

Abstract
Movement disorders including Parkinson’s disease and dystonia are caused by neurological dysfunction, typically resulting from the loss of a neuronal input within a circuit. Neuromodulation, specifically deep brain stimulation (DBS), has proven to be a critical development in the treatment of movement disorders. Continuing efforts aim to improve DBS techniques, both in how they exert their effects and in the efficacy of the mechanism involved in eliciting those effects. While optogenetic stimulation is currently infeasible in human patients, opto-DBS research provides an indispensable avenue to understand the mechanisms of DBS therapeutic and adverse effects. We review the benefits of cell-type specific manipulations in understanding the root cause of movement disorders and how DBS might optimally combat those causes. We also explore new circuit-inspired applications of DBS suggested by thorough, high-throughput optogenetic techniques. Maximizing the efficacy and outcome of DBS requires a multi-tiered approach; research employing optogenetics provides the specificity and feasibility to uncover the mechanisms that will help realize these gains in patient care.
Introduction
Parkinson’s disease (PD) is a movement disorder caused by the neurodegeneration of dopamine neurons in the substantia nigra pars compacta (SNc). The primary targets of SNc dopamine neurons are the basal ganglia – a series of subcortical nuclei that play an important role in motor control. Neurosurgical interventions targeted to the basal ganglia are highly effective at managing the motor symptoms of PD, particularly a neuromodulatory intervention called deep brain stimulation (DBS)[1][2]. DBS was first approved by the FDA for the treatment of PD for over twenty years. Since then, it has been explored as a treatment for a number of other neurological and neuropsychiatric disorders including depression, addiction, Tourette syndrome, dystonia, and obsessive-compulsive disorder [1][3].
In conventional DBS, high frequency electrical stimulation is delivered continuously to a target brain area through a surgically implanted stimulating electrode, whose power source is a battery implanted under the skin on the chest. Stimulation parameters are tuned empirically through a trial-and-error process until maximal therapeutic effects are achieved. This tuning process can take many hours to complete and is particularly challenging when applied to diseases where therapeutic benefits take weeks to appear (i.e. dystonia, depression).
The effective implementation of DBS is also compounded by that fact that its mechanisms of action remain obscure [4], although several hypotheses have been proposed [5][6]. Electrical stimulation applied during DBS excites not only the cell bodies of neurons surrounding the electrode but also any axons that happen to pass by the electrode, even if their cell bodies reside far outside the stimulation zone (Fig. 1). This means that stimulation, no matter how focal, will recruit many different circuits, and this confound has obscured past efforts to disambiguate therapeutic pathways from ineffective pathways. In fact, several hypotheses concerning the functional mechanism of DBS suggest that the benefits arise not from the stimulation of the target area, but of the fibers of passage - which may lead to widespread prodromic and antidromic changes in activity ([4-5], but see [10]).
Figure 1). Target specificity of electrical and optogenetic stimulation.
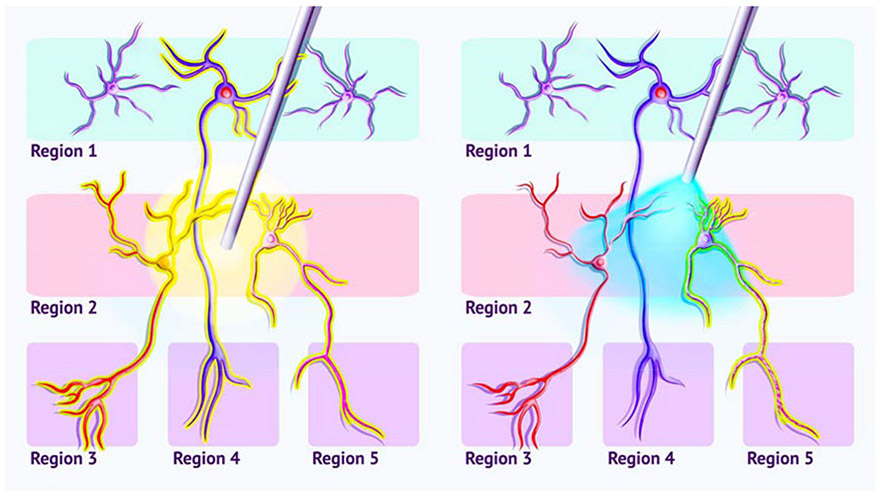
Neurons originating in two areas are depicted, each with a different downstream target region. Left) Electrical stimulation, such as that used in DBS, will drive the activity of all cells in a given area, including those whose axons only pass through the area, even though the brain region in which their cell bodies lie may be distant. These compound effects can make understanding stimulation effects difficult and may lead to off-target, adverseeffects. Right) Optogenetic stimulation targets a specific cell-type (the neuron expressing ChR2, shown as light-green dots on the neuron’s surface), sparing other tissue in the stimulated area, including fibers of passage.
