

Abstract
With its high morbidity rate and increasing resistance to treatment, methicillin-resistant Staphylococcus aureus (MRSA) is a grave concern in the medical field. In methicillin-susceptible strains, β-lactam antibiotics disable the penicillin binding proteins (PBPs) that cross-link the bacterial cell wall. However, methicillin-resistant strains have PBP2a and PBP4, which continue enzymatic activity in the presence of β-lactam antibiotics. The activity of PBP2a and PBP4 is linked to the presence of wall teichoic acid (WTA); thus, WTA has emerged as a target for antibiotic drug discovery. In this work, we disable WTA in situ using its anionic phosphodiester backbone to attract cationic branched polyethylenimine (BPEI). Data show that BPEI removes β-lactam resistance in common MRSA strains and clinical isolates. Fluorescence microscopy was used to investigate this mechanism of action. The results indicate that BPEI prevents the localization of PBP4 to the cell division septum, thereby changing the cellular morphology and inhibiting cell division. Although PBP4 is not required for septum formation, proper cell division and morphology require WTA; BPEI prevents this essential function. The combination of BPEI and β-lactams is bactericidal and synergistic. Because BPEI allows us to study the role of WTA in the cell wall without genetic mutation or altered translocation of biomolecules and/or their precursors, this approach can help revise existing paradigms regarding the role of WTA in prokaryotic biochemistry at every growth stage.
Graphical Abstract
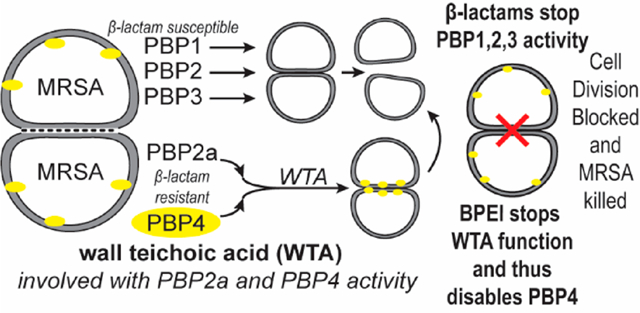
Of the antimicrobially resistant pathogens that currently threaten human health, methicillin-resistant Staphylococcus aureus (MRSA) is one of the most dangerous. Bacterial infections from MRSA can be categorized as either hospital-acquired (HA-MRSA) or community-acquired (CA-MRSA). CA-MRSA strains, which have become more prevalent in the past 30 years, are more virulent than HA-MRSA.1 The most common CA-MRSA strain is USA300; data collected by the United States Emerging Infection Program2,3 show that the mortality rate for USA300 infections is rising dramatically as the overall MRSA mortality rate remains steady. This could be partially due to the low susceptibility of MRSA USA300 to over-the-counter triple-antibiotic ointments.4–6 Nevertheless, one of seven individuals infected with MRSA dies from the infection due to treatment delays or complications, despite the availability of antibiotics of last resort.7 Survival is determined by patient age, comorbidities, the severity of the acute infection, timely treatment, and effective treatment.8 While the first two factors are beyond the control of pharmaceutical therapy, antibiotics can be used effectively via out-patient prescriptions or in-patient settings against susceptible infections.
Because they are generally recognized as safe and effective (GRASE) with high oral bioavailability, β-lactam antibiotics are a preferred antibacterial drug9–12 against methicillin-susceptible S. aureus (MSSA) infections.13 However, β-lactam antibiotics alone cannot cure MRSA infections; instead, doctors must resort to more drastic treatments, such as vancomycin or linezolid. Vancomycin must be given intravenously and has severe side effects,14–18 while linezolid causes mitochondrial toxicity over time.19 Using β-lactams to treat CA-MRSA infections requires a means of disabling resistance mechanisms, which could be done by inhibiting the penicillin binding proteins (PBPs) that continue to function in the presence of β-lactam antibiotics.20
Bacteria use PBPs to cross-link the cell wall peptidoglycan that protects vital intracellular components necessary for the survival of bacteria. All S. aureus strains have four PBPs denoted as PBP1–PBP4. Common β-lactam antibiotics, such as ampicillin, amoxicillin, and oxacillin, inhibit these PBPs by binding to the active site and preventing cell wall cross-linking, thereby weakening the cell wall in a manner that eventually leads to cell death.21 All MRSA strains have an additional PBP denoted as PBP2a. Encoded by the mecA gene, PBP2a has a low binding affinity for β-lactam antibiotics and can therefore continue cross-linking in the presence of β-lactams.22 Some fifth-generation cephalosporins, such as ceftaroline,23 have anti-PBP2a activity, but resistance has already emerged.24–26 In CA-MRSA strains, β-lactams also have a low affinity for PBP4, which grants oxacillin resistance.27 Cefoxitin, a second-generation cephalosporin, targets PBP4 and restores efficacy to penicillin-like β-lactams; however, the threat of resistance lingers.27
PBP2a’s oxacillin resistance and PBP4’s functionality require wall teichoic acid (WTA), an anionic phosphodiester biopolymer.28 Hydrophilic, cationic, 600 Da branched polyethylenimine (BPEI) can diffuse throughout the cell wall peptidoglycan and disable WTA.29 Because it targets mature WTA, 600 Da BPEI avoids the undesirable protein binding associated with WTA inhibitors that target the cytoplasmic synthesis and assembly of WTA. At high concentrations (>100 μM), 600 Da BPEI is an antimicrobial agent; at low concentrations (<10 μM), BPEI synergistically restores β-lactam efficacy. We have previously reported that combinations of 600 Da BPEI and β-lactam are effective against MRSA 700787, a vancomycin intermediate-resistant strain.29,30 Here, we show that 600 Da BPEI also displays synergy with β-lactam antibiotics against clinical isolates and laboratory reference strains of CA-MRSA USA300 and MW2.
The proposed mechanism of action relies on electrostatic binding between BPEI and WTA in the cell wall.29,30 Because WTA localizes several proteins used for cell wall synthesis, including PPB4, disabling WTA also hinders an important resistance factor in CA-MRSA.28,31–34 Data collected using the MW2 ΔtarO strain (which lacks WTA) demonstrate that BPEI potentiation requires WTA. The electrostatic binding between WTA and BPEI prevents PBP4 from accessing the WTA scaffold; consequently, it is not localized to the division septum. This mechanism is shown in fluorescence microscopy images of PBP4 labeled with yellow fluorescent protein. The data presented in this report establish the mechanism of action by which BPEI disables resistance, indicate where this process occurs, and illustrate why the combination of BPEI and β-lactam may be a crucial weapon in the battle against antibiotic resistance.
MATERIALS AND METHODS
Materials.
