

Abstract
In recent years, functional polymeric compounds have been widely used to modify the silica surface, which allows one to obtain the corresponding organomineral composites for broad application prospects. In this case, lignin—a cross-linked polyphenolic macromolecule—is of great interest according to its valuable properties and possible surplus as a by-product of pulp and paper industry and various biorefinery processes. Hybrid materials based on kraft softwood lignin and silica were obtained via the electrostatic attraction of oxidized lignin to the aminosilica surface with different porosities, which were prepared by the amination of the commercial silica gel with an average pore diameter of 6 nm, and the silica prepared in the lab with the oxidized kraft lignin and lignin–silica samples with an average pore diameter of 38 nm was investigated by physicochemical methods: two-dimensional nuclear magnetic resonance (NMR), 31P NMR, Fourier transform infrared spectroscopy, thermogravimetric analysis in nitrogen and air atmosphere, scanning electron microscopy, and adsorption methods. After oxidation, the content of carboxylic groups almost doubled in the oxidized lignin, compared to that in the native one (0.74 mmol/g against 0.44 mmol/g, respectively). The lignin content was deposited onto the surface of aminosilica, depending on the porosity of the silica material and on the content of amino groups on its surface, giving lignin–aminosilica with 20% higher lignin content than the lignin–aminosilica gel. Both types of lignin–silica composites demonstrate a high sorptive capacity toward crystal violet dye. The suggested approach is an easy and low-cost way of synthesis of lignin–silica composites with unique properties. Such composites have a great potential for use as adsorbents in wastewater treatment processes.
1. Introduction
Kraft lignin, one of the major types of technical lignins, is a biomacromolecule characterized by relatively low molecular weight, variable functional group contents, high polydispersity, and by the presence of some amount of thiol groups in the aliphatic chains.1−3 This type of technical lignin is obtained as a result of chemical fragmentation and condensation reaction occurring during kraft pulping processes.4 Kraft lignin is a prosperous material for the development of well-designed biomaterials, biodegradable films, and composites for potential industrial applications.4−10 The LignoBoost lignin process for partial removal of kraft lignin from black liquor was developed by Innventia (Sweden) together with Chalmers and has offered a pure from ash lignin material with reproducible molecular weight characteristics for various commercial applications, for example, in the building, construction, and automotive sectors, where lignin offers sustainable alternatives for phenols in plywood glues and other wood-based panels, and polyols used in foams.11,12
Despite the high content of functional groups in the lignin structure, lignin activation is needed to increase its solubility or reactivity for the development of new functional materials.13 Oxidation of kraft lignin could be used to address both the issues. Among different oxidizing agents, for example, nitric acid,14 sodium periodate,15−19 metal oxides,20 nitrobenzene,20 and oxygen with catalysts,21 hydrogen peroxide is of greatest interest as it is common and broadly available as an oxidizing agent in pulp industry.22 Recently, several works described lignin oxidation by hydrogen peroxide under different conditions.22−24 It was reported that temperature and pH play a crucial role in the oxidation process. Adjustment of these parameters led to obtaining the controlled cracking of lignin macromolecules and the needed degree of dearomatization in the modified lignin. For instance, at moderate temperatures (80–160 °C), mono- and dicarboxylic acids are the main products of the cracking reaction, with the yields of these products in the range of 30–50% of initial lignin. A change in the acidity of the reaction results in a change in the reaction mechanism and the product distribution.23
It was proved that the modified kraft lignin could be utilized effectively as a component of organic–inorganic composites, where the inorganic carrier would get the needed functionality and the lignin macromolecule could improve its physicochemical characteristics such as textural, morphological, thermal, and adsorptional abilities.15−19,25−31 Silica–lignin composites were obtained in the process of aminated silica surface modification with an oxidized lignin solution.15,16,18,28 The synthesized materials were tested as sorbents for pollutant removal from aqueous solutions.
In our previous paper, we discussed the method of kraft lignin-based hybrid material synthesis by the sol–gel method, where lignin was activated by aminosilane for increasing its reactivity and possibility to be involved into the created silica network.32 In the current paper, we present a simpler method for lignin deposition on the silica surface, namely by coating aminated silica with oxidized kraft lignin, where hydrogen peroxide was applied as the oxidizing agent. To estimate the influence of the initial silica structure on the thermal and structural characteristics of lignin–silica hybrids, two types of silica, commercial silica gel and silica synthesized from fumed silica, with different specific surface areas and pore sizes were used.
2. Materials and Methods
2.1. Materials
LignoBoost (LB) softwood kraft lignin, kindly supplied by a plant in Northern Europe, was used in this study. The molecular weight of LB was approximately 5600 Da.32 Hydrogen peroxide (35%), glacial acetic acid, ethanol, and 1,4-dioxane were purchased from Sigma-Aldrich. Silica gel was purchased from Merck. The particle size for silica gel was 0.2–0.5 mm, whereas the pore average diameter was 6 nm. Silica was obtained from fumed silica according to the procedure described in ref (33). All the chemicals were of reagent grade. Distilled water was used for aqueous solution preparations.