Understanding the elusive mechanisms underlying how (and why) DBS works is central to improving the efficacy of current methods across the patient population as well as increasing the potential for future applications. But such advances cannot come from clinical trials alone, where opportunities for experimentation are limited by a variety of factors. Instead, improvements in DBS are being driven by discoveries from research in animal models, where neural circuits can be mapped and controlled with unparalleled specificity using a technique called optogenetics. Optogenetics refers to a technique in which neural activity is controlled by opsins, light-sensitive ion channels whose expression can be directed towards neuronal populations of interest. Often these genes are delivered with the aid of adeno-associated viruses (AAV) whose pathogenic genes have been removed and replaced with genes encoding excitatory (e.g. channelrhodopsin, ChR2) or inhibitory (e.g. halorhodopsin, NpHr) opsins [11]. The use of viral vectors for gene therapy and optogenetics in humans is an emerging field of research, but its widespread use is still many years away.
In this review, we will focus on the application of optogenetics as a research tool to inspire the next generation of DBS-based therapies. We will discuss how knowledge about cell type diversity in the basal ganglia has shaped clinical approaches to treating PD. We will then describe how the use of optogenetics has provided insights into the mechanisms of DBS and the circuits underlying its therapeutic effects. Finally, we conclude the review with a perspective on the future of optogenetics as a vital partner in the development of new DBS-based therapies, in which neuromodulation will be used to repair, rather than simply mask the function of damaged circuits.
Circuit-Inspired Therapies For Parkinson’s Disease
Since the Nobel prize-winning discovery by the late Arvid Carlsson that PD symptoms could be treated with dopamine replacement therapy, levodopa (L-DOPA) has been the most commonly prescribed medication to treat the symptoms of PD. L-DOPA is initially quite effective but over time, patients require higher and higher doses of the drug and it causes a number of side effects that can become more debilitating than the disease itself. Attempts to improve the long-term management of PD have drawn inspiration from cell-based models of basal ganglia function.
The most influential of these models has been the ‘rate model’ which posits that motor symptoms of PD arise through an imbalance in the activity of two parallel, but opposing motor pathways in the basal ganglia: the pro-kinetic ‘direct’ pathway and the anti-kinetic ‘indirect’ pathway [12][13]. A landmark discovery in 1990 [14] found that the direct and indirect pathways originate from genetically dissociable populations of spiny projection neurons (SPNs) in the striatum (Fig. 2). Taking advantage of genetic differences in dopamine receptors (D1 vs. D2) in these neurons, among other genetic differences, researchers have aimed to directly test the rate model of PD. In a mouse model of PD, optogenetic stimulation of D1-SPNs activated the direct pathway and was sufficient to alleviate bradykinesia (slowness of movement), a canonical pathology of PD [15] (Fig. 2). In healthy mice, optogenetic stimulation of D2-SPNs activated the indirect pathway and was sufficient to induce bradykinesia [16]. These results illustrate how different behavioral effects can be achieved through the activation of different cell populations, even within the same brain region.
Figure 2: Summary of cell types whose relevance for PD therapeutics has been explored using optogenetics.
