

Summary
DNA double-strand break (DSB) repair is critical for cell survival and genome integrity. Upon recognition of DSBs, repair proteins are transiently upregulated to facilitate repair through homologous recombination (HR) or non-homologous end joining (NHEJ). We present evidence that PRMT5 cooperates with pICln to function as a master epigenetic activator of DNA damage response (DDR) genes involved in HR, NHEJ, and G2 arrest (including RAD51, BRCA1, and BRCA2) to upregulate gene expression upon DNA damage. Contrary to the predominant role of PRMT5 as an epigenetic repressor, our results demonstrate that PRMT5 and pICln can activate gene expression, potentially independent of PRMT5's obligate cofactor MEP50. Targeting PRMT5 or pICln hinders repair of DSBs in multiple cancer cell lines, and both PRMT5 and pICln expression positively correlates with DDR genes across 32 clinical cancer datasets. Thus, targeting PRMT5 or pICln may be explored in combination with radiation or chemotherapy for cancer treatment.
Subject Areas: Molecular Genetics, Molecular Biology, Molecular Mechanism of Gene Regulation
Graphical Abstract
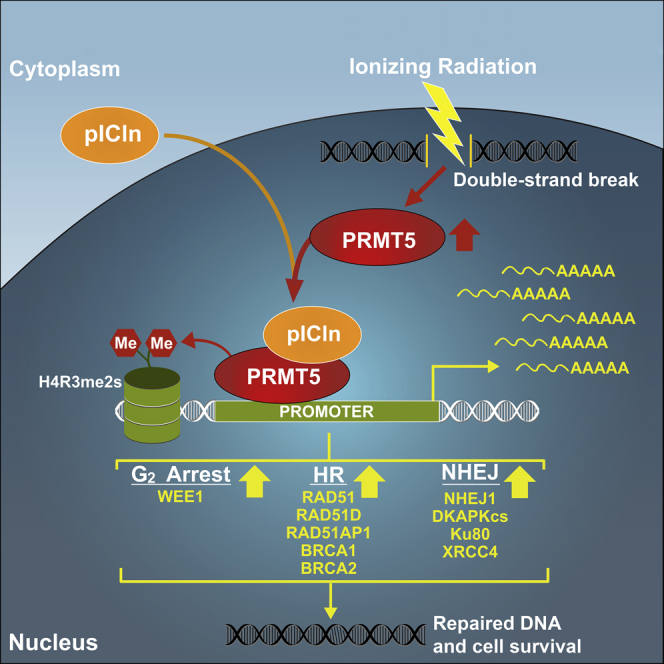
Highlights
-
•
PRMT5 activates transcription of DSB repair genes upon DNA damage
-
•
pICln cooperates with PRMT5 to activate transcription of DSB repair genes
-
•
Targeting PRMT5 is effective to sensitize multiple cancer types to radiation
-
•
PRMT5 expression positively correlates with DSB repair genes in cancer tissues
Molecular Genetics; Molecular Biology; Molecular Mechanism of Gene Regulation
Introduction
Repair of DNA double-strand breaks (DSBs), the most lethal DNA damage, is critical for cell survival and maintenance of genome integrity (Khanna and Jackson, 2001). DSBs can be induced both endogenously as well as exogenously through DNA damaging agents or ionizing radiation (IR). Upon recognition of extensive DSBs, repair proteins are upregulated (Khalil et al., 2012, Rieger, 2004, Russell et al., 2003) and recruited to the sites of damage to facilitate repair through either homologous recombination (HR) or non-homologous end joining (NHEJ) (Thompson, 2012). Although the highly regulated recruitment and action of repair proteins are well characterized, little is known about how their expression is induced upon DNA damage.
Protein arginine methyltransferase 5 (PRMT5) is an emerging epigenetic enzyme that regulates cellular processes including cell proliferation, differentiation, and cell cycle progression (Karkhanis et al., 2011, Stopa et al., 2015). PRMT5 regulates these cellular processes through changes in gene expression via symmetrical dimethylation of arginine residues in histones H4R3 (H4R3me2s), H3R2 (H3R2me2s), H3R8 (H3R8me2s), and H2AR3 (H2AR3me2s) and post-translational regulation of non-histone substrates (Stopa et al., 2015). PRMT5 activity is modulated by several interacting proteins including MEP50, which is believed to be the obligate cofactor of PRMT5 and required for PRMT5 methyltransferase activity (Burgos et al., 2015, Chen et al., 2017, Karkhanis et al., 2011, Stopa et al., 2015). Accumulating evidence suggests that PRMT5 may act as an oncogene to promote cancer cell growth (Karkhanis et al., 2011, Stopa et al., 2015). Consistent with this, PRMT5 is overexpressed in several cancers and its elevated expression correlates with disease progression and poor prognosis (Karkhanis et al., 2011, Stopa et al., 2015, Yang and Bedford, 2013). Thus, PRMT5 has been proposed as a potential therapeutic target for cancer treatment (Richters, 2017).
We have recently reported that PRMT5 is overexpressed in ∼60% of intermediate- and high-risk prostate cancer cases and that PRMT5 expression in prostate cancer tissues positively correlates with androgen receptor (AR) expression (Deng et al., 2017). Mechanistically, PRMT5 is recruited to the AR promoter to activate AR transcription through H4R3me2s in prostate cancer cells (Deng et al., 2017). Because AR drives prostate cancer development and progression, targeting AR signaling through androgen deprivation therapy (ADT) is a standard of care to treat metastatic prostate cancer (Heidenreich et al., 2014). ADT is also used as a radiosensitization approach to enhance radiation therapy (RT) for localized prostate cancer patients (Golabek et al., 2016). Our finding that PRMT5 activates AR transcription raised the possibility that PRMT5 may be a therapeutic target for prostate cancer radiosensitization.
Contrary to our expectation, we observed that targeting PRMT5 sensitized prostate cancer cells to IR independently of AR expression. Here, we present evidence that PRMT5 cooperates with pICln, independently of its canonical cofactor MEP50, to function as a master epigenetic activator of DNA damage response (DDR) genes in various cell types. Upon DNA damage, PRMT5 and pICln upregulate target genes that encode proteins involved in HR (RAD51, RAD51D, RAD51AP1, BRCA1, and BRCA2), NHEJ (NHEJ1/XLF and DNAPKcs), and G2 arrest (WEE1). Targeting PRMT5 or pICln decreases expression of these DDR genes and hinders repair of DSBs in multiple cancer cell lines suggesting that PRMT5 may play a conserved role in DDR. Thus, targeting PRMT5 or pICln may be explored as a monotherapy or in combination with radiation or chemotherapy for cancer treatment. Significantly, both PRMT5 and pICln expression positively correlates with the expression of these target genes across most of the 32 clinical cancer datasets analyzed. Although PRMT5, along with its obligate cofactor MEP50, primarily functions as an epigenetic repressor, our results demonstrate that PRMT5 together with pICln can activate gene expression and provide a potential mechanism for the transient upregulation of repair proteins upon DNA damage.
Results
Targeting PRMT5 Sensitizes Prostate Cancer Cells to IR in an AR-Independent Manner
ADT is the only approved clinical radiosensitization approach for prostate cancer treatment (Golabek et al., 2016). Because we recently identified PRMT5 as a novel epigenetic activator of AR (Deng et al., 2017), we tested whether targeting PRMT5 can mimic ADT to sensitize prostate cancer cells to IR. To this end, we established lentivirally infected stable pools with doxycycline (Dox)-inducible PRMT5 knockdown and observed that knockdown of PRMT5 sensitized AR-expressing LNCaP prostate cancer cells (LNCaP-shPRMT5 pool) to IR when compared with scramble control cell lines (LNCaP-shSC) (Figure 1A). Likewise, inhibition of PRMT5 by our inhibitor BLL3.3 (Alinari et al., 2015, Deng et al., 2017) also sensitized LNCaP cells to IR (Figure 1B). Consistent with previous findings that AR regulates several target genes involved in NHEJ (Goodwin et al., 2013, Polkinghorn et al., 2013, Tarish et al., 2015), pharmacological inhibition of PRMT5 with BLL3.3 in irradiated LNCaP cells indeed caused a decrease in AR expression and a concomitant decrease in the expression of Ku80/XRCC5, XRCC4, and DNAPKcs/PRKDC at the mRNA level (Figure 1C). Contrary to our expectation, knockdown of PRMT5 also sensitized AR-negative prostate cancer cell lines PC3 and DU145 to IR when similar Dox-inducible knockdown stable cell lines (PC3-shPRMT5 pool and DU145-shPRMT5 pool) were used (Figures 1D and 1E). However, BLL3.3 treatment had little to no effect on the expression of AR-target genes involved in NHEJ in irradiated AR-negative DU145 cells (Figure 1F). Given these results, we isolated single-cell-derived clones to develop Dox-inducible knockdown stable cell lines (LNCaP-shPRMT5 and LNCaP-shPRMT5 #2) for all subsequent studies. Dox-induced PRMT5 knockdown is shown in Figures S3B–S3F and is reported previously (Deng et al., 2017). These results suggest that the radiosensitization effect of PRMT5 targeting in prostate cancer cells is likely mediated through both AR-dependent and -independent mechanisms.
Figure 1.

Targeting PRMT5 Sensitizes Prostate Cancer Cells to IR in an AR-Independent Manner
(A, B, D, and E) Quantification of the surviving fraction via clonogenic assay immediately following the indicated dose of IR in the indicated cell lines. Dox treatment was used to express PRMT5-targeting shRNA (shPRMT5) or scramble control-targeting shRNA (shSC). BLL3.3 treatment was used to inhibit PRMT5 activity (A: LNCaP-shPRMT5/shSC, B: LNCaP + DMSO/BLL3.3, D: PC3-shPRMT5/shSC, E: DU145-shPRMT5/shSC).
(C and F) Quantification of mRNA via RT-qPCR 24 h post 2 Gy IR in LNCaP (C) and DU145 (F) cells. For each biological replicate, the value for BLL3.3 was normalized to the value for DMSO to calculate the fold change in mRNA expression upon PRMT5 inhibition.
Points in A, B, D, and E are the mean ± s.d. of three independent experiments. Bars in C and F are the mean ± s.d. of three independent experiments. Statistical analysis for A, B, D, and E comparing experimental with the control (“shSC” or “DMSO”) was performed using Welch's t test of log-transformed data, whereas statistical analysis for C and F comparing experimental with the control (“DMSO”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05, U = undetected).