The bacteria used in this work were obtained from the American Type Culture Collection (MRSA USA300, MRSA 33592, and MRSA 252). MRSA MW2 and MRSA MW2 ΔtarO (referenced in ref 31) were a generous gift from S. Walker.31 Clinical isolates were obtained from patient swabs at the University of Oklahoma Health Sciences Center and provided by C. McCloskey following institutional review board (IRB) approval. S. aureus strains encoding the fluorescent fusion of PBP2, PBP4, and FtsZ proteins are S. aureus COL BCBPR102, S. aureus COL DpbpD BCBPR1077, S. aureus COL spa::Pspac-Ftsz-CFP-BCBAJ020, S. aureus COL PBP2::sGFP-PBP2-BCBPM073, and S. aureus COL pPBP4-YFP-BCBPM162. Chemicals from Sigma-Aldrich (DMSO, low-molecular weight branched polyethylenimine, and growth media) were used as purchased. Antibiotics (oxacillin, ampicillin, amoxicillin, ceftizoxime, imipenem, and linezolid) were purchased from Gold Biotechnology.
In Vitro Checkerboard Assays.
Checkerboard assays were performed following the procedure outlined in ref 30. Briefly, the antibiotic and BPEI were added to each well and serially diluted in Mueller-Hinton broth (MHB). Each well was inoculated with bacteria to a final cell density of approximately 5 × 105 cells/mL following Clinical Practice Guidelines.35 The plates were incubated for 22 ± 2 h at 35 °C in a humidified incubator. A change in OD600, as determined from initial inoculation and postgrowth, of >0.05 was considered to be positive for growth. Minimum inhibitory concentrations (MICs) and fractional inhibitory concentration indices (FICIs) were determined. Each assay was performed as three separate trials and presented as the average change in OD600.
Clinical Isolate MIC Scans.
The MICs of oxacillin and BPEI against 16 strains of MRSA and five strains of MSSA were determined using the Clinic Practice Guidelines.35 The MICs of BPEI and oxacillin were determined by serially diluting each in 96-well plates with MHB. In separate wells, decreasing concentrations of BPEI were added to MHB with a constant amount of oxacillin (2 μg/mL). The same was done for decreasing concentrations of oxacillin with a constant amount of BPEI (4 μg/mL). Bacteria were added (5 × 105 cells/mL) to each well and grown until they reached confluency at 35 °C. The OD600 was measured before and after growth. A change in OD600 of >0.05 was considered positive for growth. Each MIC was determined in quadruplicate, and the values are presented as the average of all four trials.
Microscopy of Fluorescently Labeled Proteins.
MRSA COL strains with green fluorescent protein (GFP)-labeled PBP2, yellow fluorescent protein (YFP)-labeled PBP4, or cyan fluorescent protein (CFP)-labeled FtsZ were obtained as referenced.28,36,37 Overnight cultures of bacteria were inoculated 0.5% into MHB and incubated at 37 °C until an OD600 of 0.6. BPEI was added (64 μg/mL, the MIC of MRSA COL), and the cells were incubated for 60 min. After growth, 1 mL of culture was harvested by centrifugation, washed once in PBS, and resuspended in PBS. Next, 1 μL of resuspended cells was placed on a thin layer of 1.2% agarose in PBS. Samples were observed using a Zeiss Axio Observer microscope equipped with a Photometrics CoolSNAP HQ2 camera (Roper Scientific) and Metamorph 7.5 software (Molecular Devices). Images were analyzed using ImageJ software. To quantitatively assess the localization of the fusion proteins, the fluorescence signal at the septum and at the peripheral cell wall was determined for >100 cells with fully formed septa. Automated calculations of fluorescent ratios between septal and membrane signals were performed by eHooke software as previously described.38 Statistical analyses were performed using GraphPad Prism 7 (GraphPad Software). Unpaired Student’s t tests were used to compare fluorescence ratios between peripheral and septal wall signal intensity.
Structured Illumination Microscopy of Labeled Cell Membranes.
S. aureus JE2-derived strains were obtained from the Network of Antimicrobial Resistance in S. aureus (NARSA) program (supported under National Institute of Allergy and Infectious Diseases Contract SN272200700055C). Cells, back-diluted from an overnight culture, were grown to an OD600 of 0.6 at 37 °C while being shaken. Cells were exposed to 600 Da BPEI (at twice the MIC for MRSA JE2, 32 μg/mL) for 60 min at 37 °C. Cell membranes were labeled with Nile Red (Invitrogen) at a final concentration of 10 μg mL−1 for 5 min at room temperature, washed with PBS, and then mounted on microscope slides. Super-resolution SIM imaging was performed using an Elyra PS.1 microscope (Zeiss) with a Plan-Apochromat 63×/1.4 oil DIC M27 objective and a Pco.edge 5.5 camera. SIM images were acquired using five grid rotations, unless stated otherwise, with a 34 μm grating period for the 561 nm laser (100 mW). Reconstructions were performed using Zen software and theoretical PSF (point spread function). Quantifications and cell measurements were performed using ImageJ.
Scanning Electron Microscopy.
MRSA USA300 and MRSA MW2 cells were inoculated 0.5% from an overnight culture and grown at 37 °C while being shaken. The optical density was monitored, and growth was stopped after 2 h when the bacteria reached the late-lag phase, at an OD600 of 0.2.Growth in the presence of 600 Da BPEI was performed by adding 64 μg/mL BPEI (1× MIC) to each culture at time zero. Aliquots were fixed with a mixture of 2% glutaraldehyde and 2% formaldehyde in 0.1 M cacodylate buffer for 30 min at room temperature. The cells were washed and fixed with 1% OsO4 for 30 min at room temperature in the dark. Afterward, the cells were washed three times with water. One drop of each sample was placed on poly-L-lysine-coated coverslips. The samples immediately underwent a dehydration series by immersion in ethanol solutions (20%, 35%, 50%, 70%, 95%, and 100%) for 15 min each. The samples were dried with HMDS and then sputter-coated with AuPd. The samples were imaged on a Zeiss NEON SEM instrument. Size analysis was performed on ImageJ, and ANOVA was used to establish statistical significance.
Transmission Electron Microscopy.
Preparation for TEM was modified from the procedure of Campbell et al.31 MRSA USA300 cells were grown by inoculating TSB medium with a 0.5% culture of a frozen stock grown overnight. The inoculation was shaken at 37 °C, and the cells were harvested after growing for 2 h, at an OD600 of 0.2. BPEI-treated cells were obtained by inoculating TSB medium with a 0.5% overnight culture and adding 64 μg/mL 600 Da BPEI at time zero and harvesting after growth for 2 h. The cells were harvested by centrifugation, fixed with 2.5% glutaraldehyde and 2% formaldehyde in 0.1 M cacodylate buffer for 2 h at room temperature, and then washed three times with cacodylate buffer. The cells were stained with 1% OsO4 in 1.5% PFA for 2 h at room temperature in the dark and washed three times with water. The samples were stained with 1% uranyl acetate for 30 min at room temperature in the dark and washed three times with water. A dehydrate series with ethanol (50%, 70%, 95%, 95%, and 100%) was done for 20 min each. The samples were placed in propylene oxide (PO) for 1 h with one change of solvent at the 30 min mark. The cells were infiltrated with freshly made Epon in an increasing Epon:PO ratio (1:2, 1:1, and 2:1) for 1–2 h each. The samples were then placed in 100% Epon and left overnight at room temperature. The cells were then placed in fresh Epon and allowed to sink to the bottom of an embedding mold. The samples were embedded for 24 h at 60 °C. An ultramicrotome with a glass knife was used to thin-section the blocks to a thickness of 90 nm, and sections were placed on grids. The grids were stained for 30 min with 1% uranyl acetate. Imaging was performed on a JEOL 2000-FX TEM at an accelerating voltage of 200 kV.
