefore, their intestinal solubility will also be impacted by their solubility in gastric fluids which is considerably higher than that in FaSSIF in the majority of cases (Table 3). Most of these compounds also exhibited greatly improved solubility in FeSSIF compared to FaSSIF likely as a result of solubilization by colloidal species present in the medium. This raises the potential for a food effect if the dose is high and if enabling formulations are not used to mitigate the solubility limitations.
Given that the clinical dose for the development compounds is either unknown or not yet fixed, and the solubility properties over the full pH range of 1–7.5 have not been determined, these compounds cannot strictly be classified according to either the Biopharmaceutics Classification System (BCS [33]) or the Biopharmaceutics Drug Disposition Classification System (BDDCS [63]). However, given the available data it is likely that, with the exception of P218, all of the development compounds will fall into either Class I or II based on either the BCS (i.e. high permeability) or the BDDCS (i.e. metabolism as the predominant clearance pathway). In contrast, several of the legacy drugs (e.g. sulfamethoxazole, doxycyclin, azithromycin, proguanil, and cycloguanil) have high polarity (PSA > 75 Å2), low Log D7.4 (Log D < 0), low permeability and high solubility placing them into BCS/BDDCS Class III. This is consistent with several of these compounds being subject to predominantly renal and/or biliary clearance mechanisms.
Case studies relating physicochemical properties to current target product profiles
Current target product profiles for new anti-malarials to treat uncomplicated malaria aim to improve patient compliance through shorter treatment regimens (< 3 days and ideally with a single administration) and maximize efficacy and reduce transmission by maintaining effective concentrations for a period sufficient to achieve a 6–12 Log reduction in parasite burden [64]. This goal places a high burden on the pharmacokinetic properties to deliver the required half-life, and in many cases, this comes at the expense of good physicochemical properties. A high dose may also be required to extend the duration of pharmacological exposure (depending on potency) which further exacerbates issues related to less than ideal physiochemical properties.
One example of the impact of physicochemical properties on duration of exposure is the recently FDA approved 8-aminoquinoline, tafenoquine, designed at the Walter Reed Army Institute of Research (WRAIR) to increase the half-life of the structural analogue, primaquine ([65] and references therein). Primaquine (PQ) and tafenoquine (TQ) are the only available drugs that are effective in treating both pre-erythrocytic and erythrocytic forms of Plasmodium vivax, including the relapsing hypnozoite form. Compared to PQ which requires daily administration for 14 days, TQ achieves similar efficacy with only a single dose, representing a significant improvement with respect to dosing convenience. Physicochemical and pharmacokinetic properties of TQ and PQ are summarized in Table 10. The considerably longer half-life of TQ (~ 15 days) compared to PQ (~ 7 h) results from its higher lipophilicity, higher plasma protein binding, higher apparent oral volume of distribution, and lower apparent oral clearance (resulting from reduced free concentrations due to high binding). Not surprisingly, the solubility of TQ free base in FaSSIF is considerably lower than that for PQ, but TQ solubility increases considerably in FeSSIF, consistent with the known increase in exposure of TQ when administered with food. The absorption of TQ is not compromised by the lower solubility due to its formulation as the succinate salt to improve solubility/dissolution properties and recommendation that it is administered with food [66].
Table 10.