PRMT5 Regulates the Repair of DNA Double-Strand Breaks in Prostate Cancer Cells Independently of AR Expression
Next, we determined if the radiosensitization effect of PRMT5 targeting was due to defects in the repair of IR-induced DSBs. We first treated LNCaP cells with IR and quantified DSBs via γH2AX foci analysis to assess the formation and repair of IR-induced DSBs. The majority of DSBs were repaired within 2–6 h following IR treatment (Figures 2A and 2B). To assess if PRMT5 is required for efficient repair of IR-induced DSBs, we analyzed γH2AX foci 6 h following IR in more detail. Cells with PRMT5 knockdown retained significantly more DSBs 6 h following IR treatment than cells without knockdown, indicating a defect in DSB repair (Figures 2C and 2D). Nearly identical results were obtained using a different PRMT5-targeting shRNA (LNCaP-shPRMT5 #2) (Figures 2E and 2F). Treatment of LNCaP cells with BLL3.3 conferred the same effect as PRMT5 knockdown (Figures 2G and 2H), whereas Dox-induced expression of scramble control (SC) shRNA in LNCaP-shSC cells had no effect (Figures 2I and 2J). Cells with PRMT5 knockdown retained significantly more γH2AX foci even 24 h following IR treatment than cells without knockdown (Figures 2K and 2L), indicating a prolonged defect in DSB repair. The defects in DSB repair upon PRMT5 knockdown were unlikely an artifact of crosstalk between histone posttranslational modifications, as we observed similar results when quantifying DNA damage directly via comet assay (Figures S1A–S1D). Furthermore, knockdown of PRMT5 also hinders repair of etoposide-induced DSBs (Figures S2A and S2B), which differ in their mechanism of DSB generation and are replication dependent (Furuta et al., 2003, Montecucco and Biamonti, 2007, Treszezamsky et al., 2007), suggesting that PRMT5 may be required for repair of DSBs independently of how they are formed. Thus, the radiosensitization effect of PRMT5 targeting in prostate cancer cells is likely due to defects in the repair of IR-induced DSBs.
Figure 2.

PRMT5 Regulates the Repair of DNA Double-Strand Breaks in Prostate Cancer Cells Independently of AR Expression
(A) Time course of the formation and repair of DSBs (γH2AX foci) at the indicated minutes (m) or hours (h) post 2 Gy IR in LNCaP cells.
(B) Quantification of DSBs in each individual cell from A: “average” indicates the average number of DSBs in each cell and “0 foci” indicates the percentage of cells that do not contain any DSBs.
(C, E, G, I, and K) DSBs 6 h or 24 h post 2 Gy IR in the indicated cells (C: LNCaP-shPRMT5, E: LNCaP-shPRMT5 #2, G: LNCaP, I: LNCaP-shSC, K: LNCaP-shPRMT5) with (Dox+) and without (Dox−) PRMT5 knockdown/scramble control (SC) knockdown or with (BLL3.3) and without (DMSO) PRMT5 inhibition.
(D, F, H, J, and L) Quantification of DSBs from C, E, G, I, and K as described above in Figure 2B (C: LNCaP-shPRMT5, E: LNCaP-shPRMT5 #2, G: LNCaP, I: LNCaP-shSC, K: LNCaP-shPRMT5).
(M) LNCaP-shPRMT5 cells were co-transfected with plasmids encoding Flag-AR (AR) or empty vector (EV) and a plasmid encoding cerulean fluorescent protein (CFP). Fluorescence images acquired 6 h post 2 Gy IR are representative immunocytochemistry images in 3D where each peak is a cell and the height of each peak is the intensity of signal. Blue peaks represent transfected CFP-expressing cells (CFP+). Colors are indicated as follows: endogenous PRMT5 (red), endogenous and exogenous AR (green), and exogenous CFP (cerulean).
(N) Quantification of protein intensity from M in untransfected (CFP−) and transfected (CFP+) cells. For each biological replicate, values were normalized to the value for “Dox−/EV” to calculate the fold change in protein expression upon treatment.
(O) DSBs at 6 h post 2 Gy IR in LNCaP-shPRMT5 cells where AR expression was rescued via co-transfection with plasmids encoding Flag-AR (AR) or empty vector (EV) and a plasmid encoding Cerulean fluorescent protein (CFP) as a transfection control.
(P) Quantification of DSBs in transfected cells (defined as CFP+) from O as described above.
Fluorescence images in A, C, E, G, I, and K are representative immunocytochemistry images (blue = DAPI, green = γH2AX, red = PRMT5). Fluorescence images in M are representative immunocytochemistry images (red = PRMT5, green = AR, and blue = CFP). Fluorescence images in O are representative immunocytochemistry images (blue = DAPI, green = γH2AX, red = AR, Cerulean = CFP). All bars are the mean ± s.d. of four independent experiments. Statistical analysis for B, N, and P comparing experimental with the control (“IR−”, “Dox−”, “DMSO”, or “Dox−/EV”) was performed using Brown-Forsythe and Welch ANOVA followed by Dunnett's T3 multiple comparisons test, whereas statistical analysis for D, F, H, J, and L comparing experimental with the control (“Dox−” or “DMSO”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05).
To further confirm that PRMT5 also regulates the repair of IR-induced DSBs independently of AR, we performed rescue experiments. Although exogenously expressed AR in Dox-treated LNCaP-shPRMT5 cells fully rescued AR protein levels (Figures 2M and 2N), the repair of IR-induced DSBs was only partially rescued (Figures 2O and 2P). Thus, PRMT5 can also regulate repair of IR-induced DSBs through an AR-independent mechanism.
PRMT5 Regulates NHEJ, HR, and G2 Arrest in Response to IR
The ability to repair DSBs is mainly dependent on NHEJ and HR as well as G1 and G2 cell-cycle arrest. We next analyzed IR-induced Ku70 and RAD51 foci formation to examine if PRMT5 knockdown would affect NHEJ or HR repair, respectively. Consistent with the finding that targeting PRMT5 causes a decrease in AR-target genes involved in NHEJ, PRMT5 knockdown decreased IR-induced Ku70 foci formation (Figures 3A and 3B), indicating that PRMT5 regulates NHEJ. Interestingly, PRMT5 knockdown also decreased IR-induced RAD51 foci formation (Figures 3C and 3D), confirming that PRMT5 regulates HR repair of IR-induced DSBs as well.
Figure 3.

PRMT5 Regulates NHEJ, HR, and G2 Arrest in Response to IR
(A) NHEJ repair foci (Ku70) 1 h post 2 Gy IR in LNCaP-shPRMT5 cells with (Dox+) and without (Dox−) PRMT5 knockdown.
(B) Quantification of Ku70 foci from A as described in Figure 2B.
(C) HR repair foci (RAD51) 1 h post 2 Gy IR in LNCaP-shPRMT5 cells with (Dox+) and without (Dox−) PRMT5 knockdown.
(D) Quantification of RAD51 foci from C as described in Figure 2B.
(E) Cell-cycle analysis via flow cytometry of propidium iodide (PI) stained LNCaP-shPRMT5 cells with (Dox+) and without (Dox−) PRMT5 knockdown.
(F) Cell cycle analysis via flow cytometry of PI-stained LNCaP-shPRMT5 cells 24 h post 2 Gy IR, with (Dox+) and without (Dox−) PRMT5 knockdown.
(G) RNA-seq analysis 1 h post 2 Gy IR in irradiated (IR+) and non-irradiated (IR−) LNCaP-shPRMT5 cells with (Dox+) and without (Dox−) PRMT5 knockdown. Volcano plot shows statistical significance (false discovery rate, FDR-corrected p values) vs fold change (FC, in logarithm scale with base 2) between PRMT5 knockdown and WT in IR+ and IR− cells, respectively. Upregulated DEGs (red or green) and downregulated DEGs (blue or yellow) are indicated in color.
(H) Venn diagram indicating the overlap of DEGs between IR+ (red or blue) and IR− (green or yellow) samples.
(I) Gene ontology (GO) analysis of IR+ only DEGs that were downregulated upon PRMT5 knockdown. Groups of GO terms related to DNA repair and cell-cycle regulation were identified to be significantly enriched in the DEG set. The height of each bar represents the enrichment score for the GO term, whereas the q-value (FDR-corrected p value) in red indicates the significance of enrichment. The number in the bar indicates the number of DEGs associated with the corresponding GO annotation.
(J) Differentially regulated pathways of IR+ only DEGs that were downregulated upon PRMT5 knockdown identified by IPA. The pathways with the highest –log (p value), represented by the bars, are shown. Pathways shown in blue (negative Z score) are inhibited upon PRMT5 knockdown, whereas pathways in orange (positive Z score) are activated upon PRMT5 knockdown.
Fluorescence images in A and C are representative immunocytochemistry images (blue = DAPI, green = Ku70, red = RAD51). Bars in B and D are the mean ± s.d. of three independent experiments, whereas bars in E and F are the mean ± s.d. of four independent experiments. Graphs in E and F are representative flow traces of cells in various cell-cycle stages (green = G1, orange = S, blue = G2). Statistical analysis comparing experimental with the control (“Dox−”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05).
We next investigated whether PRMT5 knockdown has any effect on cell cycle. Consistent with previous findings (Lim et al., 2014, Scoumanne et al., 2009, Wei et al., 2012, Yang et al., 2016), cells with PRMT5 knockdown had an increase in the G1 population and a concomitant decrease in the S population in the absence of IR (Figure 3E) indicative of G1 arrest. Upon IR treatment, cells with PRMT5 knockdown retained the ability to undergo IR-induced G1 arrest (or already arrested in G1 phase due to PRMT5 knockdown prior to IR) yet failed to arrest at G2 (Figure 3F). This result suggests that PRMT5 regulates IR-induced G2 arrest but may not be required for IR-induced G1 arrest. Overall, our findings that PRMT5 can regulate repair of IR-induced DSBs through an AR-independent mechanism and that targeting PRMT5 sensitizes prostate cancer cells to IR independently of AR expression are likely due to the regulation of multiple DDR pathways by PRMT5.