2.2. Methods
2.2.1. Oxidation of Lignin
A 0.5 mL of 35% hydrogen peroxide water solution and 0.25 mL of glacial acetic acid were added to lignin solution in the dioxane/water (4:1) mixture. The mixture was heated at 50 °C for 35 min. After this time, the mixture was cooled at room temperature. The solvents were evaporated under reduced pressure. In order to evaporate water, EtOH was added and then evaporated again. Brown oil was formed, which was washed with distilled water two times (to remove acetic acid and hydrogen peroxide). The sample was dried overnight at 55 °C. The Mass yield of oxidized lignin (LB–COOH) was 350 mg.
2.2.2. Synthesis of Aminosilica
The synthesis of aminosilica was conducted as described by Tertykh and Belyakova (1991).34 Briefly, aminated silica/silica gel (aminosilica/aminosilica gel) was obtained by the silica modification with 3-aminopropyltriethoxysilane in the toluene medium. The content of amino groups was 0.0020 and 0.0047 m2/g for the aminated silica gel and silica, respectively.
2.2.3. Synthesis of Lignin–Silica Hybrid Composites
LB–COOH (1 g) after washing was dissolved in 50 mL of dioxane/water (4:1) mixture and then added to aminosilica gel or aminosilica in the lignin/aminosilica gel mass ratios of 1:1 (LSG1) and 1:2 (LGS2) and the lignin/aminosilica mass ratios of 1:1 (LS1) and 1:2 (LS2) for 2 h (Table 1). The samples were filtered and washed two times by dioxane and three times by water to remove the dioxane residues. The samples were dried at 50 °C overnight in a vacuum oven.
Table 1. Abbreviations for the Samples.
| sample/mass ratio | short name |
|---|---|
| lignin–silica gel/1:1 | LSG1 |
| lignin–silica gel/1:2 | LSG2 |
| lignin–silica/1:1 | LS1 |
| lignin–silica/1:2 | LS2 |
2.3. Analysis
2.3.1. Nuclear Magnetic Resonance Spectroscopy
31P NMR; the content of functional groups in the lignin samples was measured by 31P NMR.35 Approximately 20–30 mg of the lignin sample was weighed and phosphitylated using 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane. Endo-N-hydroxy-5-norbornene-2,3-dicarboximide (Sigma-Aldrich, 40 mg/mL) and chromium (III) acetylacetonate (Aldrich, 5 mg/mL) were used as an internal standard and a relaxation reagent, respectively. The derivatized sample was dissolved in CDCl3 prior to analysis. The 31P NMR experiment was performed with a 90° pulse angle, inverse-gated proton decoupling, and a delay time of 10 s. For analysis, 256 scans with a time delay of 6 s and a total runtime of 34 min were collected. Measurements were performed in duplicate.
2.3.2. 2D Heteronuclear Single-Quantum Coherence Nuclear Magnetic Resonance
Approximately 100 mg of the sample was acetylated for better solubility36 The residue was dissolved in 700 μL of dimethyl sulfoxide (DMSO)-d6. The two-dimensional heteronuclear single-quantum coherence (2D HSQC) nuclear magnetic resonance (NMR) spectrum was acquired using the Bruker pulse program “hsqcetgpsi”, with a relaxation delay of 1.7 s, a coupling constant of 145 Hz, an INEPT transfer delay time of 1.72 ms (d4 = 1/4J), a spectral window of 10.5 ppm in F2 and 166 ppm in F1 with 1024 × 512 increments, 240 scans per increment, and a spectral center set at 90.0 ppm in F1 and 5.3 ppm in F2. The 2D NMR data set was processed with 2k × 1k data points using a π/2-shifted sine-bell window function in both dimensions. Central DMSO (δC/δH = 39.5/2.5 ppm) was used as an internal reference according to the solvent adopted.
2.3.3. Fourier Transform Infrared Spectroscopy
IR spectra were collected using a PerkinElmer spectrophotometer [Spotlight 400 Fourier transform infrared (FTIR) imaging system, Waltham, MA, USA] equipped with a Spectac MKII Golden Gate system (Creekstone Ridge, GA, USA). The samples were analyzed in the range of 600–4000 cm–1 with 16 scans at a 4 cm–1 resolution and a 1 cm–1 interval at room temperature.
2.3.4. Thermogravimetric Analysis
Thermogravimetric analysis (TGA) was carried out on a TGA/DSC 1 (Mettler Toledo) instrument under the following operational conditions: heating rate of 10 °C min–1, dynamic atmosphere of synthetic air or nitrogen (50 mL min–1), temperature range of 30–900 °C, and sample mass of ∼2.5 mg.
