

In this study, Labi et al. examined the role of miRNAs in suppressing apoptosis by targeting miRNA-17~92 involved in regulating the pro-apoptotic protein BIM. Through conditional seed match mutagenesis the authors show that blocking miR-17~92:Bim interaction early in development phenocopied the lethal lung phenotype, produced a selective inability of B cells to resist cellular stress, and prevented B and T cell hyperplasia caused by Bim haploinsufficiency.
Keywords: apoptosis, BIM, miR-17∼92, seed match mutation, B cells, lung development
Abstract
Knockout of the ubiquitously expressed miRNA-17∼92 cluster in mice produces a lethal developmental lung defect, skeletal abnormalities, and blocked B lymphopoiesis. A shared target of miR-17∼92 miRNAs is the pro-apoptotic protein BIM, central to life-death decisions in mammalian cells. To clarify the contribution of miR-17∼92:Bim interactions to the complex miR-17∼92 knockout phenotype, we used a system of conditional mutagenesis of the nine Bim 3′ UTR miR-17∼92 seed matches. Blocking miR-17∼92:Bim interactions early in development phenocopied the lethal lung phenotype of miR-17∼92 ablation and generated a skeletal kinky tail. In the hematopoietic system, instead of causing the predicted B cell developmental block, it produced a selective inability of B cells to resist cellular stress; and prevented B and T cell hyperplasia caused by Bim haploinsufficiency. Thus, the interaction of miR-17∼92 with a single target is essential for life, and BIM regulation by miRNAs serves as a rheostat controlling cell survival in specific physiological contexts.
Mature microRNAs (miRNAs) are 22-nucleotide (nt) noncoding RNAs that regulate gene expression. They affect a wide range of cellular processes involved in organismal development and pathology (Bartel 2018). miRNAs guide the RNA-induced silencing complex (RISC) containing Argonaute (AGO) proteins to partially complementary sequences on messenger RNAs (mRNAs), leading to inhibition of translation and/or accelerated mRNA decay (Bartel 2018). The mammalian genome harbors thousands of miRNA genes, many of which are organized into transcriptionally coregulated clusters such as miR-17∼92.
The specificity of miRNA:mRNA interactions largely depends on the miRNA 6- to 8-nt “seed” region, which exhibits Watson-Crick complementarity with the “seed match” region in the cognate mRNA, located mostly in its 3′ untranslated region (UTR). The identification of conserved seed matches is the key element of in silico miRNA target predictions (Rajewsky 2006; Bartel 2009, 2018), which typically come up with hundreds of potential binding sites across the transcriptome. These predictions can be experimentally validated by measuring gene expression changes (Lim et al. 2005), reporter assays (Krek et al. 2005), directly detecting AGO binding sites and predicting seed matches within these sites (Hafner et al. 2010; Helwak et al. 2013; Grosswendt et al. 2014), or by identifying physical miRNA:target interactions as chimeric reads in AGO cross-linking and immunoprecipitation (CLIP) data (Grosswendt et al. 2014). While such data point to a multifactorial nature of miRNA control, they often fall short of identifying functionally important miRNA:target interactions in specific cellular contexts. Here, small numbers of target genes, even single targets, may be critical, as suggested by studies in the murine hematopoietic system where target gene mutation could mimic or rescue a miRNA knockout phenotype (Xiao and Rajewsky 2009). The vital importance of direct control of a single target gene by a miRNA in development was demonstrated in the nematode Caenorhabditis elegans by seed match mutagenesis (Ecsedi et al. 2015; McJunkin and Ambros 2017), extending earlier experiments in mice where seed match mutation for one particular hematopoietic miRNA, miR-155, had provided direct evidence for major functional roles of distinct single target genes in different immunological contexts (Dorsett et al. 2008; Teng et al. 2008; Lu et al. 2014, 2015). Although miR-155 and hundreds of its mRNA targets are abundantly expressed throughout the immune system, the set of transcripts physically bound by miR-155 is unique to individual immune cell subsets (Hsin et al. 2018).
