

Abstract
Background & Aim:
Acetaminophen (APAP)-induced acute liver failure is associated with substantial alterations in the hemostatic system. In mice, platelets accumulate in the liver after APAP overdose and appear to promote liver injury. Interestingly, patients with acute liver injury have highly elevated levels of the platelet-adhesive protein von Willebrand factor (VWF), but a mechanistic connection between VWF and progression of liver injury has not been established. We tested the hypothesis that VWF contributes directly to experimental APAP-induced acute liver injury.
Methods:
Wild-type mice and VWF-deficient (VWF−/−) mice were given a hepatotoxic dose of APAP (300mg/kg, i.p.) or vehicle (saline).
Results:
In wild-type mice, VWF plasma levels, high molecular weight (HMW) VWF multimers, and VWF activity decreased 24h after APAP challenge. These changes coupled to robust hepatic VWF and platelet deposition, although VWF deficiency had minimal effect on peak hepatic platelet accumulation or liver injury. VWF plasma levels were elevated 48h after APAP challenge, but with relative reductions in HMW multimers and VWF activity. Whereas hepatic platelet aggregates persisted in livers of APAP-challenged wild-type mice, platelets were nearly absent in VWF−/− mice 48h after APAP challenge. The absence of platelet aggregates was linked to dramatically accelerated repair of the injured liver. Complementing observations in VWF−/− mice, blocking VWF or the platelet integrin αIIbβ3 during development of injury significantly reduced hepatic platelet aggregation and accelerated liver repair in APAP-challenged wild-type mice.
Conclusion:
These studies are the first to suggest a mechanistic link between VWF, hepatic platelet accumulation, and liver repair. Targeting VWF in acute liver injury might provide a novel therapeutic approach to improve repair of the APAP-injured liver.
Keywords: von Willebrand factor, platelets, coagulation, acute liver injury, thrombosis
Graphical Abstract
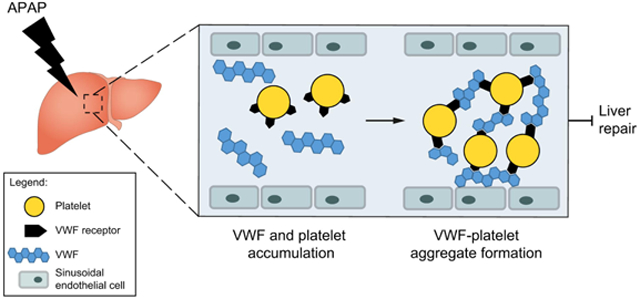
Lay Summary
Patients with acute liver injury due to acetaminophen overdose have highly elevated levels of the platelet-adhesive protein von Willebrand factor. It is not known whether von Willebrand factor plays a direct role in the progression of acute liver injury. We discovered that von Willebrand factor delays repair of the acetaminophen-injured liver in mice and that targeting von Willebrand factor, even in mice with established liver injury, accelerates liver repair.
Introduction
Acetaminophen (paracetamol, APAP) overdose is a leading cause of drug-induced acute liver injury and acute liver failure (ALF) in the western world.1 Accumulating evidence from experimental and clinical studies suggests that the hemostatic system contributes to the progression of acute liver injury after APAP overdose.2-4 Although not associated with clinically significant bleeding5, substantial alterations in the hemostatic system are evident in patients with AAP-induced liver failure, including a reduced platelet count.6,7 The ALF study group has demonstrated in a large cohort of ALF patients that a much more profound thrombocytopenia is associated with poor outcome (i.e. death or the need for a liver transplant).8 Platelets have been proposed to drive disease progression through the formation of microthrombi within the liver microvasculature.9 APAP-induced liver damage in mice causes persistent thrombocytopenia, which is coupled to accumulation of platelets in the injured liver.3 Notably, platelets have been shown to promote APAP-induced liver injury in mice.3 However, the mechanisms driving platelet accumulation in the APAP-injured liver remain to be elucidated.
The platelet-adhesive glycoprotein von Willebrand factor (VWF) is a key component of the hemostatic system. VWF synthesis is restricted to endothelial cells and megakaryocytes.10 Following vascular damage, VWF binds to subendothelial collagens, which activate VWF to serve as an adhesion molecule for platelets, thereby initiating platelet plug formation.11 VWF is a multimeric protein and its reactivity towards platelets depends on the size of its multimers. The high molecular weight (HMW) multimers are the most effective in supporting platelet adhesion.10,12 Multimeric size of VWF is regulated by ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), which proteolytically cleaves the large multimers into smaller, less active multimers.12 VWF size regulation is important for normal hemostatic function, as evidenced by patients with a congenital or acquired ADAMTS13 deficiency, who suffer from severe thrombotic episodes.13
VWF plasma levels are highly elevated in patients with ALF2,14, while ADAMTS13 levels are reduced. 2,15 It has been proposed that insufficient regulation of VWF activity could cause hepatic platelet-induced microthrombi formation that drives disease progression. .2 Although several studies have reported elevated VWF levels in patients with liver disease2,14,16, the basis for this increase is unclear. In addition, a direct mechanistic connection between VWF and the progression of liver injury has not yet been established. We tested the hypothesis that VWF contributes to experimental acute liver injury. Using genetic and antibody-mediated strategies to target VWF, we determined the contribution of VWF to both APAP-induced liver injury and repair of the injured liver. Our results establish a clear mechanistic connection between changes in VWF, hepatic platelet accumulation and liver repair, suggesting an explanation for clinical observations.
Materials and Methods
Mice
VWF−/− mice on a congenic C57Bl/6J background were obtained from Jackson Laboratory (Bar Harbor, ME) and maintained by homozygous breeding. These mice have been described previously.17 Aged and sex-matched wild-type mice on an identical C57Bl/6J background were used as control mice. A total of 140 male mice were used for these studies between the ages of 8-14 weeks. Mice were housed under a 12-h light/dark cycle and standard diet (Teklad 8940), and drinking water were provided ad libitum. The Institutional Animal Care and Use Committee of Michigan State University, East Lansing, USA, approved all animal procedures.