2.3.5. Surface Area and Average Pore Size Measurements
The specific surface areas and pore volumes were determined from the low-temperature nitrogen adsorption data (automatic sorption analyzer, ASAP 2420, Micromeritics, USA). The samples were degassed at 77 K before measurement.
2.3.6. Scanning Electron Microscopy
The structural characteristics of the fabricated samples were studied with a field-emission scanning electron microscope (S-4800, Hitachi, Japan). The samples were coated with a 1 nm thick Pt–Pd layer sputtered with a Cressington 208HR high-resolution coater.
3. Results and Discussion
3.1. Lignin Oxidation
Oxidation of lignin consists of the oxidative cracking reaction involving the cleavage of the lignin ring, aryl-ether bond, or other linkages within lignin.23 The distribution of the reaction product content could vary according to the applied conditions to the oxidizing process. It was shown by Xiang and Lee23 that 30–50% yields of mono- and dicarboxylic acids as the main products of initial lignin could be reached under strong alkaline conditions, which proceeds even at low reaction temperatures (80–90 °C). Under acidic conditions, 130–160 °C is required to attain the same degree of cracking.
In our study LB kraft lignin was successfully oxidized by hydrogen peroxide in acidic media under mild thermal conditions (50 °C). As a result, the benzene rings in lignin macromolecules were opened and transformed into carboxylate groups, resulting in the formation of muconic acid-type structure (Figure 1).22,37−40
Figure 1.

Scheme of lignin oxidation in acidic conditions.
The number of phenolic, aliphatic hydroxyl groups, and carboxyl groups in the initial and oxidized LB was determined from the 31P NMR spectra (Figure 2) according to the method presented by Argyropoulos in ref (35). Table 2 shows the total content of functional groups in the lignin samples before and after H2O2 oxidation. The obtained results confirmed the oxidation of lignin, resulting in the transformation of phenolic groups to carboxylic groups. The number of carboxyl groups increased from 0.44 to 0.74 mmol/g after oxidation. From the other side, the number of phenolic OH decreased from 4.29 to 4.00 mmol/g.
Figure 2.

31P NMR spectra of initial (a) LB kraft lignin and (b) oxidized LB lignin.
Table 2. Contents of the Functional Groups in Initial LB Kraft Lignin and Oxidized LB Measured by 31P NMR (Internal Standard Peak—151 ppm).
| content
of functional groups, mmol g–1 |
|||||||
|---|---|---|---|---|---|---|---|
| phenolic
OH |
aliphatic OH | total OH | COOH | ||||
| Sample | condensed | noncondensed | H–OH | total | |||
| LB | 1.95 | 1.92 | 0.42 | 4.29 | 1.92 | 6.21 | 0.44 |
| LB–COOH | 1.68 | 1.75 | 0.57 | 4.00 | 1.75 | 5.75 | 0.74 |
The 2D HSQC NMR method also confirmed the changes in the aromatic structure and carboxyl content in lignin before and after oxidizing. The appearance of intense signals at 1.9 (1H) and 20 ppm (13C) in the 2D NMR spectra of oxidized lignin (Figure 3b), compared to the initial one (Figure 3a), confirms the higher number of carboxylic groups in the modified macromolecule. The signal in the range of 8.5–5.5 ppm (1H) and 135–100 ppm (13C) can be assigned to the higher amount of aromatic units in the initial lignin than that in the one after oxidizing (Figure 3), confirming the successful partial dearomatization of lignin.41−43
Figure 3.

2D NMR spectra of: (a) LB initial and (b) LB oxidized.
A significant increase in the content of carbonyl group arising from esters, acids, aldehydes, and so forth, as well as the decrease in the intensity of the aromatic stretching bands in the region of 1400–1600 cm–1, was observed in the FTIR spectrum of oxidized lignin presented in Figure 4. These observations are in line with the others presented in the literature studies of lignin oxidation by peroxides.41,44,45 The ionization state of carboxylic groups can be distinguished by measuring the wavenumber separation (Δ) between the symmetric and asymmetric vibrations. Thus, the low wavenumber separation (60 cm–1) in the absorption band between 1710 and 1650 cm–1 in the oxidized lignin sample LB-COOH corresponded to the formation of nonionized carboxyl groups in lignin after the oxidation of aromatic rings. After the oxidized lignin deposition on the silica and silica gel surface, the Δ value was 140 cm–1. Such an increase in wavenumber separation confirmed the intermolecular electrostatic interaction between the dissociated carboxylic groups of oxidized lignin and protonated amino groups of aminated silica or silica gel (Figure S1). The scheme of oxidized lignin on the silica/silica gel surface is presented in Figure 5.
Figure 4.

FTIR spectra of initial and oxidized kraft lignin.
Figure 5.
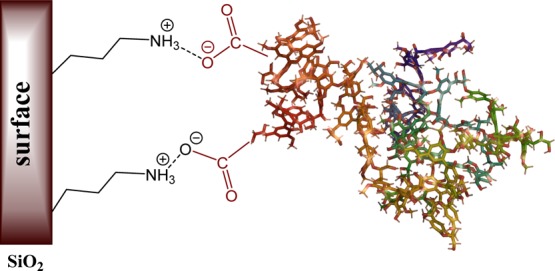
Scheme of lignin–silica composites.
