

Abstract
Immunoengineering applies quantitative and materials-based approaches for the investigation of the immune system and for the development of therapeutic solutions for various diseases, such as infection, cancer, inflammatory diseases and age-related malfunctions. The design of immunomodulatory and cell therapies requires the precise understanding of immune cell formation and activation in primary, secondary and ectopic tertiary immune organs. However, the study of the immune system has long been limited to in vivo approaches, which often do not allow multidimensional control of intracellular and extracellular processes, and to 2D in vitro models, which lack physiological relevance. 3D models built with synthetic and natural materials enable the structural and functional recreation of immune tissues. These models are being explored for the investigation of immune function and dysfunction at the cell, tissue and organ levels. In this Review, we discuss 2D and 3D approaches for the engineering of primary, secondary and tertiary immune structures at multiple scales. We highlight important insights gained using these models and examine multiscale engineering strategies for the design and development of immunotherapies. Finally, dynamic 4D materials are investigated for their potential to provide stimuli-dependent and context-dependent scaffolds for the generation of immune organ models.
Vaccines and immunotherapeutics protect the body against infections by stimulating a robust and coordinated immune response towards a specific target. By contrast, immune suppressors prevent harmful immunity occurring in autoimmune diseases and allergy. Immunotherapeutics aim at modulating immune cells, such as B and T lymphocytes, which are white blood cells that reside in the lymphatic system. B and T cells arise from haematopoietic stem cells (HSCs) (BOX 1), but their maturation, activation and function depend on signals from distinct lymphoid niches. Lymphoid niches are specialized microenvironments that are essential for B and T cell maintenance, providing distinct signals in the different lymphatic organs (FIG. 1a). Primary lymphoid organs — the thymus and bone marrow — support T and B cell formation prior to their activation in the secondary lymphoid organs (lymph nodes, spleen, tonsils, Peyer’s patches and mucosa-associated lymphoid tissue). Activated B and T cells then infiltrate the site of infection to initiate a specific response against the target. Disease states, such as infection, transplant rejection, cancer and chronic inflammation, often lead to B and T cell dysregulation, including the accumulation of both cell types in structured niches at ectopic locations1–4. The role of these structured niches (or tertiary lymphoid structures) is not yet well understood, but they might offer a potential site to counteract disease.
Box 1 |. Haematopoiesis and immune cell types.
Haematopoiesis occurs in the blood marrow and is the process by which all blood cells are formed by differentiation of haematopoietic stem cells (HSCs)28. In the bone marrow microenvironment (niche), HSCs can self-renew and differentiate into myeloid or lymphoid progenitor cells. Myeloid cells further differentiate into granulocytes — neutrophils, eosinophils and basophils — and into monocytes, which differentiate into macrophages. Macrophages are phagocytes and they form the backbone of innate immunity, which is the first yet unspecific response of the body to infections. Lymphoid progenitor cells differentiate into dendritic cells, which are antigen-presenting cells, and into lymphocytes — T cells, B cells and natural killer (NK) cells. Lymphocytes are the main mediators of adaptive immunity and respond to infection in an antigen-specific manner. Lymphocytes are also the main targets of immunotherapeutics. T cells mature in the thymus and can be subdivided into CD8+ cytotoxic T cells, which kill virally infected cells and tumours and express CD8 on their surface, and CD4+ helper T cells, which express CD4 on their surface, secrete cytokines and prime B cells. B cells mature in the bone marrow and can then differentiate into antibody-secreting plasma cells and memory B cells in secondary lymphoid organs. Once formed, plasma cells are immediately active to produce antibodies against pathogens. Memory B cells are long-living antigen-specific cells that can initiate a fast response against future infections. The lifespan of plasma cells can reach years in mice and decades to a lifetime in humans.
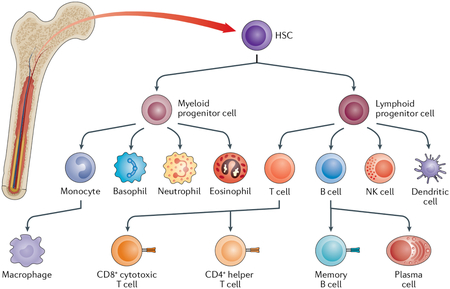
Fig. 1 |. The different levels of the immune response.

a | B and T cells originate in lymphoid organs and reside in the lymphatic system. During an immune response, Band T cells are first generated in primary lymphoid organs — the bone marrow and thymus — from haematopoietic stem cells (HSCs) and lymphoid progenitors. B and T cells then migrate to secondary lymphoid organs, such as the lymph node, where they localize in specific T cell and B cell zones. In these zones, each cell is activated by intact antigens or processed antigens presented on antigen-presenting cells (APCs), followed by differentiation into effector cells. B effector cells, such as plasma cells, and T effector cells, such as cytotoxic T cells, then migrate to sites of infection, including sites created by vaccine delivery. APCs, such as dendritic cells, encounter antigen at the infection site and present it to naive B and T cells in the lymph node for stronger and sustained responses. During disease, the normal immune response can be deregulated, leading to the formation of ectopic tertiary immune organs, which are structured immune aggregates often found near tumours. b | The immune response is regulated at the cell, tissue and organ levels. Efficient immune cell effector function is crucial for the targeting and killing of infected cells or tumour cells. Interactions between T cells and APCs (that is, the formation of immune synapses) play key roles in immune cell activation. Maturation of immune cells occurs in bone marrow niches and in the thymus. Engineering approaches are needed to recapture functionality at each biological scale. The dependency on material incorporation increases with the complexity of the immune function. MHC, major histocompatibility complex; TCR, T cell receptor.
The possibility to engineer immune cells and tissues has greatly contributed to the discovery of immunological mechanisms and targeted immunotherapeutics. Technologies to support survival, expansion and differentiation of immune cells enable the development and large-scale manufacturing of immune cell-based therapeutics. In particular, activated T cells, such as chimeric antigen receptor (CAR) T cells, and monoclonal antibodies secreted by differentiated B cells hold promise in curing many cancers5,6 and autoimmune diseases7 as well as in alleviating diabetes8–10, neural diseases11–13 and other devastating disorders7,14. In addition, engineered immune tissues could enable faster and reproducible development of materials-based infectious disease vaccines15–17.
However, many aspects of the immune system remain elusive thus far, and therapeutic cell generation is challenging owing to the lack of complexity of current engineered systems, which cannot recreate the specific niches, the various immune system components or their interplay. Current in vivo approaches to model the immune system do not yet provide highly tunable biological control of specialized immune niches. 2D in vitro models are often an oversimplification of the niche. Combining these systems with synthetic and natural materials to engineer specific immune niches at multiple timescales could ultimately bridge the gap between the high-throughput nature of in vitro systems and physiologically relevant in vivo models.
In this Review, we discuss 2D and 3D approaches to recreate key cell-level, tissue-level and organ-level immune functionalities in primary (TABLE 1), secondary (TABLE 2) and tertiary lymphoid organs, and we highlight the potential for materials science to greatly contribute to immune cell and tissue engineering (FIG. 1b). Finally, we investigate the development of 4D, stimuli-responsive and context-dependent materials to more effectively model and engineer immune cells at the cell, tissue and organ levels.
Table 1 |.
Engineering primary immune organs and cells
| Characteristics | Engineering approach | Reported findings | Limitations | Refs |
|---|---|---|---|---|
| 2D bone marrow HSC niche | ||||
| Regions with varying ECM composition and biophysical properties | Fibronectin-coated, collagen-coated and laminin-coated PA substrates; seeded with HSCs from C57Bl/6 mice | Altered HSC fate in response to varying substrate stiffness and ECM protein composition | Combinations of ECM proteins not tested; only thin gels explored | 41 |
| 2D thymus | ||||
| T cell differentiation | Murine DLL1-expressing OP9 stromal cells co-cultured with human cord blood-derived HSCs | DLL1-dependent expansion of HSCs and differentiation along T cell lineages | Requires genetic manipulation of cells; suboptimal yield of CD4 or CD8 single-positive cells | 170 |
| Magnetic polystyrene microbeads functionalized with DLL4; co-cultured with human cord blood-derived HSCs and OP9 stromal cells | Controlled density, orientation and timing of DLL4 and induction of T cell progenitors | Level of T cell development and functionality not determined | 71 | |
| Human cord blood-derived HSCs cultured on DLL1-coated plates in the presence of VCAM1 | Induction of T cell progenitors in feeder cell-free and serum-free conditions | Differentiation limited to progenitor T cells; positive and negative selection mechanisms not present | 72 | |
| 3D bone marrow HSC niche | ||||
| ECM matrix proteins that promote HSPC adhesion | Fibronectin-immobilized and collagen-immobilized PET scaffolds; seeded with human cord blood-derived HSCs | Significant increase in CD34+ cell expansion; expanded cells differentiate into multiple haematopoietic lineages in bone marrow of NOD-SCID mice | Mechanisms driving HSPC differentiation unclear | 39 |
| Structural topology of bone marrow matrix with large surface area, high porosity and mimetic stromal tissue function | Silicate or PA scaffolds with ICC geometry coated layer-by-layer with PDDA and clay; seeded with a cocktail of human stromal cells and human HSCs | Significant (P < 0.05) increase in CD34+ cell proliferation, B cell differentiation and antibody production in ICC cultures compared with 2D controls after 28 days | Contributions from specific stromal cells are unclear | 37 |
| Cell-cell interactions within the 3D vascular niche | Human femur-derived bone scaffolds; seeded with hBMSC-HUVEC feeder co-cultures and human cord blood-derived HSCs | Significant (P < 0.05) expansion of short-term and long-term repopulating CD34+ cells and increased HSC quiescence by co-cultures compared with hBMSCs or HUVECs alone after 14 days | Unclear whether HUVECs directly affect HSCs or whether effects were due to HUVEC-induced osteogenic differentiation of hBMSCs | 43 |
| Overlapping patterns of niche-specific cells and matrix constituents | Microfluidic mixing platform fabricated with gradients of cells and type I collagen hydrogel with defined densities; seeded with HSPCs from C57Bl/6 mice and MC3T3 osteoblasts | Demonstrated overlapping gradients of fluorescently tagged cells and ability to isolate subregions of the gradient for cell analysis | Modest reduction in viability of HSPCs observed after 3 days | 45 |
| Platelet production by MKs within vascular niche | Porous films of varying stiffness; topography fabricated using silk solution, PEO porogens and ECM proteins patterned onto PDMS moulds; gel-spun vascular microtubes with incorporated HMECs; seeded with HSC-derived MKs | Modulation of MK adhesion and proplatelet formation by film topography and stiffness, respectively; ECM protein-functionalization and presence of HMECs enhance these outputs | Platelet generation low relative to in vivo production | 50 |
| Platelet production by MKs within perfusable vascular niche | Perfusion bioreactor system containing porous, multichannel silk sponge; seeded with HSC-derived MKs | Proportional increase in production of functional platelets with increasing silk channels; perfusion-facilitated platelet collection | Incorporation of multiple niche-specific cells not explored with this system | 52 |
| Cell diversity and toxicological responses of HSC niche | In vivo marrow formation via subcutaneous implantation of type I collagen with bone-inducing factors in mice; engineered bone marrow explanted and maintained in microfluidic device | Morphology and architecture of engineered bone marrow mimetic of native tissue, with comparable heterogeneous niche-specific cell populations; ex vivo culture supported HSC and HSPC maintenance and function | Long-term ex vivo culture not yet evaluated | 46 |
| 3D bone marrow HSC niche (cont.) | ||||
| Long bone tissue compartments with spatially different cues | Non-mineralized and mineralized macroporous PEGDA-co-A6ACA hydrogels, mimicking skeletal and haematopoietic compartments of bone; seeded with donor murine-derived intact whole bone marrow flush or cells isolated from bone marrow flush and implanted subcutaneously in recipient mice | Abundant CD34+ cell population within inner, non-mineralized compartment and hardened bone tissue restricted to mineralized compartment after 4 weeks in vivo; migration of donor cells into circulation | Species-specific responses may not translate to human application | 56 |
| 3D thymus | ||||
| Differentiation of mature T cells from HSPCs | Murine DLL1-expressing stromal cells aggregated with HSPCs and seeded on cell culture insert | Long-term maintenance of lymphoid progenitors; T cell differentiation resembling thymopoiesis; enhanced positive selection | Bias towards CD8+ T cell maturation owing to absence of MHC class II-dependent positive selection | 74 |
| ECM composition and architecture | Thymus from B6 athymic mice decellularized and seeded with TECs and non-epithelial cells from thymic digests; engrafted in nude mice | Maintenance of ECM composition and conformation; recruitment of haematopoietic progenitors; increased epithelial cell retention and survival supporting thymopoiesis | Thymopoiesis not displayed ex vivo. When engrafted, thymocyte number does not differ from controls prepared by isolation and reaggregation of thymic cells (reaggregate thymic organ culture) | 77 |
| Maintenance of donor epithelial cells to support tolerance to allografts | Decellularized murine thymus seeded with thymic fibroblasts and epithelial cells and bone marrow progenitors transplanted in B6 nude mice | Survival of functional TECs; lymphocyte homing after implantation and induction of immune tolerance | Lack of TEC organization and compartmentalization, resulting in less diverse T cell repertoire; thymopoiesis not displayed ex vivo; decellularized matrix shows batch-to-batch variability | 58 |
| Lymphocyte-stromal cell interaction for thymopoiesis | Self-assembling amphiphilic EAK16-II and EAKIIH6 peptides modified with histidine tags; tethered with TECs using complexes of antibodies and recombinant proteins | Clusters of TECs formed, resulting in development of mature functional T cells after implantation in athymic nude mice | Lacks segregation of niche-specific thymic epithelial cells | 80 |
DLL, delta-like ligand; EAK16-II, peptide (AEAEKAKAEAEAKAK); EAKIIH6, peptide (AEAEKAKAEAEAKAKHHHHHH); ECM, extracellular matrix; hBMSC, human bone marrow-derived mesenchymal stem cell; HMEC, human microvascular endothelial cell; HSC, haematopoietic stem cell; HSPC, haematopoietic stem or progenitor cell; HUVEC, human umbilical vein endothelial cell; ICC, inverted colloidal crystal; MHC, major histocompatibility complex; MK, megakaryocyte; NOD-SCID, nonobese diabetic-severe combined immunodeficient; PA, polyacrylamide; PEGDA-co-A6ACA, polyethylene glycol-diacrylate-co-N-acryloyl 6-aminocaproic acid; PDDA, poly(diallyldimethylammonium chloride); PDMS, polydimethylsiloxane; PEO, polyethylene oxide; PET, polyethylene terephthalate; TEC, thymic epithelial cell; VCAM1, vascular cell adhesion protein 1.