Because PRMT5 is an emerging epigenetic regulator (Karkhanis et al., 2011, Stopa et al., 2015), we reasoned that PRMT5 may regulate the expression of genes involved in the repair of DSBs. We performed RNA-seq analysis of both non-irradiated (IR-) and irradiated (IR+) LNCaP-shPRMT5 cells with PRMT5 knockdown (Dox+) and without PRMT5 knockdown (Dox-). We identified 2,036 differentially expressed genes (DEGs) upon PRMT5 knockdown in IR−cells and 1,710 DEGs in IR+ cells (Figure 3G and Tables S1 and S2). Comparing the IR− and IR+ datasets, we determined that 886 genes were differentially regulated only in IR+ cells, of which 563 were downregulated (Figure 3H and Table S3). Consistent with our functional studies, multiple genes encoding repair proteins in HR and NHEJ and genes involved in G2 arrest were identified as DEGs. Using Gene Ontology (GO) analysis, we identified several GO functions and KEGG pathways associated with DDR such as “DNA damage repair” and “cell-cycle regulation” that were significantly over-represented in IR+ only DEGs (Figure 3I and Table S4). Ingenuity pathway analysis (IPA) of the IR+ only DEGs conferred similar outcomes as GO analysis and further revealed that PRMT5 likely regulates genes involved in G2 arrest as well as repair proteins such as BRCA1 and BRCA2 (Figure 3J and Table S5). Results from our RNA-seq analysis suggest PRMT5 regulates expression of DDR genes in response to IR.
PRMT5 Activates Transcription of Genes that Encode Proteins Involved in the Repair of DSBs
Next, we sought to validate a potential role for PRMT5 in regulating the transcription of genes required for DSB repair. Notably, we identified six DEGs that encode repair proteins (RAD51, RAD51D, RAD51AP1, BRCA1, BRCA2, and NHEJ1/XLF), and the regulation of these genes by PRMT5 in both irradiated and non-irradiated cells was verified by reverse transcriptase quantitative real-time PCR (RT-qPCR) (Figures 4A and S3A) and Western blot (Figures S3B–S3F) (additional genes were also individually verified at the mRNA level in Figure S3G). IR induces the expression of these genes at both the mRNA (Figure 4B) and protein level (Figures S3B–S3F) on a timescale consistent with the repair of IR-induced DSBs, suggesting that PRMT5-mediated upregulation of these genes upon IR is critical for DSB repair. The regulation of positive control genes by PRMT5 was also confirmed: knockdown of PRMT5 prevented repression of IVL expression (Saha et al., 2016) and prevented activation of AR expression (Deng et al., 2017) (Figures 4A and S3G). As expected, IR did not affect expression of IVL or AR at the mRNA level (data not shown).
Figure 4.

PRMT5 Activates Transcription of Genes that Encode Proteins Involved in the Repair of DSBs
(A and F) Quantification of mRNA via RT-qPCR 24 h post 2 Gy IR in LNCaP-shPRMT5 cells with (Dox+) and without (Dox−) PRMT5 knockdown. For each biological replicate, the value for Dox+ was normalized to the value for Dox− to calculate the fold change in mRNA expression upon PRMT5 knockdown in both irradiated (IR+) and non-irradiated cells (IR−) (See also Figure S3G).
(B) Quantification of mRNA via RT-qPCR at the indicated time points post IR in irradiated (IR+) and non-irradiated (IR−) LNCaP-shPRMT5 cells without PRMT5 KD. For each biological replicate, the value for IR+ was normalized to the value for IR− to calculate the fold change in mRNA expression upon IR treatment (see also Figure S3A for experiments with PRMT5 KD).
(C–E and G–I) Quantification of enrichment (C and G: PRMT5, D and H: H4R3me2s, and E and I: H3K9ac) at the promoter region of the indicated genes 1 h post 2 Gy IR via ChIP-qPCR in irradiated (IR+) and non-irradiated (IR−) LNCaP-shSC cells via ChIP-qPCR. For each biological replicate, the value for IP was normalized to the value for IgG to calculate the fold enrichment (See also Figure S4).
All bars are the mean ± s.d. of three independent experiments. Statistical analysis for A, B, and F comparing experimental with the control (“Dox−” or “IR−”) was performed using Brown-Forsythe and Welch ANOVA followed by Dunnett's T3 multiple comparisons test, whereas statistical analysis for C–E and G–I comparing experimental with the control (“IgG”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05).
Chromatin immunoprecipitation (ChIP)-qPCR assays using LNCaP-shSC and LNCaP-shPRMT5 cell lines confirmed that PRMT5 indeed bound to the proximal promoter regions of these six genes that encode for repair proteins (Figures 4C and S4A). IR treatment further increased the binding of PRMT5 at a time point prior to the upregulation of these genes (Figures 4C and S4A). Consistent with potential epigenetic activation of these genes by PRMT5, the promoter regions of these genes were selectively enriched with PRMT5-catalyzed H4R3me2s (but not H3R2me2s, H3R8me2s, or H2AR3me2s) as well as the activating histone modification H3K9ac, both of which were induced by IR (Figures 4D, 4E, and S4B–S4F). Consistent with our previous finding (Deng et al., 2017), PRMT5 binding and the enrichment of H4R3me2s and H3K9ac at the proximal promoter region, but not a distal region, of the AR gene were also confirmed (Figures 4C–4E and S4A–S4F). To confirm the specificity of our ChIP experiments, we repeated experiments after knocking down PRMT5. PRMT5 knockdown decreased both PRMT5 binding and enrichment of H4R3me2s and H3K9ac at the proximal promoter regions, further supporting the specificity of our ChIP assays and suggesting that PRMT5 contributes to transcriptional activation of these genes via methylation of H4R3 (Figures S4A–S4F). Collectively, these data demonstrate that PRMT5 is required to maintain basal expression of DDR genes, and PRMT5 facilitates the IR-induced transient upregulation of DDR genes by activating their transcription.
Given that PRMT5 was required for IR-induced G2 arrest, we also sought to validate putative PRMT5 target genes involved in the regulation of G2/M transition (DEGs: CCNB2, CDC20, CDC25C, CDK1, and WEE1). RT-qPCR analysis demonstrated that PRMT5 knockdown decreased their expression in both non-irradiated and irradiated cells (Figures 4F and S3G). Interestingly, ChIP-qPCR assay results suggest that out of the five putative target genes, WEE1 may be the only direct target gene of PRMT5 (Figures 4G–4I and S4A–S4F). Since CCNB2, CDC20, CDC25C, and CDK1 are typically activators of G2 progression and WEE1 is an activator of G2 arrest, it is possible that PRMT5 actively regulated WEE1 expression while the changes in CCNB2, CDC20, CDC25C, and CDK1 expression were a secondary effect. Therefore, although positive and negative regulators of G2 arrest were downregulated at the mRNA level upon PRMT5 knockdown, the net phenotypic effect is impaired IR-induced G2 arrest.
pICln Is Also Required for Transcriptional Activation of DDR Genes and for Efficient Repair of DSBs
As MEP50 is believed to be the obligate cofactor of PRMT5 and required for PRMT5 methyltransferase activity (Stopa et al., 2015, Wilczek et al., 2011, Chen et al., 2017, Burgos et al., 2015), we determined if MEP50 plays a role in regulating the expression of DDR genes. Using Dox-inducible MEP50 knockdown stable cell lines isolated from single-cell-derived clones (LNCaP-shMEP50), we unexpectedly observed that knockdown of MEP50 did not affect the expression of PRMT5 target genes involved in DDR at the mRNA (Figure 5A) or protein (Figures S5A and S5B) level in untreated or irradiated LNCaP-shMEP50 cells. However, consistent with the previous finding that PRMT5 and MEP50 represses IVL expression (Saha et al., 2016), knockdown of MEP50 caused an increase in IVL expression (Figure 5A). Furthermore, MEP50 knockdown did not affect the repair of IR-induced DSBs (Figures 5B and 5C). These results suggest that PRMT5 may not rely on MEP50 to regulate transcription of genes involved in DDR.
Figure 5.

pICln is Also Required for Transcriptional Activation of DDR Genes and for Efficient Repair of DSBs
(A) Quantification of mRNA via RT-qPCR 2 h post 2 Gy IR in LNCaP-shMEP50 cells with (Dox+) and without (Dox−) MEP50 knockdown. For each biological replicate, values were normalized to the value for “‘Dox−, IR−” (untreated) to calculate the fold change in mRNA expression upon treatment.
(B) DSBs 6 h post 2 Gy IR in LNCaP-shMEP50 cells with (Dox+) and without (Dox−) MEP50 knockdown.
(C) Quantification of DSBs in each individual cell from B as described in Figure 2B.
(D) Quantification of mRNA via RT-qPCR 2 h post 2 Gy IR in LNCaP-shpICln cells with (Dox+) and without (Dox−) pICln knockdown. For each biological replicate, values were normalized to the value for “Dox−, IR−” (untreated) to calculate the fold change in mRNA expression upon treatment.
(E) DSBs 6 h post 2 Gy IR in LNCaP-shpICln cells with (Dox+) and without (Dox−) pICln knockdown.
(F) Quantification of DSBs in each individual cell from E as described in Figure 2B.
(G and H) Quantification of enrichment (G: MEP50 and H: pICln) at the promoter region of the indicated genes 1 h post 2 Gy IR via ChIP-qPCR in irradiated (IR+) and non-irradiated (IR−) LNCaP-shSC cells via ChIP-qPCR. For each biological replicate, the value for IP was normalized to the value for IgG to calculate the fold enrichment (see also Figures S5E and S5F).
Fluorescence images in B and E are representative immunocytochemistry images (blue = DAPI, green = γH2AX, red = MEP50 or pICln). All bars are the mean ± s.d. of three independent experiments. Statistical analysis for A and D comparing experimental with the control (“Dox−, IR−”) was performed using Brown-Forsythe and Welch ANOVA followed by Dunnett's T3 multiple comparisons test, whereas statistical analysis for C, F, G, and H comparing experimental with the control (“Dox−” or “IgG”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05).
We previously performed mass spectrometry analysis of PRMT5-immunoprecipitated lysate to identify interacting proteins of PRMT5 in LNCaP cells (Zhang et al., 2016). We predictably identified MEP50 (peptide fragment ILLWDTR), but we also identified pICln (Chari et al., 2008, Friesen et al., 2001, Friesen et al., 2002, Guderian et al., 2011, Meister et al., 2001) (peptide fragment GLGTGTLYIAESR) as an interacting protein of PRMT5. We then developed Dox-inducible pICln knockdown stable cell lines isolated from single-cell-derived clones (LNCaP-shpICln) and surprisingly observed that knockdown of pICln caused a decrease in PRMT5 target gene expression at the mRNA (Figure 5D) and protein (Figures S5C and S5D) level in untreated and irradiated LNCaP-shpICln cells. Furthermore, knockdown of pICln impaired repair of IR-induced DSBs (Figures 5E and 5F) to an extent comparable to PRMT5 knockdown. Although MEP50 was present at the promoter of the control gene IVL, MEP50 was not present at the promoter of PRMT5 target genes involved in DDR while pICln was present (Figures 5G, 5H, S5E, and S5F). Upon IR, binding of pICln to the promoter of DDR genes increased (Figure 5H) and knockdown of PRMT5 almost completely abrogated enrichment of pICln (Figure S5F), suggesting that PRMT5 recruits pICln to targeted promoter regions. Additionally, other previously identified PRMT5 interacting proteins, RioK1 (Guderian et al., 2011) and COPR5 (Lacroix et al., 2008, Paul et al., 2012, Paul et al., 2015), were not found at the promoter of DDR genes in untreated or irradiated LNCaP cells (Figures S5G and S5H). These results suggest that PRMT5 may cooperate with pICln to regulate transcription of DDR genes.