In the present study, we use conditional seed match mutagenesis of a single, broadly expressed, and physiologically critical target gene, Bcl2l11, to determine the impact of its direct control by a set of equally broadly expressed, vital miRNAs, the miR-17∼92 cluster, in different cellular contexts.
Bcl2l11, encoding the pro-apoptotic protein BCL-2 interacting mediator of cell death (BIM), has evolved in its 3′ UTR seed matches for multiple members of the miR-17∼92 cluster (Koralov et al. 2008; Ventura et al. 2008). BCL-2-family proteins initiate (e.g., BIM, PUMA, BAX) or prevent (e.g., BCL-2, BCL-XL, MCL-1) mitochondrial apoptosis through complex protein interactions, thereby executing life-death decisions under stress conditions and in normal development and tissue homeostasis (Labi et al. 2006; Tuzlak et al. 2016). In mice, combined moderate reduction of MCL-1 and BCL-XL caused developmental apoptosis that could be prevented by the additional loss of a single Bim allele (Grabow et al. 2018). In Caenorhabditis elegans, repression of the BIM homolog EGL-1 through cooperative binding of miR-35 and miR-58 prevented developmental apoptosis (Sherrard et al. 2017).
The miR-17∼92 polycistronic transcript is processed to produce six mature miRNAs categorized into four families of distinct target specificity (Mogilyansky and Rigoutsos 2013); an allelic series of miR-17∼92-mutant mice demonstrated cooperation and specialization among these miRNAs (Han et al. 2015). In line with its abundance, recent work has uncovered broad functions in development and tissue homeostasis (Mogilyansky and Rigoutsos 2013; Labi et al. 2019). Thus, germ line deletion of the miR-17∼92 cluster in mice led to perinatal lethality due to multi-organ failure, prominently improper lung development (Ventura et al. 2008). On the other hand, amplification and overexpression of miR-17∼92 is frequently observed in human cancers (Ota et al. 2004; He et al. 2005; Mogilyansky and Rigoutsos 2013), and its oncogenic properties have been confirmed in mouse models of B cell pathologies (Jin et al. 2013).
BIM has repeatedly emerged as a potential critical miR-17∼92 target (Koralov et al. 2008; Ventura et al. 2008; Molitoris et al. 2011; Li et al. 2014). Thus, targeted deletion of DICER (an enzyme critically involved in miRNA processing) or miR-17∼92 in mice coincided with a strong increase in BIM, massive apoptosis, and a complete block in early B cell development (Koralov et al. 2008; Ventura et al. 2008). This phenotype could be partially rescued by BIM-deficiency or ectopic expression of BCL-2. Overexpression of miR-17∼92 in mouse lymphocytes caused fatal autoimmunity associated with reduced BIM levels (Xiao et al. 2008). However, although supported by reporter assays in vitro, none of these experimental results necessarily reflects direct miR-17∼92:Bim interactions. Other regulators of lymphocyte biology, such as Phosphatase and tensin homolog (PTEN), are also jointly targeted by the miR-17∼92 miRNAs (Mogilyansky and Rigoutsos 2013). Given that BIM transcription can be positively regulated by PTEN (Xiao and Rajewsky 2009), miR-17∼92 may control BIM levels indirectly, via repressing PTEN. Further confusing the situation is evidence from combined knockout and rescue experiments arguing against miR-17∼92-mediated control of BIM and PTEN in B cell development (Lai et al. 2016).
In order to assess the impact of direct miR-17∼92:Bim interactions beyond the B cell lineage and throughout development, we generated a system of conditional mutagenesis in the mouse, which allows exchange of the wild-type Bim 3′ UTR against a miR-17∼92 seed match mutated counterpart in a Cre-dependent manner.