Physicochemical and pharmacokinetic properties for selected anti-malarials
| Property | 8-Aminoquinolines | Peroxides | |||
|---|---|---|---|---|---|
| Primaquinea | Tafenoquinea | DHAb | OZ277b | OZ439b | |
| Log D7.4 | 0.54 | 4.24 | 2.3 | 2.6 | > 5 |
| Plasma fu | 0.26 | 0.0007 | 0.21 | 0.086 | < 0.0001 |
| FaSSGF solubility (µg/mL) | > 2000 | > 2000 | 140 | > 2000 | 79 |
| FaSSIF solubility (µg/mL) | > 2000 | 15.8 | 159 | > 2000 | 40.6 |
| FeSSIF solubility (µg/mL) | > 2000 | 1310 | 246 | > 2000 | 526 |
| Half-life (h) | 7 | 360 | 1 | 3 | > 40 |
| V/F (L) | 277 | 1600 | 385 | 929 | 1570 |
| CL/F (L/h) | 28 | 3.0 | 272 | 184 | 41 |
| Food effect | no | yes | no | no | yes |
A second example of the link between half-life and physicochemical properties is the synthetic ozonide, OZ439 [67]. Similar to the artemisinin derivatives and the first generation ozonide, OZ277 ([68]), OZ439 contains a relatively unique peroxide pharmacophore which is responsible for its potent and fast acting activity on all erythrocytic stages of P. falciparum and P. vivax. Physicochemical and pharmacokinetic properties of DHA, OZ277, and OZ439 are summarized in Table 10. Both dihydroartemisinin (DHA) and OZ277 suffer from a short in vivo half-life of approximately 1 h or 3 h, respectively, necessitating a 3-day treatment regimen for each (in combination with a longer acting partner drug). For both compounds, this short half-life is due in part to rapid breakdown of the peroxide moiety in blood as described previously [67, 69]. As a result of the substantially higher lipophilicity, higher plasma protein binding, higher apparent oral volume of distribution, and lower apparent oral clearance (resulting from a combination of higher plasma protein binding and reduced blood-mediated degradation [67]), OZ439 has a considerably longer half-life (> 40 h) than either DHA or OZ277. The improved half-life of OZ439 comes at the expense of solubility, resulting in an increase in exposure when administered with food [70] and significant formulation challenges [71]. This is further confounded by the need for a relatively high dose (> 500 mg) to achieve parasite clearance with a single administration.
Conclusions
The methods used in these studies have been designed to provide the necessary in vitro data to support PBPK modelling activities for new anti-malarials and address practical issues common to several of the assays used for this purpose. The work highlights the challenges that are often encountered with compounds that cover a wide range of physicochemical characteristics and emphasizes that for many of these platforms, it is unlikely that a single method format will be universally applicable to all compounds. Two other useful platforms that have not been included in this work are the assessment of CYP and UGT reaction phenotyping, and time-dependent CYP inhibition as both of these are important for predicting potential drug–drug interactions. Methods and conditions for these studies are well described in the literature (see reviews [72] for reaction phenotyping and [73] for time-dependent inhibition). Further work is needed to develop suitable and practical methods that can be used to estimate human intrinsic clearance and metabolic pathways for highly bound, highly stable compounds since the standard methodology is often unsuitable for this purpose.
In recent years there has been an increased focus on the discovery of anti-malarial compounds and combination treatments that can be given as a single oral dose to improve patient compliance and reduce treatment costs compared to the current 3-day dosing regimens for most anti-malarials [4, 64]. While the benefits of this goal are clear, there is an associated requirement for an extended duration of pharmacological exposure to achieve a 6–12 Log reduction in parasitaemia [64]. Furthermore, both components of a combination treatment need to have matched durations of coverage to avoid exposing parasites to suboptimal concentrations of a single agent which would facilitate the development of resistance [74]. This requirement means that compounds need to have very low clearance (as a result of low unbound intrinsic clearance rather than high protein binding) and a moderate to high volume of distribution (typically driven by increasing lipophilicity, the introduction of one or more basic centres, or a combination of the two [75]) to achieve a long half-life. Depending on the potency, a relatively high dose may also be necessary to maintain exposure for the required duration. Such a high total exposure also increases the need for a wide safety margin. Given these challenges, predictive modelling tools are likely to play an increasing role in identifying risks and developing early mitigation strategies in late stage discovery and translational development of new anti-malarial drugs [16].
As illustrated by the current data set and the two examples given, there has been a trend toward the discovery of more lipophilic compounds to drive long half-life. In contrast to several of the legacy compounds which low Log D7.4 and low to moderate permeability, only one of the development compounds was poorly permeable suggesting that neither passive permeability or transporters are likely to limit oral absorption or hepatic elimination of the development compounds [63, 76]. It is unsurprising that these physicochemical trends come at the expense of good aqueous solubility in several cases. This emphasizes that there is significant scope, and substantial need, for the development of alternative formulation and delivery approaches to address solubility-limited absorption which are cost effective, stable under the harsh environmental conditions of climatic zone 4, and which can be used across all patient populations, including children and infants. The emphasis of current discovery projects is to achieve an extended duration of exposure by maximizing potency (to maintain a low effective dose) and minimizing unbound intrinsic clearance (to extend the half-life) without compromising physicochemical properties such as solubility, and several compounds currently in clinical development fulfil these objectives.