IR Induces PRMT5 Expression, pICln Nuclear Localization, and the PRMT5:pICln Interaction in the Nucleus
Given the importance of PRMT5 in DSB repair and activation of target gene expression upon IR, we hypothesized that IR may induce PRMT5 expression. To test this, we analyzed PRMT5 protein expression via both immunocytochemistry and Western blotting at various time points following IR of LNCaP cells. Indeed, IR induced PRMT5 expression as quickly as 5 m, and the induction lasted for nearly 24 h (Figures 6A–6D). Importantly, the peak of PRMT5 protein expression (between 5 m and 1 h) coincided with the increased recruitment of PRMT5 to the promoters of DDR genes (1 h). Similarly, changes in PRMT5 expression closely mirrored the time course of DSB repair. The rapid induction of protein expression likely suggests a post-transcriptional or post-translational regulation. However, RT-qPCR analysis confirmed a small, but significant, sustained induction of PRMT5 expression at the mRNA level (Figure 6E). This result suggests that transcriptional activation of PRMT5 also contributes to prolonged elevation of PRMT5 expression. Furthermore, PRMT5 was upregulated at the protein level by etoposide treatment (Figures S2C–S2F), suggesting that DNA damage, in general, can signal the induction of PRMT5. Collectively, these results indicate that PRMT5 is upregulated upon IR to promote repair of IR-induced DSBs, and that the upregulation of DDR genes is likely facilitated by IR-induced upregulation of PRMT5.
Figure 6.

IR Induces PRMT5 Expression, pICln Nuclear Localization, and the PRMT5:pICln Interaction in the Nucleus
(A) Time course of PRMT5 expression at the indicated minutes (m) or hours (h) post 2 Gy IR in LNCaP cells.
(B) Quantification of PRMT5 expression in images from A. For each biological replicate, values were normalized to the value for “IR−” to calculate the fold change in protein expression upon IR.
(C) Representative Western blot showing the time course of protein expression at the indicated minutes (m) or hours (h) post 2 Gy IR in LNCaP cells. Values shown indicate the intensity relative to IR− for the biological replicate used as the representative Western blot.
(D) Quantification of protein expression via Western blotting from C. For each biological replicate, values were normalized to the value for “IR−” to calculate the fold change in protein expression upon IR.
(E) Time course of PRMT5 expression at the mRNA level at the indicated minutes (m) or hours (h) post 2 Gy IR in LNCaP cells via RT-qPCR. For each biological replicate, values were normalized to the value for “IR−” to calculate the fold change in mRNA expression upon IR.
(F) Time course of MEP50/pICln expression/localization at the indicated minutes (m) or hours (h) post 2 Gy IR in LNCaP cells.
(G) Quantification of MEP50 expression/localization in images from F: “Whole cell” indicates MEP50 expression in the entire cell, “Nuclear” indicates MEP50 expression in the nucleus, which was defined by DAPI staining, “Cytoplasmic” indicates MEP50 expression in the cytoplasm that was defined as staining outside DAPI, and “N:C ratio” was calculated by dividing the value for nucleus by the value for cytoplasmic for each cell individually such that an N:C ratio of 1 indicates equal expression in both the nucleus and cytoplasm.
(H) Quantification of pICln expression/localization in images from F as described above.
(I) PRMT5:MEP50 and PRMT5:pICln interaction 6 h post 2 Gy IR in irradiated (6 h) and non-irradiated (IR−) LNCaP cells via BiFC assay.
(J) Quantification of PRMT5:MEP50 BiFC intensity in images from I: “Whole cell” indicates BiFC intensity in the entire cell, “Nuclear” indicates BiFC intensity in the nucleus that was defined by NLS-CFP signal, “Cytoplasmic” indicates BiFC intensity in the cytoplasm that was defined as staining outside NLS-CFP signal, and “N:C ratio” was calculated by dividing the value for nucleus by the value for cytoplasmic for each cell individually such that an N:C ratio of 1 indicates equal interaction in both the nucleus and cytoplasm. NLS-CFP was used as a transfection control and a marker of the nucleus.
(K) Quantification of PRMT5:pICln BiFC intensity in images from I as described above.
Fluorescence images in A are representative immunocytochemistry images (red = PRMT5). Fluorescence images in F are representative immunocytochemistry images (blue = DAPI and red = MEP50 or pICln). Blue circles outline DAPI staining to allow for better visibility of expression in the nucleus. Fluorescence images in I are representative images from BiFC assay (green = PRMT5:MEP50 and PRMT5:pICln, cerulean = NLS-CFP). Blue circles outline NLS-CFP signal to allow for better visibility of expression in the nucleus. All bars are the mean ± s.d. of three independent experiments. Statistical analysis for B, D, E, G, and H comparing experimental with the control (“IR−”) was performed using Brown-Forsythe and Welch ANOVA followed by Dunnett's T3 multiple comparisons test, whereas statistical analysis for J and K comparing experimental with the control (“IR−”) was performed using Welch's t test (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05).
To gain insight into how pICln functions with PRMT5 to regulate the expression of genes involved in DDR, we first determined if IR affects expression of MEP50 or pICln. However, neither MEP50 nor pICln protein expression was affected by IR (Figures 6F–6H). We next analyzed the subcellular localization of MEP50 and pICln upon IR. Upon IR, the nuclear:cytoplasmic (N:C) ratio of MEP50 decreased, whereas the N:C ratio of pICln increased (Figures 6F–6H). This suggests that upon IR there is more pICln and less MEP50 in the nucleus to interact with PRMT5. IR-induced nuclear localization of pICln thus likely contributes to IR-induced pICln binding to the promoters of genes involved in DDR. To assess the protein-protein interaction (PPI) directly, we utilized bimolecular fluorescence complementation (BiFC) assay (Hu et al., 2002, Kodama and Hu, 2010, Kodama and Hu, 2012, Shyu and Hu, 2008), which is uniquely applicable in assessing the subcellular localization of PPIs in live cells (Pratt et al., 2016). BiFC confirmed an IR-induced PPI increase between PRMT5 and pICln and a decrease in the PRMT5:MEP50 PPI, particularly in the nucleus (Figures 6I–6K). Overall, these results suggest that the regulation of genes involved in DDR by PRMT5 is facilitated by the IR-induced PRMT5:pICln interaction in the nucleus.
The Transcriptional Regulation of DSB Repair Genes by PRMT5 Is Not Dependent on RuvBL1 or Tip60
While this study was ongoing, Clarke et al reported that PRMT5 participates in the DSB repair choice process and promotes HR through methylation of RuvBL1: Methylation of RuvBL1 by PRMT5 alters the RuvBL1:Tip60 complex, promotes Tip60-mediated acetylation of histone H4K16, demotes 53BP1 binding to DSBs, and initiates DSB repair via HR (Clarke et al., 2017). We sought to confirm that the mechanism we describe here is independent of RuvBL1 and Tip60. We determined that knockdown of RuvBL1 did not affect expression of PRMT5, RAD51, RAD51D, RAD51AP1, NHEJ1, and Tip60 at the protein level (Figures S6A and S6B), nor did it affect the efficiency of repair of IR-induced DSBs in LNCaP cells (Figures S6C and S6D). This is consistent with another study reporting that knockdown of RuvBL1 had no effect on IR-induced 53BP1 foci in hematopoietic cells (Hamard et al., 2018). Therefore, methylation of RuvBL1 by PRMT5 likely affects DSB repair choice (favoring HR over NHEJ) but not the overall efficiency of repair, whereas the transcriptional activation of DDR genes by PRMT5 is likely required for repair of DSBs.
Similarly, while this study was ongoing, Hamard et al. also reported that PRMT5 regulates DSB repair choice via splicing of Tip60 in hematopoietic cells: PRMT5 is required for appropriate splicing of Tip60, which in turn allows for normal Tip60 acetyltransferase activity, demotes 53BP1 binding to DSBs, and initiates DSB repair via HR (Hamard et al., 2018). Knockdown of Tip60 did not affect the expression of PRMT5 or the putative PRMT5 target genes we characterized (Figures S6E and S6F), suggesting that PRMT5-assoicated splicing of Tip60 does not affect the regulation of DDR genes by PRMT5. Given the role of Tip60 in DDR, PRMT5-assoicated splicing of Tip60 likely regulates DSB repair choice but not DSB repair efficiency or at least does not affect PRMT5-associated transcriptional regulation of genes involved in DDR.
PRMT5 Regulates the Expression of DDR Genes in Multiple Cancer Cell Lines, and PRMT5 Expression Positively Correlates with DDR Genes in Human Cancer Tissues
To extend our findings and determine if the role of PRMT5 in DSB repair is conserved across multiple cell types, we performed similar experiments in AR-negative prostate cancer cells (DU145), luminal breast cancer cells (MCF7), glioblastoma cells (U87MG), and non-cancerous HEK293T cells. Inhibition of PRMT5 by BLL3.3 hindered repair of IR-induced DSBs as the cells retained significantly more γH2AX foci 6 h following IR treatment (Figures 7A and S7A–S7E). Although DDR genes were generally upregulated upon IR, targeting PRMT5 also caused a decrease in the expression of PRMT5 target genes involved in DSB repair in both irradiated and non-irradiated cells (Figures 7B and S7F–S7J). These results suggest that PRMT5 may function as a key regulator of DSB repair in multiple tissue types.
Figure 7.

PRMT5 Regulates the Expression of DDR Genes in Multiple Cancer Cell Lines, and PRMT5 Expression Positively Correlates with DDR Genes in Human Cancer Tissues
(A) Quantification of DSBs 6 h post 2 Gy IR in the indicated cell lines with (BLL3.3) and without (DMSO) PRMT5 inhibition as described in Figure 2B (see also Figures S7A–S7E for representative images).