Results
An engineered Bim allele allowing conditional inactivation of miR-17∼92 seed matches
Using a computational approach (Krek et al. 2005), we scored all mouse 3′ UTRs for combinations of binding sites for miR-17∼92 miRNAs. The Bim 3′ UTR was, genome-wide, one of the top scoring 3′ UTRs with nine putative miR-17∼92 sites (Fig. 1A). To collectively disrupt miR-17∼92:Bim interactions, we introduced three point mutations into each of the predicted seed matches in a targeting vector which would allow Cre-mediated replacement of the endogenous wild-type Bim 3′ UTR by its mutant counterpart in vivo (Fig. 1B,C). Correct homologous recombination in the targeted embryonic stem cells was verified by Southern blotting (Supplemental Fig. S1A,B), and after germ line transmission, the mutant Bim locus, designated Bim3′UTRF, was combined with the hematopoietic lineage-specific Vav-cre (de Boer et al. 2003), the B lineage-specific Mb1-cre (Hobeika et al. 2006), and the germline-expressed CMV-cre (Schwenk et al. 1995) transgenes. In the latter case, CMV-cre transgene-negative mice heterozygous for the mutant (Bim3′UTRmut) allele were intercrossed to generate homozygous mutant animals (Bim3′UTRmut/mut) that are defective for miR-17∼92:Bim interactions in all body tissues. PCR on cells from Mb1-cre and Mb1-cre; Bim3′UTRF/F mice demonstrated efficient and selective Cre-mediated 3′ UTR replacement from the early pro-B cell stage on (Fig. 1D; Supplemental Fig. S1C), and Sanger sequencing confirmed the presence of all point mutations (Supplemental Fig. S1D).
Figure 1.

An engineered Bim allele allowing the conditional inactivation of miR-17∼92 seed matches. (A) Scheme depicting the nine putative miR-17∼92 binding sites (A–I) in the Bim 3′ UTR. (B) Conditional in vivo replacement strategy: Upon Cre-mediated excision, the mutated 3′ UTR (red) functionally replaces the wild-type 3′ UTR (black). Blue arrows indicate the genotyping strategy. Targeted allele F(loxed), deleted allele mut(ated), L (loxP), F (frt), BGH3xpA (3 bovine growth hormone polyadenylation sequences). (C) Alternate mutations were introduced into each of the putative Bim 3′ UTR miR-17∼92 seed matches. The mutations were chosen such as not creating de novo seed matches for any known miRNA (miRBase Release 18). Lowercase (mutated nt), black (poorly conserved), red (conserved between mouse and human). (D) Efficient and cell type-specific Cre-mediated Bim 3′ UTR replacement in vivo from early B cell development on (pro-B cells) is shown by PCR on various B cell subsets and myeloid cells FACS-sorted from bone marrow.
Finally, using AGO2 Photoactivatable-Ribonucleoside CLIP (AGO2 PAR-CLIP) technology (Hafner et al. 2010), we assessed whether the seed match mutations introduced into the Bim 3′ UTR indeed precluded interaction with the miR-17∼92 miRNAs. This analysis was done in Abelson Virus transformed pro-B cells (Abl-B cells), which we generated from wild-type and Bim3′UTRmut/mut E14.5 fetal livers knowing that these cells express both BIM and miR-17∼92, can be expanded to large numbers (Rosenberg et al. 1975), and incorporate 4-thiouridine (4SU) sufficiently well (Supplemental Fig. S2A–D). Focusing on 21-nt windows surrounding the nine putative miR-17∼92 seed matches in the Bim 3′ UTR (A-I) and excluding reads lacking T-to-C transitions, we found differential seed match coverage in the wild type, with one miR-19 and two miR-92 seed matches codominating, while in the mutant 3′ UTR miR-17∼92 binding was completely abolished (Fig. 2A,B; Supplemental Fig. S2E,F). Significant differential coverage was not found in the 3′ UTR of Pten, another top scoring “combinatorial” miR-17∼92 target gene (Fig. 2C). Bim 3′ UTR mutagenesis did not affect the concentrations of mature miRNAs including miR-17∼92 in Abl-B cells (Fig. 2D).
Figure 2.