Acetaminophen-induced acute liver injury
Mice were fasted overnight (i.e., approximately 15 hours) on new alpha-dry bedding, before administration of 300 mg/kg APAP (30 μl/g body weight) or vehicle (warm sterile saline, 0.9% sodium chloride) via i.p. injection as described previously.18 Food was returned immediately after injection. For VWF supplementation studies, VWF−/− mice were treated with APAP, and 3h later received human plasma-derived VWF (3U VWF:RCo/mouse, Humate-P, CSL Behring, obtained from MSU Clinical Center Pharmacy) or vehicle (saline) via i.p injection, as described.19 For studies in which VWF was blocked, wild-type mice were treated with APAP, and 4h and 24h later a rabbit polyclonal anti-VWF antibody (50μg/mouse, DAKO, A0082, Glostrup, Denmark, dialyzed against endotoxin-free PBS) or rabbit IgG control (50μg/mouse, Innovative Research, Novi, MI, dialyzed against endotoxin-free PBS) was injected i.p. For studies blocking the platelet integrin αIIbβ3, wild-type mice were challenged with APAP, and 12h and 24h later treated with_GR144053 (10mg/kg, R&D Systems, Minneapolis, MN) or vehicle (saline) by i.p. injection. Blood and liver samples were collected 24h, 48h or 72h after APAP administration. Blood was collected under isoflurane anesthesia by exsanguination from the inferior vena cava into a syringe containing sodium citrate (0.38% final) for the collection of plasma. Blood samples were centrifuged at 4000 x g for 10 minutes to obtain plasma and were stored at −80°C. Livers were rinsed in PBS and fixed in either 10% neutral-buffered formalin or snap frozen in liquid nitrogen.
Clearance of human VWF in APAP-challenged mice
Male VWF−/−mice were treated with 300mg/kg APAP or vehicle (saline) as described above. Twenty-four hours after APAP or vehicle administration, mice were treated with a single dose (3U VWF:RCo/mouse) of VWF concentrate (Humate-P, CSL Behring) by retro-orbital injection. Injections were performed under anesthesia using isoflurane. At 30min, 1h, 2h, 4h and 8h after injection, blood was collected into citrate as described above from separate cohorts of mice at each time. VWF:Ag was measured as described below, and expressed as percentage of injected amount, which was set at 100%.
Measurement of liver necrosis and hepatocyte proliferation.
Paraffin-embedded livers were sectioned at 5 μm and stained with hematoxylin and eosin (H&E) or labeled for proliferating cell nuclear antigen (PCNA), as described previously.20 All immunohistochemical labeling was performed by the MSU Investigative Histopathology Laboratory, a division of Human Pathology. Necrosis area was calculated as described previously.20 Serum alanine aminotransferase (ALT) activity was determined using commercial reagents (ThermoFisher, Waltham, MA).
Immunofluorescent labeling of liver sections
Fibrin(ogen), VWF and platelets were detected by immunofluorescent labeling of frozen liver sections. Briefly, frozen liver pieces were sectioned at 8 μm and fixed in 4% neutral-buffered formalin for 10min. The sections were blocked in 10% normal goat serum in PBS for 1h at room temperature, and incubated with polyclonal rabbit anti-human fibrin(ogen) antibody (DAKO, A0080, 1:2500) or polyclonal rabbit anti-human VWF (DAKO, A0082, 1:400) overnight at 4 °C. Primary antibodies were detected using Alexa Fluor 594-conjugated goat anti-rabbit IgG or Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (Jackson Immunoresearch, West Grove, PA, USA, 1:500), respectively. The sections were scanned with a VS110 Virtual Slide System. The area of positive labeling was quantified in four or five randomly selected images (captured with a 5x virtual objective in OLYVIA Software, approximately 2.75 mm2 of liver tissue per image) in an unbiased fashion using a batch macro and color de-convolution tool in Fiji (imageJ). Colocalization of VWF with platelets was examined in frozen liver tissues. Sections were cut, fixed and blocked as described above. The sections were first incubated with monoclonal rat anti-mouse CD41 (clone MWReg30, Biolegend, 1:200) overnight at 4 °C and then for 2h at room temperature with polyclonal rabbit anti-human VWF (DAKO, A0082, 1:400). Staining was detected with cross-adsorbed Alexa 594-conjugated secondary goat anti-rat antibody and Alexa 488-conjugated secondary goat anti-rabbit IgG (Jackson Immunoresearch, 1:500), respectively. Fluorescent images were captured at 100X total magnification using Olympus DP70 microscope (Olympus, Lake Success, NY). The area of positive platelet labeling was quantified in four or five randomly selected images (approximately 0.5 mm2 of liver tissue per image) from each mouse liver in an unbiased fashion using a batch macro and color de-convolution tool in Fiji (ImageJ).
Measurement of VWF plasma level, size, activity, and thrombin-antithrombin complexes.