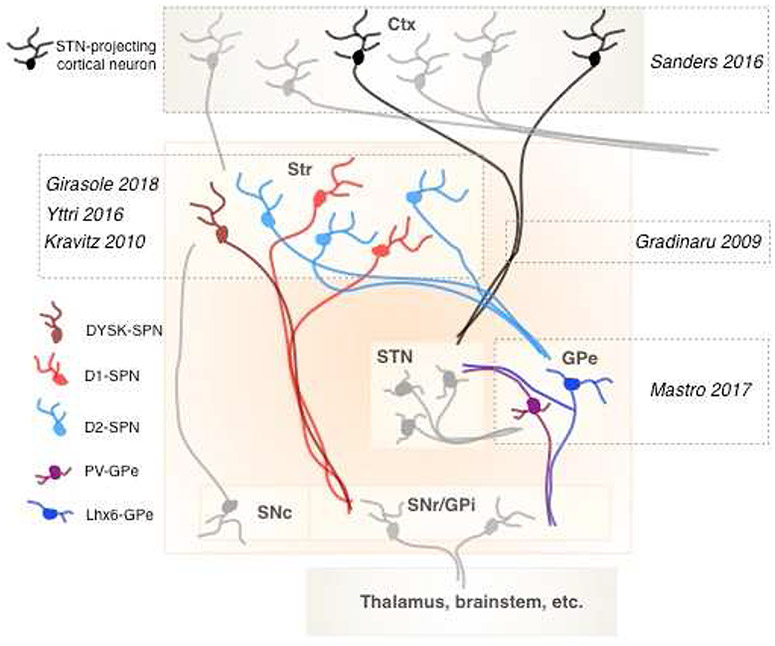
Specific neuronal subpopulations are denoted in different colors (legend), and relevant references are listed. Basal ganglia nuclei: Str, striatum; STN, subthalamic nucleus; GPe, external globus pallidus; SNc, substantia nigra compacta; SNr, substantia nigra reticulata; GPi, internal globus pallidus. Ctx, cortex.
Clinically, however, discoveries about cell type diversity in the striatum have been slow to translate into improved therapies for PD because the same neurons responsible for facilitating movement, D1-SPNs, also contribute to levodopa-induced dyskinesias (LIDs [17][18]), one of the most debilitating side effects of PD medications. Nearly 90% of patients on dopamine replacement therapy (e.g. L-DOPA) develop LIDs within 10 years [19]. Currently there are few strategies available to dissociate the therapeutic effects of medications from their dyskinetic side effects, but new approaches are being developed, driven in large part by discoveries about the cellular origins of LIDs, made possible through optogenetics. In a mouse model of LIDs, the expression of involuntary movements caused by L-DOPA administration was greatly reduced by inhibiting a small, but distinct subset of D1-SPNs (DYSK-SPNs) with NpHr [17][20] (Fig. 2). Conversely, activation DYSK-SPNs with ChR2 was sufficient to produce LIDs. These results, as well as the discovery that DYSK-SPNs exhibit a unique electrophysiological signature compared to other D1-SPNs [20], suggest that different subsets of neurons are responsible for the therapeutic vs. dyskinetic effects of medication. This discovery opens new avenues of research into drugs or dosing regimens that will be more selective for the therapeutic population of D1-SPNs, but not the dyskinetic population.
Using optogenetics to understand and improve DBS
DBS is used as a therapy in patients whose symptoms or side effects are so severe that they can no longer be controlled with medication alone. The efficacy of DBS, even under these extreme conditions, speaks to its therapeutic potential, but the route to its continued optimization will require insights into its neuromodulatory mechanisms. To this end, optogenetics provides an invaluable research tool uncover the circuits that provide maximal therapeutic benefit.
In PD, the most common target for DBS is the subthalamic nucleus (STN), so it is natural to assume that the prokinetic effects of stimulation relate to its effect on STN neurons. But this assumption remains an area of debate. An alternative hypothesis is that the therapeutic effects of STN stimulation are driven by fibers of passage – axons from other brain areas that happen to pass through the STN (Fig. 1). Although nearly impossible to differentiate with electrical stimulation, these hypotheses can be directly tested with optogenetics. In 2009, a study found that when excitation was restricted to STN neurons with ChR2, motor deficits could not be ameliorated [7] (Fig. 2). In contrast, optogenetic stimulation of cortex was highly effective. Recently, these results were confirmed and extended by a second study that showed that ChR2-mediated excitation, restricted only to those cortical neurons projecting directly to the STN, is sufficient to ameliorate motor deficits and restore healthy patterns of neural activity in both cortex and STN [8][21] (Fig. 2). These and similar optogenetic approaches provide critical insights into the neural mechanisms underlying DBS. In some cases, this could lead to the development of more effective stimulation parameters or better placements for electrode. In other cases, results could identify cellular biomarkers for calibrating DBS stimulation, or driving DBS stimulation directly (see adaptive DBS, below).