3.2. Synthesis of Lignin–Silica Composites
The synthesized silica and commercial silica gel were previously modified with 3-aminopropyltriethoxysilane to obtain a positively charged surface which will be able to interact with the dissociated carboxyl groups of the oxidized kraft lignin. The concentration of amino groups on the surfaces measured by acid/base titration was 0.67 and 0.56 mmol/g for silica gel and silica, respectively.
3.3. Characterization of Lignin–Silica Composites
TGA in nitrogen atmosphere was conducted in order to investigate the thermal characteristics of the synthesized composites and compare with the initial silicas and LB kraft lignin (Figure 6). The maximum moisture evaporation of LB kraft lignin was found at 51 °C, whereas the maximum water evaporation because of self-condensation reactions occurs at 184 °C (can occur up to 400 °C). The β–β and C–C linkages between the lignin monomeric units cleave at 275–350 °C, whereas the recombination of the formed radicals leads to the formation of guaiacyl and syringyl compounds and the cleavage of aryl ether bonds at 293 °C. At 390 °C conversion of phenols into pyrocatechols and conversion of short substituents of the benzene rings take place with further rearrangement of the backbone and carbonization at a temperature higher than 400 °C. A comprehensive analysis of the LB kraft lignin thermal decomposition is presented in our previous work.32 It was found that the degradation in the inert atmosphere of immobilized lignin occurred in the same temperature range for all synthesized composites: from 200 to 580 °C, with the maximum at 416 and 404 °C for lignin–silica gel composites and lignin–silica composites, respectively. Difference was found in the moisture content of composites based on the silica gel and silica, which, as could be seen from the TG plots, refers to the type of silica as part of the composite.
Figure 6.

TG (a,b) and differential TG (DTG) (c,d) curves of thermal decomposition in N2 atmosphere for original LB kraft lignin, silicas, aminosilicas, and lignin–silica composites.
TGA in oxygen atmosphere led to estimate the concentration of macromolecules in the final composites. For this, the TG curves of the synthesized composites, initial lignin, aminosilica (gel), and silica (gel) were compared (Figure 7). It was found that the content of lignin in the composites was 166 mg/g for LSG1, 161 mg/g for LSG2, 254 mg/g for LS1, and 124 mg/g for LS2.
Figure 7.

TG (a,b) and DTG (c,d) curves of thermal decomposition in air atmosphere for original kraft lignin, silicas, aminosilicas, and lignin–silica composites.
3.4. Textural Characteristics
Two different silicas were applied to estimate and emphasize the influence of carriers to the properties of the final composites. The main difference of the used carriers is in the textural characteristics and particle size. Nitrogen adsorption/desorption isotherms and pore size distribution (PSD) curves obtained for all the studied materials are presented in Figures 8 and 9. It is obvious that the isotherms and PSD by the area and volume of silica gel (Figure 8a) and silica (Figure 8b) have quite divergent shapes. This indicates the difference in the silica structure, which is preserved after each step of modification.
Figure 8.

Nitrogen adsorption/desorption isotherms for (a) lignin–silica gel composites, (b) lignin–silica composites and initial materials.
Figure 9.

Pore size distribution by area and volume for (a) lignin–silica gel composites, (b) lignin–silica composites and initial materials.
The isotherms of both types of materials are characterized by the visible hysteresis loops formed at intermediate (silica gel-based materials) and high (silica-based materials) pressures, which confirm the significant contributions of mesopores to their porosity. The differences in the isotherm shape and the amount of nitrogen adsorbed suggest the essential differences in the pore surface area, pore volume, and pore diameter.46 According to PSD (Figures 9, S2), the porous structure of materials is also quite different. For instance, PDSs for silicas and the corresponding aminosilicas are almost the same. However, after the deposition of oxidized lignin onto the carrier in a mass ratio of 1:1, the lowest size pores of the carrier were probably blocked by lignin, according to the decrease and slight shift of peaks with the maxima at 3.3 and 4.3 to 3.5 and 4.5 nm (Figure 9a), respectively, and peaks with maxima at 3.3, 5.0, 6.3 to 2.0 and 6.5 nm (Figure 9b). In case of the 1:2 lignin–silica gel composite, the range of PDS was shifted to 8.7–25.6 nm with the maxima at 13.0 and 21.2 nm (pore area) and to 8.0–25.0 nm with the maxima at 12.7 and 21 nm (pore volume).