Plasma VWF antigen levels were determined by ELISA, as previously described.21 Pooled normal mouse plasma or pooled normal human plasma was used to develop a standard curve. The Hydragel 5 VWF multimers kit (H5VWM, PN4359 and VWF multimer visualization kit, PN4747, Sebia) was used with Hydrasys 2 Scan instrumentation to perform semi-automated agarose gel electrophoresis using preformed 2% agarose gels, direct immunofixation, and visualization with peroxidase-labelled antibody as recommended by the manufacturer.22 In order to enhance the intensity of the murine VWF multimer signal, dilution was adjusted from the standard 1:6 for human plasma samples to a 1:1 dilution for mouse samples. A pooled normal mouse plasma was included on each gel. VWF multimer analysis was performed at the Thrombosis Center of the Cincinnati Children’s Hospital. The first 4 bands were considered low-molecular weight (LMW) multimers, while the other bands were considered high molecular weight (HMW) multimers. Gels were scanned using the Hydrasys 2 Scan, and analyzed with the ImageJ gel analysis tool. Data were expressed as the percentage of HMW multimers per total VWF multimers. The ability of VWF to bind to immobilized collagen (VWF:CBA) was assessed using a VWF:CBA assay, as described.23 In brief, collagen type III (Sigma Type X, C4407) was dissolved in 0.05M acetic acid to 1mg/ml. Plates were coated with collagen overnight (diluted to 10μg/ml in 50mM carbonate buffer, pH 9.6, 115 μl/well), and after washing the plate was blocked for 1h at 37 °C with blocking buffer (3% bovine serum albumin, 0.1% Tween20, in PBS). A standard curve was generated by serial dilution of normal pooled mouse plasma (1:5 to 1:320 in blocking buffer). Plasma samples (100 μl/well) were incubated for 2h at 37 °C, and after washing the plate, bound VWF was detected using a peroxidase-conjugated anti-human VWF antibody (DAKO, P0226) (1:1000 in blocking buffer, 100 μl/well, 1.5h incubation at 37 °C). Peroxidase activity was assessed using TMB substrate set (421101, Biolegend) according to the manufacturer’s instruction. Plasma thrombin-antithrombin (TAT) levels were determined using a commercially available enzyme-linked immunosorbent assay kit (Siemens Health Care Diagnostics, Deerfield, IL).
RNA isolation, cDNA synthesis and quantitative real-time PCR (qPCR)
Small sections (~20 mg) of the liver lobes were collected for RNA isolation and snap frozen in liquid nitrogen. RNA isolation, cDNA synthesis and qPCR were performed as described.18 Detection and quantification of select mRNAs by SYBR Green qPCR were performed using primers described previously.24 The expression of each gene was normalized to the geometric mean Ct of individual housekeeping genes, i.e. Hprt and Gapdh, and relative fold change was determined with the ΔΔCt method. All fold changes were normalized relative to expression in wild-type mice. A complete list of all gene names and primer sequences is provided in Supplementary Table 1.
Statistics
Statistical analyses were performed with GraphPad Prism v.5 (San Diego, CA, USA) software package. Clearance data was calculated as previously described.25 Continuous variables are presented as mean + SEM. Comparison of two groups was performed using Student’s t test. Comparison of three or more groups was performed using one- or two-way analysis of variance (ANOVA), as appropriate, with the Student-Newman-Keuls (SNK)post-hoc test. Differences were considered significant if the P-value was < 0.05.
Results
Impact of APAP-induced acute liver injury on plasma and hepatic VWF.
Plasma VWF antigen (VWF:Ag) levels initially decreased 24h after APAP overdose relative to saline-treated control animals, but were increased 48h after APAP challenge and remained elevated 72h after APAP administration (Fig. 1A), in agreement with the elevated VWF:Ag levels in ALF patients2,14. The increase in VWF:Ag plasma levels was coupled to a slight increase in hepatic VWF mRNA expression 24h after APAP administration, but hepatic mRNA levels returned to baseline at 48h and 72h after APAP overdose (Fig. 1B). The initial decrease in VWF:Ag plasma levels at 24 hours was paralleled by robust hepatic accumulation of VWF in livers of APAP-treated mice (Fig. 1C). The amount of hepatic VWF deposition decreased over time but remained significantly elevated compared to livers of saline-treated mice (Fig. 1D).
Figure 1. Impact of APAP-induced acute liver injury on plasma and hepatic VWF.

Male wild-type mice were given 300mg/kg APAP or saline (i.p.) and samples were collected 24h, 48h, and 72h later. (A) VWF plasma antigen (VWF:Ag) levels were determined by ELISA.. Levels are expressed as % of normal pooled mouse plasma which was set at 100%. (B) Hepatic mRNA levels of VWF in wild-type-challenged mice. (C) Representative images of hepatic VWF immunofluorescent labeling (green) (D) Quantification of hepatic VWF deposition, expressed as percentage of positive pixel count. For E, male VWF−/− mice were challenged with saline (black dashed line) or 300mg/kg APAP (i.p.) (red line) and 24h after challenge, the mice were given 3U of Humate-P® by intravenous injection. Human VWF:Ag levels were determined by ELISA various times after VWF administration, and expressed as % of infused VWF amount set to 100%. Bars represent mean ± standard error of mean (n = 5-12 mice per group for panel A-D, n=3-6 mice per time point for panel E) *p <0.05 compared to saline-treated animals.
VWF clearance is impaired after APAP-induced acute liver injury.
As VWF is mainly cleared by the liver26, it has been proposed that the elevated VWF levels observed in ALF patients could be a consequence of impaired hepatic clearance. However, this concept has not yet been tested experimentally. We therefore assessed clearance of VWF using VWF−/− mice. VWF−/− mice were challenged with APAP or saline, and 24h after challenge received a single intravenous injection of plasma-derived human VWF. Liver injury was confirmed by increased ALT levels (data not shown). Pharmacokinetic analysis showed that the mean residence time (MRT) of infused VWF was longer in APAP-challenged mice than in saline-treated mice (MRT of 194.7 min versus MRT of 107.2 min), indicating that VWF clearance is indeed impaired in APAP-induced acute liver injury (Fig. 1E).
Impact of APAP-induced acute liver injury on VWF multimer distribution and activity.