Long-lasting therapeutic effects
A significant limitation of existing PD therapies is that they treat the symptoms of the disease, but do not correct the underlying circuit dysfunction responsible for the symptoms. As a result, symptoms rapidly return once DBS stimulation is turned off. The need for constant stimulation creates a drain on battery power and increased risk of side effects. Achieving the next level in therapeutic efficacy will require the development of strategies to repair, not simply mask, circuit dysfunction. Evidence that such a goal can be obtained comes from the development of a refined DBS protocol, coordinated reset (CR-DBS), developed based on theoretical predictions about how synaptic learning rules can be leveraged to train a network out of a pathologically synchronized state[22-25]. Desynchronizing electrical pulses delivered to distributed sites within the STN produced long-lasting prokinetic effects that persisted for days, and possibly weeks after stimulation in both monkeys [23,25] and human PD patients [24]. Although CR-DBS is not yet in widespread use clinically, its early success demonstrates the feasibility of translating discoveries from basic research into improved therapies for PD.
An important lesson from CR-DBS is that it is possible to recover motor function, despite the continued absence of dopamine. Discovering the cellular and circuit mechanisms that support this long-lasting rescue is of obvious therapeutic value. Recently, the use of optogenetics has uncovered populations of neurons within the external globus pallidus (GPe), where targeted interventions reliably induce long-lasting motor rescue in a mouse model of PD [26] (Fig. 2). The GPe is reciprocally coupled to the STN and under conditions of low dopamine, this coupling has been hypothesized to generate pathological oscillations that impair motor function [27]. The GPe contains a number of different cell types [28-31], but the relevance of this neuronal diversity for therapeutic applications had not been well explored. Optogenetic perturbations in the GPe that globally increased or decreased the activity of all neurons simultaneously had no effect on movement [26]. However, restricting optogenetic interventions to particular neuronal subpopulations in the GPe produced dramatically different results. Optogenetic stimulation of a subset of GPe neurons, enriched in parvalbumin (PV-GPe), induced a long-lasting recovery of movement that persisted until the end of experiments, over 3 hours post stimulation. A similarly long-lasting behavioral rescue was induced by optogenetic inhibition of a different subset of GPe neurons, enriched in lim homeobox 6 (Lhx6-GPe) (Fig. 2). These results suggest that neuronal subpopulations in the GPe are important therapeutic nodes in the basal ganglia circuit, where targeted interventions have the potential to induce longer-lasting motor rescue than what is currently possible using existing strategies. Such cell-specific modulation might be challenging to obtain with electrical stimulation alone, but numerous approaches to obtain greater cell-type specificity are being developed including stimulation combined with pharmacology [32], cell-specific targeting of pharmacological compounds [33], and improved viral vectors to deliver genes to specific cell types without pre-existing genetic modifications [11].
Re-training damaged circuits
Although PD is caused by the loss of dopamine neurons, its symptoms are thought to reflect aberrant activity in circuits. Thus, fixing incorrect patterns of neural activity should provide a solution. The brain is adept at adjusting patterns of activity through synaptic plasticity. Research across human patients and model organisms suggests that while individual cell types may often conform to a particular role, more nuanced and therapeutic potency may lie in plasticity mechanisms [34] [35]. Dopamine, and the neurons and neurotransmitters it modulates, play an important role in synaptic plasticity [36,37]. This critical function, responsible for action selection and performance, is lost in PD [36,38]. Recent work examined the effects of plasticity in the striatal projection neurons. It was discovered that bradykinesia can be induced through cell-type specific optogenetic stimulation-mediated plasticity in the striatum[39] (Fig. 2). Indirect pathway neurons were stimulated during only the fastest reaches performed by a mouse.
Cumulatively, and in a dopamine-dependent manner, the average reach speed decreased and remained decreased for tens of minutes following removal of the stimulation condition. To verify that this bradykinetic effect was the result of reinforcing plasticity mechanism, the stimulation paradigm was switched such that stimulation occurred on the slowest, not fastest reaches. In this case, indirect pathway stimulation induced a steady increase in reach speed, while direct pathway stimulation led to a bradykinetic slowing of reach speed that was indistinguishable from that observed previously following indirect pathway stimulation.