RESULTS AND DISCUSSION
Methicillin was introduced in 1959 to treat penicillin-resistant infections, but reports of methicillin resistance began appearing just two years later.39 Today, improved β-lactam antibiotics have replaced methicillin. Some β-lactams, such as oxacillin, ampicillin, and ceftizoxime, are used to treat staphylococcal infections in out-patient settings, but the increased prevalence of MRSA has made vancomycin a first-line antibiotic against nosocomial infections. Vancomycin cannot cross the stomach lining and therefore must be given intravenously, which often requires hospitalization and can cause severe side effects, such as hearing loss, allergic reactions, and kidney damage.40 Furthermore, cases of vancomycin-resistant S. aureus (VRSA) have already been documented.41,42
BPEI Has in Vitro Synergy with β-Lactam Antibiotics.
BPEI combined with oxacillin showed synergy against multiple MRSA strains (Table 1). The combination of BPEI and oxacillin against MRSA USA300, a common clinical strain, had a fractional inhibitory concentration index (FICI) of 0.266 (Figure 1A). FICI values that are less than 0.5 indicate synergy; values between 0.5 and 1.0 indicate additivity, and values between 1.0 and 4.0 indicate antagony. Another common strain, MRSA MW2, had an FICI of 0.250 for the combination of BPEI and oxacillin (Figure 1B). Against MRSA USA300 [FICI = 0.313 (Figure 1C)], 4 μg/mL BPEI decreased the MIC of ceftizoxime from 256 to 8 μg/mL. Similarly, against MRSA MW2 [FICI = 0.156 (Figure 1D)], 8 μg/mL BPEI decreased the MIC of ceftizoxime from 256 to 16 μg/mL. Imipenem and BPEI also exhibited synergy against both MRSA USA300 [FICI = 0.375 (Figure 1E)] and MRSA MW2 [FICI = 0.25 (Figure 1F)], as did amoxicillin, meropenem, and piperacillin with BPEI (Figure S1). The synergistic effect was also present against HA-MRSA 252 and HA-MRSA 33592 (Table 1). Against MRSA 252, 600 Da BPEI had synergy with ampicillin, ceftizoxime, and cephalothin; against MRSA 33592, BPEI had synergy with oxacillin, amoxicillin, imipenem, and cephalothin. However, BPEI did not decrease the MICs of vancomycin (a glycopeptide that targets cell wall synthesis) or linezolid (an oxazolidinone that inhibits protein synthesis) against any of the tested strains. For both MRSA USA300 and MRSA MW2, the FICI for vancomycin and BPEI was 1.00 (Figure S2A,B), while the FICI for linezolid and BPEI was 0.5 (Figure S2C,D). This result is not surprising because WTA, the BPEI target, is not involved in the mechanism of action for vancomycin or linezolid. Overall, BPEI and oxacillin showed the best efficacy against the tested MRSA panel
Table 1.
Minimum Inhibitory Concentrations and Fractional Inhibitory Values of 600 Da BPEI in Combination with Various Antibiotics against MRSAa
| MRSA MW2 | MRSA USA300 | MRSA 252 | MRSA 33592 | |||||
|---|---|---|---|---|---|---|---|---|
| MIC | FICI | MIC | FICI | MIC | FICI | MIC | FICI | |
| 600-Da BPEI | 64 | - | 32 | - | 32 | - | 32 | - |
| oxacillin | 32 | 0.250 | 32 | 0.266 | >256 | 0.501 | 256 | 0.281 |
| ampicillin | 128 | 0.188 | 2 | 0.516 | 256 | 0.375 | 256 | 0.516 |
| amoxicillin | 128 | 0.250 | 4 | 0.5 | 256 | 0.500 | 256 | 0.375 |
| piperacillin | >256 | 0.266 | 16 | 0.313 | >256 | 0.501 | >256 | 0.501 |
| imipenem | 0.5 | 0.375 | 0.25 | 0.25 | 64 | 0.508 | 32 | 0.281 |
| meropenem | 2 | 0.313 | 4 | 0.250 | 32 | 0.504 | 8 | 0.563 |
| ceftizoxime | >256 | 0.156 | 256 | 0.313 | >256 | 0.254 | >256 | 0.5 |
| cephalothin | 4 | 0.5 | 2 | 0.5 | 32 | 0.281 | 32 | 0.375 |
| vancomycin | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 0.504 |
| linezolid | 2 | 0.5 | 1 | 0.5 | 1 | 0.5 | 1 | 0.5 |
Each value is the average of three separate trials.
Figure 1.

Checkerboard assays show synergy between BPEI and β-lactams on MRSA USA300 and MRSA MW2. BPEI had synergy with (A and B) oxacillin, (C and D) ceftizoxime, and (E and F) imipenem against (A, C, and E) MRSA USA300 and (B, D, and F) MRSA MW2. Each assay was performed as three separate trials, and the presented data are shown as the average change in OD600.
Our previous work showed that BPEI had synergy with oxacillin and ampicillin against MRSA 700787, a vancomycin intermediate-resistant strain.29,30 Using checkerboard assays, we have demonstrated synergy between 600 Da BPEI and β-lactams against five commercial reference strains of MRSA. The USCAST definition of β-lactam resistance is an oxacillin MIC of >2 μg/mL; the cutoffs for other β-lactams are referenced to this value.43 The 600 Da BPEI also eliminates resistance to cephalosporins and imipenem. Furthermore, we observed that the oxacillin MIC is the same in pure CAMHB and in 50% fetal bovine serum (FBS), which indicates that serum proteins do not hinder potentiation from BPEI.