Table 2 |.
Engineering secondary immune organs and cells
| Characteristics | Engineering approach | Reported findings | Limitations | Refs |
|---|---|---|---|---|
| 2D activated T cells | ||||
| Immune synapse: T cell-APC interface | Glass-supported planar bilayer with pMHC and an adhesion ligand; ICAM1 for imaging | Immune synapse formation between a T cell and the synthetic surface | Quantitative, qualitative and pattern shape studies cannot be achieved | 171 |
| Alternative patterns of GPI-linked pMHC and ICAM1 into a supported lipid bilayer membrane using electron-beam lithography on silica substrates | Chromium barriers enable investigation of basic mechanisms of immunological synapse formation; membrane lipid mobility allows molecular motion in lipid-linked T cell ligands to generate alternatively patterned synapse | Quantitative and qualitative studies cannot be achieved | 86 | |
| Aqueous solvent-based photoresist and polyelectrolyte bilayers for printing multi-biotinylated protein arrays | Altered secretion of IL-2 and IFNγ from CD4+ T cells by spatial patterns; PKCθ exhibits aberrant clustering, which resulted in reduced production of IFNγ if patterns of activation sites preclude centralized clustering of TCR ligands | Complex procedure for fabricating multicomponent patterns | 92,172 | |
| Selective patterns of the immune synapse generated by incorporating multiple rounds of microcontact printing using a topological PDMS mould | Increased IL-2 and IFNγ production by CD4+ T cells if CD28 receptor clusters are segregated from the cSMAC-localized CD3 receptor and located in the periphery of an artificial immune synapse | Low degree of control over quantity of transferred protein; protein drying process required prior to stamping | 93 | |
| Intercellular mechanical forces: T cell-APC interface | Elastomeric PDMS micropillar arrays for regulation of the T cell mechanical force under CD3 and CD28 co-stimulation | CD28 binding increases traction forces associated with CD3 by stimulating a signalling pathway involving PI3K, causing pillar array deformation | Manufacturing process may lead to pillar defects, resulting in low accuracy of intercellular force measurements | 173 |
| Polyacrylamide hydrogels (Matrigen) and PDMS micropillars to measure force exertion across the synapse and the kinetics of perforin pore formation in the target cell | CD8+ cell-mediated cytotoxic activity; increased tunnelling of T cells through APC membranes on stiffer substrate; exerted forces across the synapse affect kinetics of perforin pore formation, enhancing cytotoxic capabilities | 2D environment cannot fully capture 3D aspects; therefore, it is unclear whether force exertion is solely responsible for cytotoxic activity | 96 | |
| 2D germinal centre B cells | ||||
| Memory B cell and plasma cell formation | Murine spleen-derived B cells cultured with BALB/c 3T3 fibroblasts expressing CD40 ligand and BAFF in the presence of IL-4 | Induction and expansion of germinal centre B cells; B cell receptor class switching and differentiation into memory B cells; additional culture with IL-21 leads to plasma cell formation | Unaltered immunoglobulin genes in germinal centre B cells after stimulation with IL-21 and CD40L | 123 |
| Antigen-specific B cell expansion | Polyvalent presentation of CD40 ligand, complexed with HA peptide, on iron oxide microbeads coated with anti-HA antibody | Expansion of antigen-specific B cells; class switching; germinal centre B cell differentiation in the absence of feeder cells | Influence of microbead properties unknown | 125 |
| Antigen-specific B cell responses | Multi-layered lipid vesicles covalently crosslinked and maleimide-functionalized for presentation of malaria antigen | Germinal centre formation; improved strength and breadth of antibody response relative to conventional adjuvants; induction of antigen-specific follicular helper T cells | Unknown whether a wide range of clinically relevant adjuvants, except monophosphoryl lipid A, is effective with lipid vesicle vaccine platform | 174 |
| 3D artificial APCs | ||||
| T cell activation and expansion | Chemically treated bundles of SWNTs embedded with anti-CD3 | Chemical treatment induced surface defects lead to increased anti-CD3 absorption; increased IL-2 release following culture with T cells | Incorporation of stimuli for T cell activation was not explored | 100 |
| Carbon nanotube-PLGA composite containing magnetite and IL-2, as a platform for T cell stimuli delivery | Magnetite and IL-2 incorporation increases cytotoxic T cell population through maximizing cell isolation and enrichment; adoptive therapy for murine melanoma by transferring CD8+ T cells delays tumour growth | Difficult to remove carbon nanotube residues from isolated T cells; nanocomposite is cytotoxic | 101 | |
| Paramagnetic iron-dextran nanoparticles system for activation and expansion of the naive T cell repertoire | Iron-dextran nanoparticles conjugated with an MHC-immunoglobulin dimer and anti-CD28 increases target T cell population by positive selection from the naive T cell pool, inducing expansion of target cell | Incorporation of multiple signals for T cell expansion not explored; suboptimal T cell expansion | 106 | |
| Composite of mesoporous silica microrods and supportive lipid bilayers with soluble IL-2 and anti-T cell receptors | Higher efficiency in polyclonal and antigen-specific T cell expansion than commercial T cell expansion beads; antitumour efficacy in a model of Burkitt’s lymphoma | Density, quantity or spatial distribution of factors for T cell activation cannot be controlled | 102 | |
| 3D B cell follicle | ||||
| Germinal centre B cell differentiation | RGD peptide-functionalized gelatin hydrogel with silicate nanoparticles, splenic B cells from C57BL6 mice and stromal cells presenting CD40 and BAFF; stimulated with IL-4 | Enhanced, rapid B cell proliferation and differentiation, with antibody class switching | B cell responses not antigen-specific | 127 |
| Degradable and tuneable maleimide-crosslinked polyethylene glycol hydrogels presenting integrin ligands; seeded with murine splenic B cells and stromal cells presenting CD40 and BAFF; stimulated with IL-4 | Modulation of B cell differentiation; antibody class switching; enrichment of antigen-specific B cells | Additional niche-specific signals required to mimic complexity of germinal centre reactions | 131 | |
APC, antigen-presenting cell; BAFF, B cell activating factor; CD40L, CD40 ligand; cSMAC, central supramolecular activation cluster; GPI, glycosylphosphatidylinositol; HA peptide, human influenza haemagglutinin tag (YPYDVPDYA); ICAM1, intercellular adhesion molecule 1; IFNγ, interferon-γ; IL, interleukin; MHC, major histocompatibility complex; PDMS, polydimethylsiloxane; PI3K, phosphoinositide 3-kinase; PKCθ, protein kinase Cθ; PLGA, poly(lactide-co-glycolide); pMHC, peptide-MHC; RGD, Arg-Gly-Asp; SWNT, single-walled carbon nanotube; TCR, T cell receptor.
Primary lymphoid organ engineering
The bone marrow and thymus are primary lymphoid organs because they play a crucial role in generating and maintaining immune cells. The bone marrow is the main site for the production and maintenance of all haematopoietic cells18, which give rise to all blood cells through a process called haematopoiesis (BOX 1). Bone marrow is also the primary site of B cell maturation. By contrast, T cell progenitors produced in the bone marrow migrate to the thymus, where T cell maturation occurs. The specific tissue structure and biological cues of the bone marrow and thymus are key to supporting and regulating these processes19–22.
Engineering bone marrow
HSCs are multipotent stem cells that can self-renew and differentiate. Therefore, healthy HSCs from donor tissue can be transplanted to regenerate damaged haematopoietic tissue of immune cell-depleted recipient patients23–25 (FIG. 2a). HSC transplantation is used not only as a therapeutic strategy for treating bone marrow diseases, cancer and immune cell dysfunction but also as a tool for modulating the immune response following whole organ transplantation to minimize graft rejection26. Despite the widespread clinical use of HSC transplantation, there are serious risks associated with it, including graft-versus-host disease, organ failure, prolonged immunodeficiency and infection27. Moreover, the population of HSCs in bone marrow, which is the main source of HSCs, is small and susceptible to impacts of genetic diseases and ageing28,29.
Fig. 2 |. Engineering bone marrow niches.

a | Transplantation of haematopoietic stem cells (HSCs) is one of the most widely used cell therapies. Donor HSCs are collected from bone marrow or blood, purified and transferred to a conditioned recipient, b | Bone marrow comprises distinct HSC niches, including the endosteum and vascular niches, with distinct molecular and cellular compositions. Niche-specific factors drive HSC fate. HSC niches can be designed by engineering a specific extracellular matrix (ECM) and by modelling cell-cell interactions. The vascular niche can be engineered for platelet production, and entire bone marrow tissue can be obtained by implanting materials in animal models to trigger bone marrow formation. These tissues can then be explanted and maintained in a bioreactor for continuous HSC expansion.
Engineered bone marrow can serve as a model to investigate HSC behaviour in healthy and diseased tissues. Moreover, it can provide a predictive platform to improve patient therapies and for the generation of large quantities of immune cells for HSC transplantation. However, native bone marrow has a complex microenvironment with distinct yet dynamic regions (niches) that guide the self-renewal and differentiation of HSCs. The specific bone marrow niches are the endosteal surface of bone, where bone-forming cells reside; arteriolar blood vessels, which control the resting state of HSCs (quiescence) to support lifelong self-renewal; and sinusoidal blood vessels that support HSC activation and differentiation30,31. Importantly, haematopoiesis occurs in the extravascular space between HSC niches21,22 and is controlled by complex physical and biochemical interactions between the stroma, HSCs and the extracellular matrix (ECM), which are not yet fully understood.
Therefore, to engineer bone marrow and its niches for HSC expansion, the effects of the specific microenvironmental cues on haematopoiesis need to be investigated. Expansion of HSCs in static 2D cultures is difficult because they cannot provide complex microenvironmental regulation. By contrast, 3D biomaterials-based culture systems can be used to replicate aspects of the native microenvironment and to provide spatiotemporal control of multiplexed cell, biophysical and biomolecular signals (FIG. 2b; TABLE 1). The composition, architecture, topology and mechanical properties of materials can be tailored and decoupled to parse the impact of the different biochemical and biophysical factors on HSC fate to improve the yield of HSCs32–38 ex vivo.
The surface of 3D material scaffolds can be modified with ECM proteins to mimic niche-specific tissue composition for improved HSC expansion39 (FIG. 2b). Thin polyacrylamide matrices with varying stiffness can also be coated with a combination of different ECM proteins to recreate transitions in the microenvironment reminiscent of discrete endosteal and vascular niches40, which can alter HSC fate41. Surface modification with ECM proteins does not fully capture the complexity of the bone marrow microenvironment, but it provides a tool to mimic local biophysical and biochemical niche properties to control and modulate HSC differentiation.
Interactions between HSCs and stromal cells also play a crucial role in HSC fate. Models that recreate the topology of bone marrow and include stromal constituents can be applied to investigate these cellular interactions. For example, a 3D polyacrylamide hydrogel matrix can be fabricated with inverted colloidal crystal (ICC) geometry using a sol-gel approach; in this model, the matrix displays hexagonally ordered lattices of spherical cavities37. To promote stromal cell adhesion and enable tunability of the scaffold stiffness, the ICC surface can be modified by a layer-by-layer approach to coat the scaffold with sequential layers of negatively charged silicate nanoparticles and positively charged poly(diallyldimethylammonium chloride). This approach can be applied to create a high degree of order, similar to that seen in native bone marrow, and provide control over tissue porosity and spatial organization for modulating cell interactions and migration42. Human bone marrow stromal cells and HSCs can then be co-cultured on the modified ICC scaffolds to promote the development of B cells ex vivo37. Cell-cell interactions within vascular HSC niches can also be modelled by co-culturing human bone marrow mesenchymal stem cells (MSCs) and human endothelial cells on 3D bone scaffolds derived from human femurs; the native bone architecture coupled with cellular interactions triggers early differentiation of MSCs into bone-forming cells, which promote the expansion of repopulating and quiescent HSCs43.
Bone-marrow-on-a-chip platforms have been developed to address the complex structure and functions of the bone marrow. In contrast to traditional 2D surfaces and 3D matrices, on-chip platforms44 allow the controlled integration of fluids, which can be used to manipulate mass transport kinetics of drugs and to incorporate molecular and cellular gradients. These systems are particularly useful for designing tissue interfaces and for modelling human physiology in vitro. On-chip platforms can be combined with 3D matrices to mimic ECM composition and architecture. For example, a microfluidic mixing platform with staggered herringbone features, which induce chaotic advection in the channel, can be used to generate 3D hydrogels with tuneable and defined opposing gradients of cells and matrix constituents that are inspired by the HSC niche45 (FIG. 2b). This approach allows the dissection of the effect of the different microenvironments within the bone marrow niche on HSC fate.