Supplementary information
Acknowledgements
The advice and input from Drs. David Wesche and Nathalie Gobeau are gratefully acknowledged. We also acknowledge the Pharma partners including Sanofi, Merck KGaA, Novartis, Zydus Pharmaceuticals, Shin Poong Pharmaceutical Co., Ltd. and Jacobus Pharmaceutical Company and project leaders and teams for supporting the publication of this work.
Abbreviations
- ACT
artemisinin-based combination therapy
- PBPK
physiologically-based pharmacokinetic
- ICH
International Conference on Harmonization
- VIS
volunteer infection study
- pKa
ionization constant
- Log D7.4
Logarithm of the octanol/pH 7.4 buffer partition coefficient
- FaSSIF
fasted-state simulated intestinal fluid
- FeSSIF
fed-state simulated intestinal fluid
- FaSSGF
fasted-state simulated gastric fluid
- PBS
phosphate buffered saline
- SD
standard deviation
- S.E.
standard error
- Papp
apparent permeability coefficient
- Peff
effective human jejunal permeability coefficient
- V
fluid volume of small intestine
- SSI
solubility in small intestine
- Mp
permeability multiplier
- An
absorption number
- tres
residence time in small intestine
- R
radius of small intestine
- SLAD
solubility limited absorbable dose
- DMEM
Dulbecco’s modified Eagle’s medium
- UC
ultracentrifugation
- RED
rapid equilibrium dialysis
- fu
fraction unbound
- Ctotal
total concentration in plasma or medium
- Cunbound
unbound concentration in plasma or medium
- k
apparent first order rate constant for substrate depletion
- HLM
human liver microsome
- CLint
intrinsic clearance
- CYP
cytochrome P450
- IC50
concentration resulting in 50% inhibition of CYP activity
- Km
Michaelis–Menten constant
- Ki
enzyme inhibition constant
Authors’ contributions
SAC analysed and interpreted data and wrote the manuscript; NA and JNB reviewed data and contributed to manuscript preparation and review; JJM, LA, MD and DAS contributed to manuscript preparation and review; AA and HB measured ionization constants; HB measured partition coefficients and solubility; SB and ER conducted microsomal stability assays; AC, EC, RP, JS developed and validated LC–MS methods and analysed samples; MC developed methods, measured physicochemical properties and interpreted data; FCKC developed and validated bioanalytical methods, reviewed and interpreted data; GC and KK developed methods for permeability and binding; KK, GC and TP conducted permeability, binding and CYP inhibition studies; JM developed and validated LC–MS methods and conducted solubility experiments; DMS developed methods, analysed and interpreted data and contributed to manuscript preparation; KLW collated and reviewed data and contributed to manuscript preparation and review. All authors read and approved the final manuscript.
Funding
This work was supported by the Medicines for Malaria Venture donors. The Centre for Drug Candidate Optimisation is partially supported by the Monash University Technology Research Platform network and Therapeutic Innovation Australia (TIA) through the Australian Government National Collaborative Research Infrastructure Strategy (NCRIS) program.
Availability of data and materials
All data and additional information are provided in the manuscript or additional information.
Ethics approval and consent to participate
Protocols using human liver microsomes (from a commercial source) and human blood and plasma were reviewed by the Monash University Human Research Ethics Committee and granted exemption on the basis that donors and associated data were non-identifiable. Consent to participate was not required.
Consent for publication
No personal data for any individual is included in the manuscript.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12936-019-3075-5.