Differential AGO2 PAR-CLIP-sequencing shows loss of miR-17∼92:Bim interactions and reveals the dominant interaction sites. (A) PAR-CLIP on wild-type (top) and Bim3′UTRmut/mut (bottom) Abl-B cells confirms loss of miR-17∼92:Bim interactions in seed match mutated cells. Each of the nine 21-nt seed match windows (A–I) represents one Bim 3′ UTR miR-17∼92 binding site. Colored nucleotides (blue, red) represent seed matches, surrounding nucleotides are gray. 151 and 35 indicate maximum coverage of the respective nt, part of a candidate miR-24-3p binding site that overlaps with the seed match window of site E; the significance of the differential coverage of this nt is uncertain. Small RNAseq read coverage correlates with AGO2 binding. One of two biological replicates per genotype is shown. (B) Scatterplot depicts coverage statistics for 41-nt windows surrounding miR-17∼92 seed matches found within unique 3′ UTRs of protein-coding genes. Red dots indicate significantly differentially covered windows (P < 0.05), enlarged red dots miR-17∼92:Bim seed matches. Combined analysis of both biological replicates is shown. (C) PAR-CLIP confirms unaltered miR-17∼92:Pten interactions in Bim 3′ UTR seed match mutated cells. Each of the six 21-nt seed match windows depicts one putative miR-17∼92 seed match in the Pten 3′ UTR. Analysis of the biological replicate depicted in A is shown. (D) The Bim3′UTRmut/mut (n = 5) Abl-B cells show no change in the abundance of mature miRNAs as compared to wild-type (n = 5) Abl-B cells (nCounter gene expression analysis, Pearson correlation).
Together, these results demonstrate functionality and efficiency of our mouse model for in vivo conditional seed-match inactivation.
Disruption of miR-17∼92:Bim 3′ UTR interactions in the germ line leads to neonatal lethality
When mice carrying one mutant Bim 3′ UTR allele in the germ line were inter-crossed, no homozygous mutant pups were identified at weaning, suggesting that the disruption of miR-17∼92:Bim interactions early in development is lethal. Timed mating between Bim3′UTRmut/+ animals resulted in embryonic genotypes consistent with Mendelian ratios until E18.5 (Supplemental Fig. S3A), indicating that Bim3′UTRmut/mut pups die during or after birth. We therefore recovered E18.5 embryos by Caesarean section (CS). In six litters examined, all wild-type and heterozygous animals established rhythmic breathing, lively movement, and a pink skin color within minutes. In contrast, Bim3′UTRmut/mut neonates took short, gasping breaths, showed continuous signs of respiratory distress, quickly became cyanotic, and died within the first 30 postnatal minutes. Overall, taking 21 litters of timed heterozygous intercrosses into account, from a total of 101 offspring at weaning only four mice were Bim3′UTRmut/mut, of which one died at the age of 123 d for unknown reasons (Fig. 3A,B; Supplemental Fig. S3A). The three survivors displayed no obvious gross defects except for their small size (and correspondingly small-size organs; not shown), a kinky tail, and slightly decreased red blood cell numbers at the age of 8 wk (Fig. 3C). They harbored normal numbers of immune cells of various subsets in lymphoid tissues (Supplemental Table S1).
Figure 3.

Disruption of miR-17∼92:Bim 3′ UTR interactions in the germ line leads to neonatal lethality. (A) Expected and observed numbers and fractions (in parentheses) of offspring from Bim3′UTRmut/+ intercrosses. (B) E18.5 neonates 30 min after Caesarean recovery. Bim3′UTRmut/mut neonates show marked cyanosis. (C) Bim3′UTRmut/mut survivors were analyzed at 8 wk. Photograph is representative (n = 3–4 mice per genotype). (D) Bim3′UTRmut/mut E18.5 embryos display reduced body weight as compared to their siblings (n = 15–30 mice per genotype). (E) Representative photographs of Alcian blue- and Alizarin red-stained skeletons of E18.5 wild-type and Bim3′UTRmut/mut embryos (n = 4–5 mice per genotype). (F) E18.5 Bim3′UTRmut/mut embryos display reduced lung weight and lung/body weight ratio compared to their siblings (n = 15–30 mice per genotype). (**) P < 0.01, (***) P < 0.005, (****) P < 0.0001. Data are presented as mean ± SD.