Microfluidic chips can further be used to maintain bone marrow explants for continuous HSC expansion. For example, a polydimethylsiloxane (PDMS) cylinder filled with a type I collagen gel and specific stimuli can be subcutaneously implanted to induce bone marrow formation in vivo46 (FIG. 2b). The engineered bone marrow closely resembles the composition and architecture of native bone marrow tissue 8 weeks after implantation and can then be explanted and maintained for 7 days in a microfluidic chip device to support bone marrow viability and function. The on-chip platform promotes the expansion of a higher number of long-term HSCs than do static stroma-supported control cultures46, demonstrating the importance of HSC interactions with the tissue microenvironment.
Fluid flow plays an integral role in tissue homeostasis47 and can be implemented in bone marrow models using bioreactors. Materials-based systems with fluid flow have advanced our understanding of platelet generation by megakaryocytes48–52 and of the progression of different disease states, such as multiple myeloma53–55. For example, using a template made of polyethylene oxide (PEO), silk-based films can be fabricated with a porous architecture50. The films can then be casted on patterned PDMS moulds and modified with different bone marrow ECM proteins to model the heterogeneity of the composition, architecture and stiffness of HSC niches. The niche vasculature and platelet formation can further be incorporated by gel-spinning solutions containing silk, PEO and ECM proteins around a wire, which result in hollow microtube structures, which can be filled with endothelial cells and perfused in a bioreactor chamber50 (FIG. 2b). Similarly, increasing the number of silk channels, or vessels, within silk sponges leads to a proportional increase in platelet production, which could have a profound clinical impact on platelet transfusions52.
HSC transplantations require patient conditioning with radiation or chemotherapy, which can potentially be prevented using engineered bone marrow. For example, bone tissues with a functional bone marrow can be engineered in vivo using modular synthetic matrices with spatially defined cues56. In this approach, poly(methyl methacrylate) microspheres are used as templates for macroporous polyethylene glycol-diacrylate-co-N-acryloyl 6-aminocaproic acid hydrogels, which can then be immersed in simulated body fluid to create calcium phosphate moieties. By modular assembly of mineralized and non-mineralized hydrogels, dual-compartment matrices can be engineered that represent bone and central bone marrow. Notably, subcutaneous implantation of these matrices in mice results in mature dual-compartment structures with similar cell and ECM composition to native bone tissue-containing marrow56, suggesting that this approach can be used to provide surrogate ectopic bone marrow to increase survival of donor cells after HSC transplantation. Finally, injectable bone marrow-like macroporous scaffolds made of alginate-polyethylene glycol (PEG) can enhance T cell immunity after HSC transplantation57.
Engineering thymus tissue
The thymus is the primary site for the generation of a self-tolerant T cell repertoire necessary for adaptive immunity19,58. However, the thymus is a vulnerable organ, and its function can be compromised by several factors, including viral infection, irradiation, environmental insults and ageing59. Thymic dysfunction can result in thymic involution, or shrinking, and consequently a decrease in naive T cell output and weakened immune responses59 (FIG. 3a). Dysfunction can further lead to unresponsiveness, autoimmunity or malignancies60. Therefore, there is interest in using materials to model and recreate thymic functionality to understand thymic regeneration and rejuvenation and to engineer an implantable thymus58. Thymic transplants have the potential to help patients with decreased thymic function resulting from thymic involution or congenital immune deficiencies. Transplants developed with a mixture of donor and recipient cells can be used to induce tolerance in the donor, preventing transplant rejection and graft-versus-host disease. Thymic models can further be applied to generate T cells from renewable cell sources, such as from induced pluripotent stem cells, for adoptive T cell transfer. Adoptive T cell transfer is a clinically used personalized treatment strategy in which a patients T cells are reintroduced into the body to act as a living drug for the treatment of haematological malignancies61,62, intracellular pathogens and autoimmunity63,64 (FIG. 3a).
Fig. 3 |. Engineering thymus tissue.

a | Thymic dysfunction leads to thymic involution, causing a decrease in the production of T cells, b | A schematic of thymic structure is presented, showing key cell types and microenvironmental cues, including thymic epithelial cells (TECs), haematopoietic stem cells (HSCs), thymocytes (that is, lymphocytes in the thymus), cortical epithelial cells and Notch ligands (delta-like ligand 4 (DLL4)). Negative and positive selection processes, as determined by the reactivity of thymocytes to self-peptides on the major histocompatibility complexes (MHCs) of cortical epithelial cells, are required to establish a diverse set of MHC-restricted yet self-tolerant mature T cells, c | Progenitor cells can be differentiated into T cells using different strategies: Notch signalling can be activated by co-culturing HSCs and bone marrow stromal cells (OP9), which are genetically engineered to present the Notch ligand DLL1. Magnetic polystyrene beads can be functionalized with DLL4 at a specific density and orientation. A stromal cell-free thymic niche can be designed by culturing HSCs with both DLL1 and vascular cell adhesion protein 1 (VCAM1). d | Organoid cultures can be engineered by aggregating and centrifuging HSCs and MS5-hDLL1 cells — a genetically engineered bone marrow stromal cell line expressing human DLL1 (hDLL1). Decellularized extracellular matrices (ECMs) can be used to culture TECs and HSCs. Self-assembling hydrogels based on amphiphilic peptides can be applied to encapsulate HSCs and TECs, enabling precise control of the microenvironment. Anti-histidine (His) and TEC-specific anti-epithelial cell adhesion molecule (EpCAM) antibodies can be bound to protein A/G adaptor complexes to promote binding to histidinylated amphiphilic peptides and for 3D aggregation of TECs, respectively. TCR, T cell receptor.
For thymic transplantation and T cell generation, thymic models must contain the key structural, cellular and microenvironmental cues required for positive and negative T cell selection. The thymus is an epithelial organ characterized by two compartments: an outer cortex region that supports early-stage T cell development and an inner medulla, where later stages of differentiation occur19. The compartments are separated by a vascular corticomedullary zone, and they contain distinct populations of stromal and thymic epithelial cells arranged in a complex 3D network (FIG. 3b). This cellular network produces homing signals to recruit lymphocyte progenitors from the bone marrow The epithelial cells provide maturation and differentiation signals for the generation of a diverse major histocompatibility complex (MHC)-restricted (that is, the T cell can bind and respond to an antigen only when it is bound to a particular MHC molecule) yet self-tolerant population of mature T cells. T cell receptors usually recognize antigenic peptides only when they are displayed by MHC molecules; however, recognition of self-peptides or of antigens not bound to MHC can cause autoimmune disease. Therefore, to select for T cells with T cell receptors that are self-tolerant and restricted to binding peptides presented by MHCs, cortical epithelial cells in the thymus present self-MHC-self-peptide complexes to differentiated T cell precursors (thymocytes). Thymocytes that fail to bind or weakly bind the self-MHC-self-peptide complex undergo apoptosis because they are not restricted to binding peptides on MHCs and can theoretically react to any peptide. Similarly, thymocytes that strongly bind the complex are potentially autoreactive and undergo apoptosis. Therefore, MHC-restricted, self-tolerant T cells are positively selected. Thymocyte differentiation and selection are also controlled by microenvironmental cues that direct their migration through the different regions of the thymus19,58.
Early approaches to generate T cells and model the thymus were limited to explant cultures of embryonic thymic lobes. These cultures were then modified by disassociating these lobes and reaggregating purified T cell precursors with stromal cells65. Although restricted by donor availability, fetal thymic organ cultures enabled the investigation of important pathways and structural cues in T cell development; for example, the Notch ligand delta-like ligand 1 (DLL1) affects T cell differentiation in a density-dependent and orientation-dependent manner66–68. Thus, DLL1-presenting bone marrow stromal cells (OP9-DLL1) have been generated to differentiate haematopoietic and T progenitor cells into early T cells in vitro69,70. However, using OP9-DLL1 cells, the density and orientation of DLL1 cannot be controlled. This issue can be addressed by using magnetic polystyrene beads to present an alternative Notch ligand (DLL4) (FIG. 3c), which allows control of the density, orientation and timing of ligand presentation. In combination with OP9 cells, the DLL4-modified beads induce the differentiation of bone marrow HSCs into T cell progenitor cells in insert-based or mixed stromal cell culture71. This bead-based approach relies on the incorporation of stromal cells. Alternatively, a stromal cell-free thymic niche can be designed by coating plates with defined densities of DLL4-Fc and vascular cell adhesion protein 1 (VCAM1)-Fc (FIG. 3c). Using this plate-based system, VCAM1 was identified as an important cue that potently enhances Notch activation and T cell induction. Moreover, the thymic niche model can be used to generate progenitor cells, which mature into cytokine-producing mature T cells in vivo72.
Notch ligand-based approaches using DLL presentation lack efficiency and negative and positive cell selection, which are necessary for adaptive immunity and tolerance. Monolayer models of OP9-DLL1 cells further show suboptimal DLL1 expression with time. Therefore, 3D architectures similar to the thymic microenvironment are required73. For example, a compaction reaggregation technique can be applied using centrifugation to form an aggregate of HSCs, progenitor cells and genetically engineered bone marrow stromal cells expressing human DLL1. The aggregate can then be plated on a cell culture insert at the air-fluid interface (FIG. 3d), resulting in a serum-free artificial thymic organoid culture platform. This platform enables long-term maintenance of lymphoid progenitors, phenotypic progression closely resembling human thymopoiesis, improved positive selection and efficient differentiation of progenitor and receptor-engineered T cells74.
To overcome the use of genetically engineered stromal cells for thymic transplantation, thymic epithelial cells can be used, which also provide the necessary signals for the recruitment of lymphocyte progenitors from the bone marrow, thymocyte differentiation and thymocyte maturation75. However, thymic epithelial cells need to form a sponge-like 3D architecture to function76. Thymic ECMs, which can be prepared by chemical or mechanical decellularization, which preserve native ECM composition and architecture, can be used as a natural polymer microenvironment for the culture of thymic epithelial cells. Decellularized thymic ECMs seeded with thymic epithelial cells can be implanted into athymic nude mice to trigger the formation of naive T cells77. Decellularized ECMs can further be seeded with both thymic epithelial cells and bone marrow-derived lymphocyte progenitors and transplanted into athymic mice (FIG. 3d). These grafts facilitate homing of lymphoid progenitors, thymopoiesis, T cell support for antigen-specific humoral responses upon immunization and rejection of skin allografts displaying non-self-antigen. Most importantly, donor-derived thymic epithelial cells provide immunological tolerance against donor-derived skin grafts58.
Decellularization and approaches based on natural ECMs can improve the self-organizing capacity of progenitor cells78. However, in natural ECMs, the protein composition varies between batches, resulting in a lack of reproducibility79. Synthetic materials offer more precise control of the microenvironment and are not limited by donor availability For example, a self-assembling hydrogel platform based on the co-assembly of amphiphilic or self-assembling peptides can be used to tether thymic epithelial cells through protein adaptor complexes, leading to the formation of mini-clusters (FIG. 3d). This platform supports the development of functional T cells after transplantation into athymic nude mice80. Incorporating synthetic materials may also enable the compartmentalization of niche-specific thymic epithelial cells to generate a diverse T cell repertoire required for effective thymic transplants and T cell generation.
Secondary lymphoid organ engineering
Secondary lymphoid organs — lymph nodes, spleen, tonsils, Peyer’s patches, and mucosa-associated lymphoid tissue — are strategically placed at sites where antigens are likely to be encountered. The primary function of these organs is to enable lymphocyte activation and induction of the adaptive immune response81. The function of secondary lymphoid organs is modulated by a microarchitecture that is broadly defined by three zones: an outer antigen-sampling zone, a T cell activation zone and a B cell activation zone (FIG. 1a). The outer antigen-sampling zone is rich in antigen-presenting cells (APCs) that can limit the spread of pathogens and deliver antigens to the B and T cell zones. Both the T and B cell zones then provide a niche to mediate interactions with other immune cells, surrounding stromal cells and extracellular proteins present in the lymphoid microenvironment, enabling B and T cell differentiation into immune effector cells81.
Engineering activated T cells
T cell activation and expansion are needed for adoptive T cell therapy but currently suffer from suboptimal activation rates82 and the production of T cells with undesirable functionality83. T cell therapies usually involve the extraction of T cells from blood or a tumour biopsy Prior to re-infusion, all types of T cells are activated and expanded. In addition, T cells from the blood can be edited to become specific for disease-causing cells64 (FIG. 4a).
Fig. 4 |. Engineering activated T cells.

a | For adoptive T cell transfer, T cell receptors (TCRs) can be modified ex vivo to make them specific for disease-causing cells by incubation with disease-causing cells (through an interaction between TCR and antigen bound to a major histocompatibility complex (MHC)) or by inducing expression of a chimeric antigen receptor (CAR), which includes a specified antigen-binding domain directly linked to downstream signalling domains, b | The immune synapse between a T cell and an antigen-presenting cell (APC) has a bull’s eye pattern, with a central supramolecular activation cluster (cSMAC), a peripheral SMAC (pSMAC) and a distal SMAC (dSMAC). This structure can be mimicked using 2D substrates, c | 2D approaches, such as lithography, contact printing and polymer pillar arrays, can be applied to study immune synapse organization and mechanosignalling pathways. In lithography, a biotinylated photoresist that dissolves in aqueous buffers upon UV exposure enables control of protein presentation through iterative UV exposure, dissolution and conjugation steps. Microcontact printing can be used to functionalize a substrate with multiple ligands by applying repeated rounds of contact between several coated polydimethylsiloxane (PDMS) stamps and the substrate. Micropillar arrays modified with anti-T cell ligands can be applied to measure mechanical forces during T cell activation on the basis of the dimension, composition and deflection of the pillars, d | 3D approaches can be applied to develop artificial APCs for T cell activation and expansion. Carbon nanotubes can be functionalized with neutravidin, which is bound to biotinylated anti-CD28 antibodies, MHC class I and poly(lactide-co-glycolide) (PLGA) nanoparticles encapsulating interleukin-2 (IL-2). Paramagnetic iron-dextran nanoparticles can be functionalized with anti-CD28 and MHC to select and expand specific T cells. Mesoporous silica microrods can be functionalized with IL-2 and coated with biotinylated liposomes bound to peptide-bound MHC, anti-CD3 and anti-CD28 to design a modular APC-mimetic scaffold, for example, for CAR T cell expansion. Yeast cells can be engineered to display cohesion domains that bind to dockerin-fused proteins for the design of protein scaffolds. The yeast-based scaffolds can be used to control the stoichiometric and spatial organization of T cell ligands. APC-mimicking and T cell-stimulating microparticles can be incorporated into implantable and degradable biomaterials to deliver T cell therapies to the tumour microenvironment. CNT, carbon nanotube; DSPE-PEG, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(amino(polyethylene glycol)); ICAM1, intercellular adhesion molecule 1; LFA1, lymphocyte function-associated antigen 1; pMHC, peptide-MHC.