References
- 1.WHO . World malaria report 2015. Geneva: World Health Organization; 2015. [Google Scholar]
- 2.WHO . World malaria report 2018. Geneva: World Health Organization; 2018. [Google Scholar]
- 3.Alonso P, Noor AM. The global fight against malaria is at crossroads. Lancet. 2017;390:2532–2534. doi: 10.1016/S0140-6736(17)33080-5. [DOI] [PubMed] [Google Scholar]
- 4.Wells TN, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 5.Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–169. doi: 10.1007/s40495-016-0059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247–262. doi: 10.1002/cpt.37. [DOI] [PubMed] [Google Scholar]
- 7.Jones HM, Dickins M, Youdim K, Gosset JR, Attkins NJ, Hay TL, et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica. 2012;42:94–106. doi: 10.3109/00498254.2011.627477. [DOI] [PubMed] [Google Scholar]
- 8.Jones RD, Jones HM, Rowland M, Gibson CR, Yates JW, Chien JY, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, Part 2: comparative assessment of prediction methods of human volume of distribution. J Pharm Sci. 2011;100:4074–4089. doi: 10.1002/jps.22553. [DOI] [PubMed] [Google Scholar]
- 9.Marshall S, Madabushi R, Manolis E, Krudys K, Staab A, Dykstra K, et al. Model-informed drug discovery and development: current industry good practice and regulatory expectations and future perspectives. CPT Pharmacomet Syst Pharmacol. 2019;8:87–96. doi: 10.1002/psp4.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poulin P, Jones RD, Jones HM, Gibson CR, Rowland M, Chien JY, et al. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, Part 5: prediction of plasma concentration-time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J Pharm Sci. 2011;100:4127–4157. doi: 10.1002/jps.22550. [DOI] [PubMed] [Google Scholar]
- 11.FDA briefing document: Pharmaceutical science and clinical pharmacology advisory committee meeting. Brief. Doc. Silver Spring: U.S. Food and Drug Administration; 2017.
- 12.Clinical pharmacology. Guidance for Industry. Physiologically based pharmacokinetic analyses—format and content. Silver Spring: U.S. Food and Drug Administration; 2018.
- 13.Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. London: European Medicines Agency; 2018.
- 14.Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the US Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108:21–25. doi: 10.1016/j.xphs.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 15.Marshall SF, Burghaus R, Cosson V, Cheung SY, Chenel M, DellaPasqua O, et al. Good practices in model-informed drug discovery and development: practice, application, and documentation. CPT Pharmacomet Syst Pharmacol. 2016;5:93–122. doi: 10.1002/psp4.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews KA, Wesche D, McCarthy J, Mohrle JJ, Tarning J, Phillips L, et al. Model-informed drug development for malaria therapeutics. Annu Rev Pharmacol Toxicol. 2018;58:567–582. doi: 10.1146/annurev-pharmtox-010715-103429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jimenez-Diaz MB, Mulet T, Viera S, Gomez V, Garuti H, Ibanez J, et al. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. 2009;53:4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engwerda CR, Minigo G, Amante FH, McCarthy JS. Experimentally induced blood stage malaria infection as a tool for clinical research. Trends Parasitol. 2012;28:515–521. doi: 10.1016/j.pt.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy JS, Marquart L, Sekuloski S, Trenholme K, Elliott S, Griffin P, et al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrob Agents Chemother. 2016;60:3669–3675. doi: 10.1128/AAC.02883-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy JS, Sekuloski S, Griffin PM, Elliott S, Douglas N, Peatey C, et al. A pilot randomised trial of induced blood-stage Plasmodium falciparum infections in healthy volunteers for testing efficacy of new antimalarial drugs. PLoS ONE. 2011;6:e21914. doi: 10.1371/journal.pone.0021914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanisic DI, McCarthy JS, Good MF. Controlled human malaria infection: applications, advances, and challenges. Infect Immun. 