We also tested clinical isolates of MRSA from patients at the University of Oklahoma College of Medicine. The MICs for oxacillin and BPEI were determined for each isolate (Table 2). Every clinical isolate showed oxacillin resistance (MIC > 2 μg/mL), but 600 Da BPEI disabled resistance in all of them. The amount of BPEI required to achieve an oxacillin MIC of 2 μg/mL varied (BPEI = 1–32 μg/mL); 600 Da BPEI did not show synergy with oxacillin in isolates with an oxacillin MIC of 256 μg/mL (isolates 1, 8, and 11), but it did show synergy in isolates with an oxacillin MIC of 128 μg/mL (isolates 5 and 7). These data demonstrate that the combination of 600 Da BPEI and oxacillin displays broad efficacy against clinical strains of MRSA. The isolates with no synergy of BPEI and oxacillin (isolates 1, 8, and 11) may be more responsive to BPEI combined with other β-lactams.
Table 2.
Treatment of Clinical Isolates of MRSAa
| removing resistance | |||||
|---|---|---|---|---|---|
| strain | oxacillin MIC (μg/mL) | [oxacillin] (μg/mL) | [BPEI] (μg/mL) | BPEI MIC (μg/mL) | FICI |
| 1 | 256 | 2 | 16 | 32 | 0.508 |
| 2 | 8 | 2 | 1 | 32 | 0.281 |
| 3 | 64 | 2 | 16 | 16 | 1.03 |
| 4 | 64 | 2 | 8 | 32 | 0.281 |
| 5 | 128 | 2 | 16 | 64 | 0.266 |
| 6 | 32 | 2 | 8 | 64 | 0.188 |
| 7 | 128 | 2 | 16 | 64 | 0.266 |
| 8 | 256 | 2 | 8 | 8 | 1.01 |
| 9 | 32 | 2 | 16 | 64 | 0.313 |
| 10 | 16 | 2 | 32 | 256 | 0.25 |
| 11 | 256 | 2 | 32 | 64 | 0.508 |
| 12 | 64 | 2 | 32 | 128 | 0.281 |
| 13 | 16 | 2 | 4 | 8 | 0.625 |
| 14 | 32 | 2 | 8 | 64 | 0.188 |
| 15 | 16 | 2 | 2 | 256 | 0.133 |
| 16 | 32 | 2 | 2 | 256 | 0.125 |
Data show the concentration of BPEI required to reduce the MIC of oxacillin to the susceptibility cutoff of 2 μg/mL. Each value is the average of four trials.
WTA Is Involved in the Mechanism of Action of BPEI and Oxacillin against MRSA.
The cationic amines of BPEI electrostatically interact with the anionic phosphate groups of WTA29,30 to form a complex that sterically hinders WTA’s interactions with nearby cell wall proteins. Fluorescence laser scanning confocal microscopy (LSCM) images indicate that the highest concentrations of BPEI are in the septa of cells undergoing division.29 Nuclear magnetic resonance (NMR) spectroscopy data depict BPEI binding directly to WTA. A direct physical interaction between WTA phosphates and BPEI amines caused a chemical shift perturbation in the 31P NMR spectra.29 To evaluate the role of WTA in the synergetic antibacterial properties between oxacillin and BPEI, we examined MRSA MW2 ΔtarO, a WTA-deficient mutant.31 Confirming previous literature reports,31 the MIC of oxacillin was 64-fold lower against MRSA MW2 ΔtarO (0.5 μg/mL) than against wild-type MRSA MW2 (32 μg/mL). These data suggest that WTA is vital for β-lactam resistance in MRSA. If BPEI targets WTA, the combination of BPEI and oxacillin should cause little to no change in the oxacillin MIC against MRSA MW2 ΔtarO. Against wild-type MW2, oxacillin and BPEI have synergy (Figure 1B); 32 μg/mL 600 Da BPEI reduces the oxacillin MIC 128-fold (from 64 to 0.5 μg/mL). However, against the WTA-absent mutant, BPEI and oxacillin have additivity instead of synergy (Figure 2A), with 32 μg/mL BPEI decreasing the oxacillin MIC only 2-fold (FICI = 0.531). Similarly, in a checkerboard assay, sublethal amounts of tunicamycin (a WTA synthesis inhibitor)31 (0.250 μg/mL) decreased the MIC of oxacillin to 0.25 μg/mL (128-fold) against wild-type MRSA MW2 (Figure S3). Furthermore, adding 0.250 μg/mL tunicamycin to each well in the checkerboard assay of oxacillin and BPEI removed synergy against MRSA MW2 (Figure 2B). The same assay against MRSA MW2 ΔtarO also showed no synergy. These results suggest that WTA is essential for synergy between BPEI and oxacillin, making it BPEI’s most likely target. Additionally, if WTA is present but unrelated to β-lactam activity, BPEI should not affect the oxacillin MIC against methicillin-susceptible S. aureus (MSSA). Indeed, for MSSA ATCC 25923, the oxacillin MIC is 0.064 μg/mL with and without 600 Da BPEI.30
Figure 2.

Mechanism by which BPEI makes MRSA susceptible via WTA. BPEI and oxacillin have synergy against MRSA MW2. Synergy is lost against (A) MRSA MW2 ΔtarO and (B) MRSA MW2 with sublethal tunicamycin (0.250 μg/mL). Each assay was performed as three separate trials, and the presented data are shown as the average change in OD600.
BPEI Prevents the Proper Localization of PBP4.
Previously, we showed that BPEI localizes in the cell wall of Gram-positive bacteria (including MRSA), where it electrostatically binds to WTA.29 While more than 30 genes are required for oxacillin resistance in staphylococci, no direct link between WTA and PBP2a has been established. Nevertheless, the fact that genetic modification and chemical inhibition of WTA synthesis restore MRSA’s susceptibility to β-lactams suggests WTA’s involvement in resistance mechanisms.28,31,34 In contrast, PBP4, which confers resistance in CA-MRSA,27 does have an identified link with WTA, thereby leading us to hypothesize that BPEI prevents the proper localization of PBP4 by sterically hindering WTA. Two major pieces of evidence support this paradigm: (1) fluorescence microscopy studies showing delocalization of PBPs in WTA-absent mutants37 and (2) reports of small molecule drugs potentiating β-lactams by disrupting PBP4 localization.44 The in situ binding of WTA with BPEI has not been previously investigated, but several studies indicate that chemical inhibition of WTA synthesis combined with β-lactams effectively kills MRSA.31,34,45–48 Fluorescence microscopy images of PBP4 linked to yellow fluorescent protein (YFP) have demonstrated the localization of PBP4 to the cell division septum. However, when WTA synthesis is inhibited, PBP4-YFP delocalizes from the septum and disperses throughout the cell wall.28 Similarly, when FtsZ (membrane-associated filaments that form the Z-ring of cell division) is altered, PBP2 tagged with green fluorescent protein (sfGFP-PBP2) delocalizes.46 These fluorescence studies can be used to investigate the mode of action for BPEI’s synergy with β-lactams. MRSA COL sfGFP-PBP2 (strain referenced by Tan et al.),37 MRSA COL PBP4-YFP (strain referenced by Atilano et al.),28 and MRSA COL FtsZ-CFP (strain referenced by Pereira et al.)36 were used to examine BPEI-induced enzyme delocalization. PBP4 localization is linked to WTA, while PBP2 localization and FtsZ localization are not. Thus, we expected BPEI to prevent the proper localization of PBP4-YFP by electrostatically binding to WTA while leaving the localization patterns of sfGFP-PBP2 and FtsZ-CFP unaltered. In the MRSA COL PBP4-YFP bacterial construct described by Atilano et al., BPEI prevents the proper localization of PBP4 to the division septum (Figure 3). By comparing the fluorescence content of the septal area of the cell wall to the nonseptal (S/L) area, we found that the proportion of septal fluorescence intensity is significantly higher for untreated cells (S/L = 2.95 ± 0.06) than treated cells (S/L = 2.15 ± 0.05). sfGFP-PBP2 showed no significant difference in the fluorescence ratio between untreated (S/L = 2.59 ± 0.04) and treated cells [S/L = 2.50 ± 0.05 (Figure S4)]. This result corroborates prior studies in which other PBPs, such as PBP1 and PBP2, had normal localization in the ΔtarO mutant.28 We also did not see a quantitatively significant difference of the FtsZ-CFP localization patterns between the treated and untreated cells (Figure S5). These data indicate that a more specific interaction, rather than broad steric hindrance of cell wall proteins, allows BPEI to delocalize PBP4-YFP.