In Table 3, the textural characteristics of the synthesized composites and initial lignin, oxidized lignin, and silica are summarized. It was found that the surface area decreased after each step of silica or silica gel modification (Figure 8). Thus, the initial specific surface area [Brunauer–Emmett–Teller (BET)] of silica gel was 428 m2/g, which decreased up to 323 m2/g after amination and to 202.6–201.3 m2/g after oxidized lignin deposition. The same tendency was observed for silica, where the initial specific surface area was 141.5; it was 119.5 m2/g for aminosilica gel and 80.1–84.8 m2/g for the hybrids based on the silica and oxidized lignin.
Table 3. Textural Characteristics of the Obtained Hybrids and Initial Kraft Lignin.
| parameter |
||||
|---|---|---|---|---|
| sample | SBET, m2/g | Spores, m2/g | Vpores, cm3/g | Dpores, nm |
| LB | 17.8 | 20.9 | 0.102 | 19.5 |
| aminosilica gel | 323.0 | 481.2 | 0.617 | 5.1 |
| silica gel | 428.6 | 566.8 | 0.701 | 4.9 |
| aminosilica | 119.5 | 125.3 | 1.200 | 38.2 |
| silica | 141.5 | 133.4 | 1.278 | 38.3 |
| LSG1 | 202.6 | 283.0 | 0.436 | 6.2 |
| LSG2 | 201.3 | 283.3 | 0.428 | 6.0 |
| LS1 | 80.1 | 89.0 | 0.825 | 37.1 |
| LS2 | 84.8 | 95.4 | 0.874 | 36.6 |
Based on the data obtained from the TGA curves and nitrogen adsorption/desorption isotherms, the concentration of lignin per gram and square meter of samples was calculated (Table 4). As it could be seen from the obtained data, the lignin concentration in LSG1 and LSG2 is almost the same, which could confirm reaching of the maximal lignin saturation on the silica gel surface. In case of silica, where the pores are much larger (more than 6 times), the surface capacity toward lignin macromolecules is higher. The higher value of oxidized lignin adsorption on the aminosilica surface could also be explained by the higher number of amino groups on the surface compared to aminosilica gel.
Table 4. Lignin Concentration in Synthesized Composites.
| sample | lignin concentration, mg/g | lignin concentration, mg/m2 |
|---|---|---|
| LSG1 | 166 | 0.82 |
| LSG2 | 161 | 0.80 |
| LS1 | 254 | 3.17 |
| LS2 | 124 | 1.46 |
The size of the synthesized composite particles was determined from the scanning electron microscopy (SEM) images (Figure S3). It was found that the size of the particles of hybrid composites based on commercial silica gel as the inorganic carrier was 0.2–0.5 mm, as for the initial silica gel. The samples based on prepared silica had a bigger average size of 0.4–1.0 mm and also refer to size of initial silica material.
3.5. Morphology
The morphologies of the synthesized composites were characterized via SEM analysis. Figure 10 presents the surface structures of the silica gel (Figure 10a–f) and silica (Figure 10g–l) coated by oxidized kraft lignin. It could be observed from the SEM micrographs that the surface morphology of composites based on one type of silica is quite similar; however, the composites with lower lignin content have more regular surface, which could be an evidence of more uniform kraft lignin deposition and creation of monolayer on the support surface. Nevertheless, all synthesized materials were characterized by the rough surface with holes, which was also confirmed by the studied textural characteristics.
Figure 10.

SEM micrographs of LB–silica gel 1 (a–c), LB–silica gel 2 (d–f), LB–silica 1 (g–i), and LB–silica 2 (j–l).
The synthesized composites were tested as sorbents for the common synthetic dye in textile production—crystal violet. As it could be seen from the diagram (Figure 11), the capacity of LB–silica samples was higher (108–110 mg/g) than that of LB–silica gel samples (90–95 mg/g) for the selected dye. The initial silica gel and silica have showed activity for the crystal violet dye adsorption, but the capacity was lower: 75 mg/g for silica gel and 100 mg/g for silica. Thus, the obtained materials have high capacity toward the selected dye, which confirmed the high potential of these composites as sorbents for the treatment of the textile industry effluents as well as dye recovery for further reuse.
Figure 11.

Adsorption capacity of the synthesized composites toward the crystal violet dye.
4. Conclusions
The synthesized lignin–aminosilica composites contain up to 20 wt % of lignin, proving that the developed approach is efficient for lignin deposition on charged silica surfaces. The concentrations of the amino groups on the silica surface, as well as the pore sizes and volume, play an important role for lignin deposition. In case of larger pores (silica-based samples), lignin seems to be adsorbed as spherical particles, whereas on silica gel materials, with more narrow pores and higher surface areas, lignin seems to form a uniformly distributed thin and flat layer. The adsorption capacity of immobilized lignin toward crystal violet dye was found to be relatively high and the capacity of initial silicas and lignin expanded greatly. This can be explained by the more “extended” conformation of lignin with a better accessibility of various functional groups, when it is adsorbed on the surface of charged silica materials. The developed lignin–aminosilica composite materials may have great potential for the removal of dyes from wastewater in a textile industry.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03222.