In the body, T cells become activated once they bind processed antigen presented by an APC, which leads to the formation of an immune synapse. The immune synapse is a complex microscale structure at the T cell-APC interface characterized by a canonical bull’s eye-type pattern (FIG. 4b). This pattern consists of three primary clusters: the central supramolecular activation cluster (cSMAC) with T cell receptor-MHC complexes; the peripheral supramolecular activation cluster (pSMAC) rich in intercellular adhesion molecule 1 (ICAM1)-lymphocyte function-associated antigen 1 (LFA1) clusters; and the distal supramolecular activation cluster (dSMAC) enriched in CD43 and CD45 (REFS84,85). The specific spatiotemporal distribution of these cues, which can vary depending on the type of T cell and APC, leads to T cell activation and expansion through T cell receptor stimulation, co-stimulation and the expression and secretion of pro-survival cytokines. The bull’s eye pattern and its dynamic geometry are key parameters for T cell activation and for the manufacturing of T cells. To better understand immune synapses formed between T cells and APCs, various 2D platforms can be used to model and modulate the topological pattern and density of key molecules (FIG. 4c; TABLE 2). Multicomponent materials-based 3D systems have further been designed to more accurately mimic APCs for T cell activation (FIG. 4d; TABLE 2).
2D materials to mimic the immune synapse interface.
Several mechanisms in the activation of T cells remain unclear thus far, including the role of the physical structure of the immune synapse, which is defined by ligand presence, density and distribution. The sub-micrometre topological scale and slow rate of immune synapse formation preclude in situ 3D imaging to provide the exact structure between T cells and APCs. Moreover, the inherent fluidic nature of membrane lipids limits the controllability of cell-based approaches. Materials-based strategies enable spatiotemporal control of key T cell activating cues, allowing a quantitative investigation of cell-intrinsic modes of interaction.
Immune synapses can be modelled by mimicking APCs with a planar substrate that presents an array of antigenic and co-stimulatory signals, which can be fabricated by lithography (FIG. 4c). For example, silica substrates can be coated with a continuous, fluid lipid bilayer. Glycolipid-linkages can then be used to functionalize the substrate with antigenic MHCs and ICAM1, which is a ligand for the integrin LFA1 (REF.86). Geometric constraints for the generation of different patterned synapses can then be introduced by varying the configurations of chromium lines on the silica substrate using electron-beam lithography. Using 2D APC-mimicking substrates, it could be demonstrated that T cell receptors form an immune synapse by first engaging MHCs and then clustering into microclusters; the microclusters are then directly transported across the peripheral regions of the synapse to form the cSMAC86. Geometric constraints, such as chromium barriers, enable the recreation of specific distances between the T cell receptor cluster and the centre of the synapse to study their effect on synapse formation and disassembly and the consequent changes in T cell receptor signalling and T cell activation. Interestingly, the radial position of T cell receptors impacts the signalling activity of the receptors, showing prolonged signalling by receptor microclusters that are mechanically trapped in the peripheral regions of the synapse86. This effect can possibly be related to cytoskeletal regulation processes that alter the spatial organization of membrane proteins and, thus, downstream signalling and T cell activation. Similarly, the control of geometry and protein density by spatial patterning of 2D surfaces has also proved efficient in elucidating mechanisms of cell adhesion87–91, for example, to identify the ECM area threshold for focal adhesion assembly and force transmission, which is regulated by integrin activation and cytoskeleton tension88,91. Therefore, this approach can also be potentially applied to the immune cells.
Lithography can also be used to present multiple protein ligands in defined physical patterns (FIG. 4c). For example, a substrate with a defined array of tethered anti-CD3 antibodies can mimic T cell receptor activation sites; immobilizing adhesive ICAM1 on the substrate further enables cell attachment and migration92. Such arrays can be engineered using a biotinylated, pH-sensitive photoresist copolymer (methacrylate-r-poly-ethylene glycol methacrylate (PNMP)) cast on cationic aminosilane glass substrates. Upon UV exposure, the photoresist polymer can be dissolved in aqueous buffers, leaving behind a thin biotinylated layer bound to the cationic substrate. Through repeated steps of photo-masking, UV exposure, dissolution and conjugation of proteins on the newly exposed biotin, multiple protein ligands can be presented (FIG. 4c). Such ligand-modified substrates also allow the temporal monitoring of T cell signalling events, which occur within seconds to hours after seeding, at the cell population and single-cell level. Using substrates with distinct patterns and types of ligands, it could be demonstrated that T cell activity is sensitive to spatial patterns and depends on focal clustering of T cell receptor ligands. Therefore, patterning approaches can be applied to elucidate the molecular assembly of T cell synapses and effects of the microscale organization of stimuli.
However, independent control of the geometry of multiple ligands, against a background of IC AMI, is difficult to achieve using lithography. Independent control is particularly important to study the impact of the microscale organization of additional T cell co-stimulatory signals, such as CD28, in relation to cSMAC, on T cell activation. To address this question, separate patterns of anti-CD3 and anti-CD28 antibodies can be printed on a substrate by repeated rounds of contact printing using antibody-coated PDMS stamps moulded from separate topological masters (FIG. 4c). The patterns can then be coated with ICAM1. The antibodies can be printed in a way to mimic the canonical bull’s eye synapse pattern (FIG. 4b) with a central activating cluster of anti-CD3 antibodies. Anti-CD28 can either be colocalized with anti-CD3 or printed as peripheral satellite ligands. Peripheral presentation of anti-CD28 increases T cell activity and cytokine production, which can be related to changes in NF-κB translocation and protein kinase B (PKB) signalling93. However, microcontact printing suffers from a lack of precise control over the quantity of transferred protein, and it requires drying of the protein prior to stamping, which can lead to local heterogeneities of protein density at the microscale88.
In addition to the organization and presence of bio-molecular cues, microscale and nanoscale physical and topological patterns have an effect on the mechanosignalling of cells and, thus, on cell behaviour, morphology, differentiation and activation. Microscale elastomer pillar patterns can be designed to measure the subcellular distribution of contraction forces on the basis of the magnitude and direction of pillar deformation91,94,95 (FIG. 4c). At cell-cell interfaces, such as at immune synapses, the mechanical forces created by cytoskeletal and membrane movements in one cell can induce physical changes in the adjacent cell, thereby modulating intercellular communication. These mechanical forces are also present in a synapse formed between a cytotoxic T cell and an infected target cell. The cytotoxic T cell secretes toxic proteins, such as the membrane pore-forming protein perforin, causing the infected target cell to undergo apoptosis96. Using a micropillar array that presents immobilized antigen and ICAM1, it could be shown that cytotoxic T cells exert forces on their target cell, which are dependent on T cell myosin activity96. Partial knockdown of the myosin heavy chain in T cells results in a consistent, subtle killing defect without compromising the release of toxic proteins or T cell receptor activation and signalling. Interestingly, exertion of mechanical forces potentiates cytotoxicity by causing tension in the infected target cell, which leads to an increase in the pore-forming activity of perforin. Thus, the mechanical properties of materials can potentially be exploited to alter T cell activity and differentiation.
3D artificial antigen-presenting cells for T cell activation and expansion.
T cell expansion rate and function are controlled by interactions with APCs, which stimulate T cell receptors, express co-stimulatory signals for amplification and secrete pro-survival cytokines, such as interleukin-2 (IL-2). T cells can be expanded in vitro using commercial non-degradable microbead systems, such as Dynabeads, which can be functionalized with CD3 and CD28 ligands and used in combination with exogenous cytokines. The beads are separated prior to reinfusion, which presents an additional costly cell production step. T cells produced with beads show a suboptimal expansion rate and functionality owing to the low number of specific activation signals and the differences in organization and presentation between the microbeads and natural APCs. This lack of similarity can be addressed by artificial APCs, which can be engineered using various different materials, including biodegradable materials97–99, carbon-based100,101 and silicon-based structures102 and magnetic beads98,99,103 (FIG. 4d).
APCs facilitate synapse formation through T cell receptor clustering and activation by presenting a high density of antigens. Materials with high surface areas, such as carbon nanotubes, are particularly suited for functionalization with a high density of antigens. For example, in a carbon nanotube-poly(lactide-co-glycolide) (PLGA) nanoparticle composite, defects can be introduced into the nanotubes to enable protein absorption. Neutravidin — a deglycosylated version of avidin — can then be absorbed onto the surface to conjugate biotinylated anti-CD28 and peptide-loaded MHCs to the material. Such biotinylated PLGA nanoparticles can be used to encapsulate IL-2 and magnetite to trigger paracrine cytokine signalling and for the magnetic separation of the composite material (that is, carbon nanotube with PLGA nanoparticle) from the cells101 (FIG. 4d). This composite system can be applied to achieve a higher expansion efficiency of cytotoxic T cells than that of soluble IL-2, which is currently standard in the clinic, and the expanded, purified T cells can be injected to reduce tumour growth in a murine melanoma model101 because CD8+ T cells can kill tumour cells through cell-cell contact83.
However, carbon nanotube-based materials often contain residues of degraded and debundled carbon nanotubes, which are difficult to completely separate from T cells and can cause cytotoxicity at the injection site104,105. To provide better biocompatibility, paramagnetic iron-dextran nanoparticles can be used as artificial nanoscale APCs106 (FIG. 4d). Iron-dextran nanoparticles can be injected in vivo, are biocompatible and accumulate more efficiently in target (tumour) tissues than microparticle-based approaches. The particle diameters range from 50 nm to 100 nm, which is large enough to cluster T cell receptors and, therefore, to robustly activate naive T cells. These particles can further be functionalized with antigen-coupled MHCs to activate antigen-specific T cell receptor signalling pathways and with anti-CD28 antibodies to provide co-stimulation. The functionalized nanoparticles can be used to bind antigen-specific cells from a pool of naive CD8+ T cells, which can then be positively selected by using a magnetic column106 (FIG. 4d). Enrichment of antigen-specific T cells provides a proliferative advantage and, thus, enables expansion with high efficiency Expanded T cells can initiate antitumour responses by responding to shared tumour antigens that result from overexpression or aberrant expression of non-mutated proteins. They can also respond to computationally predicted neoepitopes, highlighting the possibility to customize artificial APCs for patient-specific mutations106. However, iron-dextran nanoparticles do not allow control of the stoichiometric and spatial organization of T cell ligands.
Alternatively, yeast cells can be engineered to display cohesion units, which strongly bind their cognate dockerins. The density, valency and clustering sites of the cohesion units can be tuned to direct the supramolecular assembly of MHCs and ICAM1, both fused to a dockerin domain, which binds to its cognate protein cohesin107 (FIG. 4d). Using this system, the minimum MHC density required for T cell activation could be determined, which is greater than the one assessed with 2D systems owing to differences in sustained interfacial contact. Co-assembly of MHC with adhesive ICAM1 can lead to increased intercellular contact and, thereby, increase T cell sensitivity to antigens, which depends on the stoichiometric ratio between adhesive and stimulatory peptides. This increase in sensitivity can be up to sixfold that of MHC alone and is comparable to 2D substrates. Therefore, the stoichiometric ratio and the spatial organization of peptides are key factors for the design of artificial APCs for T cell activation.
Even more efficient T cell activation can be achieved by mimicking the membrane of natural APCs, in which ligands are mobile and free to cluster108. For example, an APC-mimetic scaffold can be designed with a fluid lipid bilayer, which can present T cell stimulatory and co-stimulatory cues, supported by cytokine-emitting mesoporous silica microrods. IL-2 is first adsorbed on the mesoporous silica, which can then be coated with biotinylated liposomes functionalized with peptide-bound MHC, anti-CD3 and anti-CD28 antibodies by using a streptavidin intermediate102 (FIG. 4d). In culture, the mesoporous rods settle and randomly stack together, forming 3D scaffolds, which can be infiltrated and remodelled by T cells. The large size and surface area of the scaffold promote T cell cluster formation. The size, number and persistence of generated clusters depend on the surface cue density and the amount of total material. However, owing to the use of a fluid membrane, only a relatively low number of surface cues is required to initiate T cell receptor clustering, resulting in the generation of more functional T cells through prevention of T cell overstimulation and exhaustion109, dysfunctional T cells with hierarchical loss of functional properties and expression of multiple inhibitory receptors on T cells. Therefore, these artificial APC scaffolds can be applied to promote increased efficiency of expansion of polyclonal and rare antigen-specific T cells, as compared with the current clinical standard (CD3 and/or CD28 super-paramagnetic microbeads (Dynabeads)). The scaffold is biodegradable, biocompatible and cost effective and thus has the potential to expedite the clinical use of T cell therapeutics and artificial APCs. The modularity of the system further enables spatiotemporal control of biomolecular cues through tuning of the degradation of the mesoporous silica rods and of lipid formulation. Additional surface and soluble cues can potentially be incorporated to further optimize T cell expansion, functionality and phenotype for adoptive cell transfer.