To assess whether APAP-induced liver injury affects VWF multimer distribution, we performed VWF multimer analysis using a semi-automated VWF multimer kit. Analysis of plasma VWF multimers in healthy wild-type mice using the Hydrasys 2 Scan instrument revealed a multimer pattern consistent with the conventional analysis approach.27 Specificity was documented by the absence of detectable signal in plasma from VWF−/− mice (Supplementary Fig 1). VWF multimer analysis was next performed in plasma from APAP-challenged wild-type mice. High molecular weight (HMW) VWF multimers were reduced 24 h after APAP challenge (Fig. 2A-C), corresponding to a reduction in plasma VWF collagen binding activity (VWF:CB) (Fig. 2D) The increase in VWF:Ag levels at 48 hours and 72 hours after APAP challenge (Fig. 1A) was also evident by multimer analysis (Fig. 2A). Although total VWF antigen levels were higher at 48 and 72 hours, the fraction of plasma VWF with high activity (i.e., HMW multimers) at these times was reduced compared to saline-treated mice (Fig. 2C). Thus, although total plasma VWF:CB activity 48 and 72 hours after APAP challenge exceeded that of saline control mice, the ratio of VWF:CB activity to VWF antigen (VWF:Ag) displayed a relative reduction (Fig. 2E). Notably, these observations are consistent with what has been described for ALF patients.2
Figure 2. Impact of APAP-induced acute liver injury on VWF multimer distribution and VWF activity.

Male wild-type mice were given 300mg/kg APAP or saline (i.p.) and samples were collected 24h, 48h, and 72h later. (A) Representative images of VWF multimer analysis from normal pooled mouse plasma (NPP) (first lane) and plasma from saline-treated (second lane) and APAP-challenged wild-type mice (third to fifth lane) using a semi-automated gel electrophoresis method. (B) Representative densitometry tracings of VWF multimers comparing normal pooled mouse plasma (grey line) to challenged mice (pink line). (C) The proportion of HMW VWF multimers (relative to total VWF) in plasma was quantified by software-assisted morphometric analysis. (D) VWF collagen binding activity (VWF:CB) in plasma samples from wild-type APAP-challenged mice, shown as a percentage compared to normal pooled mouse plasma, set to 100%. (E) Ratio of VWF:CB and VWF:Ag (VWF:CB/VWF:Ag) in plasma samples from APAP-challenged wild-type mice. LMW = Low molecular weight. HMW = High molecular weight. Bars represent mean + standard error of the mean (n=3 mice per group for panel C, n= 5-12 mice per group for panel D-E), * p <0.05 compared to saline-treated animals.
VWF deficiency does not affect coagulation activation after APAP-induced acute liver injury.
As we have previously shown that APAP overdose in mice is associated with activation of the coagulation cascade4,28, we examined whether the deposition of VWF in the injured liver was connected to coagulation activation after APAP administration. Consistent with previous studies3,4,28, we observed a significant increase in plasma thrombin anti-thrombin (TAT) levels, indicative of coagulation activation, in wild-type mice after APAP administration (Fig 3A). However, there was no significant difference in TAT levels between wild-type or VWF−/− mice 24h after APAP challenge (Fig 3A). TAT plasma levels subsequently decreased over time in both wild-type and VWF−/− mice, showing no difference between genotypes (Fig 3A). Intrahepatic deposition of fibrin(ogen) in the livers of APAP-treated mice at 24h was primarily within the areas of hepatocellular necrosis (Fig 3C). VWF deficiency had no effect on the amount of hepatic fibrin(ogen) deposition 24h after APAP challenge (Fig. 3B-C). These results suggest that coagulation activation and deposition of fibrin(ogen) in the APAP-injured liver are not mechanistically connected to the deposition of VWF in the injured liver.
Figure 3. VWF deficiency does not affect coagulation cascade activation after APAP-induced acute liver injury in mice.

Wild-type (WT) and VWF−/− (VWF KO) mice were given 300mg/kg APAP or saline (i.p.) and samples were collected 24h, 48h, and 72h later. (A) Thrombin-antithrombin complex (TAT) plasma levels in wild-type (black bars) and VWF−/− (grey bars) mice were determined by ELISA. (B) Quantification of hepatic fibrin(ogen) immunofluorescent labeling, expressed as positive pixel count in wild-type (black bars) and VWF−/− (grey bars) mice challenged with saline or APAP at 24h. (C) Representative images of hepatic fibrin(ogen) immunolabeling (red) in DAPI (blue)-counterstained liver sections. Scale bars = 100 μm. Bars represent mean + standard error of the mean (n = 5-7 mice per group).
VWF prolongs platelet accumulation in the APAP-injured liver.
APAP overdose in mice leads to rapid hepatic platelet accumulation.3 As VWF serves as an adhesion molecule for platelets, we speculated that hepatic platelet influx after APAP overdose is mediated by VWF. APAP overdose caused platelet accumulation in the injured livers of wild-type mice, predominantly within areas of hepatocellular necrosis (Fig 4A,C). Focal colocalization of VWF and platelets, particularly in the liver sinusoids, persisted in livers of wild-type mice 48 hours after APAP challenge (Fig. 4A,C). Surprisingly, VWF deficiency did not significantly affect hepatic platelet accumulation 24h after APAP administration, as we observed abundant platelet accumulation in VWF−/− mice at this time (Fig 4B,C). Whereas platelets remained in the liver 48 hours after APAP challenge in wild-type mice, platelets were nearly absent in livers of VWF−/− mice 48h and 72h after APAP administration, suggesting a more rapid removal of accumulating platelets in the absence of VWF (Fig 4B,C).
Figure 4. VWF prolongs platelet deposition in the APAP-injured liver.

Wild-type (WT) and VWF−/− (VWF KO) mice were given 300mg/kg APAP or saline (i.p.) and samples were collected 24h, 48h, and 72h later. Representative images showing immunolabeling of CD41 (platelets; red) and VWF (green) in DAPI (blue)-counterstained liver sections from wild-type mice (A) or VWF KO mice (B), colocalization of VWF with platelets is indicated by yellow/orange. Original magnification is 100x. (C) Quantification of platelet deposition in wild-type (black bars) mice and VWF−/− (grey bars) mice, expressed as percentage of positive pixel count at indicated time points. Bars represent mean + standard error of the mean (n = 5-7 mice per group) * p <0.05
VWF deficiency accelerates liver repair after APAP-induced acute liver injury.