(B) Quantification of mRNA via RT-qPCR 6 h post 2 Gy IR in the indicated cell lines with (BLL3.3) and without (DMSO) PRMT5 inhibition. For each biological replicate, values were normalized to the value for “DMSO/IR−” (untreated) to calculate the fold change in mRNA expression upon treatment (see also Figures S7F–S7J for statistical analysis).
(C) Violin plots representing Spearman correlations comparing the mRNA expression level between PRMT5 and DDR genes (DDR), PRMT5 and AR (AR), or PRMT5 and all other genes (All other) across 32 clinical cancer datasets from TCGA. The gene set for DDR genes was defined as RAD51, RAD51D, RAD51AP1, NHEJ1, BRCA1, BRCA2, WEE1, DNAPKcs, Ku70, Ku80, an XRCC4 (see also Figure S8).
(D) Violin plots representing Spearman correlations comparing the mRNA expression level between DDR genes and PRMT5. Cancer types were stratified by the correlation coefficient (c.c.) between PRMT5 and AR: positively correlated (c.c. > 0 & p < 0.01) (Positive), negatively correlated (c.c. < 0 & p < 0.01) (Negative), or not correlated (p > 0.01) (Non).
(E) Violin plot representation of Spearman correlation values between DDR genes and either PRMT5, CLNS1A (pICln), or WDR77 (MEP50).
Bars in A and values used in the heatmap in B are the mean of three independent experiments. Box-and-whiskers plots in C–E show the median value (line) and interquartile range between the first and third quartiles (box). The upper whisker extended to the largest value no further than “1.5 x interquartile range” and the lower whisker extended to the smallest value at most “1.5 x interquartile range”. Outliers beyond the whiskers are shown as dots. Statistical analysis in A comparing experimental to the control (“DMSO”) was performed using Welch's t test (**p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). Statistical analysis in C–E was performed using Wilcoxon test, and the p values are displayed.
To assess potential clinical significance of our findings, we analyzed mRNA expression in 32 clinical cancer datasets achieved from The Cancer Genome Atlas (TCGA) Pan-Cancer analysis (The Cancer Genome Atlas Research Network et al., 2013). We focused on PRMT5, pICln, MEP50, AR, and DDR genes, which were identified as primary target genes of both PRMT5 and AR. The expression of PRMT5 correlated positively with DDR genes in almost all cancers (Figures 7C and S8A). As a control, we assessed if PRMT5 generally correlated with the expression of all genes in the transcriptome. As expected, there was no correlation across the cancer datasets (Figure 7C). PRMT5 expression generally did not correlate with AR and varied significantly for individual cancer types (Figures 7C and S8A). However, the strength of correlations between PRMT5 and several AR target genes involved in DDR seemed independent of the correlation between PRMT5 and AR (Figure S8A). To assess this further, we sorted the cancer types into three groups based on the correlation coefficient between PRMT5 and AR: positively correlated, negatively correlated, and not correlated. We observed no difference in the correlation between PRMT5 and DDR genes when the cancer types were stratified (Figure 7D), indicating that PRMT5 correlates positively with DDR genes independently of any correlation with AR. This leaves the possibility that AR target genes involved in DDR may also be primary target genes of PRMT5. However, similar RT-qPCR and ChIP-qPCR experiments revealed that only DNAPKcs is likely a target gene of PRMT5 (Figures S4A–S4F). DNAPKcs also had the highest correlation with PRMT5 across the 32 clinical cancer datasets (Figure S8A), further implicating DNAPKcs as a PRMT5 target gene.
As our data suggested that pICln, but not MEP50, cooperates with PRMT5 to regulate DDR genes, we also analyzed the correlations between MEP50 or pICln and the same DDR genes. Although MEP50 expression positively correlated with some DDR genes in some cancers (Figures 7E and S8A), pICln exhibited significantly stronger correlations with these DDR genes than MEP50 in almost all cancers (Figures 7E and S8A). Furthermore, PRMT5 correlated more significantly with pICln than MEP50 (Figure S8B). Collectively, our results suggest that PRMT5 and pICln play a conserved role in activating expression of genes required for the repair of IR-induced DSBs.
Discussion
PRMT5 Functions as an Epigenetic Activator to Regulate the Repair of DSBs
Upon recognition of DNA DSBs, repair proteins (such as RAD51, BRCA1, and BRCA2) are transiently upregulated to facilitate repair through HR or NHEJ (Khalil et al., 2012, Rieger, 2004, Russell et al., 2003). Although this transient upregulation is required for cell survival following genotoxic stresses, there is a long-standing question of how proteins are quickly upregulated to promote repair of DNA damage. Here, we present evidence that PRMT5 functions as a master epigenetic activator of DDR genes to facilitate the repair of DSBs.
In this study, we determined that PRMT5 activated transcription of multiple genes that encode well-characterized repair proteins involved in HR (RAD51, RAD51AP1, RAD51D, BRCA1, and BRCA2) and NHEJ (NHEJ1 and DNAPKcs). DSB repair occurs in three phases: (1) recognition of DSBs via sensor proteins, (2) initiation of repair by repair proteins, and (3) resolution of repair (Thompson, 2012). Our studies suggest that PRMT5 primarily regulates the expression of repair proteins as opposed to DNA damage sensors or proteins involved in the resolution of repair. Since we also confirmed that PRMT5 regulated the expression of several other genes involved in various phases of DDR by RT-qPCR (Figure S3G), it remains to be determined whether these genes are also target genes of PRMT5.
The epigenetic regulation of genes is cell type, temporal, and context dependent. Our data suggest that PRMT5 likely activates transcription of DDR genes in a variety of cell types. However, we cannot rule out the possibility that PRMT5 regulates different DDR genes in different cells. For example, although we determined that PRMT5 activated transcription of RAD51, Clarke et al. demonstrated that depletion of PRMT5 impaired HR (reduced IR-induced RAD51 and BRCA1 foci) without affecting the expression of RAD51 or changes in cell cycle (Clarke et al., 2017). There are a few possible explanations: (1) their experiment was performed in HeLa-shPRMT5 stable cell lines and were not performed in inducible knockdown lines. It is possible that these cells compensated for depleted PRMT5 although all experiments were conducted on low passage cells to minimize effects of chronic PRMT5 depletion. (2) PRMT5 may not regulate RAD51 expression in HeLa cells. (3) HeLa cells may respond differently to depleted PRMT5 as evidenced by a lack of cell-cycle changes in HeLa cells that we and others have observed in other cell lines (Lim et al., 2014, Scoumanne et al., 2009, Wei et al., 2012, Yang et al., 2016). As an epigenetic regulator, it is possible that PRMT5 may not regulate the same cohort of DDR genes in every tissue but rather regulates the same pathways such as HR, NHEJ, and G2 arrest.
During preparation of this manuscript, Braun et al reported that PRMT5 post-transcriptionally regulates the splicing out of detained introns (DIs) of genes to modulate gene expression (Braun et al., 2017). However, our analysis of their data showed that the majority of DEGs we identified either do not contain DIs or DI splicing of our DEGs was not affected by PRMT5 targeting. Additionally, Tan et al. reported that PRMT5 is required for appropriate splicing in hematopoietic stem cells and that targeting PRMT5 causes increased intron retention and exon skipping events (Tan et al., 2019). In their study, they performed functional enrichment analysis on alternative splicing events upon PRMT5 knockdown and they identified that genes associated with “DNA repair” were enriched. However, there was little overlap between the genes we validated as PRMT5 target genes (RAD51, RAD51AP1, RAD51D, BRCA1, BRCA2, NHEJ1, DNAPKcs, and WEE1) and genes they identified as splicing targets. Interestingly, they functionally validated five splicing targets (FANCA, FANCG, MUTYH, RTEL1, and RAD52), and we identified both FANDCA and FANCG as “IR+ only” downregulated DEGs in our RNA-seq analysis. However, we did not pursue further validation of these genes. Overall, our findings are likely independent of potential splicing changes upon PRMT5 knockdown, and the role of PRMT5 in transcriptional regulation of DDR genes likely mediates the transient upregulation of repair proteins upon DNA damage.
PRMT5 Is Required for Efficient Repair of DSBs
We demonstrate that PRMT5 is required for efficient repair of DSBs. Interestingly, knockdown of PRMT5 alone caused an increase in spontaneous DSBs independent of external DNA damage inducers (Figures S2A and S2B), indicating that PRMT5 is required to repair endogenous DSBs. Two recent studies have also demonstrated that PRMT5 is required for efficient repair of DSBs in additional cell lines (Clarke et al., 2017, Hamard et al., 2018). We provide evidence that the activation of gene expression by PRMT5 is essential to DSB repair efficiency, whereas the regulation of RuvBL1 and Tip60 by PRMT5 likely only affects DSB repair choice. Specifically, PRMT5-catalyzed methylation of RuvBL1 and PRMT5-associated splicing of Tip60 may promote HR over NHEJ, yet the DSB can be repaired regardless of pathway choice. Therefore, observation that PRMT5 is required for efficient repair of IR-induced DSBs is most likely explained by our finding that PRMT5 activates transcription of DDR genes. Because Tip60 is required for ATM activation (Bhoumik et al., 2008, Sun et al., 2005) and ATM phosphorylates H2AX to form γH2AX foci and signals the initiation of DSB repair (Burma et al., 2001), we did not perform γH2AX foci analysis to assess repair of IR-induced DSBs. Because Tip60 has been shown to be essential for γH2AX foci formation (Ikura et al., 2000, Murr et al., 2006), we would have observed a decrease in γH2AX regardless if knockdown of Tip60 would affect repair of IR-induced DSBs.
PRMT5 Regulates DSB Repair Independently of AR
We recently reported that PRMT5 is an epigenetic activator of AR (Deng et al., 2017). In prostate cancer cells, AR has been reported to regulate DSB repair via HR and NHEJ (Asim et al., 2017, Goodwin et al., 2013, Li et al., 2017, Polkinghorn et al., 2013, Spratt et al., 2015, Tarish et al., 2015). Upon IR, AR is recruited to the promoter of DDR genes to activate their expression (Goodwin et al., 2013) (∼8–24 h post IR), albeit at a much later time point than the recruitment of PRMT5 to the promoter of DDR genes (∼1 h post IR). Knockdown or inhibition of AR signaling has also been shown to directly impair HR (Asim et al., 2017, Polkinghorn et al., 2013), and recent studies suggest that AR may be essential for HR particularly in castration-resistant prostate cancer (CRPC) (Asim et al., 2017, Goodwin et al., 2013, Li et al., 2017). However, AR's role in regulating NHEJ is more established in that AR transcriptionally activates genes involved in NHEJ (Goodwin et al., 2013, Polkinghorn et al., 2013, Spratt et al., 2015, Tarish et al., 2015).