2018;86:e00479–e00517. doi: 10.1128/IAI.00479-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krause A, Dingemanse J, Mathis A, Marquart L, Mohrle JJ, McCarthy JS. Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion-451840 in an induced blood stage malaria study in healthy subjects. Br J Clin Pharmacol. 2016;82:412–421. doi: 10.1111/bcp.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarthy James S., Baker Mark, O'Rourke Peter, Marquart Louise, Griffin Paul, Hooft van Huijsduijnen Rob, Möhrle Jörg J. Efficacy of OZ439 (artefenomel) against earlyPlasmodium falciparumblood-stage malaria infection in healthy volunteers. Journal of Antimicrobial Chemotherapy. 2016;71(9):2620–2627. doi: 10.1093/jac/dkw174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarthy JS, Lotharius J, Ruckle T, Chalon S, Phillips MA, Elliott S, et al. Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human Phase 1a/1b randomised study. Lancet Infect Dis. 2017;17:626–635. doi: 10.1016/S1473-3099(17)30171-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy JS, Ruckle T, Djeriou E, Cantalloube C, Ter-Minassian D, Baker M, et al. A Phase II pilot trial to evaluate safety and efficacy of ferroquine against early Plasmodium falciparum in an induced blood-stage malaria infection study. Malar J. 2016;15:469. doi: 10.1186/s12936-016-1511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FDA Guidance for Industry . Clinical drug interaction studies - study design, data analysis and clinical implications. Silver Spring: Food Drug Administration; 2017. [Google Scholar]
- 27.Wagner C, Pan Y, Hsu V, Grillo JA, Zhang L, Reynolds KS, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54:117–127. doi: 10.1007/s40262-014-0188-4. [DOI] [PubMed] [Google Scholar]
- 28.Wagner C, Zhao P, Pan Y, Hsu V, Grillo J, Huang SM, Sinha V. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacomet Syst Pharmacol. 2015;4:226–230. doi: 10.1002/psp4.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albert A, Serjeant EP. The determination of ionization constants. London: Chapman and Hall; 1984. [Google Scholar]
- 30.Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25:1663–1676. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 31.Katneni K, Pham T, Saunders J, Chen G, Patil R, White KL, et al. Using human plasma as an assay medium in Caco-2 studies improves mass balance for lipophilic compounds. Pharm Res. 2018;35:210. [DOI] [PMC free article] [PubMed]
- 32.Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci. 2010;99:4940–4954. doi: 10.1002/jps.22217. [DOI] [PubMed] [Google Scholar]
- 33.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 34.Dahlgren D, Roos C, Sjogren E, Lennernas H. Direct in vivo human intestinal permeability (Peff) determined with different clinical perfusion and intubation methods. J Pharm Sci. 2015;104:2702–2726. doi: 10.1002/jps.24258. [DOI] [PubMed] [Google Scholar]
- 35.Lennernas H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica. 2007;37:1015–1051. doi: 10.1080/00498250701704819. [DOI] [PubMed] [Google Scholar]
- 36.Nakai D, Kumamoto K, Sakikawa C, Kosaka T, Tokui T. Evaluation of the protein binding ratio of drugs by a micro-scale ultracentrifugation method. J Pharm Sci. 2004;93:847–854. doi: 10.1002/jps.20012. [DOI] [PubMed] [Google Scholar]
- 37.Plise EG, Tran D, Salphati L. Semi-automated protein binding methodology using equilibrium dialysis and a novel mixed-matrix cassette approach. J Pharm Sci. 2010;99:5070–5078. doi: 10.1002/jps.22188. [DOI] [PubMed] [Google Scholar]
- 38.Shackleford DM, Jamsen KM. Quantifying uncertainty in the ratio of two measured variables: a recap and example. J Pharm Sci. 2016;105:3462–3463. doi: 10.1016/j.xphs.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 39.Di L, Breen C, Chambers R, Eckley ST, Fricke R, Ghosh A, et al. Industry perspective on contemporary protein-binding methodologies: considerations for regulatory drug–drug interaction and related guidelines on highly bound drugs. J Pharm Sci. 2017;106:3442–3452. doi: 10.1016/j.xphs.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Riccardi K, Cawley S, Yates PD, Chang C, Funk C, Niosi M, et al. Plasma protein binding of challenging compounds. J Pharm Sci. 2015;104:2627–2636. doi: 10.1002/jps.24506. [DOI] [PubMed] [Google Scholar]