Previous studies have shown increased VWF levels in patients with acute liver disease.2,14 We therefore investigated whether VWF contributes to APAP-induced acute liver injury and liver repair. Increases in serum ALT activity, hepatocellular necrosis, and hepatic hemorrhage and congestion were not different between wild-type mice and VWF−/− mice 24h after APAP challenge, suggesting VWF does not contribute to initial APAP-induced liver injury (Fig. 5A-D). Minimal repair of the liver was evident in wild-type mice at 48h after APAP challenge (Fig. 5A-D). This persistent necrosis in wild-type mice was associated with hemorrhage/congestion both within the necrotic areas and neighboring parenchymal tissue (Fig. 5C,D). However, the area of hepatic necrosis was significantly reduced between 24h and 48h in APAP-challenged VWF−/− mice (Fig 5B,D). In contrast to wild-type mice, hemorrhage and congestion was almost completely absent in VWF−/− mice at this time point (Fig. 5C,D). Whereas hepatic necrosis was essentially absent by 72h in APAP-treated VWF−/− mice, wild-type mice still displayed marked necrosis (Fig. 5B,D). Survival also improved in APAP-challenged VWF−/− mice, consistent with improved repair of the injured liver (Supplemental Figure 2). The more rapid resolution of necrosis in VWF−/− mice was coupled to an increase in hepatocyte proliferation, as the number of PCNA-positive hepatocyte nuclei was significantly greater in APAP-treated VWF−/− mice at 24h (Fig. 5E,F). Hepatic cyclin gene expression tended to increase in APAP-challenged VWF−/− mice (Fig. 5G), but these changes trailed the observed increase in hepatocyte PCNA labeling. Collectively, the results indicate that VWF deficiency facilitates repair of the APAP-injured liver.
Figure 5. VWF deficiency accelerates liver repair after APAP-induced acute liver injury in mice.

Wild-type (WT) and VWF−/− (VWF KO) mice were given 300mg/kg APAP or saline (i.p.). (A) Serum alanine aminotransferase (ALT) activity. (B) Area of hepatocellular necrosis and (C) Area of hepatic congestion and hemorrhage were determined 24h, 48h, and 72h after APAP challenge. (D) Representative photomicrographs of H&E-stained liver sections depict hepatocellular necrosis (closed triangle) and congestion/hemorrhage (asterisk). (E) Representative photomicrographs of proliferating cell nuclear antigen (PCNA) labeled liver sections various times after APAP challenge. (F) PCNA-positive hepatocytes were quantified as the sum of positive cells in 5 random 200X fields. (G) Hepatic mRNA levels of cell cycle genes. Data are expressed as means + standard error of the mean (n = 5-12 mice per group) * p<0.05.
Supplementation of VWF in VWF−/− mice restores hepatic VWF and platelet deposition that impacts necrosis resolution in APAP-challenged mice.
To validate observations in VWF−/− mice and confirm loss of VWF as the basis for accelerated liver repair in VWF−/− mice, we supplemented VWF−/− mice with human plasma-derived VWF (Humate-P). Specifically, VWF−/− mice were challenged with APAP and then received either VWF or saline as control 3h after APAP challenge. Compared to APAP-challenged VWF−/− mice given saline, administration of VWF 3h after APAP restored abundant deposition of VWF and dramatically increased platelet accumulation 48h after APAP challenge (Fig 6A,B). Consistent with the notion that VWF inhibits repair of the APAP-injured liver, the restoration of hepatic VWF in APAP-challenged VWF−/− mice was associated with a restoration of platelet deposits and more hepatic necrosis 48h after APAP challenge (Fig 6D,E).
Figure 6. VWF supplementation restores hepatic platelet accumulation and necrosis in APAP-challenged VWF−/− mice.

APAP-challenged VWF−/− mice were supplemented with human plasma-derived VWF (3U VWF:RCo/mouse) or saline (i.e., control) 3h after APAP administration and samples were collected 48h after APAP challenge. Representative images showing immunolabeling of CD41 (platelets; red) and VWF (green) in DAPI (blue)-counterstained liver sections of saline-treated (A) and VWF-supplemented mice (B). Colocalization of VWF with platelets is indicated by yellow/orange. Original magnification is 100x. (C) Quantification of hepatic platelet accumulation in saline-treated (black bar) mice and VWF-supplemented (grey bar) mice, expressed as percentage of positive pixel count. (D) Representative photomicrographs of H&E-stained liver sections depict hepatocellular necrosis (closed triangle) and congestion/hemorrhage (asterisk). (E) Area of hepatocellular necrosis. Bars represent mean + standard error of the mean (n = 5-6 mice per group). * p<0.05 compared to saline-supplemented mice.
Blocking VWF or platelet integrin αIIbβ3_accelerates liver repair and decreases hepatic platelet deposition after APAP overdose.
To evaluate the effect of therapeutic VWF inhibition on hepatic platelet accumulation and liver repair after APAP overdose, we blocked VWF in wild-type mice with a polyclonal antibody against VWF, with previously established specificity.29-33 Wild-type mice were challenged with APAP and received either the VWF blocking antibody or purified rabbit IgG control, beginning 4h after APAP overdose and again 24h after APAP challenge. In agreement with our observations in VWF−/− mice, administration of the VWF blocking antibody significantly reduced hepatocellular necrosis 48h after APAP challenge compared with mice receiving the control antibody (Fig 7A,B). Moreover, in agreement with observations in VWF−/− mice, a dramatic reduction in hepatic platelet accumulation 48h after APAP challenge was observed in mice that were treated with the VWF antibody (Fig 7C).
Figure 7. Blocking VWF function or platelet integrin αIIbβ3 accelerates liver repair after APAP-induced liver injury in mice.