A variety of materials are also being explored for the activation and expansion of T cells in vivo to overcome challenges associated with adoptive cell transfer, such as T cell reinfusion, for the treatment of solid tumours. Current reinfusion methods show poor trafficking to the tumour site, followed by inadequate expansion of T cells in the immunosuppressive tumour microenvironment110. Implantable biopolymers111,112 can be used to increase targeting of engineered T cells and their antitumour activity For example, degradable alginate functionalized with collagen-mimetic peptides can be applied for the co-delivery of therapeutic T cells and T cell-stimulating APC-mimicking lipid-coated porous silica microparticles (FIG. 4d). The microparticles support high-capacity encapsulation and sustained release of biomolecules. The lipid coatings further mimic cell membranes. IL-15 can be encapsulated in the porous silica microparticles, which can be surface-functionalized with CD3, CD28 and CD 137 for T cell stimulation and expansion. The resulting implantable biopolymer reduces tumour relapse in comparison with injections received intravenously or directly into the resection bed. The platform also triggers tumour regression in mouse models112 with advanced-stage unresectable tumours, in which adoptive T cell therapies are not effective. Moreover, the biopolymer can be implemented with other adoptive cell transfer therapies and incorporate supplementary signals, such as checkpoint inhibitors, to modulate the immunosuppressive environment. This technology holds promise as an intraoperative tool and showcases how materials can be used to activate and expand T cells in vivo.
Engineering activated B cells
The B cell activation zone in secondary lymphoid organs provides a niche for B cells to encounter antigens and differentiate into antibody-secreting cells, such as plasma cells and memory B cells (FIG. 1a). Antibodies are Y-shaped molecules that specifically bind antigens with high affinity to flag them for neutralization, blocking, opsonization, antibody-dependent cellular cytotoxicity or complement activation113–115 (FIG. 5a).
Fig. 5 |. Engineering the germinal centre.

a | Antibodies contain a variable region that binds antigens and a constant region that binds immune cells, which leads to specific cell responses: neutralization, receptor blocking, opsonization, antibody-dependent cellular cytotoxicity (ADCC) and complement activation. ADCC can be carried by out several immune effector cells, including natural killer (NK) cells. Complement activation can either mark an antigen for opsonization or lead to complement-mediated cytotoxicity (CMC), which causes cell lysis through the formation of a membrane attack complex (MAC), b | In the germinal centre, antigen-specific plasma cells, memory B cells and antibodies are produced. B cells are activated once an antigen crosslinks a B cell receptor (BCR), which leads to antigen processing and major histocompatibility complex (MHC) presentation. Moreover, the centre becomes compartmentalized into two zones (dark and light zones). After receiving co-stimulatory signals, such as CD40 ligand (CD40L) from T follicular helper (TFH) cells, which binds to the CD40 receptor on the B cell surface, B cells in the dark zone undergo clonal expansion followed by somatic hypermutation of the antigen-binding antibody variable region. In the light zone, B cells presenting high-affinity BCRs for the antigen are selected to survive and undergo class-switch recombination of the antibody constant region to define the downstream immune response. The germinal centre reaction is completed by terminal differentiation of B cells into antigen-specific antibody-secreting cells, such as plasma cells or memory B cells, c | 2D approaches can be applied for immune cell support. Engineered stromal cells (40LB cells) or iron oxide magnetic microbeads presenting both CD40L and antigen can be used to bind naive B cells, d | The lymphoid microenvironment can be modelled using 3D hydrogels to recreate the events during high-affinity antibody generation. Gelatin can be reinforced using 2D nanosilicates to form a hydrogel for the creation of germinal centre organoids ex vivo. Alternatively, a polyethylene glycol (PEG)-maleimide hydrogel can be functionalized with protein or peptide ligands to trigger an antigen-specific immune response. FDC, follicular dendritic cell; TCR, T cell receptor.
Monoclonal antibodies are specific for a single epitope and have become one of the fastest growing therapeutics. By the end of 2017, 57 monoclonal antibodies and 11 biosimilars were in clinical use for various diseases, including cancer, inflammatory diseases and renal pathologies, and exceeded $98 billion in sales116. Monoclonal antibodies are usually produced using hybridomas — hybrid immortalized antibody-secreting cells generated by fusion of myeloma cells and antibody-secreting splenocytes, which are isolated from immunized mice117. The need for immunized mice leads to long production times and high costs and can often result in the production of low-affinity antibodies. Alternatively, in vitro display technologies, for example, phage display, can be applied to iteratively screen large combinatorial libraries of antibody sequences to select specific antigen-binding clones. However, the expression of high-affinity antibodies can negatively affect cell growth, and thus high-affinity cells are progressively depleted and outcompeted by cells that produce lower-affinity antibodies118. Therefore, in vitro platforms for B cell differentiation are being explored for the production of high-affinity therapeutic antibodies.
Germinal centre.
The specificity and effector function of antibodies are a result of affinity maturation, including somatic hypermutation of the antigen-binding variable region and class-switch recombination of the immune cell-binding constant region (FIG. 5a). Affinity maturation occurs in the germinal centre, which is a transient micro-anatomical structure that forms in the B cell zone once activated B cells begin to proliferate. The germinal centre consists of two cellular compartments (the light zone and the dark zone), which can be clearly distinguished using imaging techniques113–115 (FIG. 5b). The prevailing view is that B cells are activated once antigens engage and crosslink a B cell receptor. The B cell processes the antigen and presents it on MHCs to receive costimulatory and survival signals from T follicular helper cells and follicular dendritic cells. B cells then undergo clonal expansion and subsequent somatic hypermutation, during which the immunoglobulin variable genes are rearranged. In the light zone, mutated B cells then compete for antigen; B cells with a high affinity for the antigen receive survival signals, whereas B cells with low-affinity or autoreactive receptors undergo apoptosis119. B cells can also cycle between the dark and the light zone for repeated rounds of somatic hypermutation and selection to increase the affinity for a specific antigen. Selected B cells then interact with T follicular helper cells to undergo class-switch recombination of the immune cell-binding antibody constant region prior to their final differentiation into plasma cells or memory B cells (FIG. 5b). The efficiency of the germinal centre reaction and therefore antibody production is also dependent on the support of specialized cells, including follicular dendritic cells and T follicular helper cells, as well as of the lymphoid microenvironment. Materials offer the opportunity to provide immune cell support and to mimic the lymphoid microenvironment for the investigation of the germinal centre reaction and for the production of antibody-based therapeutics.
Activated B cells are often present in only small numbers, difficult to isolate and prone to apoptosis without immune cell support. In particular, CD40 ligand (CD40L) presented by T follicular helper cells is a crucial factor for germinal centre induction, and blocking of the CD40L-CD40 interaction inhibits germinal centre formation. Follicular dendritic cells also present B cell activating factors, and they contain immune complexes120, which can be explored to mediate in vitro somatic hypermutation. CD40L and various other B cell stimulatory signals require membrane-bound (instead of soluble) ligand presentation to be functional. Therefore, CD40L-presenting stromal cells have been explored for the activation and/or differentiation of B cells in vitro121–123 (TABLE 2). For example, CD40L-transfected L cells in combination with cytokines can be used to induce differentiation of B cells with a human germinal centre phenotype into cells resembling plasma and memory B cells122. Similarly, fibroblasts can be engineered to present CD40L and secrete B cell activating factor (BAFF) to improve B cell proliferation, isotype switching and induction of the germinal centre phenotype122,124 (FIG. 5c).
However, stromal cells are a variable and thus a potential barrier for the reproducible clinical translation of engineered immune cells and immune cell-derived therapeutics. Replacing stromal cells with synthetic materials could offer a possibility to study B cell biology in vitro, to create multiplex screening platforms for immunomodulatory drugs and therapeutics and for the development of antibodies. For example, a synthetic stroma-free germinal centre niche (FIG. 5c) can be designed by functionalizing iron oxide microbeads with CD40L and antigen, which can be used for the production of therapeutic cells and antibodies125. However, most of these approaches rely on the phenotypic evaluation of a limited subset of B cell markers, whereas analysis of transcriptome or somatic hypermutation similarities to in vivo B cells provides a more detailed assessment of cell production124.
Immune organoids mimicking the germinal centre.
Cell-cell interactions have been the main focus of prior in vitro models of germinal centres122; however, germinal centres are also affected by cell-microenvironmental interactions, in particular, cell-ECM interactions. Therefore, to account for the role of the microenvironment and to create germinal centres ex vivo, we applied an engineering approach to mimic the lymphoid microenvironment (FIG. 5d). We first examined protein and gene expression in germinal centre cells, which were isolated from immunized mice, and analysed the expression of various integrin receptors on germinal centre B cells126. We then engineered nanocomposite hydrogels of gelatin, reinforced with polyionic silicate nanoparticles, to encapsulate murine naive B cells and 40LB stromal cells (which present CD40L and secrete BAFF) in the presence of cytokines124,126,127. Incorporation of silicate nanoparticles in the hydrogels allows for the tuning of the stiffness, porosity and architecture of the scaffold to partly resemble those of native lymphoid tissue128 and support the functioning of encapsulated cells. Arg-Gly-Asp (RGD) motifs present in gelatin further mimic integrin-binding sites of native ECM and thus enable the binding of the scaffold to αvβ3 integrin on murine germinal centre B cells. Through imitating the lymphoid microenvironment, these ‘immune organoids achieve an approximately tenfold increase in the production of germinal centre B cells compared with 2D cultures and induced antibody class switching.
The organoid system can also be used to investigate the signalling and epigenetic events crucial to the success of the immune response. For example, the histone methyltransferase enhancer of zeste homologue 2 (EZH2) controls the exuberant pace of germinal centre B cell proliferation through repression of cyclin-dependent kinase inhibitor CDKN1A124 — a signalling pathway that cannot be demonstrated in immunized mouse models because mice lacking EZH2 do not form germinal centres, and B cells in mice with wild-type or depleted CDKN1A have a heterogeneous cell cycle state owing to their migration in the dark and light zones of B cell follicles. In immune organoids, this limitation is overcome by synchronizing the cell cycle state of all germinal centre B cells while recapitulating the transcriptome, apoptotic signature and somatic hypermutation of B cells of immunized mice. Remarkably, the germinal centre organoids exhibit a similar germinal centre-like genetic signature, including epigenetic modifications, to genetically engineered mouse models and enable the study of EZH2 and transcriptional feedback loops regulating the pace of germinal centres. Thus, immune organoids provide a powerful tool to investigate epigenetic modifications of germinal centres. In addition, materials-based organoids can be used to synchronize the proliferative states of germinal centre cells ex vivo, which is not possible in vivo owing to the continuous migration of cells across lymph node zones.
To more precisely investigate receptor-ligand interactions in B cell differentiation, we engineered designer immune organoids using four-arm maleimide-functionalized polyethylene glycol (PEG-MAL) (FIG. 5d). Naive B and 40LB cells can be encapsulated in the PEG-MAL organoids, which are conjugated with thiolated adhesive peptides at defined densities to mimic the ECM in B cell follicles. The peptides were designed to specifically bind to αvβ3 or α4β1 integrins, which are highly or differentially expressed on germinal centre B cells and bind to the ECM proteins vitronectin and VCAM1 on follicular dendritic cells. Compared with αvβ3 integrin-specific organoids, α4β1 integrin-specific organoids result in a greater number of early germinal centre-like B cells in a ligand concentration-dependent manner126. The lymphoid microenvironment mimics can further be used to study malignant cancerous B cells (lymphomas)129,130. For B cell differentiation and in B cell malignancies, changes in cell phenotype are triggered by specific integrins, partly owing to the role of integrins in mechanosignalling128,131–133. Therefore, the use of mechanically dynamic materials has the potential to decipher the role of the mechanical properties of the microenvironment in B cell differentiation and disease. Moreover, mechanomodulation of the B cell receptor could also be implemented in the organoids because the strength of the B cell receptor-antigen bond can be used for the selection of high-affinity antigen-specific B cells.
The question remains whether antigen-specific B cells can be made ex vivo. PEG-MAL germinal centre organoids with encapsulated B cells from immune tissues of a transgenic B1–8hi mutant mouse can be used to produce antigen-specific B cells and antibodies in a dish131. B1–8hi mutant mice have been used because of their restricted antibody repertoire. The mice possess a variable region of a recombined antibody derived from a 4-hydroxy-3-nitrophenylacetyl hapten binding antibody (B1–8), and when exposed to hapten, they produce high-affinity antibodies against 4-hydroxy-3-nitrophenylacetyl hapten. Ex vivo, the high-affinity, hapten-specific response is achieved by further incorporating a nonspecific cell elimination process using FAS ligand, which is a T cell signal. In vivo, FAS ligand binding can induce apoptosis in non-hapten-specific and autoreactive B cells by binding to the death receptor CD95 (REF.134). However, FAS is not essential for B cell elimination or inactivation, and alternative approaches such as the incorporation of follicular dendritic cells and T helper cells in the organoids maybe required to mimic the selection process for complex antigens135. Various PEG-MAL hydrogel parameters, such as hydrogel size, affect the B cell phenotype in terms of light zone induction131. Upon antigen addition, B cell receptor crosslinking causes the expression of plasma cell-like hallmark transcription factors and changes in receptor signalling. Therefore, the combination of materials with specific properties, T cell signals and antigens enables the selective enrichment of antigen-specific, high-affinity B cells and secreted antibodies in an integrin-dependent manner131. Immune organoids are a modular platform that could be applied for the production of antigen-specific cells and antibody-based therapeutics. However, combination of this technology with hydrogels, organ-on-chip platforms with chemokine gradients, follicular T helper cells, stromal cells and lymphatic fluid flow characteristics is needed to promote the affinity maturation and T cell selection required for the generation of antigen-specific B cells and antibodies against complex antigens.