As we reported that targeting PRMT5 decreases AR expression (Deng et al., 2017), the requirement of PRMT5 for efficient repair of DSBs could be mediated through the regulation of AR. Indeed, we demonstrated that targeting PRMT5 caused a decrease in AR expression and concomitant decrease in the expression of AR target genes involved in NHEJ (Ku80, XRCC4, and DNAPKcs). However, several pieces of evidence in our study suggest that PRMT5 also regulates repair of DSBs independently of AR: (1) PRMT5 targeting sensitized both AR-positive and AR-negative prostate cancer cells to IR; (2) exogenous expression of AR only partially rescued the impairment of IR-induced DSB repair by PRMT5 knockdown; (3) targeting PRMT5 hindered the repair of IR-induced DSBs in AR-negative DU145 cells and several other cancer cell lines with varying AR expression level; and (4) in clinical cancer datasets, PRMT5 expression was positively correlated with the expression of DDR target genes regardless of its correlation with AR. Collectively, these data strongly suggest that although targeting PRMT5 may mimic targeting AR to sensitize prostate cancer cells to IR, PRMT5 can regulate DSB repair independently of AR expression.
PRMT5 Likely Regulates the Repair of IR-Induced DSBs via Multiple Mechanisms
In this study, we determined that PRMT5 is required for efficient repair of DSBs via activation of DDR genes. Additionally, there are several reports suggesting that PRMT5 may regulate the repair of DNA damage via multiple mechanisms. As detailed above, recent reports show that PRMT5 regulates the DSB repair choice process and promotes HR through methylation of RuvBL1 (Clarke et al., 2017) and altered splicing of Tip60 (Hamard et al., 2018). Indeed, RuvBL1 was identified in our mass-spec analysis (peptide fragment TISHVIIGLK) as a potential interacting protein of PRMT5 in LNCaP cells. Therefore, our observation that PRMT5 knockdown decreased HR-associated RAD51 foci may be partially explained by these previous results.
PRMT5 can also regulate protein expression via splicing (Braun et al., 2017, Tan et al., 2019). These two studies identified some PRMT5 splicing targets involved in DDR. For example, PRMT5 is required to maintain appropriate expression of functional RAD52 (Tan et al., 2019), and recent reports demonstrate the importance of RAD52 to HR (Hanamshet et al., 2016, Mahajan et al., 2019, Manthey et al., 2017). Future studies may determine if PRMT5-associated splicing directly affects the repair of IR-induced DSBs.
PRMT5 has also been shown to methylate and regulate several proteins associated with DDR: p53 (Du et al., 2016, Durant et al., 2009, Jansson et al., 2008, Scoumanne et al., 2009), E2F1 (Cho et al., 2012, Wu et al., 2015, Zheng et al., 2013), FEN1 (Guo et al., 2010, Guo et al., 2012), RAD9 (He et al., 2011), KLF4 (Hu et al., 2015), and TDP1 (Rehman et al., 2018). As detailed in these studies, DNA damage via etoposide, hydroxyurea, doxorubicin, and UV can induce PRMT5-catalyzed methylation of these non-histone substrates, which alters the cellular response to DNA damage. For example, PRMT5-catlyzed methylation of p53 altered binding to p53 target genes, which promoted cell-cycle arrest and inhibited apoptosis (Jansson et al., 2008). However, these studies did not identify a direct role for PRMT5 in the repair of DSBs or in the response to IR. For example, PRMT5-catalyzed methylation of RAD9 was essential to the cellular response to hydroxyurea but did not play a significant role in the cellular response to IR. It is likely that there are unknown PRMT5 substrates involved in DDR, and future studies may determine if PRMT5-catalyzed methylation of these proteins directly affects DSB repair efficiency.
In various yeast species, the PRMT5 homologue Hsl7 was shown to interact with and promote degradation of WEE1 homologues to promote G2 progression independently of its methyltransferase activity (Cid et al., 2001, Theesfeld et al., 2003, Yamada et al., 2004). Although it is unknown if PRMT5 interacts with WEE1 protein in human tissues, the studies in yeast contrast with our study in which PRMT5 activated transcription of WEE1 and promoted DNA damage-induced G2 arrest. The opposing post-translational and transcriptional regulation of WEE1 by PRMT5 may be modulated by PRMT5 methyltransferase activity and/or the absence or presence of DNA damage. In fact, the PRMT5-mediated changes in cell cycle in the Xenopus egg are independent of transcription because Xenopus egg extracts can cycle without de novo mRNA transcription (Yamada et al., 2004). Therefore, PRMT5 likely modulates WEE1-mediated cell-cycle changes in multiple ways.
pICln May Function as a Cofactor of PRMT5 to Epigenetically Regulate Gene Expression Independently of MEP50
There is a long-standing view in the field that the cofactor MEP50 is required for PRMT5 methyltransferase activity and epigenetic function (Burgos et al., 2015, Chen et al., 2017, Karkhanis et al., 2011, Stopa et al., 2015). In solution, PRMT5 can exist as a homodimer or homotetramer. With MEP50, PRMT5 forms a heterooctomeric complex (PRMT54:MEP504) (Antonysamy et al., 2012). Consistent with these structural studies, biochemical studies have provided evidence that purified PRMT5:MEP50 complex can catalyze dimethylation of various histone substrates including H4R3 (Burgos et al., 2015, Wang et al., 2014). However, our data suggest that PRMT5 works with pICln for the transcriptional activation of DDR genes via H4R3me2s. This is inconsistent with a previous report where Pesiridis et al showed that titration of pICln decreased H3 and H4 methylation by PRMT5 in an in vitro methylation assay (Pesiridis et al., 2009). However, as PRMT5 functions in a larger complex, the in vitro assay using proteins from a bacterial expression system might not recapitulate the biochemical and cellular conditions required for H4R3me2s in vivo. In our study, knockdown of MEP50 did not affect the expression of PRMT5 target genes involved in DDR, and MEP50 was not present at any of the target gene promoters characterized. Instead, pICln was present at the promoter regions of PRMT5 target genes along with H4R3me2s, and knockdown of pICln caused a decrease in PRMT5 target gene expression and impaired IR-induced DSB repair. Thus, it is likely that pICln may function as a cofactor of PRMT5 to activate transcription of DDR genes. As the epigenetic regulation of gene expression likely involves formation of a larger protein complex in a gene-specific manner, future characterization of PRMT5 and its cofactors or interacting proteins in vivo will provide mechanistic insight into the regulation of expression of PRMT5 target genes.
Although PRMT5-catalyzed histone methylation is predominantly repressive (Stopa et al., 2015), recent studies show PRMT5 can function as an activator of gene expression (Deng et al., 2017, Chen et al., 2017, Tarighat et al., 2016). Activation or repression is not likely dependent solely on PRMT5-catalyzed histone methylation, as H4R3me2s has been shown to be both a repressive (Chen et al., 2017) and active chromatin mark (Deng et al., 2017). Therefore, additional factors are required to mediate the positive or negative epigenetic regulation by PRMT5. Because PRMT5 does not contain a DNA binding domain, additional proteins that recruit PRMT5 to sites on the genome may play a role in mediating the epigenetic function of PRMT5. Future studies will elucidate the full structure and interactome of PRMT5 on DNA and will determine differences between its active and repressive complexes. This will also provide an answer for how the same PRMT5-catalyzed histone modifications can mediate gene repression and activation. As we reported that PRMT5 functions as an epigenetic activator of AR expression (Deng et al., 2017), future work will determine if this is dependent on pICln. It is possible that pICln promotes activation by PRMT5, whereas other cofactors, such as MEP50, may promote repression by PRMT5.
PRMT5 Targeting May Be Explored for Cancer Treatment
According to the American Cancer Society, over half of all cancer patients receive RT. RT induces DSBs in DNA, which are lethal to cells if not repaired. Although potentially curative, tumors can still regrow following RT. For example, 10% of prostate cancer patients with low-risk disease and 30-50% of patients with high-risk disease treated with RT still experience tumor recurrence (Boorjian et al., 2011, D'Amico et al., 2008). Thus, identification of novel therapeutic targets to enhance RT will likely reduce cancer mortality.
PRMT5 is overexpressed in many cancers, and its overexpression correlates with poor prognosis (Karkhanis et al., 2011, Stopa et al., 2015, Yang and Bedford, 2013). Our findings suggest that PRMT5 overexpression may increase the efficiency of DSB repair and confer survival advantages particularly following DNA-damaging treatments. For example, upregulation of RAD51, a putative PRMT5 target gene, has been shown to promote resistance to DNA damaging agents (Petermann et al., 2010, Schild and Wiese, 2010) and decreasing RAD51 expression sensitizes cancer cells to IR (Hayman et al., 2012). Because targeting DSB repair is a validated therapeutic approach for cancer treatment (Gavande et al., 2016), our findings that PRMT5 expression positively correlates with multiple DDR genes across clinical cancer datasets strongly suggests that PRMT5 targeting may be explored as a monotherapy or in combination with RT or chemotherapy for cancer treatment. PRMT5 may also be a particularly attractive therapeutic target for prostate cancer patients because targeting PRMT5 decreases AR expression (Deng et al., 2017) and targeting AR signaling via ADT enhances RT for prostate cancer patients (Golabek et al., 2016).
One criticism of PRMT5 targeting is potential systemic side effects as epigenetic regulators typically have essential roles in various tissues. Although targeting PRMT5 does not affect the growth of AR-negative DU145 and PC3 cells as well as normal prostate RWPE-1 cells (Deng et al., 2017), we do find that targeting PRMT5 inhibits repair of IR-induced DSBs in non-cancerous HEK293T cells. It is reasonable to suspect that targeting PRMT5 may also sensitize adjacent normal tissue to RT. Given advances in RT, the amount of adjacent normal tissue that is irradiated is minimalized. Thus, it is likely that the combination of PRMT5 targeting and RT will allow for either a lower dose of drug or IR to limit side effects. Alternatively, targeted delivery of PRMT5 inhibitors as radiosensitizers will circumvent systemic toxicity. This can be effectively achieved through prostate specific membrane antigen-based delivery (Rowe et al., 2016). Nevertheless, our findings here provide convincing evidence that PRMT5 functions as a master epigenetic regulator to activate transcription of DNA damage repair genes and is a potential therapeutic target to enhance RT or chemotherapy for cancer treatment.