There is a long history of using electrical stimulation to induce synaptic plasticity. Adjusting stimulation frequency in many cases can dictate whether synaptic strengths are potentiated or depressed. Therefore, an important use of DBS may be to reinforce healthy patterns of circuit activity in order to put the brain back into a healthy operating state, or to prevent it from engaging in a pathological state. Such a strategy is already being implemented to some extent by closed-loop DBS paradigms where timing and patterns of electrical stimulation are driven by the neural activity itself. Adaptive, or responsive DBS (aDBS) promises to improve treatment outcome while decreasing battery use and negative side effects[40-42]. This nascent field seeks to apply stimulation in closed-loop with physiological or behavioral signals, although it is unclear what hallmarks will provide the greatest benefit[43,44]. In line with the series of studies outlined above, it has been proposed that closed-loop aDBS could make use of synaptic plasticity mechanisms. For instance, selective stimulation of motor cortex could alleviate bradykinesia by invoking cortico-indirect pathway neuron synaptic plasticity [37,45]. Others have agreed that normalization of striatal plasticity mechanisms may yield significant improvements in disease outcome[36,46,47]. Moreover, improving plasticity may benefit the management of symptoms that are poorly understood and treated, like PD-related dementia[34,48]. Applied in this manner, plasticity-focused neuromodulation could move beyond the masking of maladaptive activity to the reinforcement of beneficial neural patterns, thereby correcting the root circuit dysfunctions.
Conclusion
Circuits are a function of different cell types interacting with each other. As the fields of PD and neuromodulation research progress, we are reminded that more and better information about how the components of those circuits interact can improve our application of neuromodulation therapies to PD. The inability to apply optogenetics in patients is therefore not a bust, but a boon – enabling the examination of how DBS alleviates symptoms and how unintended effects may be reduced. Therefore, optogenetics provides an invaluable research tool to guide clinical and translation research towards solutions that will repair, not merely mask, damaged circuits, for more sustainable, effective therapies in a wide array of disorders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wichmann T, Delong MR: Deep brain stimulation for neurologic and neuropsychiatric disorders. Neuron 2006, 52:197–204. [DOI] [PubMed] [Google Scholar]
- 2.Olanow CW, Brin MF, Obeso JA: The role of deep brain stimulation as a surgical treatment for Parkinson’s disease. Neurology 2000, 55:S60–6. [PubMed] [Google Scholar]
- 3.Krack P, Hariz MI, Baunez C, Guridi J, Obeso JA: Deep brain stimulation: from neurology to psychiatry?. Trends Neurosci 2010, 33:474–484. [DOI] [PubMed] [Google Scholar]
- 4.Hammond C, Ammari R, Bioulac B, Garcia L: Latest view on the mechanism of action of deep brain stimulation. Mov Disord 2008, 23:2111–2121. [DOI] [PubMed] [Google Scholar]
- 5.Herrington TM, Cheng JJ, Eskandar EN: Mechanisms of deep brain stimulation. J Neurophysiol 2016, 115:19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum R, Zimnik A, Zheng F, Turner RS, Alzheimer C, Doiron B, Rubin JE: Axonal and synaptic failure suppress the transfer of firing rate oscillations, synchrony and information during high frequency deep brain stimulation. Neurobiol Dis 2014, 62:86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K: Optical deconstruction of parkinsonian neural circuitry. Science 2009, 324:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders TH, Jaeger D: Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiol Dis 2016. 95:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Ke Y, Chan DCW, Qian Z-M, Yung KKL, Ko H, Arbuthnott GW, Yung W-H: Therapeutic deep brain stimulation in Parkinsonian rats directly influences motor cortex. Neuron 2012, 76:1030–1041. [DOI] [PubMed] [Google Scholar]