E18.5 Bim3′UTRmut/mut embryos displayed moderately reduced body weight compared to their wild-type siblings (Fig. 3D) and a kinky tail phenotype with 100% penetrance (Fig. 3E), an abnormality that we also found in the rare survivors (Fig. 3C). Cell counts of liver, thymus, platelets, white and red blood cells, and hemoglobin levels were normal, suggesting normal blood formation and oxygenation before birth (Supplemental Fig. S3B,C) and in adult survivors (Supplemental Table S1). Because of the respiratory distress of Bim3′UTRmut/mut newborns, we paid special attention to the innervation of the diaphragm and brainstem respiratory neurons but failed to detect any alterations (Supplemental Fig. S3D,E). As E18.5 Bim3′UTRmut/mut embryos had significantly reduced lung weight (Fig. 3F), we turned our attention to the lungs themselves.
miR-17∼92:Bim interactions are essential for lung development
Consistent with the respiratory distress and reduced organ weight, lungs from Bim3′UTRmut/mut E18.5 embryos were of hypoplastic appearance and showed collapsed alveolar spaces and increased tissue density (Fig. 4A; Supplemental Fig. S4A). In E18.5 Bim3′UTRmut/mut hearts, dilatation of the atria and hypertrophy of the ventricles were noticed (Supplemental Fig. S4A). This phenotype might be caused by hypertension in the pulmonary circulation due to the lack of lung expansion. When placed in PBS 15 min after CS, lungs isolated from E18.5 wild-type neonates floated whereas lungs from mutant animals sank, indicating that no aeration had occurred (Supplemental Fig. S4B). Consistently, lungs from Bim3′UTRmut/mut neonates did not show evidence of air in the distal airways (Fig. 4B).
Figure 4.

miR-17∼92:Bim interactions are essential for lung development. (A) Representative H&E-stained sections of E18.5 wild-type and Bim3′UTRmut/mut lungs (n = 3–4 mice per genotype). Scale bar: 200 µm. (B) Fetal lungs isolated from E18.5 neonates 15 min upon exteriorization. Wild-type embryos aerate their lungs (left), Bim3′UTRmut/mut embryos insufficiently aerate their lungs (right). Representative photographs are shown (n = 3 mice per genotype). Scale bar: 1 mm. (C) qRT-PCR analysis for Bim mRNA expression in total organ extracts of fetal liver and lung of wild-type and Bim3′UTRmut/mut E18.5 littermate embryos (n = 4 mice per genotype). (D,E) Immunoblot for BIM and PTEN protein in lung extracts of wild-type and Bim3′UTRmut/mut E18.5 littermate embryos (n = 4–5 mice per genotype). (F) E18.5 Bim3′UTRmut/mut lungs (n = 5) show no change in the global levels of mature miRNAs as compared to wild-type (n = 4) lungs (nCounter gene expression analysis, Pearson correlation). (*) P < 0.05, (**) P < 0.01. Data are presented as mean ± SD.
Proteomic analysis revealed, among others, an underrepresentation of CC10 (P = 0.0003) (Stripp et al. 1992), surfactant protein A1 (P = 0.0271) (Korfhagen et al. 1996), and SLC34A2 (P = 0.0174) (Feild et al. 1999), indicative of distal airway cell depletion, specifically of club cells and epithelial type II pneumocytes in Bim3′UTRmut/mut lungs (Supplemental Table S2).
If miR-17∼92-mediated BIM repression is fundamental to the observed lung phenotype, BIM levels should be higher in E18.5 Bim3′UTRmut/mut lungs as compared to controls. Indeed, Bim mRNA expression was elevated in the mutant lungs, a difference not observed in fetal liver, an organ without apparent phenotypic alterations (Fig. 4C). This correlated with elevated BIM protein in Bim3′UTRmut/mut lungs (Fig. 4D,E), whereas expression of miR-17∼92 miRNAs remained unchanged (Fig. 4F). Immunohistochemistry revealed a moderate, albeit not significant, increase in the number of apoptotic cells in the pulmonary parenchyma and the bronchial epithelium of E18.5 Bim3′UTRmut/mut lungs (Supplemental Fig. S4A).