FTIR spectra of the synthesized lignin–silica composites; pore size distribution by area and volume for initial lignin, silica, aminosilica, and lignin–silica composites; and SEM images of LB–silica gel and LB–silica composites (PDF)
The research leading to these results was financed by the Lars-Erik Thunholms Foundation, and EU 7th FP7/2007–2013/REA no: PIRSES-GA-2013-612484 and ÅForsk Foundation (ref. no 19-676). The Knut and Alice Wallenberg Foundation in connection WWSC Program, the Wood and Pulping Chemistry Research Network (WPCRN) at KTH, the Olle Engkvists Stiftelse (grant no 198-0329), COST Action LignoCOST (CA17128) are greatly acknowledged for financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Lin X.; Zhou M.; Wang S.; Lou H.; Yang D.; Qiu X. Structure, and Dispersion Property of a Novel Lignin-Based Polyoxyethylene Ether from Kraft Lignin and Poly(Ethylene Glycol). ACS Sustainable Chem. Eng. 2014, 2, 1902–1909. 10.1021/sc500241g. [DOI] [Google Scholar]
- Argyropoulos D. S.; Sadeghifar H.; Cui C.; Sen S. Synthesis and Characterization of Poly(Arylene Ether Sulfone) Kraft Lignin Heat Stable Copolymers. ACS Sustainable Chem. Eng. 2014, 2, 264–271. 10.1021/sc4002998. [DOI] [Google Scholar]
- Huang C.; He J.; Narron R.; Wang Y.; Yong Q. Characterization of Kraft Lignin Fractions Obtained by Sequential Ultrafiltration and Their Potential Application as a Biobased Component in Blends with Polyethylene. ACS Sustainable Chem. Eng. 2017, 5, 11770–11779. 10.1021/acssuschemeng.7b03415. [DOI] [Google Scholar]
- Naseem A.; Tabasum S.; Zia K. M.; Zuber M.; Ali M.; Noreen A. Lignin-Derivatives Based Polymers, Blends and Composites: A Review. Int. J. Biol. Macromol. 2016, 93, 296–313. 10.1016/j.ijbiomac.2016.08.030. [DOI] [PubMed] [Google Scholar]
- Duval A.; Lawoko M. A Review on Lignin-Based Polymeric, Micro-and Nano-Structured Materials. React. Funct. Polym. 2014, 85, 78–96. 10.1016/j.reactfunctpolym.2014.09.017. [DOI] [Google Scholar]
- Gioia C.; Lo Re G.; Lawoko M.; Berglund L. Tunable Thermosetting Epoxies Based on Fractionated and Well-Characterized Lignins. J. Am. Chem. Soc. 2018, 140, 4054–4061. 10.1021/jacs.7b13620. [DOI] [PubMed] [Google Scholar]
- Lora J.Industrial Commercial Lignins: Sources, Properties and Applications. Monomers, Polymers and Composites from Renewable Resources; Elsevier, 2008; pp 225–241. [Google Scholar]
- Zhao Y.; Tagami A.; Dobele G.; Lindström M. E.; Sevastyanova O. The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (Cnf)-Starch Composite Films. Polymers 2019, 11, 538. 10.3390/polym11030538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Tang S.; Zhang W.; Tao Y.; Lai C.; Li X.; Yong Q. Unveiling the Structural Properties of Lignin–Carbohydrate Complexes in Bamboo Residues and Its Functionality as Antioxidants and Immunostimulants. ACS Sustainable Chem. Eng. 2018, 6, 12522–12531. 10.1021/acssuschemeng.8b03262. [DOI] [Google Scholar]
- Huang C.; Wang X.; Liang C.; Jiang X.; Yang G.; Xu J.; Yong Q. A Sustainable Process for Procuring Biologically Active Fractions of High-Purity Xylooligosaccharides and Water-Soluble Lignin from Moso Bamboo Prehydrolyzate. Biotechnol. Biofuels 2019, 12, 189. 10.1186/s13068-019-1527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohman F.; Theliander H.; Tomani P.; Axegard P.. Method for Separating Lignin from Black Liquor, a Lignin Product, and Use of a Lignin Product for the Production of Fuels or Materials. U.S. Patent 9,382,389 B2, 2008.
- Öhman F.; Theliander H.; Tomani P.; Axegard P., Method for Separating Lignin from Black Liquor. U.S. Patent 8,486,224 B2, 2004.