- 41.Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 42.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 43.Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32:647–660. doi: 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- 44.Settimo L, Bellman K, Knegtel RM. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm Res. 2014;31:1082–1095. doi: 10.1007/s11095-013-1232-z. [DOI] [PubMed] [Google Scholar]
- 45.Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, et al. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res. 2002;19:1400–1416. doi: 10.1023/A:1020483911355. [DOI] [PubMed] [Google Scholar]
- 46.van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 47.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- 48.Crowe A, Ilett KF, Karunajeewa HA, Batty KT, Davis TM. Role of P glycoprotein in absorption of novel antimalarial drugs. Antimicrob Agents Chemother. 2006;50:3504–3506. doi: 10.1128/AAC.00708-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Senarathna SM, Page-Sharp M, Crowe A. The interactions of P-Glycoprotein with antimalarial drugs, including substrate affinity, inhibition and regulation. PLoS ONE. 2016;11:e0152677. doi: 10.1371/journal.pone.0152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 51.Zor T, Selinger Z. Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal Biochem. 1996;236:302–308. doi: 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- 52.Redgrave TG, Roberts DC, West CE. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal Biochem. 1975;65:42–49. doi: 10.1016/0003-2697(75)90488-1. [DOI] [PubMed] [Google Scholar]
- 53.McIntosh MP, Porter CJ, Wasan KM, Ramaswamy M, Charman WN. Differences in the lipoprotein binding profile of halofantrine in fed and fasted human or beagle plasma are dictated by the respective masses of core apolar lipoprotein lipid. J Pharm Sci. 1999;88:378–384. doi: 10.1021/js980152g. [DOI] [PubMed] [Google Scholar]
- 54.Curran RE, Claxton CR, Hutchison L, Harradine PJ, Martin IJ, Littlewood P. Control and measurement of plasma pH in equilibrium dialysis: influence on drug plasma protein binding. Drug Metab Dispos. 2011;39:551–557. doi: 10.1124/dmd.110.036988. [DOI] [PubMed] [Google Scholar]
- 55.Jones HM, Houston JB. Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos. 2004;32:973–982. doi: 10.1124/dmd.104.000125. [DOI] [PubMed] [Google Scholar]
- 56.Gleeson MP, Hersey A, Montanari D, Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 58.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 59.Meanwell NA. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24:1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- 60.Waring MJ. Lipophilicity in drug discovery. Expert Opin Drug Discov. 2010;5:235–248. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- 61.Smith DA, Allerton C, Kalgutkar AS, van de Waterbeemd H, Walker DK, editors. Pharmacokinetics and metabolism in drug design. 3. Weinheim: Wiley; 2012. [Google Scholar]
- 62.Rosenberger J, Butler J, Dressman J. A refined developability classification system. J Pharm Sci. 2018;107:2020–2032. doi: 10.1016/j.xphs.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 63.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 64.Burrows JN, Duparc S, Gutteridge WE, Hooft van Huijsduijnen R, Kaszubska W, Macintyre F, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017;16:26. [DOI] [PMC free article] [PubMed]
- 65.Frampton JE. Tafenoquine: first global approval. Drugs. 2018;78:1517–1523. doi: 10.1007/s40265-018-0979-2. [DOI] [PubMed] [Google Scholar]
- 66.GlaxoSmithKline. KRINTAFEL (tafenoquine) tablets, for oral use: US prescrbing information. 2018.https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210795s000lbl.pdf. Access 20 Oct 2019.