Figure 3.

(A and B) Fluorescence images of MRSA COL PBP4-YFP show that PBP4 primarily localizes at the division septum of dividing cells. (C and D) BPEI delocalizes PBP4 from the division septum. (E) Quantitative analysis of the fluorescence intensity at the division septum vs fluorescence intensity at peripheral cell wall regions was performed for untreated and BPEI-treated samples. Analysis was performed for >100 cells of each sample that showed a visible closed septum. The PBP4 protein in untreated cells (S/L = 2.95 ± 0.06) localized more at the division septum than in the BPEI-treated samples (S/L = 2.15 ± 0.05). The scale bar is 1 μm.
Delocalization of PBP4 in a WTA-deficient mutant impedes peptidoglycan cross-linking.28 Further analysis revealed that TagO, a membrane protein responsible for the early stages of WTA synthesis, localizes at the division septum before PBP4. During the subsequent WTA assembly, PBP4 is recruited to the septum.28 Atilano et al. found that defects in the TagO protein produced WTA knockout mutants and caused PBP4 delocalization throughout the cell wall,28 an observation confirmed by Gautam et al.49 Additional data reveal a direct correlation between the amount of WTA present and the amount of PBP4 recruited to the division septum.28 PBP4 is a transpeptidase that performs the final cross-linking step of nascent peptidoglycan, the assembly of which involves transglycosylation by PBP2 and transpeptidation by PBP2a enzymes when β-lactams are present. Because PBP2 contributes to the activity of PBP2a, drug combinations that disable PBP4 and PBP2 kill CA-MRSA.50 For instance, because cefoxitin (which targets PBP4) restores oxacillin activity, it must disable PBP2a. However, for MRSA COL, disabling PBP4 did not restore susceptibility to oxacillin.27 Our data are consistent with these results. While checkerboard assays of BPEI and oxacillin against MRSA COL display no synergy (Figure S6), we observed oxacillin potentiation against HA-MRSA strain 33592 and CA-MRSA strains USA300 and MW2. Although 600 Da BPEI did not show synergy with oxacillin against HA-MRSA strain 252, it did show synergy with ampicillin, ceftizoxime, and cephalothin.
BPEI Causes Morphological Changes to MRSA Cells.
Because WTA is important for cell morphology, altering WTA activity with BPEI affects the size and shape of MRSA. Previously, we found that MRSA 700787 had a larger cell diameter upon treatment with BPEI.30 Given the ubiquitous role of WTA in cell division, other strains of MRSA should exhibit similar morphological changes when grown in BPEI. Accordingly, we observed that MRSA USA300 cells treated with BPEI have difficulty dividing. Although SEM images of treated and untreated cells do not show drastic changes in cell morphology, TEM images depict differences in septal formation (Figure 4).
Figure 4.

SEM of MRSA USA300 shows multiple septa formations when treated with BPEI. (A) Untreated cells have a single septal formation (black arrows). (B) Some BPEI-treated cells have multiple septa formations (white arrows). The scale bar is 1 μm.
For each sample, TEM was used to analyze 200 cells. Treated cells had visibly thicker division septa and lower electron density around the septa compared to the control cells (Figure 5).
Figure 5.

TEM images of MRSA USA300 cells do not show drastic changes in division septa. Cells treated with (B and D) BPEI have thicker division septa in comparison to (A and C) those of the untreated cells. Black arrows indicate complete septa formation. White arrows indicate incomplete septa formation. The scale bar is 500 nm.
Additionally, only approximately one-quarter of the untreated cells had visible septa whereas approximately one-half of the BPEI-treated cells had visible septa. These data suggest that BPEI prevents cells from dividing properly, which may explain why higher BPEI concentrations have antibiotic properties even in the absence of β-lactams. This observation was confirmed by performing fluorescence microscopy on membranes labeled with Nile Red dye (Figure 6). In the untreated MRSA JE2 cells (N = 743), 56.5% did not have a septum, 27% had a partial septum, and 16.5% had a complete septum. Exposure to 64 μg/mL BPEI changed the distribution; of the 2495 analyzed cells,40% had no septum, 36.6% had a partial septum, and 22.9% had a complete septum after BPEI exposure for 1 h at twice the MIC. Because BPEI disrupts WTA activity, its effects on cell morphology and division are similar to those of WTA-absent MRSA mutants. Whether WTA is disabled or absent altogether, cells cannot divide properly and form asymmetrical, abnormal, and multiple division septa. Our results confirm previous findings. Schlag et al. used TEM to show that WTA-deficient knockout mutants of S. aureus were larger and had rougher surfaces, cell wall degradation, and lower electron density in the cell walls,32 and Campbell et al. found similar defects in MRSA MW2 ΔtarO.31
Figure 6.

Fluorescence images of MRSA JE2 bacteria showing that BPEI binds to the peripheral cell wall and the division septum of dividing cells. The intensity of the Nile Red dye is used to count the number of cells with no septum (denoted as P1), a partial septum (denoted as P2), or a complete septum (denoted as P3).