- Laurichesse S.; Avérous L. Chemical Modification of Lignins: Towards Biobased Polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. 10.1016/j.progpolymsci.2013.11.004. [DOI] [Google Scholar]
- Couch R. L.; Price J. T.; Fatehi P. Production of Flocculant from Thermomechanical Pulping Lignin Via Nitric Acid Treatment. ACS Sustainable Chem. Eng. 2016, 4, 1954–1962. 10.1021/acssuschemeng.5b01129. [DOI] [Google Scholar]
- Klapiszewski L.; Madrawska M.; Jesionowski T. Preparation and Characterisation of Hydrated Silica/Lignin Biocomposites. Physicochem. Probl. Miner. Process. 2012, 48, 463–473. 10.5277/ppmp120212. [DOI] [Google Scholar]
- Klapiszewski Ł.; Nowacka M.; Milczarek G.; Jesionowski T. Physicochemical and Electrokinetic Properties of Silica/Lignin Biocomposites. Carbohydr. Polym. 2013, 94, 345–355. 10.1016/j.carbpol.2013.01.058. [DOI] [PubMed] [Google Scholar]
- Klapiszewski Ł.; Nowacka M.; Szwarc-Rzepka K.; Jesionowski T. Advanced Biocomposites Based on Silica and Lignin Precursors. Physicochem. Probl. Mi. 2013, 49, 497. [Google Scholar]
- Nowacka M.; Klapiszewski Ł.; Norman M.; Jesionowski T. Dispersive Evaluation and Surface Chemistry of Advanced, Multifunctional Silica/Lignin Hybrid Biomaterials. Open Chem. 2013, 11, 1860–1873. 10.2478/s11532-013-0322-4. [DOI] [Google Scholar]
- Klapiszewski Ł.; Bartczak P.; Wysokowski M.; Jankowska M.; Kabat K.; Jesionowski T. Silica Conjugated with Kraft Lignin and Its Use as a Novel’green’sorbent for Hazardous Metal Ions Removal. Chem. Eng. J. 2015, 260, 684–693. 10.1016/j.cej.2014.09.054. [DOI] [Google Scholar]
- Villar J. C.; Caperos A.; García-Ochoa F. Oxidation of Hardwood Kraft-Lignin to Phenolic Derivatives. Nitrobenzene and Copper Oxide as Oxidants. J. Wood Chem. Technol. 1997, 17, 259–285. 10.1080/02773819708003131. [DOI] [Google Scholar]
- Villar J. C.; Caperos A.; García-Ochoa F. Oxidation of Hardwood Kraft-Lignin to Phenolic Derivatives with Oxygen as Oxidant. Wood Sci. Technol. 2001, 35, 245–255. 10.1007/s002260100089. [DOI] [Google Scholar]
- He W.; Gao W.; Fatehi P. Oxidation of Kraft Lignin with Hydrogen Peroxide and Its Application as a Dispersant for Kaolin Suspensions. ACS Sustainable Chem. Eng. 2017, 5, 10597–10605. 10.1021/acssuschemeng.7b02582. [DOI] [Google Scholar]
- Xiang Q.; Lee Y. Y. Oxidative Cracking of Precipitated Hardwood Lignin by Hydrogen Peroxide. Appl. Biochem. Biotechnol. 2000, 84, 153–162. 10.1385/abab:84-86:1-9:153. [DOI] [PubMed] [Google Scholar]
- Evstigneev E. I. Oxidation of Hydrolysis Lignin with Hydrogen Peroxide in Acid Solutions. Russ. J. Appl. Chem. 2013, 86, 258–265. 10.1134/s1070427213020201. [DOI] [Google Scholar]
- Barrett E. P.; Joyner L. G.; Halenda P. P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. 10.1021/ja01145a126. [DOI] [Google Scholar]
- Ciesielczyk F.; Bartczak P.; Jesionowski T. Removal of Cadmium (Ii) and Lead (Ii) Ions from Model Aqueous Solutions Using Sol–Gel-Derived Inorganic Oxide Adsorbent. Adsorption 2016, 22, 445–458. 10.1007/s10450-015-9703-7. [DOI] [Google Scholar]
- Ciesielczyk F.; Bartczak P.; Wieszczycka K.; Siwińska-Stefańska K.; Nowacka M.; Jesionowski T. Adsorption of Ni (Ii) from Model Solutions Using Co-Precipitated Inorganic Oxides. Adsorption 2013, 19, 423–434. 10.1007/s10450-012-9464-5. [DOI] [Google Scholar]
- Klapiszewski Ł.; Bartczak P.; Wysokowski M.; Jankowska M.; Kabat K.; Jesionowski T. Silica Conjugated with Kraft Lignin and Its Use as a Novel ‘Green’sorbent for Hazardous Metal Ions Removal. Chem. Eng. J. 2015, 260, 684–693. 10.1016/j.cej.2014.09.054. [DOI] [Google Scholar]
- Klapiszewski Ł.; Wysokowski M.; Majchrzak I.; Szatkowski T.; Nowacka M.; Siwińska-Stefańska K.; Szwarc-Rzepka K.; Bartczak P.; Ehrlich H.; Jesionowski T. Preparation and Characterization of Multifunctional Chitin/Lignin Materials. J. Nanomater. 2013, 2013, 425726. 10.1155/2013/425726. [DOI] [Google Scholar]