Ectopic tertiary lymphoid structures
Secondary lymphoid organs mediate the immune response to antigenic stimuli; however, non-resolving inflammation leads to the formation of tertiary lymphoid structures, which organize the response to disease-causing antigens. These structures are ectopic aggregates of immune cells that assemble postnatally in the periphery of, within or near the antigen-hosting tissue136. Tertiary lymphoid tissues are structurally similar to secondary lymphoid organs, for example, they may contain segregated B and T cell zones, follicular dendritic cells networks and high endothelial venules, but they are less ordered and can contain different cell types136–138, such as pro-inflammatory macrophages and T helper 17 (TH17) cells136,139. B and T cells also show chronic hyperactivation. As in primary and secondary lymphoid organs, the spatiotemporal regulation of biochemical and biophysical factors within the microenvironment plays a key role in tertiary lymphoid organogenesis136–138.
Tertiary lymphoid structures have been detected in tissues affected by autoimmune disease139,140, cancer141 and infection142 and during allograft and implant rejection143,144 and other inflammatory conditions145; however, it is not yet clear whether they have protective or detrimental effects on disease progression because their often transient nature makes their characterization challenging136. Different disease conditions can vary widely in their capacity to induce tertiary lymphoid structures. Evidence suggests that tertiary lymphoid structures can function as immune inductive sites for protective immunity in infectious diseases and contribute to pathology in autoimmunity and cancers146,147. Owing to the complexity, heterogeneity and undefined nature of these ectopic lymph node-like structures and limited in vivo models2, engineered models of tertiary lymphoid organs are required to not only delineate their immunological functions but also inform the design of immunotherapies.
Ectopic transplantable bone marrow niches can be engineered by fabricating β-tricalcium phosphate (β-TCP) scaffolds with controlled, interconnected porous geometries using slip casting. Collagen type I and III or Matrigel can then be incorporated to recreate the bone marrow ECM148. Co-culture of HSCs, progenitor cells and MSCs on the β-TCP-matrix scaffolds and subsequent subcutaneous transplantation in C57Bl/6 mice support ectopic haematopoiesis and bone deposition, enabling the study of haematopoietic-mesenchymal interactions.
Cell-free artificial tertiary lymphoid structures can be generated to eliminate the use of genetically engineered stromal cells and to produce transplantable tissues that are more clinically translatable than tissues containing virally transduced cells. For example, lymphoid organogenesis-stimulating factors can be loaded into slow-releasing Medgel beads, which can then be trapped in a collagen sponge149. Three weeks after transplantation of the sponge into the renal subcapsular space of pre-immunized BALB/c mice, artificial tertiary lymphoid organs are formed. The lymphoid structures display organized B and T cell zones, a functional vasculature and fibroblastic reticular cell and follicular dendritic cell networks characteristic of native ectopic lymphoid tissue. Importantly, excising and re-transplanting the artificial lymphoid tissue into immune-deficient mice lacking the normal haematopoietic microenvironment lead to an increase in the production of antigen-specific high-affinity antibody-forming cells 1 week after immunization149.
Alternatively, to target the immune response in vivo, a macroporous PLGA matrix can be functionalized with granulocyte-macrophage colony-stimulating factor (GM-CSF), which is a potent stimulator of dendritic cell recruitment, danger signals, such as CpG oligodeox-ynucleotide (CpG-ODN) sequences, which are uniquely expressed in bacterial DNA, and tumour-derived antigens150. Upon subcutaneous implantation of the matrix in mice, dendritic cells are recruited and reprogrammed in situ at the implant site, which triggers an increase in dendritic cell homing to the lymph nodes, resulting in 90% animal survival in mice that would otherwise die from cancer in less than 25 days150. This approach demonstrates that functionalized materials can be used to target immune cells to ectopic sites of immune activation. Similarly, in situ crosslinkable hydrogels carrying chemokines and microparticle vaccines can be used as a depot for the recruitment and priming of immune cells at ectopic sites, such as muscles. Such hydrogel-based approaches for delivering factors that attract immature dendritic cells and that drive T cell responses towards tumour-suppressive activation states in lymphoma151 provide a tool for the development of vaccines against cancer and infectious diseases.
The potential of 4D materials
3D systems can recreate certain structural and functional aspects of immune tissues and have advanced our understanding of immune processes. However, static 3D materials are limited by irreversible immune reactions and intercellular signalling events. By contrast, native lymphoid organs are dynamic and respond to changes in the biochemical, physicochemical and biophysical properties of the microenvironment. Therefore, 4D systems that change with space and/or time and thus allow the tissue to dynamically respond to microenvironmental changes offer more accurate physiological models to precisely mimic and recreate lymphoid structures. An artificial system should address several features of the native tissue: secretion of proteins and signalling molecules in the ECM; migration and localization of cells in the ECM; temporal and spatial control of cellular events; and mechanical and biological changes of microenvironments over time. To address these challenges, dynamic matrix systems need to be engineered that enable spatiotemporal control over cell adhesion, migration, biomolecule distribution and mechanical properties. For example, stimuli-responsive materials allow modulation of the microenvironment and of biological cues. Similarly, cells can be used as material building blocks (FIGS 6,7).
Fig. 6 |. Temperature-responsive, photo-responsive and magnetoresponsive 4D materials.

a | Temperature-responsive polymers with a defined sol-gel transition can be used to create patterned cell sheet layers, which can be temporally activated. 0, a, b and c represent different time points and t represents time, b | Light-responsive materials can be applied to produce spatially and temporally controlled peptide patterns based on the location of switchable crosslinkers and irradiation. Controlling the intensity, exposure time and location of irradiation enables the engineering of a material with defined zones, c | Magnetic beads can be conjugated to cell-specific antibodies to trigger cell migration and isolation upon application of a magnetic field. Magnetic beads can also be functionalized with a major histocompatibility complex (MHC) or anti-CD3 antibody to bind to the T cell receptor (TCR) or CD3 receptor on a single T cell. The distance of these beads can be quantitatively measured by Forster resonance energy transfer (FRET). Upon application of a magnetic field, the fluorescence intensity can be related to the strength of the TCR-MHC bond to identify autoreactive T cells.
Fig. 7 |. Electroresponsive, pH-responsive and cellular 4D materials.

a | Electroresponsive porous materials provide a conductive platform to facilitate electroporation of cells at specified time points for the intake of biomolecules. The conductive matrix can also include a channel for cell input and be attached to an electrode to sense changes in conduction. Upon changes in conduction, drugs or genetic material can be released into electroporated cells, leading to cellular transformation, b | pH-responsive materials can include latent caged peptides, which can be sequentially activated by subtle changes in pH provided by the user or the environment to induce a specific biological effect. pH-responsive materials can further be applied to activate antitumour moieties for cancer cell apoptosis. c | Cells can be genetically engineered by introducing circuits that can sense particular inputs and result in corresponding outputs. To mimic the sequential processes of immune cell formation, activation and response, a target immune cell can be engineered to respond to a number of activating signals and recruitment signals from interacting cells. The interacting cells then respond to the environment or user-based cues. The target cell is modified at each step, leading to new functionalities.
Stimuli-responsive materials
Cell-cell and cell-environment interactions regulate the differentiation, adhesion, migration and activation of B and T cells. Similarly, interactions between chemokines, which are expressed on the surfaces of osteoblasts152 and perivascular endothelial cells153 in the bone marrow, and their cognate receptors on the surfaces of HSCs are crucial for recruitment, retention and maintenance of HSCs.
Temperature-responsive materials.
Dynamic interactions in heterogeneous HSC niches can be modelled using temperature-responsive systems, such as poly(N-isopropylacrylamide) (PNIPAM), which has been applied for the regeneration of various tissues, including the cornea, oesophageal epithelia, myocardium and liver154. Owing to intrinsic changes in surface hydrophilicity and hydrophobicity, resulting from the energy of conformational transformation, PNIPAM undergoes sol-gel transitions at varying temperatures. Thus, this material exhibits compact chain conformations at physiological temperature and loose confirmations at room temperature. By grafting PNIPAM on cell culture surfaces with set geometries, the sol-gel transition of this material can be exploited to detach cell monolayers of defined topographies while preserving cell membranes and cell-cell adhesions to generate cell sheets for tissue-engineered organs154 (FIG. 6a).
The same approach can be used to pattern different bone marrow HSC niches on multiple PDMS substrates (FIG. 6a). In contrast to conventional or encapsulation-based co-cultures, interactions between the niches and cell populations can then be temporally controlled through reversibly adding or removing cell monolayers by changing the temperature as well as by the use of multiple substrates. Similarly, B and T cell layers could be fabricated for investigating synapse formation.
Photo-responsive materials.
Stimuli-responsive systems can also be used to generate germinal centres in a stepwise manner to investigate B cell activation. The cycling of B cells through the dark and light zones of the germinal centre is driven by ON-OFF cyclic interactions with the microenvironment and other cells. The spatiotemporal presentation of these signals can be controlled in vitro using photo-responsive systems, which enable irreversible uncaging (FIG. 6b). Photo-assisted dynamic systems provide cell culture platforms for the spatiotemporal control over cell-surface interactions155. Moreover, multiple biochemical cues can be incorporated within the same network by applying photo-coupling reactions155–157 and by using ligand-switchable crosslinkers. The extent of patterning is then controlled by regulating light intensity and exposure time. Therefore, photo-responsive systems can be tailored to uncage different signals at user-controlled times to present antigens, modulate B cell cycling and control selection within the germinal centre.
Magnetoresponsive materials.
Magnetoresponsive materials can be used to shed light on T cell formation, differentiation and activation. For example, magnetic particles can be incorporated into alginate microbeads, making the beads responsive to magnetic fields. This property can be explored to control cell migration in T cell development by magnetotaxis resembling chemo-taxis of cells158. The hydrogel-based magnetic microbeads can be loaded with cytokines (for example, IL-2), which act as stimulators of T cell proliferation, therefore enabling proliferation, activation and cell migration using one system.
Magnetic beads could also be coated with antibodies targeting CD3 or with MHCs and self-antigen. Additional functionalization with a Forster resonance energy transfer (FRET) pair then allows the assessment of T cell separation (FIG. 6c). Application or withdrawal of a magnetic field can then be used to control separation of T cell-bound beads and serve as a checkpoint for the auto-reactivity of manufactured T cells based on the relationship between fluorescence, bond strength and the applied magnetic field (FIG. 6c).
Electroresponsive materials.
Specific T cell populations can be manufactured in a controlled fashion using gene editing technologies, such as CRISPR159,160 and microRNA161. The CRISPR-Cas9 technology can be applied to activate transcription factors associated with specific T cell subsets. For example, the T-box transcription factor TBX21 and the transcription factor GATA3 can be delivered to drive TH1 cell and TH2 cell responses, respectively. CRISPR-based manipulation of T cells could also prioritize new targets for therapeutic discoveries; however, large-scale CRISPR screens in primary human immune cells remain challenging159. Nanomaterials coupled with electroresponsive platforms, such as carbon nanotubes embedded in gelatin-based hydrogels162 or alginate hydrogels loaded with conductive gold nanowires163, can be used for electroporation and temporal control of RNA delivery161 (FIG. 7a). For example, an electric field applied to a microfluidic device channel can create a potential difference within the hydrogel and induce electroporation of cells that deliver genetic material161.
pH-responsive materials.
pH-responsive systems can be applied to increase the yield of immune cell manufacturing, for example, T cell manufacturing. Rapid proliferation of immune cells can be achieved by changing the pH of the culture medium164; for example, ex vivo, T cells proliferate more at pH 7.0–7.2 than at pH 7.4. pH also regulates the kinetics of IL-2 receptor expression on T cells165. T cells proliferate rapidly in vitro, and thus changes in the pH of the cell culture medium can be used to reduce T cell proliferation and functionality. pH-sensitive systems can respond to changes in pH with the release of growth factors, nutrients and buffers, which causes more T cell proliferation, creating a positive feedback loop to increase the yield of engineered immune cells for cell therapy. pH-sensitive systems can also be explored for the treatment of malignant immune cells and tumours. A decrease in pH is among the most important characteristics of the microenvironment of growing tumours, and it affects ECM remodelling in vivo. Thus, subtle pH changes in growing tumours can be used as a trigger for pH-sensitive systems, for example, to release pH-sensitive caged peptides (FIG. 7b). Changes in the pH cause inert protective groups to be cleaved, activating effector ligands that can bind to cancer cells and induce apoptosis.
Multi-responsive materials.
Coupling multiple stimuli-responsive strategies allows the control of both migration and compartmentalization of cells. For example, stimuli-responsive porogens of gelatin, alginate and hyaluronic acid can be used to design dynamic hydrogels with tuneable macroporosity166. The hydrogels can be combined in a scaffold, in which macropore formation can be controlled by temperature, chelating or enzymatic digestion. Similarly, structures resembling secondary lymphoid tissues can be designed in vitro and implanted subcutaneously to create artificial tertiary lymphoid organs.
Engineered scaffolds with multi-responsive controllable features can serve as powerful in vitro models to examine cell responses to the dynamic milieu often found in healthy and pathological tissues. Smart materials in combination with integrated fluidics can be applied to capture the dynamic signalling events of immune tissues and cells at the molecular level and to precisely mimic the micro structure of lymphoid organs. Therefore, we developed a lymphoid-on-chip that mimics the lymphatics-like fluid flow characteristics of the subcapsular sinus of lymph nodes. This technology enables the investigation of B cell receptor crosstalk with integrins in malignant B cells133. The B cell receptor expression pattern on malignant B cells cultured on the chip is comparable to that of tumour xenografts of immunocompromised mice, highlighting the power of dynamic, immune-engineered ex vivo systems.