CONCLUSION
Because WTA is essential for resistance mechanisms that involve penicillin binding proteins (PBPs), WTA is an “Achilles heel” of MRSA’s β-lactam resistance. However, our data show that this vulnerability is strain-dependent because hospital-acquired MRSA (HA-MRSA) and community-acquired MRSA (CA-MRSA) employ different PBPs in their resistance mechanisms.27,51 HA-MRSA relies on only PBP2a, while CA-MRSA utilizes both PBP2a and PBP4.52 Fortunately, Fishovitz et al. recently inhibited PBP2a in MRSA COL using one molecule of ceftaroline bound to the allosteric site, which opened the active site of PBP2a to other β-lactams;53 however, resistance has been reported.24–26 Additionally, WTA recruits PBP4 to the cell division septum, which allows PBP4 to participate in β-lactam resistance.2,3,31,54
Because WTA synthesis is essential for a robust cell wall architecture,55 previous studies that disrupt WTA biosynthesis with mutants were not accurately representing the MRSA cell wall. Furthermore, disrupting WTA biosynthesis causes biomolecular precursors and Lipid II fragments to accumulate, which downregulates vital biochemical pathways.55 To avoid these confounding variables, we used wild-type MRSA cells (which have a completely intact envelope composition and architecture) to investigate the role of WTA in cell division and β-lactam resistance. We found that hydrophilic, amine-terminated, 600 Da branched polyethylenimine (BPEI) binds to and disables WTA within wild-type MRSA cells.8,29,30 Here, we have shown that BPEI delocalizes PBP4 but does not affect other essential cell division enzymes, such as PBP2 and FtsZ. We also hypothesized that, if WTA function is similar between strains, the same amount of BPEI should theoretically decrease the oxacillin MIC below 2 μg/mL (the resistance threshold) in each strain. This hypothesis was incorrect; different strains required varying amounts of BPEI (from 1 to 32 μg/mL) to impair resistance. These results suggest that, in some MRSA strains, WTA either is less important to resistance or is harder to disable than in other strains. Future studies will explore cell wall changes with SEM and TEM imaging of different MRSA strains treated with BPEI.
Even if BPEI is an effective potentiator, it is cationic, and cationic compounds (such as aminoglycosides and polymyxins) can lead to nephrotoxicity.56,57 However, according to Wiegand et al., even though high-molecular weight BPEIs (>25000 Da) are toxic, 600 Da BPEI has high biocompatibility and a low likelihood for mutagenesis.58 This report demonstrates BPEI’s safety, biocompatibility, and antimicrobial properties but does not evaluate BPEI’s synergy with antibiotics against antibiotic-resistant bacteria. To confirm the literature reports of low toxicity, an in vitro nephrotoxicity assay, which detects the release of the metabolic enzyme lactate dehydrogenase (LDH), was performed using primary human renal proximal tubule epithelial cells (hRPTECs).30 A low level of LDH release suggests that the membrane is not damaged and that the test agent does not cause in vitro nephrotoxicity. Exposure to 600 Da BPEI caused minimal release of LDH (~1% at 8 μg/mL, 16% and 31 μg/mL, 3.5% at 62 μg/mL, and 8% at 125 μg/mL). As per the literature, these values are much lower than the LDH release values for cationic, nephrotoxic colistin (1% at 8 μg/mL, 2.3% at 16 μg/mL, 18% at 31 μg/mL, 26% at 62 μg/mL, and 28% at 125 μg/mL). However, the LDH assays also showed that 1200, 1800, and 10000 Da BPEIs are more toxic than 600 Da BPEI.30 Hydrophilicity is likely responsible for the differences in toxicity between low and high-molecular weight BPEI. Because 600 Da BPEI is miscible with water and does not contain regions of hydrophobic character (as seen with cationic peptides, aminoglycosides, and polymyxins), it lacks the energetic force that drives hydrophobic compounds into lipid membranes. On the other hand, 25000–1000000 Da BPEI molecules possess hydrophobic interiors that increase their lipophilicity and lead to membrane penetration. Furthermore, 600 Da BPEI is not toxic toward colon, kidney, or HeLa cells unless the concentration is orders of magnitude higher than the concentration required for potentiation; the in vitro effective concentration is 1–8 μg/mL, whereas the IC50 > 300 μg/mL for 600 Da BPEI.30 Because of its low cytotoxicity, 600 Da BPEI holds promise as a lead potentiator in drug development. However, further assessment of in vitro and in vivo toxicity requires additional preclinical trials.
Supplementary Material
ACKNOWLEDGMENTS
This work was possible due to the kindness and contributions of Daniel Glatzhofer, Ph.D., Robert Cichewicz, Ph.D., Scott Russel, Ph.D., and Preston Larson, Ph.D. The authors also thank Cindy McCloskey, M.D., for the clinical isolates and acknowledge Dr. Suzanne Walker (Harvard University, Cambridge, MA) for providing MRSA MW2 and the ΔtarO mutant.
Funding
Funding provided by the National Institutes of Health (C.V.R., R03AI142420-01), the Oklahoma Center of Advancement of Science and Technology (C.V.R., HR16-084-3), and the European Research Council (M.G.P., ERC-2017-CoG-771709).
ABBREVIATIONS
- MRSA
methicillin-resistant S. aureus
- MSSA
methicillin-susceptible S. aureus
- BPEI
branched polyethylenimine
- NMR
nuclear magnetic resonance
- TEM
transmission electron microscopy
- SEM
scanning electron microscopy
- WTA
wall teichoic acid
- LTA
lipoteichoic acid
- PBP
penicillin binding protein
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- FICI
fractional inhibitory concentration index
- MIC
minimum inhibitory concentration
- MBC
minimum bactericidal concentration
- AuPd
gold palladium
- HMDS
hexamethyldisilazane
- OD600
optical density at 600 nm
- TSB
tryptic soy broth
- MHB
Mueller-Hinton broth
- ANOVA
analysis of variance
- USCAST
United States Committee on Antimicrobial Susceptibility Testing
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00523.