The inability of Bim3′UTRmut/mut neonates to aerate their lungs to a functional level phenocopies the lung hypoplasia and resulting lethality in miR-17∼92−/− neonates (Ventura et al. 2008; Han et al. 2015). Altogether, our findings provide in vivo evidence that repression of BIM by miR-17∼92 is critical for proper lung development and of vital importance.
Hematopoiesis including B lymphopoiesis is not detectably affected upon disruption of miR-17∼92:Bim 3′ UTR interactions in vivo
BIM limits the survival of hematopoietic stem/progenitor cells during transplantation or cytokine deprivation (Nordigården et al. 2009; Labi et al. 2013; Kollek et al. 2017). However, restricting seed match mutagenesis to the hematopoietic system using a Vav-cre transgene, we failed to detect phenotypic alterations in hematopoietic progenitors or their progeny (Fig. 5A–C), in accord with our observations on the rare survivors of miR-17∼92 seed match mutation in the germ line (Supplemental Table S1). PCR on cells FACS-sorted from Vav-cre;Bim3′UTRF/+ and Vav-cre;Bim3′UTRF/F mice demonstrated efficient and selective Cre-mediated 3′ UTR replacement in hematopoietic progenitors (LSK cells), pro-B cells, and mature B and T cells (Fig. 5D; Supplemental Fig. S5A). These results are in line with previous reports suggesting no major role for miR-17∼92 in hematopoiesis, with the exception of the severe block of B cell development accompanied by massive BIM upregulation, in miR-17∼92 knockouts (Koralov et al. 2008; Ventura et al. 2008).
Figure 5.

Hematopoiesis is not detectably affected upon disruption of miR-17∼92:Bim interactions. (A) Flow cytometry analysis of bone marrow of Vav-cre and Vav-cre;Bim3′UTRF/F mice: LSK (Ter119−CD11b−B220−NK1.1−CD3−Sca-1+cKit+), CLP (Ter119−CD11b−B220−NK1.1−CD3−Sca-1intcKitint), CMP (Ter119−CD11b−B220−NK1.1−CD3−Sca-1−cKit+CD34+CD16/32−), GMP (Ter119−CD11b−B220−NK1.1−CD3−Sca-1−cKit+CD34+CD16/32+), MEP (Ter119−CD11b−B220−NK1.1−CD3−Sca-1−cKit+CD34−CD16/32−), MPP (Ter119−CD11b−B220−NK1.1−CD3−Sca-1+cKit+CD34+CD135+), ST-HSC (Ter119−CD11b−B220−NK1.1−CD3−Sca-1+cKit+CD34+CD135−), LT-HSC (Ter119−CD11b−B220−NK1.1−CD3−Sca-1+cKit+CD34−CD135−), HSC SLAM (Ter119−CD11b−B220−NK1.1−CD3−Sca-1+cKit+CD48−CD150+); (n = 4–6 mice per genotype). (B) Flow cytometry analysis of thymocyte subsets of Vav-cre and Vav-cre;Bim3′UTRF/F mice: DN1 (Ter119−CD11b−B220−NK1.1−gδTCR−CD4−CD8−CD25−CD44+), DN2 (Ter119−CD11b−B220−NK1.1−gδTCR−CD4−CD8−CD25+CD44+), DN3 (Ter119−CD11b−B220−NK1.1−gδTCR−CD4−CD8−CD25+CD44−), DN4 (Ter119−CD11b−B220−NK1.1−gδTCR−CD4−CD8−CD25−CD44−), DP (CD4+CD8+), CD4SP (CD4+CD8−), CD8SP (CD4−CD8+); (n = 4–6 mice per genotype). (C) Flow cytometry analysis of spleens of Vav-cre and Vav-cre;Bim3′UTRF/F mice: B cells (B220+CD19+), CD4+ T cells (TCRβ+NK1.1−CD8−CD4+), CD8+ T cells (TCRβ+NK1.1−CD4−CD8+), natural killer (NK) cells (TCRβ−NK1.1+), myeloid (MY) cells (CD11b+); (n = 4–6 mice per genotype). (D) FACS-sorted LSK and pro-B cells were analyzed by PCR genotyping (two mice/genotype). LSK (Lineage−Sca1+cKit+), CLP (common lymphoid progenitors), CMP (common myeloid progenitors), GMP (granulocyte-monocyte progenitors), MEP (megakaryocyte-erythroid progenitors), MPP (multipotent progenitors), ST-HSC (short-term hematopoietic stem cells), LT-HSC (long-term HSC), HSC SLAM (long-term HSC; SLAM markers), DN (double negative), DP (double positive), SP (single positive). Eight- to 12-wk-old mice were analyzed. Data are presented as mean ± SD.