Limitations of the Study
We present evidence that PRMT5 cooperates with pICln to function as a master epigenetic activator of DDR genes in various cell types. Although this potentially explains the long-standing question of how repair proteins are quickly upregulated to promote the repair of DNA damage, the precise molecular mechanisms on how PRMT5 and pICln function together to activate gene expression remain to be determined. Our findings also suggest that the regulatory role of PRMT5 in the activation of DDR genes is independent of its canonical cofactor MEP50: MEP50 was not present at the promoter of DDR genes, and knockdown of MEP50 did not affect expression of DDR genes nor did it affect repair of IR-induced DSBs. Although we provide several lines of evidence suggesting that pICln, but not MEP50, may participate in transcriptional regulation of DDR genes by PRMT5, it is possible that our knockdown of MEP50 was not sufficient to prevent the regulation of DDR gene expression. Thus, future studies with CRISPR-based knockout or PRMT5:MEP50 protein-protein interaction inhibitors may be needed to further evaluate a potential role for MEP50 in the regulation of PRMT5 target genes involved in DDR. Given that previous biochemical assays demonstrate that PRMT5 requires MEP50 for methyltransferase activity, further biochemical assays of PRMT5 with its cofactors in the presence of nucleosomes may provide mechanistic insight into the modulation of PRMT5 catalytic activity by its cofactors in the context of transcriptional regulation. These studies combined with both structural analysis of the PRMT5 complex at the promoter of target genes involved in DDR and genome-wide analyses such as ChIP-seq and ATAC-seq will likely provide mechanistic evidence for how PRMT5 functions with pICln and/or other cofactors to regulate transcription of DDR target genes.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was partially supported by grants from U.S. Army Medical Research Acquisition Activity, Prostate Cancer Research Program (PC11190, PC120512, PC150697) and NCI RO1CA212403, and Purdue University Center for Cancer Research Small Grants. Primary RNA-seq analysis and TCGA data analysis were performed by the Collaborative Core for Cancer Bioinformatics (C3B) shared by Indiana University Simon Cancer Center (P30CA082709) and Purdue University Center for Cancer Research (P30CA023168) with support from the Walther Cancer Foundation. DNA sequencing was conducted by the Genomic Core Facility supported by NCI CCSG CA23168 to Purdue University Center for Cancer Research. We would like to thank Indiana University Precision Health Initiative. J.L.O. is a recipient of the Indiana Clinical and Translational Sciences Institute (CTSI) Pre-Doctoral Fellowship, which was made possible with partial support from Grant Numbers TL1 TR001107, TL1 TR002531, UL1 TR001108, and UL1 TR002529 (A. Shekhar, PI) from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. We would also like to thank Dr. Liang Cheng and Dr. Jeannie Poulson Plantenga for providing their mentoring to J.L.O. and insightful comments on the study.
Author Contributions
Conceptualization: J.L.O., J.H. (consultations for clinical aspects), and C.D.H.; Methodology: J.L.O., X.D., and E.B.; Validation: J.L.O., E.B., S.L.T., A.M.A., and X.D.; Formal Analysis: J.L.O., S.L., and J.W.; Investigation: J.L.O., E.B., S.L.T., A.M.A., S.L., and X.D.; Resources: X.D.; Data Curation: J.L.O.; Writing—Original Draft: J.L.O.; Writing—Review & Editing: J.L.O., E.B., S.L.T., A.M.A., J.W., and C.D.H.; Visualization: J.L.O., S.L., and J.W.; Supervision: J.W. and C.D.H.; Project Administration: J.L.O. and C.D.H.; Funding Acquisition: J.H., C.L., J.W., and C.D.H.
Declaration of Interests
The authors declare no competing interests.
Published: January 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.100750.
Contributor Information
Jun Wan, Email: junwan@iu.edu.
Chang-Deng Hu, Email: hu1@purdue.edu.
Data and Code Availability
RNA-seq datasets generated in this study are available at the Gene Expression Omnibus (GEO) under accession number GSE111620.
Supplemental Information
References
- Alinari L., Mahasenan K.V., Yan F., Karkhanis V., Chung J.-H., Smith E.M., Quinion C., Smith P.L., Kim L., Patton J.T. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood. 2015;125:2530–2543. doi: 10.1182/blood-2014-12-619783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S., Bonday Z., Campbell R.M., Doyle B., Druzina Z., Gheyi T., Han B., Jungheim L.N., Qian Y., Rauch C. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U S A. 2012;109:17960–17965. doi: 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asim M., Tarish F., Zecchini H.I., Sanjiv K., Gelali E., Massie C.E., Baridi A., Warren A.Y., Zhao W., Ogris C. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017;8:374. doi: 10.1038/s41467-017-00393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoumik A., Singha N., O’Connell M.J., Ronai Z.A. Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 2008;283:17605–17614. doi: 10.1074/jbc.M802030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorjian S.A., Karnes R.J., Viterbo R., Rangel L.J., Bergstralh E.J., Horwitz E.M., Blute M.L., Buyyounouski M.K. Long-term survival after radical prostatectomy versus external-beam radiotherapy for patients with high-risk prostate cancer. Cancer. 2011;117:2883–2891. doi: 10.1002/cncr.25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun C.J., Stanciu M., Boutz P.L., Patterson J.C., Calligaris D., Higuchi F., Neupane R., Fenoglio S., Cahill D.P., Wakimoto H. Coordinated splicing of regulatory detained introns within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell. 2017;32:411–426.e11. doi: 10.1016/j.ccell.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E.S., Wilczek C., Onikubo T., Bonanno J.B., Jansong J., Reimer U., Shechter D. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem. 2015;290:9674–9689. doi: 10.1074/jbc.M115.636894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Chari A., Golas M.M., Klingenhäger M., Neuenkirchen N., Sander B., Englbrecht C., Sickmann A., Stark H., Fischer U. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135:497–509. doi: 10.1016/j.cell.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Chen H., Lorton B., Gupta V., Shechter D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene. 2017;36:373–386. doi: 10.1038/onc.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E.-C., Zheng S., Munro S., Liu G., Carr S.M., Moehlenbrink J., Lu Y.-C., Stimson L., Khan O., Konietzny R. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012;31:1785–1797. doi: 10.1038/emboj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid V.J., Shulewitz M.J., McDonald K.L., Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol. Biol. Cell. 2001;12:1645–1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T.L., Sanchez-Bailon M.P., Chiang K., Reynolds J.J., Herrero-Ruiz J., Bandeiras T.M., Matias P.M., Maslen S.L., Skehel J.M., Stewart G.S. PRMT5-Dependent methylation of the TIP60 coactivator RUVBL1 is a key regulator of homologous recombination. Mol. Cell. 2017;65:900–916.e7. doi: 10.1016/j.molcel.2017.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Shao G., Zhang H.T., Li C., Zhang D., Cheng L., Elzey B.D., Pili R., Ratliff T.L., Huang J. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene. 2017;36:1223–1231. doi: 10.1038/onc.2016.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W., Amarachintha S., Erden O., Wilson A., Pang Q. The Fanconi anemia pathway controls oncogenic response in hematopoietic stem and progenitor cells by regulating PRMT5-mediated p53 arginine methylation. Oncotarget. 2016;7:60005–60020. doi: 10.18632/oncotarget.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant S.T., Cho E.C., La Thangue N.B. p53 methylation—the Arg-ument is clear. Cell Cycle. 2009;8:801–802. doi: 10.4161/cc.8.6.7850. [DOI] [PubMed] [Google Scholar]
- D'Amico A.V., Chen M.-H., Renshaw A.A., Loffredo B., Kantoff P.W. Risk of prostate cancer recurrence in men treated with radiation alone or in conjunction with combined or less than combined androgen suppression therapy. J. Clin. Oncol. 2008;26:2979–2983. doi: 10.1200/JCO.2007.15.9699. [DOI] [PubMed] [Google Scholar]
- Friesen W.J., Paushkin S., Wyce A., Massenet S., Pesiridis G.S., Van Duyne G., Rappsilber J., Mann M., Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell. Biol. 2001;21:8289–8300. doi: 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen W.J., Wyce A., Paushkin S., Abel L., Rappsilber J., Mann M., Dreyfuss G. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 2002;277:8243–8247. doi: 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- Furuta T., Takemura H., Liao Z.-Y., Aune G.J., Redon C., Sedelnikova O.A., Pilch D.R., Rogakou E.P., Celeste A., Chen H.T. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 2003;278:20303–20312. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- Gavande N.S., VanderVere-Carozza P.S., Hinshaw H.D., Jalal S.I., Sears C.R., Pawelczak K.S., Turchi J.J. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol. Ther. 2016;160:65–83. doi: 10.1016/j.pharmthera.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golabek T., Belsey J., Drewa T., Kołodziej A., Skoneczna I., Milecki P., Dobruch J., Słojewski M., Chłosta P.L. Evidence-based recommendations on androgen deprivation therapy for localized and advanced prostate cancer. Cent. Eur. J. Urol. 2016;69:131–138. doi: 10.5173/ceju.2016.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin J.F., Schiewer M.J., Dean J.L., Schrecengost R.S., de Leeuw R., Han S., Ma T., Den R.B., Dicker A.P., Feng F.Y. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guderian G., Peter C., Wiesner J., Sickmann A., Schulze-Osthoff K., Fischer U., Grimmler M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011;286:1976–1986. doi: 10.1074/jbc.M110.148486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Zheng L., Xu H., Dai H., Zhou M., Pascua M.R., Chen Q.M., Shen B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 2010;6:766–773. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z., Kanjanapangka J., Liu N., Liu S., Liu C., Wu Z., Wang Y., Loh T., Kowolik C., Jamsen J. Sequential posttranslational modifications program FEN1 degradation during cell-cycle progression. Mol. Cell. 2012;47:444–456. doi: 10.1016/j.molcel.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamard P.-J., Santiago G.E., Liu F., Karl D.L., Martinez C., Man N., Mookhtiar A.K., Duffort S., Greenblatt S., Verdun R.E. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep. 2018;24:2643–2657. doi: 10.1016/j.celrep.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamshet K., Mazina O., Mazin A. Reappearance from obscurity: mammalian Rad52 in homologous recombination. Genes. 2016;7:63. doi: 10.3390/genes7090063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman T.J., Williams E.S., Jamal M., Shankavaram U.T., Camphausen K., Tofilon P.J. Translation initiation factor eIF4E is a target for tumor cell radiosensitization. Cancer Res. 2012;72:2362–2372. doi: 10.1158/0008-5472.CAN-12-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W., Ma X., Yang X., Zhao Y., Qiu J., Hang H. A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Res. 2011;39:4719–4727. doi: 10.1093/nar/gkq1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich A., Bastian P.J., Bellmunt J., Bolla M., Joniau S., van der Kwast T., Mason M., Matveev V., Wiegel T., Zattoni F. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 2014;65:467–479. doi: 10.1016/j.eururo.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Hu C.-D., Chinenov Y., Kerppola T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- Hu D., Gur M., Zhou Z., Gamper A., Hung M.-C., Fujita N., Lan L., Bahar I., Wan Y. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 2015;6:8419. doi: 10.1038/ncomms9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T., Ogryzko V.V., Grigoriev M., Groisman R., Wang J., Horikoshi M., Scully R., Qin J., Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Jansson M., Durant S.T., Cho E.-C., Sheahan S., Edelmann M., Kessler B., La Thangue N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008;10:1431–1439. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Karkhanis V., Hu Y.-J., Baiocchi R.A., Imbalzano A.N., Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011;36:633–641. doi: 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H., Tummala H., Zhelev N. ATM in focus: a damage sensor and cancer target. BioDiscovery. 2012;5:1–60. [Google Scholar]