Additional figures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Gordon RJ, and Lowy FD (2008) Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis 46, S350–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pasquina LW, Santa Maria JP, and Walker S (2013) Teichoic acid biosynthesis as an antibiotic target. Curr. Opin. Microbiol 16, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Swoboda JG, Campbell J, Meredith TC, and Walker S (2010) Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 11, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Carrel M, Perencevich EN, and David MZ (2015) USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000–2013. Emerging Infect. Dis 21, 1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, and Hiramatsu K (2002) Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827. [DOI] [PubMed] [Google Scholar]
- (6).Suzuki M, Yamada K, Nagao M, Aoki E, Matsumoto M, Hirayama T, Yamamoto H, Hiramatsu R, Ichiyama S, and Iinuma Y (2011) Antimicrobial ointments and methicillin-resistant Staphylococcus aureus USA300. Emerging Infect. Dis 17, 1917–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, and Fridkin S (2013) National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Intern. Med 173, 1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guillamet MCV, Vazquez R, Deaton B, Shroba J, Vazquez L, and Mercier RC (2018) Host-Pathogen-Treatment Triad: Host Factors Matter Most in Methicillin-Resistant Staphylococcus aureus Bacteremia Outcomes. Antimicrob. Agents Chemother 62, No. e01902–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mermel LA, Allon M, and Bouza E (2010) Clinical practice guidelines for the diagnosis and management of intravascular catheter related infection: 2009 update by the Infectious Diseases Society of America (vol 49, pg 1, 2009). Clin. Infect. Dis 50, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baddour LM, Wilson WR, Bayer AS, Fowler VG Jr., Tleyjeh IM, Rybak MJ, Barsic B, Lockhart PB, Gewitz MH, Levison ME, Bolger AF, Steckelberg JM, Baltimore RS, Fink AM, O’Gara P, and Taubert KA (2015) Infective Endocarditis in Adults: Diagnosis, Antimicrobial Therapy, and Management of Complications: A Scientific Statement for Healthcare Professionals From the American Heart Association. Circulation 132, 1435–1486. [DOI] [PubMed] [Google Scholar]
- (11).McDanel JS, Roghmann MC, Perencevich EN, Ohl ME, Goto M, Livorsi DJ, Jones M, Albertson JP, Nair R, O’Shea AMJ, and Schweizer ML (2017) Comparative Effectiveness of Cefazolin Versus Nafcillin or Oxacillin for Treatment of Methicillin-Susceptible Staphylococcus aureus Infections Complicated by Bacteremia: A Nationwide Cohort Study. Clin. Infect. Dis 65, 100–106. [DOI] [PubMed] [Google Scholar]
- (12).Blumenthal KG, and Shenoy ES (2016) Editorial Commentary: Fortune Favors the Bold: Give a Beta-Lactam! Clin. Infect. Dis 63, 911–913. [DOI] [PubMed] [Google Scholar]
- (13).Li J, Echevarria KL, and Traugott KA (2017) beta-Lactam Therapy for Methicillin-Susceptible Staphylococcus aureus Bacteremia: A Comparative Review of Cefazolin versus Antistaphylococcal Penicillins. Pharmacotherapy 37, 346–360. [DOI] [PubMed] [Google Scholar]
- (14).Bosso JA, Nappi J, Rudisill C, Wellein M, Bookstaver PB, Swindler J, and Mauldin PD (2011) Relationship between vancomycin trough concentrations and nephrotoxicity: a prospective multicenter trial. Antimicrob. Agents Chemother 55, 5475–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Koppula S, Ruben S, Bangash F, and Szerlip HM (2015) Pitfalls in dosing vancomycin. Am. J. Med. Sci 349, 137–139. [DOI] [PubMed] [Google Scholar]
- (16).Bruniera FR, Ferreira FM, Saviolli LRM, Bacci MR, Feder D, Pedreira MDG, Peterlini MAS, Azzalis LA, Junqueira VBC, and Fonseca FLA (2015) The use of vancomycin with its therapeutic and adverse effects: a review. European Review for Medical and Pharmacological Sciences 19, 694–700. [PubMed] [Google Scholar]
- (17).Kurosu M, Siricilla S, and Mitachi K (2013) Advances in MRSA drug discovery: where are we and where do we need to be? Expert Opin. Drug Discovery 8, 1095–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wilke MH (2010) Multiresistant bacteria and current therapy the economical side of the story. Eur. J. Med.Res 15, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Velez JC, and Janech MG (2010) A case of lactic acidosis induced by linezolid. Nat. Rev. Nephrol 6, 236–242. [DOI] [PubMed] [Google Scholar]
- (20).Sax P, and Plank R (2017) Antibiotics All-Stars Draft: Dr. Paul Sax Faces off with Dr. Rebecca Plank. Open Forum Infect. Dis 4, oxf107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Waxman DJ, and Strominger JL (1983) Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics1. Annu. Rev. Biochem 52, 825–869. [DOI] [PubMed] [Google Scholar]
- (22).Fuda C, Suvorov M, Vakulenko SB, and Mobashery S (2004) The basis for resistance to beta-lactam antibiotics by penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. J. Biol. Chem 279, 40802–40806. [DOI] [PubMed] [Google Scholar]
- (23).Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, and Mobashery S (2008) Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus: potent inhibition of PBP 2a by two anti-MRSA beta-lactam antibiotics. J. Am. Chem. Soc 130, 9212–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Strommenger B, Layer F, Klare I, and Werner G (2015) Pre-Use Susceptibility to Ceftaroline in Clinical Staphylococcus aureus Isolates from Germany: Is There a Non-Susceptible Pool to be Selected? PLoS One 10, No. e0125864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Long SW, Olsen RJ, Mehta SC, Palzkill T, Cernoch PL, Perez KK, Musick WL, Rosato AE, and Musser JM (2014) PBP2a mutations causing high-level Ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother 58, 6668–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gonzales PR, Pesesky MW, Bouley R, Ballard A, Biddy BA, Suckow MA, Wolter WR, Schroeder VA, Burnham CA, Mobashery S, Chang M, and Dantas G (2015) Synergistic, collaterally sensitive beta-lactam combinations suppress resistance in MRSA. Nat. Chem. Biol 11, 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Memmi G, Filipe SR, Pinho MG, Fu Z, and Cheung A (2008) Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother 52, 3955–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Atilano ML, Pereira PM, Yates J, Reed P, Veiga H, Pinho MG, and Filipe SR (2010) Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A 107, 18991–18996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Foxley MA, Friedline AW, Jensen JM, Nimmo SL, Scull EM, King JB, Strange S, Xiao MT, Smith BE, Thomas KJ, Glatzhofer DT, Cichewicz RH, and Rice CV (2016) Efficacy of ampicillin against methicillin-resistant Staphylococcus aureus restored through synergy with branched poly(ethylenimine). J. Antibiot 69, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Foxley MA, Wright SN, Lam AK, Friedline AW, Strange SJ, Xiao MT, Moen EL, and Rice CV (2017) Targeting Wall Teichoic Acid in Situ with Branched Polyethylenimine Potentiates beta-Lactam Efficacy against MRSA. ACS Med. Chem. Lett 8, 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Campbell J, Singh AK, Santa Maria JP Jr., Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, and Walker S (2011) Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol 6, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, and Gotz F (2010) Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol 75, 864–873. [DOI] [PubMed] [Google Scholar]
- (33).Qamar A, and Golemi-Kotra D (2012) Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob. Agents Chemother 56, 3797–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Farha MA, Leung A, Sewell EW, D’Elia MA, Allison SE, Ejim L, Pereira PM, Pinho MG, Wright GD, and Brown ED (2013) Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to beta-lactams. ACS Chem. Biol 8, 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak MJ, Talan DA, and Chambers HF (2011) Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis 52, No. e18. [DOI] [PubMed] [Google Scholar]
- (36).Pereira AR, Hsin J, Krol E, Tavares AC, Flores P, Hoiczyk E, Ng N, Dajkovic A, Brun YV, VanNieuwenhze MS, Roemer T, Carballido-Lopez R, Scheffers DJ, Huang KC, and Pinho MG (2016) FtsZ-Dependent Elongation of a Coccoid Bacterium. mBio 7, e00908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Monteiro JM, Pereira PM, Elsen NL, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillancourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Ramtohul Y, Langlois E, Landry F, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Pinho MG, Soisson SM, and Roemer T (2012) Restoring methicillin-resistant Staphylococcus aureus susceptibility to beta-lactam antibiotics. Sci. Transl. Med 4, No. 126ra35. [DOI] [PubMed] [Google Scholar]
- (38).Monteiro JM, Pereira AR, Reichmann NT, Saraiva BM, Fernandes PB, Veiga H, Tavares AC, Santos M, Ferreira MT, Macario V, VanNieuwenhze MS, Filipe SR, and Pinho MG (2018) Peptidoglycan synthesis drives an FtsZ-treadmillingindependent step of cytokinesis. Nature 554, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jevons MP (1961) Celbenin” resistant Staphylococci. British Medical Journal 1, 124–125. [Google Scholar]
- (40).Mellor JA, Kingdom J, Cafferkey M, and Keane CT (1985) Vancomycin toxicity: a prospective study. J. Antimicrob. Chemother 15, 773–780. [DOI] [PubMed] [Google Scholar]
- (41).Appelbaum PC (2006) The emergence of vancomycinintermediate and vancomycin-resistant Staphylococcus aureus. Clin. Microbiol. Infect 12, 16–23. [DOI] [PubMed] [Google Scholar]
- (42).Sievert DM, Rudrik JT, Patel JB, McDonald LC, Wilkins MJ, and Hageman JC (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis 46, 668–674. [DOI] [PubMed] [Google Scholar]
- (43).The United States Committee on Antimicrobial Susceptibility Testing (2017) Breakpoint tables for interpretation of MICs and zone diameters, version 3.0. http://www.uscast.org.