- Wysokowski M.; Klapiszewski Ł.; Moszyński D.; Bartczak P.; Szatkowski T.; Majchrzak I.; Siwińska-Stefańska K.; Bazhenov V.; Jesionowski T. Modification of Chitin with Kraft Lignin and Development of New Biosorbents for Removal of Cadmium (Ii) and Nickel (Ii) Ions. Mar. Drugs 2014, 12, 2245–2268. 10.3390/md12042245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podkościelna B.; Goliszek M.; Sevastyanova O. New Approach in the Application of Lignin for the Synthesis of Hybrid Materials. Pure Appl. Chem. 2017, 89, 161–171. 10.1515/pac-2016-1009. [DOI] [Google Scholar]
- Budnyak T.; Aminzadeh S.; Pylypchuk I.; Riazanova A.; Tertykh V.; Lindström M.; Sevastyanova O. Peculiarities of Synthesis and Properties of Lignin–Silica Nanocomposites Prepared by Sol-Gel Method. Nanomaterials 2018, 8, 950. 10.3390/nano8110950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnyak T. M.; Tertykh V. A.; Yanovska E. S.; Kołodyńska D.; Bartyzel A. Adsorption of V (V), Mo (Vi) and Cr (Vi) Oxoanions by Chitosan–Silica Composite Synthesized by Mannich Reaction. Adsorpt. Sci. Technol. 2015, 33, 645–657. 10.1260/0263-6174.33.6-8.645. [DOI] [Google Scholar]
- Tertykh V.; Belyakova L.. Chemical Reactions on the Silica Surface; Naukova Dumka: Kyiv, 1991; p 264. [Google Scholar]
- Argyropoulos D. Quantitative Phosphorus-31 Nmr Analysis of Lignins, a New Tool for the Lignin Chemist. J. Wood Chem. Technol. 1994, 14, 45–63. 10.1080/02773819408003085. [DOI] [Google Scholar]
- Lin S. Y.Dence CW Methods in Lignin Chemistry; Springer, Berlin/Verlag, 1992. [Google Scholar]
- Hobbs G. C.; Abbot J. Peroxide Bleaching Reactions under Alkaline and Acidic Conditions. J. Wood Chem. Technol. 1991, 11, 225–246. 10.1080/02773819108050272. [DOI] [Google Scholar]
- Kadla J. F.; Chang H.. The Reactions of Peroxides with Lignin and Lignin Model Compounds; ACS symposium series; American Chemical Society: Washington, DC, 1999; pp 108–129. [Google Scholar]
- Evtuguin D. V.; Robert D. The Detection of Muconic Acid Type Structures in Oxidized Lignins by 13 C Nmr Spectroscopy. Wood Sci. Technol. 1997, 31, 423–431. 10.1007/bf00702564. [DOI] [Google Scholar]
- Evtuguin D. V.; Rocha G.; Goodfellow B. J. Detection of Muconic Acid Type Structures in Oxidised Lignins Using 2d Nmr Spectroscopy 10th Ewlp, Stockholm, Sweden, August 25–28, 2008. Holzforschung 2009, 63, 675–680. 10.1515/hf.2009.083. [DOI] [Google Scholar]
- Wiermans L.; Schumacher H.; Klaaßen C.-M.; Domínguez de María P. Unprecedented Catalyst-Free Lignin Dearomatization with Hydrogen Peroxide and Dimethyl Carbonate. RSC Adv. 2015, 5, 4009–4018. 10.1039/c4ra13113d. [DOI] [Google Scholar]
- Huang C.; Su Y.; Shi J.; Yuan C.; Zhai S.; Yong Q. Revealing the Effects of Centuries of Ageing on the Chemical Structural Features of Lignin in Archaeological Fir Woods. New J. Chem. 2019, 43, 3520–3528. 10.1039/c9nj00026g. [DOI] [Google Scholar]
- Huang C.; He J.; Du L.; Min D.; Yong Q. Structural Characterization of the Lignins from the Green and Yellow Bamboo of Bamboo Culm (Phyllostachys Pubescens). J. Wood Chem. Technol. 2016, 36, 157–172. 10.1080/02773813.2015.1104544. [DOI] [Google Scholar]
- Brasileiro L. B.; Colodette J. L.; Piló-Veloso D. A Utilização De Perácidos Na Deslignificação E No Branqueamento De Polpas Celulósicas. Quim. Nova 2001, 24, 819–829. 10.1590/s0100-40422001000600020. [DOI] [Google Scholar]
- Gierer J. The Chemistry of Delignification-a General Concept-Part Ii. Holzforschung 1982, 36, 55–64. 10.1515/hfsg.1982.36.2.55. [DOI] [Google Scholar]
- Blachnio M.; Budnyak T. M.; Derylo-Marczewska A.; Marczewski A. W.; Tertykh V. A. Chitosan–Silica Hybrid Composites for Removal of Sulfonated Azo Dyes from Aqueous Solutions. Langmuir 2018, 34, 2258–2273. 10.1021/acs.langmuir.7b04076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.