For A-C, APAP-challenged wild-type mice were treated with a polyclonal rabbit anti-human VWF antibody (α-VWF, i.p. 50 μg/mouse) or control IgG,4h and 24h after APAP administration and samples were collected 48h after APAP challenge. (A) Representative photomicrographs of H&E-stained liver sections depict hepatocellular necrosis (closed triangles). (B) Area of hepatocellular necrosis. (C) Representative images showing immunolabeling of CD41 (platelets; red) in liver sections from control mice (upper left panel) or α-VWF treated mice (lower left panel) and quantification of platelet deposition in control-treated (black bars) mice and α-VWF treated (grey bars) mice (right panel), expressed as percentage of positive pixel count. For D-F, APAP-challenged wild-type mice were treated with a platelet αIIbβ3 antagonist (GR144053, i.p. 10mg/kg) or vehicle (saline), 12h after APAP administration and samples were collected 48h after APAP challenge. (D) Representative photomicrographs of H&E-stained liver sections depict hepatocellular necrosis (closed triangles) and congestion/hemorrhage (asterisk). (E) Area of hepatocellular necrosis. (F) Representative images showing immunolabeling of CD41 (platelets; red) in DAPI (blue)-counterstained liver sections from vehicle mice (upper left panel) or GR144053-treated mice (lower left panel), and quantification of platelet deposition in vehicle-treated (black bar) mice and GR144053-treated (grey bar) mice (right panel), expressed as percentage of positive pixel count. Bars represent mean + standard error of the mean (n = 5-10 mice per group). * p<0.05 compared to vehicle/control antibody-treated mice.
Because blocking VWF reduced platelet accumulation, we next determined whether targeting platelets directly would also affect liver repair after APAP challenge. Specifically, wild-type mice were challenged with APAP and received either an αIIbβ3 antagonist (GR144053) or vehicle (saline) 12h after APAP challenge. Analogous to VWF deficiency or inhibition, administration of GR144053 significantly reduced hepatocellular necrosis 48h after APAP challenge compared to vehicle-treated mice (Fig 7D,E). Moreover, and in agreement with our other observations, GR144053 treatment reduced hepatic platelet accumulation 48h after APAP overdose (Fig 7F). Collectively, these results indicate that blocking VWF or the platelet integrin receptor αIIbβ3,_even during development of injury, reduces hepatic platelet accumulation and promotes liver repair after APAP overdose in mice.
Discussion
Accumulating evidence from experimental and clinical studies suggests that the hemostatic system contributes to the progression of acute liver injury.2-5 We have previously observed that platelets accumulate in the APAP-injured liver and contribute to acute liver injury in mice,3 and ALF patients who received platelet transfusions have a worse outcome than those who do not.5 Furthermore, patients with ALF have an imbalance between VWF and its cleaving protease ADAMTS13, which is associated with poor outcome.2 Here, we provide the first direct mechanistic evidence linking VWF to the progression of ALF. Specifically, we discovered that although VWF does not contribute to initial APAP hepatotoxicity, VWF substantially delays repair of the injured liver. Moreover, our studies provide mechanistic insight into VWF regulation after APAP overdose and document the potential of VWF inhibition as a potential approach to stimulate liver repair.
We observed a reduction in plasma HMW VWF multimers and VWF antigen levels 24h after APAP challenge, which was coupled to deposition of VWF within the APAP-injured liver. In agreement with the loss of the most active VWF (i.e. HMW multimers) from plasma, we found that the ability of plasma VWF to bind to collagen was significantly reduced 24h after APAP challenge. Taken together, these results suggest that the HMW multimers in plasma might be deposited in the liver after APAP overdose. Interestingly, a small study showed intense VWF deposits in areas of necrosis in liver fragments from ALF patients34, similar to our observations in mice. In mice, VWF levels recovered to achieve higher than normal plasma levels 48h and 72h after APAP challenge, but the relative proportion of HMW multimers was reduced at these times, also consistent with the reduced proportion of HMW multimers seen in ALF patients.2
VWF collagen binding (VWF:CB) activity was increased at later times after APAP overdose, in line with the observed increase in VWF plasma levels. However, VWF activity did not increase to the same extent as antigen, indicated by a reduced VWF:CB/VWF:Ag ratio, another observation consistent with observations in ALF patients.2 In patients, this change could be due to administration of N-acetyl cysteine, which is routinely given to APAP overdose patients and has been shown to reduce VWF multimer size.35,36 The reduction in HMW VWF multimers could also be driven by cleavage of VWF by other proteases such as plasmin.37 Our observation that deposition of VWF in the injured liver coincides with a reduced proportion of HMW VWF multimers in plasma suggests an additional possible explanation for the relative loss of highly active VWF from the plasma after APAP overdose.
The mechanisms controlling elevated VWF levels in APAP-challenged mice and in patients with ALF are not fully understood, but could be attributed to endothelial cell activation38,39, increased hepatic expression38,39 and/or reduced clearance.2,16. We observed a small increase in VWF hepatic mRNA expression, suggesting that VWF is not oversynthesized by liver endothelial cells after APAP overdose. Neubauer et al. also reported minimal change in VWF gene expression in liver fragments collected from ALF patients.34 These results suggest that increased hepatic VWF expression cannot fully explain the increased plasma levels after APAP overdose. Endothelial cell activation could also lead to increased VWF plasma levels by release of VWF stored in Weibel-Palade bodies. It is possible that hepatic and non-hepatic sources contribute to release of VWF after APAP overdose in response either to extrahepatic APAP metabolism or to acute phase mediators. Impaired hepatic VWF clearance by the injured liver may also lead to increased VWF plasma levels. Hepatocytes and Kupffer cells play an important role in clearing VWF26,40, and thus, hepatocellular injury and liver macrophage dysfunction41 following APAP overdose is speculated to affect VWF clearance. Indeed, we demonstrate here that APAP overdose causes delayed VWF clearance, evidenced by prolonged MRT of infused VWF in APAP-challenged VWF−/− mice compared with saline-challenged VWF−/− mice. Defining how impaired VWF clearance links with increased VWF expression and release from various cellular sources to cause elevated VWF levels after APAP overdose requires further study.