- 67.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci USA. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 69.Lindegardh N, Hanpithakpong W, Kamanikom B, Pattayaso J, Singhasivanon P, White NJ, Day NP. Quantification of dihydroartemisinin, artesunate and artemisinin in human blood: overcoming the technical challenge of protecting the peroxide bridge. Bioanalysis. 2011;3:1613–1624. doi: 10.4155/bio.11.158. [DOI] [PubMed] [Google Scholar]
- 70.Moehrle JJ, Duparc S, Siethoff C, van Giersbergen PLM, Craft JC, Arbe-Barnes S, et al. First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol. 2013;75:524–548. doi: 10.1111/j.1365-2125.2012.04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salim M, Khan J, Ramirez G, Clulow AJ, Hawley A, Ramachandruni H, Boyd BJ. Interactions of artefenomel (OZ439) with milk during digestion: insights into digestion-driven solubilization and polymorphic transformations. Mol Pharm. 2018;15:3535–3544. doi: 10.1021/acs.molpharmaceut.8b00541. [DOI] [PubMed] [Google Scholar]
- 72.Zientek MA, Youdim K. Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos. 2015;43:163–181. doi: 10.1124/dmd.114.058750. [DOI] [PubMed] [Google Scholar]
- 73.Grimm SW, Einolf HJ, Hall SD, He K, Lim HK, Ling KH, et al. The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America. Drug Metab Dispos. 2009;37:1355–1370. doi: 10.1124/dmd.109.026716. [DOI] [PubMed] [Google Scholar]
- 74.White NJ. Pharmacokinetic and pharmacodynamic considerations in antimalarial dose optimization. Antimicrob Agents Chemother. 2013;57:5792–5807. doi: 10.1128/AAC.00287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith DA, Beaumont K, Maurer TS, Di L. Volume of distribution in drug design. J Med Chem. 2015;58:5691–5698. doi: 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- 76.Smith DA, Beaumont K, Maurer TS, Di L. Clearance in drug design. J Med Chem. 2019;62:2245–2255. doi: 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- 77.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Phillips MA, White KL, Kokkonda S, Deng X, White J, El Mazouni F, et al. A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect Dis. 2016;2:945–957. doi: 10.1021/acsinfecdis.6b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Phillips Margaret A., Lotharius Julie, Marsh Kennan, White John, Dayan Anthony, White Karen L., Njoroge Jacqueline W., El Mazouni Farah, Lao Yanbin, Kokkonda Sreekanth, Tomchick Diana R., Deng Xiaoyi, Laird Trevor, Bhatia Sangeeta N., March Sandra, Ng Caroline L., Fidock David A., Wittlin Sergio, Lafuente-Monasterio Maria, Benito Francisco Javier Gamo, Alonso Laura Maria Sanz, Martinez Maria Santos, Jimenez-Diaz Maria Belen, Bazaga Santiago Ferrer, Angulo-Barturen Iñigo, Haselden John N., Louttit James, Cui Yi, Sridhar Arun, Zeeman Anna-Marie, Kocken Clemens, Sauerwein Robert, Dechering Koen, Avery Vicky M., Duffy Sandra, Delves Michael, Sinden Robert, Ruecker Andrea, Wickham Kristina S., Rochford Rosemary, Gahagen Janet, Iyer Lalitha, Riccio Ed, Mirsalis Jon, Bathhurst Ian, Rueckle Thomas, Ding Xavier, Campo Brice, Leroy Didier, Rogers M. John, Rathod Pradipsinh K., Burrows Jeremy N., Charman Susan A. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Science Translational Medicine. 2015;7(296):296ra111–296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guidance for Industry . Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Silver Spring: U.S. Food and Drug Administration; 2015. [Google Scholar]
- 81.O’Neill PM, Amewu RK, Charman SA, Sabbani S, Gnadig NF, Straimer J, et al. A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance. Nat Commun. 2017;8:15159. doi: 10.1038/ncomms15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brueckner RP, Lasseter KC, Lin ET, Schuster BG. First-time-in-humans safety and pharmacokinetics of WR 238605, a new antimalarial. Am J Trop Med Hyg. 1998;58:645–649. doi: 10.4269/ajtmh.1998.58.645. [DOI] [PubMed] [Google Scholar]
- 83.Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenridge AM. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br J Clin Pharmacol. 1984;17:441–446. doi: 10.1111/j.1365-2125.1984.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Na-Bangchang K, Krudsood S, Silachamroon U, Molunto P, Tasanor O, Chalermrut K, et al. The pharmacokinetics of oral dihydroartemisinin and artesunate in healthy Thai volunteers. Southeast Asian J Trop Med Public Health. 2004;35:575–582. [PubMed] [Google Scholar]
- 85.Saha N, Moehrle JJ, Zutshi A, Sharma P, Kaur P, Iyer SS. Safety, tolerability and pharmacokinetic profile of single and multiple oral doses of arterolane (RBx11160) maleate in healthy subjects. J Clin Pharmacol. 2014;54:386–393. doi: 10.1002/jcph.232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and additional information are provided in the manuscript or additional information.