Cells as materials
Synthetic biology can be applied to engineer cells with levels of functionality higher than the ones observed in nature. The implementation of gene circuits in cells enables precise control of the initiation, interruption or termination of gene expression at the DNA, RNA and protein levels. The circuits can be multiplexed and spatiotempo-rally controlled by several stimuli, including light and small molecules. Synthetic cells can be made even more complex by implementing elements that endow the circuits with memory, enabling cells to remember previous inputs. Such gene circuits can also be created in immune cells, for example, to create CAR T cells167–169 (FIG. 7c).
By applying synthetic biology, cells can be designed to be their own time-dependent and context-dependent responsive material, which can serve as a building block for the generation of complex biological constructs. Many immune processes are sequential and depend on interactions with other cells. Therefore, synthetic biology could be used for the multiscale engineering of immune cells to coordinate their formation, activation and response. For example, a target cell could be engineered to respond to recruitment and action signals from other cells. The cells would secrete a recruitment signal, for example, a chemokine, at a certain time to initiate interaction with the target cell and then supply an action signal to trigger changes in the same cell, for example, proliferation, differentiation or protein secretion. Interacting cells can further be designed to respond to cues in the environment to enable autonomous control or to exogenous small molecules to allow user-based control (FIG. 7c). The sequential additive changes in the target cell resemble assembly line manufacturing approaches used to create multifaceted products. The combination of 4D materials with genetically engineered cells has the potential to implement user-control in immune tissue engineering to study immune cell formation, activation and response.
Conclusion
The complex 3D microenvironment of lymphoid organs plays a fundamental role in regulating immunity. Various 2D and 3D models have provided important insight into lymphocyte development and activation. However, the highly organized, adaptable nature of these tissues can be fully recreated through only the use of dynamic systems. Artificial 4D microenvironments offer a powerful tool for the development of better clinical models with precise and modular control to address unmet clinical needs. In combination with scalable technologies, such as 3D printing or computational modelling, 4D systems can serve as models for the investigation and development of immunotherapies for autoimmunity, infection, cancer and other chronic inflammatory diseases. However, many challenges remain to be overcome. In particular, the implementation of reversibility will be required to enable dynamic exchange between materials and cells, mimicking in vivo tissues and processes. By introducing futuristic material design principles and mass production technologies, ex vivo generation of immune cells and tissues will become a key player in the development of immunotherapies for incurable diseases.
Acknowledgements
The authors acknowledge financial support from the National Institute of Allergy and Infectious Diseases of the US National Institutes of Health (1R01AI132738-01A1 awarded to A.S.), the Innovative Molecular Analysis Technology programme of the US National Cancer Institute (NIH R33-CA212968–01 awarded to A.S.), a US National Science Foundation CAREER award (DMR-1554275 awarded to A.S.), a US Department of Defense Congressionally Directed Medical Research Program (CDMRP) cancer career development award (W81XWH-17-1-0215 awarded to A.S.) and the 3M Non-Tenured Faculty Award (awarded to A.S.).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Joshi NS et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress antitumor T cell responses. Immunity 43, 579–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W et al. Bronchus-associated lymphoid tissue-resident Foxp3+ T lymphocytes prevent antibody-mediated lung rejection. J. Clin. Invest 129, 556–568 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Hernandez ML et al. A unique cellular and molecular microenvironment is present in tertiary lymphoid organs of patients with spontaneous prostate cancer regression. Front. Immunol 8, 563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eddens T et al. Pneumocystis-driven inducible bronchus-associated lymphoid tissue formation requires Th2 and Th17 immunity. Cell Rep 18, 3078–3090(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS et al. Chimeric antigen receptor T cell therapy — assessment and management of toxicities. Nat. Rev. Clin. Oncol 15, 47–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this Review, the authors discuss cellular immunotherapy and provide recommendations for monitoring, grading and managing acute toxicities that can occur in patients.
- 6.Weiner GJ Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15, 361–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan AC & Carter PJ Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol 10, 301–316 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Burak MF et al. Development of a therapeutic monoclonal antibody that targets secreted fatty acid-binding protein aP2 to treat type 2 diabetes. Sci. Transl Med 7, 319ra205 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Harrison C Autoimmune disease: targeting IL-7 reverses type 1 diabetes. Nat. Rev. Drug Discov 11, 599 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Penaranda C et al. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc. Natl Acad. Sci. USA 109, 12668–12673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W et al. A neutralizing anti-Nogo66 receptor monoclonal antibody reverses inhibition of neurite outgrowth by central nervous system myelin. J. Biol. Chem 279, 43780–43788 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Mothe AJ et al. RGMa inhibition with human monoclonal antibodies promotes regeneration, plasticity and repair, and attenuates neuropathic pain after spinal cord injury. Sci. Rep 7, 10529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran HT et al. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep 7, 2054–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephenson R & Singh A Drug discovery and therapeutic delivery for the treatment of B and T cell tumors. Adv. DrugDeliv. Rev 114, 285–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokatlian T et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 363, 649–654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson DS et al. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater 18, 175–185(2019). [DOI] [PubMed] [Google Scholar]
- 17.Mosquera MJ et al. Immunomodulatory nanogels overcome restricted immunity in a murine model of gut microbiome-mediated metabolic syndrome. Sci. Adv 5,eaav9788(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crane GM, Jeffery E & Morrison SJ Adult haematopoietic stem cell niches. Nat. Rev. Immunol 17, 573–590 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Gordon J & Manley NR Mechanisms of thymus organogenesis and morphogenesis. Development 138, 3865–3878(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison SJ & Scadden DT The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the current understanding and controversies regarding the cellular composition and localization of haematopoietic stem cell niches within the bone marrow.
- 21.Nagasawa T Microenvironmental niches in the bone marrow required for B cell development. Nat. Rev. Immunol 6, 107–116 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Wilson A &Trumpp A Bone-marrow haematopoietic-stem-cell niches. Nat. Rev. Immunol 6, 93–106 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Chabannon C et al. Hematopoietic stem cell transplantation in its 60s: a platform for cellular therapies. Sci. Transl Med 10, eaap9630 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Cornelissen JJ et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat. Rev. Clin. Oncol 9, 579–590 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Atkins HL et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet 388, 576–585 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kawai T et al. Long-term results in recipients of combined HLA-mismatched kidney and bone marrow transplantation without maintenance immunosuppression. Am. J. Transplant 14, 1599–1611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogonek J et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front. Immunol 7, 507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bello AB, Park H & Lee SH Current approaches in biomaterial-based hematopoietic stem cell niches. Acta Biomater 72, 1–15 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Chinn IK, Blackburn CC, Manley NR & Sempowski GD Changes in primary lymphoid organs with aging. Semin. Immunol 24, 309–320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itkin T et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunisaki Y et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braccini A et al. Three-dimensional perfusion culture of human bone marrow cells and generation of osteoinductive grafts. Stem Cells 23, 1066–1072 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Ferreira MS et al. Cord blood-hematopoietic stem cell expansion in 3D fibrin scaffolds with stromal support. Biomaterials 33, 6987–6997 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Leisten I et al. 3D co-culture of hematopoietic stem and progenitor cells and mesenchymal stem cells in collagen scaffolds as a model of the hematopoietic niche. Biomaterials 33, 1736–1747 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Miyoshi H, Murao M, Ohshima N &Tun, T. Three-dimensional culture of mouse bone marrow cells within a porous polymer scaffold: effects of oxygen concentration and stromal layer on expansion of haematopoietic progenitor cells. J. Tissue Eng. Regen. Med 5, 112–118 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Mortera-Blanco T, Mantalaris A, Bismarck A, Aqel N & Panoskaltsis N Long-term cytokine-free expansion of cord blood mononuclear cells in three-dimensional scaffolds. Biomaterials 32, 9263–9270 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Nichols JE et al. In vitro analog of human bone marrow from 3D scaffolds with biomimetic inverted colloidal crystal geometry. Biomaterials 30, 1071–1079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raic A, Rodling L, Kalbacher H & Lee-Thedieck C Biomimetic macroporous PEG hydrogels as 3D scaffolds for the multiplication of human hematopoietic stem and progenitor cells. Biomaterials 35, 929–940 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Feng Q, Chai C, Jiang XS, Leong KW & Mao HQ Expansion of engrafting human hematopoietic stem/progenitor cells in three-dimensional scaffolds with surface-immobilized fibronectin. J. Biomed. Mater. Res. A 78, 781–791 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilsson SK et al. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J. Histochem. Cytochem 46, 371–377 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Choi JS & Harley BA Marrow-inspired matrix cues rapidly affect early fate decisions of hematopoietic stem and progenitor cells. Sci. Adv 3, e1600455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotov NA et al. Inverted colloidal crystals as three-dimensional cell scaffolds. Langmuir 20, 7887–7892 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Huang X et al. Co-cultured hBMSCs and HUVECs on human bio-derived bone scaffolds provide support for the long-term ex vivo culture of HSC/HPCs. J. Biomed. Mater. Res. A 104, 1221–1230 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Ronaldson-Bouchard K & Vunjak-Novakovic G Organs-on-a-Chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22, 310–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahadik BP, Wheeler TD, Skertich LJ, Kenis PJ & Harley BA Microfluidic generation of gradient hydrogels to modulate hematopoietic stem cell culture environment. Adv. Healthc. Mater 3, 449–458 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Torisawa YS et al. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 11, 663–669 (2014). [DOI] [PubMed] [Google Scholar]; In this study, the authors report an engineered bone-marrow-on-chip that recapitulates organ-level toxicity responses and protective effects of radiation countermeasure drugs.
- 47.Rodling L et al. 3D models of the hematopoietic stem cell niche under steady-state and active conditions. Sci. Rep 7, 4625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourgine PE et al. In vitro biomimetic engineering of a human hematopoietic niche with functional properties. Proc. Natl Acad. Sci. USA 115, E5688–E5695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Buduo CA et al. Modular flow chamber for engineering bone marrow architecture and function. Biomaterials 146, 60–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Buduo CA et al. Programmable 3D silk bone marrow niche for platelet generation ex vivo and modeling of megakaryopoiesis pathologies. Blood 125, 2254–2264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shepherd JH et al. Structurally graduated collagen scaffolds applied to the ex vivo generation of platelets from human pluripotent stem cell-derived megakaryocytes: enhancing production and purity. Biomaterials 182, 135–144 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Tozzi L et al. Multi-channel silk sponge mimicking bone marrow vascular niche for platelet production. Biomaterials 178, 122–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braham MVJ et al. Endosteal and perivascular subniches in a 3D bone marrow model for multiple myeloma. Tissue Eng. Part C Methods 24, 300–312 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Braham MVJ et al. Cellular immunotherapy on primary multiple myeloma expanded in a 3D bone marrow niche model. Oncoimmunology 7, e1434465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reagan MR et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood 124, 3250–3259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shih YR et al. In vivo engineering of bone tissues with hematopoietic functions and mixed chimerism. Proc. Natl Acad. Sci. USA 114, 5419–5424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shah NJ et al. An injectable bone marrow-like scaffold enhances T cell immunity after hematopoietic stem cell transplantation. Nat. Biotechnol 10.1038/s41587-019-0017-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan Y et al. Bioengineering thymus organoids to restore thymic function and induce donor-specific immune tolerance to allografts. Mol. Ther 23, 1262–1277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tajima A, Pradhan I, Trucco M & Fan Y Restoration of thymus function with bioengineered thymus organoids. Curr. Stem Cell Rep 2, 128–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer DB The effect of age on thymic function. Front. Immunol 4, 316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neelapu SS et al. Kte-C 19 (anti-CD 19 CAR T Cells) induces complete remissions in patients with refractory diffuse large B-cell lymphoma (DLBCL): results from the pivotal phase 2 zuma-1. Blood 128, LBA-6 (2016). [Google Scholar]
- 62.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischbach MA, Bluestone JA & Lim WA Cell-based therapeutics: the next pillar of medicine. Sci. Transl Med 5, 179ps177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hare KJ, Jenkinson EJ & Anderson G In vitro models of T cell development. Semin. Immunol 11, 3–12 (1999). [DOI] [PubMed] [Google Scholar]
- 66.Dallas MH, Varnum-Finney B, Delaney C, Kato K & Bernstein ID Density of the Notch ligand Delta 1 determines generation of B and T cell precursors from hematopoietic stem cells. J. Exp. Med 201, 1361–1366 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dallas MH, Varnum-Finney B, Martin PJ & Bernstein ID Enhanced T cell reconstitution by hematopoietic progenitors expanded ex vivo using the Notch ligand Delta 1. Blood 109, 3579–3587 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varnum-Finney B et al. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J. Cell Sci 23, 4313–4318 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Schmitt TM et al. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat. Immunol 5, 410–417 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Schmitt TM & Zuniga-Pflucker JC Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17, 749–756 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Taqvi S, Dixit L & Roy K Biomaterial-based notch signaling for the differentiation of hematopoietic stem cells into T cells. J. Biomed. Mater. Res. A 79, 689–697 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Shukla S et al. Progenitor T cell differentiation from hematopoietic stem cells using Delta-like-4 and VCAM-1. Nature Methods 14, 531–538 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Mohtashami M & Zuniga-Pflucker JC Three-dimensional architecture of the thymus is required to maintain delta-like expression necessary for inducing T cell development. J. Immunol 176, 730–734 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Seet CS et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 14, 521–530 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the authors report on artificial thymic organoids for directing human HSCs along the T cell lineage.