A prior study demonstrated that platelet depletion significantly reduced liver injury after APAP overdose.3 Thus, one mechanism whereby VWF deficiency could protect from APAP-induced liver injury is by reducing hepatic platelet accumulation. However, we found that VWF deficiency had no significant effect on early hepatic platelet accumulation or necrosis (i.e. 24h after APAP). Interestingly, this suggests that the transient increase in hepatocyte proliferation in APAP-challenged VWF−/− mice is driven by a platelet-independent mechanism. Moreover, these studies suggest the mechanisms driving initial platelet accumulation in the APAP-injured liver are VWF-independent, perhaps including engagement of platelets by other components of the extracellular matrix (e.g., fibrin(ogen), fibronectin) or other soluble platelet activators (e.g., thrombin). However, despite the important role of VWF in shielding factor VIII from proteolytic inactivation, coagulation activation and hepatic fibrin(ogen) deposition were not affected by VWF deficiency. Overall, it appears that VWF is not a component of the mechanism driving the initial intrahepatic accumulation of platelets in the APAP-injured liver. Additional studies are required to define the mechanisms linking VWF and hepatocyte proliferation in the early phase of APAP-induced liver injury.
Although early platelet accumulation after APAP overdose was independent of VWF, we noted that hepatic platelet accumulation was nearly absent in VWF−/− mice 48h after APAP challenge. This result suggests that VWF could stabilize platelet aggregates in the injured liver and that this interaction impedes liver repair after APAP overdose. Indeed, a previous study found that VWF is required to form stable, occlusive thrombi in injured vessels,42 and hepatic platelet-induced microthrombi formation by inadequate regulation of VWF activity has been proposed to drive disease progression.2 We present multiple lines of evidence linking VWF-platelet aggregate formation to inhibition of the APAP-injured liver. Faster removal of platelet aggregates in the VWF−/− mice after APAP overdose is consistent with unstable thrombi formed in the absence of VWF. Restoration of plasma VWF in VWF−/− mice after APAP overdose returned hepatic platelet-VWF aggregates 48h after APAP challenge, and this was coupled to a reduction in liver repair. Consistent with these results, antibody-mediated inhibition of VWF or inhibition of the platelet integrin αIIbβ3 in APAP-challenged mice, even after the development of early hepatotoxicity, reduced hepatic platelet accumulation and accelerated liver repair 48h after APAP challenge. Although we cannot exclude a role for VWF in secondary accumulation of platelets in the injured liver, we observed that after the initial hepatic influx (~6 hours after APAP overdose), blood platelet counts are relatively stable out to 36h (data not shown). Overall, the results suggest that VWF-stabilized platelet thrombi in the diseased liver might delay liver repair, and provide the proof-of-concept that a VWF-targeted therapy with low bleeding risk43 could improve repair of the injured liver. Future studies defining the precise mechanisms whereby VWF-platelet aggregates inhibit liver repair may identify additional therapeutic targets.
In summary, we identified that acute liver damage in mice caused deposition of VWF and platelets in the injured liver. This was followed by a persistent elevation in VWF plasma levels, changes consistent with observations in patients with ALF. Despite identical peak liver injury, liver repair was dramatically accelerated in VWF−/− mice in association with reduced platelet aggregates in the liver microvasculature. Conversely, restoration of VWF deposits in VWF−/− mice dramatically increased hepatic platelet accumulation. Analogous to VWF−/− mice, administration of a VWF blocking antibody or a platelet-specific integrin antagonist_significantly reduced hepatic platelet aggregates and accelerated liver repair in mice with established APAP-induced hepatotoxicity. The results suggest a connection between hepatic VWF-platelet aggregate formation and impaired liver repair after APAP overdose. These studies are the first to provide a mechanistic link between VWF and liver repair, suggesting an explanation for the clinical observations regarding VWF in patients with ALF.
Supplementary Material
Highlights.
APAP-induced acute liver injury is associated with hepatic VWF deposition.
APAP-induced acute liver injury is associated with elevation of VWF plasma levels.
VWF clearance is impaired in experimental APAP-induced acute liver injury
VWF contributes to persistent platelet accumulation in the APAP-injured liver.
VWF deficiency or inhibition accelerates repair of the APAP-injured liver.