- (44).Nair DR, Monteiro JM, Memmi G, Thanassi J, Pucci M, Schwartzman J, Pinho MG, and Cheung AL (2015) Characterization of a Novel Small Molecule That Potentiates beta Lactam Activity against Gram-Positive and Gram-Negative Pathogens. Antimicrob. Agents Chemother 59, 1876–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Farha MA, Koteva K, Gale RT, Sewell EW, Wright GD, and Brown ED (2014) Designing analogs of ticlopidine, a wall teichoic acid inhibitor, to avoid formation of its oxidative metabolites. Bioorg. Med. Chem. Lett 24, 905–910. [DOI] [PubMed] [Google Scholar]
- (46).Lee SH, Wang H, Labroli M, Koseoglu S, Zuck P, Mayhood T, Gill C, Mann P, Sher X, Ha S, Yang SW, Mandal M, Yang C, Liang L, Tan Z, Tawa P, Hou Y, Kuvelkar R, DeVito K, Wen X, Xiao J, Batchlett M, Balibar CJ, Liu J, Xiao J, Murgolo N, Garlisi CG, Sheth PR, Flattery A, Su J, Tan C, and Roemer T (2016) TarO-specific inhibitors of wall teichoic acid biosynthesis restore beta-lactam efficacy against methicillin-resistant staphylococci. Sci. Transl. Med 8, No. 329ra32. [DOI] [PubMed] [Google Scholar]
- (47).Mandal M, Tan Z, Madsen-Duggan C, Buevich AV, Caldwell JP, Dejesus R, Flattery A, Garlisi CG, Gill C, Ha SN, Ho G, Koseoglu S, Labroli M, Basu K, Lee SH, Liang L, Liu J, Mayhood T, McGuinness D, McLaren DG, Wen X, Parmee E, Rindgen D, Roemer T, Sheth P, Tawa P, Tata J, Yang C, Yang SW, Xiao L, Wang H, Tan C, Tang H, Walsh P, Walsh E, Wu J, and Su J (2017) Can. We Make Small Molecules Lean? Optimization of a Highly Lipophilic TarO Inhibitor. J. Med. Chem 60, 3851–3865. [DOI] [PubMed] [Google Scholar]
- (48).Yang SW, Pan J, Yang C, Labroli M, Pan W, Caldwell J, Ha S, Koseoglu S, Xiao JC, Mayhood T, Sheth PR, Garlisi CG, Wu J, Lee SH, Wang H, Tan CM, Roemer T, and Su J (2016) Benzimidazole analogs as WTA biosynthesis inhibitors targeting methicillin resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett 26, 4743–4747. [DOI] [PubMed] [Google Scholar]
- (49).Gautam S, Kim T, and Spiegel DA (2015) Chemical probes reveal an extraseptal mode of cross-linking in Staphylococcus aureus. J. Am. Chem. Soc 137, 7441–7447. [DOI] [PubMed] [Google Scholar]
- (50).Reed P, Atilano ML, Alves R, Hoiczyk E, Sher X, Reichmann NT, Pereira PM, Roemer T, Filipe SR, Pereira-Leal JB, Ligoxygakis P, and Pinho MG (2015) Staphylococcus aureus Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance. PLoS Pathog 11, No. e1004891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Leski TA, and Tomasz A (2005) Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: Evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J. Bacteriol 187, 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fishovitz J, Rojas-Altuve A, Otero LH, Dawley M, Carrasco-Lopez C, Chang M, Hermoso JA, and Mobashery S (2014) Disruption of allosteric response as an unprecedented mechanism of resistance to antibiotics. J. Am. Chem. Soc 136, 9814–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fishovitz J, Taghizadeh N, Fisher JF, Chang M, and Mobashery S (2015) The Tipper-Strominger Hypothesis and Triggering of Allostery in Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus aureus (MRSA). J. Am. Chem. Soc 137, 6500–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Swoboda JG, Meredith TC, Campbell J, Brown S, Suzuki T, Bollenbach T, Malhowski AJ, Kishony R, Gilmore MS, and Walker S (2009) Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem. Biol 4, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Schaefer K, Owens TW, Kahne D, and Walker S (2018) Substrate Preferences Establish the Order of Cell Wall Assembly in Staphylococcus aureus. J. Am. Chem. Soc 140, 2442–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Gallardo-Godoy A, Muldoon C, Becker B, Elliott AG, Lash LH, Huang JX, Butler MS, Pelingon R, Kavanagh AM, Ramu S, Phetsang W, Blaskovich MA, and Cooper MA (2016) Activity and Predicted Nephrotoxicity of Synthetic Antibiotics Based on Polymyxin B. J. Med. Chem 59, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Huang JX, Kaeslin G, Ranall MV, Blaskovich MA, Becker B, Butler MS, Little MH, Lash LH, and Cooper MA (2015) Evaluation of biomarkers for in vitro prediction of druginduced nephrotoxicity: comparison of HK-2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol. Res. Perspect 3, No. e00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wiegand C, Bauer M, Hipler UC, and Fischer D (2013) Poly(ethyleneimines) in dermal applications: biocompatibility and antimicrobial effects. Int. J. Pharm 456, 165–174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.