- 75.Nitta T, Ohigashi I, Nakagawa Y & Takahama Y Cytokine crosstalk for thymic medulla formation. Curr. Opin. Immunol 23, 190–197 (2011). [DOI] [PubMed] [Google Scholar]
- 76.van Ewijk W et al. Thymic microenvironments, 3D versus 2D? Semin. Immunol 11, 57–64 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Hun M et al. Native thymic extracellular matrix improves in vivo thymic organoid T cell output, and drives in vitro thymic epithelial cell differentiation. Biomaterials 118, 1–15 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Hussey GS, Dziki JL & Badylak SF Extracellular matrix-based materials for regenerative medicine. Nat. Rev. Mater 3, 159–173 (2018). [Google Scholar]
- 79.Shah SB & Singh A Cellular self-assembly and biomaterials-based organoid models of development and diseases. Acta Biomater 53, 29–45 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Tajima A et al. Bioengineering mini functional thymic units with EAK16-II/EAKIIH6 self-assembling hydrogel. Clin. Immunol 160, 82–89 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Junt T, Scandella E & Ludewig B Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat. Rev. Immunol 8, 764–775 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Zappasodi R et al. The effect of artificial antigen-presenting cells with preclustered anti-CD28/-CD3/-LFA-1 monoclonal antibodies on the induction of ex vivo expansion of functional human antitumor T cells. Haematologica 93, 1523–1534 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Li Y & Kurlander RJ Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J. Transl Med 8, 104 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dustin ML The immunological synapse. Cancer Immunol. Res 2, 1023–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huppa JB & Davis MM T cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol 3, 973–983 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Mossman KD, Campi G, Groves JT & Dustin ML Altered TCR signaling from geometrically repatterned immunological synapses. Science 310, 1191–1193 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Thery M et al. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol 7, 947–953 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Coyer SR et al. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J. Cell Sci 125, 5110–5123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coyer SR, Delamarche E & Garcia AJ Protein tethering into multiscale geometries by covalent subtractive printing. Adv. Mater 23, 1550–1553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh A et al. Adhesion strength-based, label-free isolation of human pluripotent stem cells. Nat. Methods 10, 438–444 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dumbauld DW et al. How vinculin regulates force transmission. Proc. Natl Acad. Sci. USA 110, 9788–9793 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Doh J & Irvine DJ Immunological synapse arrays: patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proc. Natl Acad. Sci. USA 103, 5700–5705 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the authors report on lithographically defined patterns of T cell receptor ligands surrounded by a field of tethered ICAM1 to mimic T cell-APC interactions.
- 93.Shen K, Thomas VK, Dustin ML & Kam LC Micropatterning of costimulatory ligands enhances CD4+ T cell function. Proc. Natl Acad. Sci. USA 105, 7791–7796 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tan JL et al. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl Acad. Sci. USA 100, 1484–1489 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fu J et al. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat. Methods 7, 733–736 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Basu R et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 165, 100–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Steenblock ER & Fahmy TM A comprehensive platform for ex vivo T cell expansion based on biodegradable polymeric artificial antigen-presenting cells. Mol. Ther 16, 765–772 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Prakken B et al. Artificial antigen-presenting cells as a tool to exploit the immune ‘synapse’. Nat. Med 6, 1406 (2000). [DOI] [PubMed] [Google Scholar]
- 99.Zitvogel L et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med 4, 594–600 (1998). [DOI] [PubMed] [Google Scholar]
- 100.Fadel TR et al. Enhanced cellular activation with single walled carbon nanotube bundles presenting antibody stimuli. Nano Lett 8, 2070–2076 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Fadel TR et al. A carbon nanotube-polymer composite for T cell therapy. Nat. Nanotechnol 9, 639–647 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Cheung AS, Zhang DKY, Koshy ST & Mooney DJ Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol 36, 160–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oelke M et al. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat. Med 9, 619 (2003). [DOI] [PubMed] [Google Scholar]
- 104.Cui HF, Vashist SK, Al-Rubeaan K, Luong JH & Sheu FS Interfacing carbon nanotubes with living mammalian cells and cytotoxicity issues. Chem. Res. Toxicol 23, 1131–1147 (2010). [DOI] [PubMed] [Google Scholar]
- 105.Sato Y et al. Influence of length on cytotoxicity of multi-walled carbon nanotubes against human acute monocytic leukemia cell line THP-1 in vitro and subcutaneous tissue of rats in vivo. Mol. Biosyst 1, 176–182 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Perica K et al. Enrichment and expansion with nanoscale artificial antigen presenting cells for adoptive immunotherapy. ACS Nano 9, 6861–6871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smith MR, Tolbert SV & Wen F Protein-scaffold directed nanoscale assembly of T cell ligands: artificial antigen presentation with defined valency, density, and ratio. ACS Synth. Biol 7, 1629–1639 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Koffeman E, Keogh E, Klein M, Prakken B & Albani S Identification and manipulation of antigen specific T cells with artificial antigen presenting cells. Methods Mol. Med 136, 69–86 (2007). [DOI] [PubMed] [Google Scholar]
- 109.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lamers CH et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther 21, 904–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Smith TT et al. Biopoiymers codelivcellering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest 127, 2176–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stephan SB et al. Biopolymer implants enhance the efficacy of adoptive T cell therapy. Nat. Biotechnol 33, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Allen CD, Okada T & Cyster JG Germinal-center organization and cellular dynamics. Immunity 27, 190–202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Silva NS & Klein U Dynamics of B cells in germinal centres. Nat. Rev. Immunol 15, 137–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McHeyzer-Williams M, Okitsu S, Wang N & McHeyzer-Williams L Molecular programming of B cell memory. Nat. Rev. Immunol 12, 24–34 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this Review, the authors discuss each phase of antigen-specific B cell development and affinity maturation of memory B cells.
- 116.Grilo AL & Mantalaris A The increasingly human and profitable monoclonal antibody market. Trends Biotechnol 37, 9–16 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Yokoyama WM et al. Production of monoclonal antibodies. Curr. Protoc. Immunol 102, 2.5.1–2.5.29 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Georgiou G et al. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol 32, 158–168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rathmell JC et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature 376, 181–184 (1995). [DOI] [PubMed] [Google Scholar]
- 120.Wu Y et al. Immune complex-bearing follicular dendritic cells deliver a late antigenic signal that promotes somatic hypermutation. J. Immunol 180, 281–290 (2008). [DOI] [PubMed] [Google Scholar]
- 121.Pound JD & Gordon J Maintenance of human germinal center B cells in vitro. Blood 89, 919–928 (1997). [PubMed] [Google Scholar]
- 122.Arpin C et al. Generation of memory B cells and plasma cells in vitro. Science 268, 720–722 (1995). [DOI] [PubMed] [Google Scholar]
- 123.Nojima T et al. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat. Commun 2, 465 (2011). [DOI] [PubMed] [Google Scholar]
- 124.Beguelin W et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat. Commun 8, 877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors develop immune organoids to reveal the epigenetic mechanism behind B cell hyperproliferation in response to infection.
- 125.Roh KH et al. A synthetic stroma-free germinal center niche for efficient generation of humoral immunity ex vivo. Biomaterials 164, 106–120 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Purwada A & Singh A Immuno-engineered organoids for regulating the kinetics of B cell development and antibody production. Nat. Protoc 12, 168–182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Purwada A et al. Ex vivo engineered immune organoids for controlled germinal center reactions. Biomaterials 63, 24–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Apoorva FNU et al. Lymph node stiffness-mimicking hydrogels regulate human B cell lymphoma growth and cell surface receptor expression in a molecular subtype-specific manner. J. Biomed. Mater. Res. A 105, 1833–1844 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Cayrol F et al. Integrin αvβ3 acting as membrane receptor for thyroid hormones mediates angiogenesis in malignant T cells. Blood 125, 841–851 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tian YF et al. Integrin-specific hydrogels as adaptable tumor organoids for malignant B and T cells. Biomaterials 73, 110–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Purwada A et al. Ex vivo synthetic immune tissues with T cell signals for differentiating antigen-specific, high affinity germinal center B cells. Biomaterials 198, 27–36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, the authors demonstrate that ex vivo 3D immune organoids can form antigen-specific B cells in a dish.
- 132.Purwada A, Shah SB, Beguelin W, Melnick AM & Singh A Modular immune organoids with integrin ligand specificity differentially regulate ex vivo B cell activation. ACS Biomater. Sci. Eng 3, 214–225 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Apoorva F et al. How biophysical forces regulate human B cell lymphomas. Cell Rep 23, 499–511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hoch S, Boyd M, Malone B, Gonye G & Schwaber J Fas-mediated apoptosis eliminates B cells that acquire self-reactivity during the germinal center response to NP. Cell. Immunol 203, 103–110 (2000). [DOI] [PubMed] [Google Scholar]
- 135.Meyer-Hermann M et al. A theory of germinal center B cell selection, division, and exit. Cell Rep 2, 162–174 (2012). [DOI] [PubMed] [Google Scholar]
- 136.Jones GW, Hill DG & Jones SA Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front. Immunol 7, 401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mueller CG, Nayar S, Gardner D & Barone F Cellular and vascular components of tertiary lymphoid structures. Methods Mol. Biol 1845, 17–30 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Nerviani A & Pitzalis C Role of chemokines in ectopic lymphoid structures formation in autoimmunity and cancer. J. Leukoc. Biol 104, 333–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rangel-Moreno J et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol 12, 639–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dorraji SE et al. Mesenchymal stem cells and T cells in the formation of tertiary lymphoid structures in lupus nephritis. Sci. Rep 8, 7861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Figenschau SL et al. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci. Rep 8, 11720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vu Van D et al. Local T/B cooperation in inflamed tissues is supported by T follicular helper-like cells. Nat. Commun 7, 10875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cheng J et al. Ectopic B cell clusters that infiltrate transplanted human kidneys are clonal. Proc. Natl Acad. Sci. USA 108, 5560–5565 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mittal S et al. Lymphoid aggregates that resemble tertiary lymphoid organs define a specific pathological subset in metal-on-metal hip replacements. PLOS ONE 8, e63470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Akhavanpoor M et al. Adventitial tertiary lymphoid organ classification in human atherosclerosis. Cardiovasc. Pathol 32, 8–14 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Neyt K, Perros F, GeurtsvanKessei CH, Hammad H & Lambrecht BN Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol 33, 297–305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yin C, Mohanta S, Maffia P & Habenicht AJ Editorial: tertiary lymphoid organs (TLOs): powerhouses of disease immunity. Front. Immunol 8, 228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ventura Ferreira MS et al. An engineered multicomponent bone marrow niche for the recapitulation of hematopoiesis at ectopic transplantation sites. J. Hematol. Oncol 9, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kobayashi Y & Watanabe T Gel-trapped lymphorganogenic chemokines trigger artificial tertiary lymphoid organs and mount adaptive immune responses in vivo. Front. Immunol 7, 316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ali OA, Huebsch N, Cao L, Dranoff G & Mooney DJ Infection-mimicking materials to program dendritic cells in situ. Nat. Mater 8, 151–158(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Singh A et al. An injectable synthetic immune-priming center mediates efficient T cell class switching and T-helper 1 response against B cell lymphoma. J. Control. Release 155, 184–192 (2011). [DOI] [PubMed] [Google Scholar]
- 152.Calvi LM et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003). [DOI] [PubMed] [Google Scholar]
- 153.Ding L & Morrison SJ Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Matsuda N, Shimizu T, Yamato M & Okano T Tissue engineering based on cell sheet technology. Adv. Mater 19, 3089–3099 (2007). [Google Scholar]
- 155.DeForest CA & Anseth KS Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem 3, 925–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.DeForest CA, Polizzotti BD & Anseth KS Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater 8, 659–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors demonstrate direct fabrication of biologically functionalized gels with photopatterned structures in the presence of cells — a first step towards 4D cultures.
- 157.Kloxin AM et al. Responsive culture platform to examine the influence of microenvironmental geometry on cell function in 3D. Integ. Biol 4, 1540–1549 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hasani-Sadrabadi MM et al. Mechanobiological mimicry of helper T lymphocytes to evaluate cell-biomaterials crosstalk. Adv. Mater 30, 1706780 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Shifrut E et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dudda JC et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 38, 742–753 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shin SR et al. Carbon nanotube reinforced hybrid microgels as scaffold materials for cell encapsulation. ACS Nano 6, 362–372 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dvir T et al. Nanowired three-dimensional cardiac patches. Nat. Nanotechnol 6, 720–725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lardner A The effects of extracellular pH on immune function. J. Leukoc. Biol 69, 522–530 (2001). [PubMed] [Google Scholar]
- 165.Carswell KS & Papoutsakis ET Extracellular pH affects the proliferation of cultured human T cells and their expression of the interleukin-2 receptor. J. Immunother 23, 669–674 (2000). [DOI] [PubMed] [Google Scholar]
- 166.Patel A et al. Highly elastomeric poly(glycerol sebacate)-co-poly(ethylene glycol) amphiphilic block copolymers. Biomaterials 34, 3970–3983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Roybal KT & Lim WA Synthetic immunology: hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol 35, 229–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wu CY, Rupp LJ, Roybal KT & Lim WA Synthetic biology approaches to engineer T cells. Curr. Opin. Immunol 35, 123–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xie M & Fussenegger M Designing cell function: assembly of synthetic gene circuits for cell biology applications. Nat. Rev. Mol. Cell Biol 19, 507–525 (2018). [DOI] [PubMed] [Google Scholar]
- 170.La Motte-Mohs RN, Herer E & Zuniga-Pflucker JC Induction of T cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 105, 1431–1439 (2005). [DOI] [PubMed] [Google Scholar]
- 171.Grakoui A et al. The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221 (1999). [DOI] [PubMed] [Google Scholar]; This work demonstrates that immunological synapse formation is a multistage process, in which T cell activation is reconstituted by interacting with MHC-peptide and the adhesion ligand ICAM1.
- 172.Doh J & Irvine DJ Photogenerated polyelectrolyte bilayers from an aqueous-processible photoresist for multicomponent protein patterning. J. Am. Chem. Soc 126, 9170–9171(2004). [DOI] [PubMed] [Google Scholar]
- 173.Bashour KT et al. CD28 and CD3 have complementary roles in T cell traction forces. Proc. Natl Acad. Sci. USA 111, 2241–2246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Moon JJ et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl Acad. Sci. USA 109, 1080–1085 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]