- 10.Neumann W-J, Staub F, Horn A, Schanda J, Mueller J, Schneider G-H, Brown P, Kuhn AA: Deep Brain Recordings Using an Implanted Pulse Generator in Parkinson’s Disease. Neuromodulation 2016, 19:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedbrook CN, Deverman BE, Gradinaru V: Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu Rev Neurosci 2018, 41:323–348. [DOI] [PubMed] [Google Scholar]
- 12.Albin RL, Young AB, Penney JB: The functional anatomy of basal ganglia disorders. Trends Neurosci 1989, 12:366–375. [DOI] [PubMed] [Google Scholar]
- 13.DeLong MR: Primate models of movement disorders of basal ganglia origin. Trends Neurosci 1990, 13:281–285. [DOI] [PubMed] [Google Scholar]
- 14.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJJ, Sibley DR: D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250:1429–1432. [DOI] [PubMed] [Google Scholar]
- 15.Kravitz A V, Freeze BS, Parker PRL, Kay K, Thwin MT, Deisseroth K, Kreitzer AC: Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 2010, 466:622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panigrahi B, Martin KA, Li Y, Graves AR, Vollmer A, Olson L, Mensh BD, Karpova AY, Dudman JT: Dopamine Is Required for the Neural Representation and Control of Movement Vigor. Cell 2015, 162:1418–1430. [DOI] [PubMed] [Google Scholar]
- 17.Girasole AE, Lum MY, Nathaniel D, Bair-Marshall CJ, Guenthner CJ, Luo L, Kreitzer AC, Nelson AB: A Subpopulation of Striatal Neurons Mediates Levodopa-Induced Dyskinesia. Neuron 2018, 97:787–795.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picconi B, De Leonibus E, Calabresi P: Synaptic plasticity and levodopa-induced dyskinesia: electrophysiological and structural abnormalities. J Neural Transm 2018, 125:1263–1271. [DOI] [PubMed] [Google Scholar]
- 19.Lanza K, Meadows SM, Chambers NE, Nuss E, Deak MM, Ferré S, Bishop C: Behavioral and cellular dopamine D1 and D3 receptor-mediated synergy: Implications for L-DOPA-induced dyskinesia. Neuropharmacology 2018, 138:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan MB, Bair-Marshall C, Nelson AB: Aberrant Striatal Activity in Parkinsonism and Levodopa-Induced Dyskinesia. Cell Rep 2018, 23:3438–3446.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanders TH: Stimulation of Cortico-Subthalamic Projections Amplifies Resting Motor Circuit Activity and Leads to Increased Locomotion in Dopamine-Depleted Mice. Front Integr Neurosci 2017, 11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hauptmann C, Tass PA: Therapeutic rewiring by means of desynchronizing brain stimulation. Biosystems 2007, 89:173–181. [DOI] [PubMed] [Google Scholar]
- 23.Tass PA, Qin L, Hauptmann C, Dovero S, Bezard E, Boraud T, Meissner WG: Coordinated reset has sustained aftereffects in Parkinsonian monkeys. Ann Neurol 2012, 72:816–820. [DOI] [PubMed] [Google Scholar]
- 24.Adamchic I, Hauptmann C, Barnikol UB, Pawelczyk N, Popovych O, Barnikol TT, Silchenko A, Volkmann J, Deuschl G, Meissner WG, et al. : Coordinated reset neuromodulation for Parkinson’s disease: proof-of-concept study. Mov Disord 2014, 29:1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Nebeck S, Muralidharan A, Johnson MD, Vitek JL, Baker KB: Coordinated Reset Deep Brain Stimulation of Subthalamic Nucleus Produces Long-Lasting, Dose-Dependent Motor Improvements in the 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine Non-Human Primate Model of Parkinsonism. Brain Stimul 2016, 9:609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mastro KJ, Zitelli KT, Willard AM, Leblanc KH, Kravitz AV, Gittis AH: Cell-specific pallidal intervention induces long-lasting motor recovery in dopamine-depleted mice. Nat Neurosci 2017, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ: Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci 2002, 25:525–531. [DOI] [PubMed] [Google Scholar]