- Khanna K.K., Jackson S.P. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Kodama Y., Hu C.-D. An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. BioTechniques. 2010;49:793–805. doi: 10.2144/000113519. [DOI] [PubMed] [Google Scholar]
- Kodama Y., Hu C.-D. Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. BioTechniques. 2012;53:285–298. doi: 10.2144/000113943. [DOI] [PubMed] [Google Scholar]
- Lacroix M., El Messaoudi S., Rodier G., Le Cam A., Sardet C., Fabbrizio E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008;9:452–458. doi: 10.1038/embor.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Karanika S., Yang G., Wang J., Park S., Broom B.M., Manyam G.C., Wu W., Luo Y., Basourakos S. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017;10:480. doi: 10.1126/scisignal.aam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.-H., Lee Y.-M., Lee G., Choi Y.-J., Lim B.-O., Kim Y.J., Choi D.-K., Park J.-W. PRMT5 is essential for the eIF4E-mediated 5′-cap dependent translation. Biochem. Biophys. Res. Commun. 2014;452:1016–1021. doi: 10.1016/j.bbrc.2014.09.033. [DOI] [PubMed] [Google Scholar]
- Mahajan S., Raina K., Verma S., Rao B.J. Human RAD52 protein regulates homologous recombination and checkpoint function in BRCA2 deficient cells. Int. J. Biochem. Cell Biol. 2019;107:128–139. doi: 10.1016/j.biocel.2018.12.013. [DOI] [PubMed] [Google Scholar]
- Manthey G.M., Clear A.D., Liddell L.C., Negritto M.C., Bailis A.M. Homologous recombination in budding yeast expressing the human RAD52 gene reveals a Rad51-independent mechanism of conservative double-strand break repair. Nucleic Acids Res. 2017;45:1879–1888. doi: 10.1093/nar/gkw1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G., Eggert C., Bühler D., Brahms H., Kambach C., Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001;11:1990–1994. doi: 10.1016/s0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- Montecucco A., Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2007;252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Murr R., Loizou J.I., Yang Y.-G., Cuenin C., Li H., Wang Z.-Q., Herceg Z. Histone acetylation by Trrap–Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- Paul C., Sardet C., Fabbrizio E. The histone- and PRMT5-associated protein COPR5 is required for myogenic differentiation. Cell Death Differ. 2012;19:900–908. doi: 10.1038/cdd.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C., Sardet C., Fabbrizio E. The Wnt-target gene Dlk-1 is regulated by the Prmt5-associated factor Copr5 during adipogenic conversion. Biol. Open. 2015;4:312–316. doi: 10.1242/bio.201411247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesiridis G.S., Diamond E., Van Duyne G.D. Role of pICLn in methylation of Sm proteins by PRMT5. J. Biol. Chem. 2009;284:21347–21359. doi: 10.1074/jbc.M109.015578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication Forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkinghorn W.R., Parker J.S., Lee M.X., Kass E.M., Spratt D.E., Iaquinta P.J., Arora V.K., Yen W.-F., Cai L., Zheng D. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt E.P.S., Owens J.L., Hockerman G.H., Hu C.-D. Bimolecular fluorescence complementation (BiFC) analysis of protein–protein interactions and assessment of subcellular localization in live cells. Methods Mol. Biol. 2016;1474:153–170. doi: 10.1007/978-1-4939-6352-2_9. [DOI] [PubMed] [Google Scholar]
- Rehman I., Basu S.M., Das S.K., Bhattacharjee S., Ghosh A., Pommier Y., Das B.B. PRMT5-mediated arginine methylation of TDP1 for the repair of topoisomerase I covalent complexes. Nucleic Acids Res. 2018;46:5601–5617. doi: 10.1093/nar/gky291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A. Targeting protein arginine methyltransferase 5 in disease. Future Med. Chem. 2017;9:2081–2098. doi: 10.4155/fmc-2017-0089. [DOI] [PubMed] [Google Scholar]
- Rieger K.E. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res. 2004;32:4786–4803. doi: 10.1093/nar/gkh783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S.P., Drzezga A., Neumaier B., Dietlein M., Gorin M.A., Zalutsky M.R., Pomper M.G. Prostate-specific membrane antigen-targeted radiohalogenated PET and therapeutic agents for prostate cancer. J. Nucl. Med. 2016;57:90S–96S. doi: 10.2967/jnumed.115.170175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J.S., Brady K., Burgan W.E., Cerra M.A., Oswald K.A., Camphausen K., Tofilon P.J. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. 2003;63:7377–7383. [PubMed] [Google Scholar]
- Saha K., Adhikary G., Eckert R.L. MEP50/PRMT5 reduces gene expression by histone arginine methylation and this is reversed by PKCδ/p38δ signaling. J. Invest. Dermatol. 2016;136:214–224. doi: 10.1038/JID.2015.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild D., Wiese C. Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability. Nucleic Acids Res. 2010;38:1061–1070. doi: 10.1093/nar/gkp1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoumanne A., Zhang J., Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009;37:4965–4976. doi: 10.1093/nar/gkp516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu Y.J., Hu C.-D. Fluorescence complementation: an emerging tool for biological research. Trends Biotechnol. 2008;26:622–630. doi: 10.1016/j.tibtech.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Spratt D.E., Evans M.J., Davis B.J., Doran M.G., Lee M.X., Shah N., Wongvipat J., Carnazza K.E., Klee G.G., Polkinghorn W. Androgen receptor upregulation mediates radioresistance after ionizing radiation. Cancer Res. 2015;75:4688–4696. doi: 10.1158/0008-5472.CAN-15-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopa N., Krebs J.E., Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015;72:2041–2059. doi: 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Jiang X., Chen S., Fernandes N., Price B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D.Q., Li Y., Yang C., Li J., Tan S.H., Chin D.W.L., Nakamura-Ishizu A., Yang H., Suda T. PRMT5 modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells. Cell Rep. 2019;26:2316–2328.e6. doi: 10.1016/j.celrep.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Tarighat S.S., Santhanam R., Frankhouser D., Radomska H.S., Lai H., Anghelina M., Wang H., Huang X., Alinari L., Walker A. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2016;30:789–799. doi: 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarish F.L., Schultz N., Tanoglidi A., Hamberg H., Letocha H., Karaszi K., Hamdy F.C., Granfors T., Helleday T. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci. Transl. Med. 2015;7:312re11. doi: 10.1126/scitranslmed.aac5671. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theesfeld C.L., Zyla T.R., Bardes E.G.S., Lew D.J. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol. Biol. Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat. Res. Mutat. Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Treszezamsky A.D., Kachnic L.A., Feng Z., Zhang J., Tokadjian C., Powell S.N. BRCA1- and BRCA2-deficient cells are sensitive to etoposide-induced DNA double-strand breaks via topoisomerase II. Cancer Res. 2007;67:7078–7081. doi: 10.1158/0008-5472.CAN-07-0601. [DOI] [PubMed] [Google Scholar]
- Wang M., Fuhrmann J., Thompson P.R. Protein arginine methyltransferase 5 catalyzes substrate dimethylation in a distributive fashion. Biochemistry. 2014;53:7884–7892. doi: 10.1021/bi501279g. [DOI] [PubMed] [Google Scholar]
- Wei T.-Y.W., Juan C.-C., Hisa J.-Y., Su L.-J., Lee Y.-C.G., Chou H.-Y., Chen J.-M.M., Wu Y.-C., Chiu S.-C., Hsu C.-P. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012;103:1640–1650. doi: 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczek C., Chitta R., Woo E., Shabanowitz J., Chait B.T., Hunt D.F., Shechter D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 2011;286:42221–42231. doi: 10.1074/jbc.M111.303677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Yang X., Duan X., Cui L., Li G. Exogenous expression of marine lectins DlFBL and SpRBL induces cancer cell apoptosis possibly through PRMT5-E2F-1 pathway. Sci. Rep. 2015;4:4505. doi: 10.1038/srep04505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A., Duffy B., Perry J.A., Kornbluth S. DNA replication checkpoint control of Wee1 stability by vertebrate Hsl7. J. Cell Biol. 2004;167:841–849. doi: 10.1083/jcb.200406048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Bedford M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Yang H., Zhao X., Zhao L., Liu L., Li J., Jia W., Liu J., Huang G. PRMT5 competitively binds to CDK4 to promote G1-S transition upon glucose induction in hepatocellular carcinoma. Oncotarget. 2016;7:72131–72147. doi: 10.18632/oncotarget.12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.-T., Zeng L.-F., He Q.-Y., Tao W.A., Zha Z.-G., Hu C.-D. The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim. Biophys. Acta Mol. Cell Res. 2016;1863:335–346. doi: 10.1016/j.bbamcr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S., Moehlenbrink J., Lu Y.-C., Zalmas L.-P., Sagum C.A., Carr S., McGouran J.F., Alexander L., Fedorov O., Munro S. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell. 2013;52:37–51. doi: 10.1016/j.molcel.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq datasets generated in this study are available at the Gene Expression Omnibus (GEO) under accession number GSE111620.