Acknowledgments
Financial Support
This research was supported by grants from the National Institutes of Health (NIH) to JPL (R01 DK105099, DK120289), T32 GM092715, John A. Penner Endowed Research Assistantship to KSB, support from the USDA National Institute of Food and Agriculture, and an EHA Research Grant from the European Hematology Association (EHA) to DG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Lee WM. Acetaminophen-related acute liver failure in the United States. Hepatol Res. 2008;38 Suppl 1:S3–8. [DOI] [PubMed] [Google Scholar]
- 2.Hugenholtz GC, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT, Lisman T. An unbalance between von Willebrand factor and ADAMTS13 in acute liver failure: implications for hemostasis and clinical outcome. Hepatology. 2013;58(2):752–761. [DOI] [PubMed] [Google Scholar]
- 3.Miyakawa K, Joshi N, Sullivan BP, et al. Platelets and protease-activated receptor-4 contribute to acetaminophen-induced liver injury in mice. Blood. 2015;126(15):1835–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganey PE, Luyendyk JP, Newport SW, et al. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46(4):1177–1186. [DOI] [PubMed] [Google Scholar]
- 5.Stravitz RT, Ellerbe C, Durkalski V, et al. Bleeding complications in acute liver failure. Hepatology. 2018;67(5):1931–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010;116(6):878–885. [DOI] [PubMed] [Google Scholar]
- 7.Lisman T, Stravitz RT. Rebalanced Hemostasis in Patients with Acute Liver Failure. Semin Thromb Hemost. 2015;41(5):468–473. [DOI] [PubMed] [Google Scholar]
- 8.Stravitz RT, Ellerbe C, Durkalski V, Reuben A, Lisman T, Lee WM. Thrombocytopenia Is Associated With Multi-organ System Failure in Patients With Acute Liver Failure. Clin Gastroenterol Hepatol. 2016;14(4):613–620.e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lisman T, Luyendyk JP. Platelets as Modulators of Liver Diseases. Semin Thromb Hemost. 2018;44(2):114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furlan M Von Willebrand factor: molecular size and functional activity. Ann Hematol. 1996;72(6):341–348. [DOI] [PubMed] [Google Scholar]
- 11.De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121(2):270–277. [DOI] [PubMed] [Google Scholar]
- 12.Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98(6):1662–1666. [DOI] [PubMed] [Google Scholar]
- 13.Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008;112(1):11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langley PG, Hughes RD, Williams R. Increased factor VIII complex in fulminant hepatic failure. Thromb Haemost. 1985;54(3):693–696. [PubMed] [Google Scholar]
- 15.Takaya H Yoshiji H, Kawaratani H, et al. Decreased activity of plasma ADAMTS13 are related to enhanced cytokinemia and endotoxemia in patients with acute liver failure. Biomedical reports. 2017;7(3):277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisman T, Bongers TN, Adelmeijer J, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44(1):53–61. [DOI] [PubMed] [Google Scholar]
- 17.Denis C, Methia N, Frenette PS, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95(16):9524–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopec AK, Joshi N, Cline-Fedewa H, et al. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017;66(4):787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Q, Kuether EL, Schroeder JA, Fahs SA, Montgomery RR. Intravascular recovery of VWF and FVIII following intraperitoneal injection and differences from intravenous and subcutaneous injection in mice. Haemophilia. 2012;18(4):639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi N, Kopec AK, Ray JL, et al. Fibrin deposition following bile duct injury limits fibrosis through an αMβ2-dependent mechanism. Blood. 2016;127(22):2751–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groeneveld DJ, Alkozai EM, Adelmeijer J, Porte RJ, Lisman T. Balance between von Willebrand factor and ADAMTS13 following major partial hepatectomy. Br J Surg. 2016;103(6):735–743. [DOI] [PubMed] [Google Scholar]
- 22.Bowyer AE, Goodfellow KJ, Seidel H, et al. Evaluation of a semi-automated von Willebrand factor multimer assay, the Hydragel 5 von Willebrand multimer, by two European Centers. Res Pract Thromb Haemost. 2018;2(4):790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lisman T, Raynal N, Groeneveld D, et al. A single high-affinity binding site for von Willebrand factor in collagen III, identified using synthetic triple-helical peptides. Blood. 2006;108(12):3753–3756. [DOI] [PubMed] [Google Scholar]
- 24.Poole LG, Pant A, Baker KS, et al. Chronic liver injury drives non-traditional intrahepatic fibrin(ogen) crosslinking via tissue transglutaminase. J Thromb Haemost. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groeneveld DJ, van Bekkum T, Cheung KL, et al. No evidence for a direct effect of von Willebrand factor's ABH blood group antigens on von Willebrand factor clearance. J Thromb Haemost. 2015;13(4):592–600. [DOI] [PubMed] [Google Scholar]
- 26.Lenting PJ, Westein E, Terraube V, et al. An experimental model to study the in vivo survival of von Willebrand factor. Basic aspects and application to the R1205H mutation. J Biol Chem. 2004;279(13):12102–12109. [DOI] [PubMed] [Google Scholar]
- 27.O'Regan N, Gegenbauer K, O'Sullivan JM, et al. A novel role for von Willebrand factor in the pathogenesis of experimental cerebral malaria. Blood. 2016;127(9):1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan BP, Kopec AK, Joshi N, et al. Hepatocyte tissue factor activates the coagulation cascade in mice. Blood. 2013;121(10):1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirschbaum M, Jenne CN, Veldhuis ZJ, et al. Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int. 2017;37(11):1731–1737. [DOI] [PubMed] [Google Scholar]
- 30.Zhu X, Cao Y, Wei L, et al. von Willebrand factor contributes to poor outcome in a mouse model of intracerebral haemorrhage. Sci Rep. 2016;6:35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolaczkowska E, Jenne CN, Surewaard BG, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. 2013;14(8):785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hillgruber C, Steingräber AK, Pöppelmann B, et al. Blocking von Willebrand factor for treatment of cutaneous inflammation. J Invest Dermatol. 2014;134(1):77–86. [DOI] [PubMed] [Google Scholar]
- 34.Baruch Y, Neubauer K, Ritzel A, Wilfling T, Lorf T, Ramadori G. Von Willebrand gene expression in damaged human liver. Hepatogastroenterology. 2004;51(57):684–688. [PubMed] [Google Scholar]
- 35.Prescott LF, Park J, Ballantyne A, Adriaenssens P, Proudfoot AT. Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine. Lancet. 1977;2(8035):432–434. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Reheman A, Gushiken FC, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121(2):593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Federici AB, Berkowitz SD, Lattuada A, Mannucci PM. Degradation of von Willebrand factor in patients with acquired clinical conditions in which there is heightened proteolysis. Blood. 1993;81(3):720–725. [PubMed] [Google Scholar]
- 38.Ferro D, Quintarelli C, Lattuada A, et al. High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: relationship to endotoxemia. Hepatology. 1996;23(6):1377–1383. [DOI] [PubMed] [Google Scholar]
- 39.Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost. 2004;91(2):267–275. [DOI] [PubMed] [Google Scholar]
- 40.Casari C, Lenting PJ, Wohner N, Christophe OD, Denis CV. Clearance of von Willebrand factor. J Thromb Haemost. 2013;11 Suppl 1:202–211. [DOI] [PubMed] [Google Scholar]
- 41.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13(3):316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109(6):2424–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: an emerging target in stroke therapy. Stroke. 2012;43(2):599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.