- 28.Mastro KJJ, Bouchard RSS, Holt HAKA, Gittis AHH: Transgenic mouse lines subdivide external segment of the globus pallidus (GPe) neurons and reveal distinct GPe output pathways. J Neurosci 2014, 34:2087–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernandez VM, Hegeman DJ, Cui Q, Kelver DA, Fiske MP, Glajch KE, Pitt JE, Huang TY, Justice NJ, Chan CS: Parvalbumin+ Neurons and Npas1+ Neurons Are Distinct Neuron Classes in the Mouse External Globus Pallidus. J Neurosci 2015, 35:11830–11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gittis AH, Berke JD, Bevan MD, Chan CS, Mallet N, Morrow MM, Schmidt R: New roles for the external globus pallidus in basal ganglia circuits and behavior. J Neurosci 2014, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, Magill PJ: Dichotomous organization of the external globus pallidus. Neuron 2012, 74:1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creed M, Pascoli VJ, Luscher C: Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science 2015, 347:659–664. [DOI] [PubMed] [Google Scholar]
- 33.Shields BC, Kahuno E, Kim C, Apostolides PF, Brown J, Lindo S, Mensh BD, Dudman JT, Lavis LD, Tadross MR: Deconstructing behavioral neuropharmacology with cellular specificity. Science (80- ) 2017, 356. [DOI] [PubMed] [Google Scholar]
- 34.Calabresi P, Picconi B, Parnetti L, Di Filippo M: A convergent model for cognitive dysfunctions in Parkinson’s disease: the critical dopamine-acetylcholine synaptic balance. Lancet Neurol 2006, 5:974–983. [DOI] [PubMed] [Google Scholar]
- 35.Bamford NS, Wightman RM, Sulzer D: Dopamine’s Effects on Corticostriatal Synapses during Reward-Based Behaviors. Neuron 2018, 97:494–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grogan JP, Tsivos D, Smith L, Knight BE, Bogacz R, Whone A, Coulthard EJ: Effects of dopamine on reinforcement learning and consolidation in Parkinson’s disease. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yttri EA, Dudman JT: A proposed circuit computation in basal ganglia: History-dependent gain. Mov Disord 2018, doi: 10.1002/mds.27321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, et al. : Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45:489–496. [DOI] [PubMed] [Google Scholar]
- 39.Yttri EA, Dudman JT: Opponent and bidirectional control of movement velocity in the basal ganglia. Nature 2016, 533:402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Little S, Pogosyan A, Neal S, Zavala B, Zrinzo L, Hariz M, Foltynie T, Limousin P, Ashkan K, FitzGerald J, et al. : Adaptive deep brain stimulation in advanced Parkinson disease. Ann Neurol 2013, 74:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swann NC, de Hemptinne C, Thompson MC, Miocinovic S, Miller AM, Gilron R, Ostrem JL, Chizeck HJ, Starr PA: Adaptive deep brain stimulation for Parkinson’s disease using motor cortex sensing. J Neural Eng 2018, 15:46006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosin B, Slovik M, Mitelman R, Rivlin-Etzion M, Haber SN, Israel Z, Vaadia E, Bergman H: Closed-loop deep brain stimulation is superior in ameliorating parkinsonism. Neuron 2011, 72:370–384. [DOI] [PubMed] [Google Scholar]
- 43.Arlotti M, Rosa M, Marceglia S, Barbieri S, Priori A: The adaptive deep brain stimulation challenge. Park Relat Disord 2016, 28:12–17. [DOI] [PubMed] [Google Scholar]
- 44.Starr PA, Ostrem JL: Commentary on “Adaptive deep brain stimulation in advanced Parkinson disease”. Ann Neurol 2013, 74:447–448. [DOI] [PubMed] [Google Scholar]
- 45.Brown MJN, Macerollo A, Kilner JM, Chen R: Is Closed-Loop, Time-Locked Primary Motor Cortex Stimulation an Ideal Target for Improving Movements in Neurological Disorders? Mov Disord 2016, 31:1341. [DOI] [PubMed] [Google Scholar]
- 46.Deffains M, Bergman H: Striatal cholinergic interneurons and cortico-striatal synaptic plasticity in health and disease. Mov Disord 2015, 30:1014–1025. [DOI] [PubMed] [Google Scholar]
- 47.Calabresi P, Mercuri NB, Di Filippo M: Synaptic plasticity, dopamine and Parkinson’s disease: one step ahead. Brain 2009, 132:285–287. [DOI] [PubMed] [Google Scholar]
- 48.Gratwicke J, Jahanshahi M, Foltynie T: Parkinson’s disease dementia: a neural networks perspective. Brain 2015, 138:1454–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]