To follow up on this discrepancy, we performed a detailed analysis of B lymphopoiesis in mice in which miR-17∼92:Bim 3′ UTR interaction was selectively ablated in the B cell lineage through the Mb1-cre transgene. Young Mb1-cre;Bim3′UTRF/F mice showed no gross defects in steady-state B cell development as suggested by normal percentage (Fig. 6A; Supplemental Fig. S5B) and number (Supplemental Fig. S5B) of pro-B, pre-B, and immature IgM+ bone marrow B cells. Similarly, peripheral maturation (transitional T1 and T2 B cells) and homeostasis of mature B cells (follicular [FO] B cells and marginal zone [MZ] B cells) in the spleen (Fig. 6B; Supplemental Fig. S5C) as well as recirculating mature B cells in the bone marrow (Fig. 6A; Supplemental Fig. S5B) were unaffected. A similar situation was encountered in aged Mb1-cre;Bim3′UTRF/F mice (Supplemental Fig. S5D,E). Although we could not detect significant changes of Bim mRNA levels in various B cell subsets in comparison to control cells (Fig. 6C), BIM protein abundance determined by flow cytometry was moderately increased in pro- and pre-B cells ex vivo (Fig. 6D; Supplemental Fig. S6A). However, this did not translate into significantly more apoptotic cells (Supplemental Fig. S5F).
Figure 6.

Specific disruption of miR-17∼92:Bim interactions does not detectably affect B cells. (A,B) Flow cytometry plots depict bone marrow B cell subsets: total (B220+CD19+), mature recirculating (B220hiCD19+AA4.1−IgM+), immature IgM+ (B220loCD19+AA4.1+IgM+), pre-B (B220loCD19+AA4.1+IgM−CD25+ckit−), and pro-B (B220hiCD19+AA4.1+IgM−CD25−ckit+); and splenic B cell subsets: total B2 (B220+CD19+), follicular (FO) (B220+CD19+AA4.1−IgMlow/+CD1dlow/+), marginal zone (MZ) (B220+CD19+AA4.1−IgMhighCD1dhigh), transitional 1 (T1) (B220+CD19+AA4.1+IgMhighCD23−), and transitional 2 (T2) (B220+CD19+AA4.1+IgMhighCD23+). Red arrows show gating strategy, numbers adjacent to regions represent percent within parent population. Plots are representative of 4–10 mice per genotype. (C) qRT-PCR for Bim expression in FACS-sorted pro-B, large pre-B (FSC-Ahigh), small pre-B (FSC-Alow), and immature IgM+ B cells from control (either Mb1-cre or Vav-cre) and either Mb1-cre;Bim3′UTRF/F or Vav-cre;Bim3′UTRF/F mice (n = 6–10 mice per genotype). (D) Flow cytometry analysis of BIM protein in B cell subsets of the indicated genotypes. Colors represent samples that were processed as a mixture with the same wild-type control cells carrying a hCD2 reporter (n = 3 mice per genotype). (MFI) Mean fluorescence intensity. (E) Pro-B/pre-B ratio is based on flow cytometry analysis of mice of the indicated genotypes (n = 4–7 mice per genotype). (F) Number of splenic FO B, CD4+, and CD8+ T cells of wild-type, germline heterozygous Bim3′UTR mutant (Bim3′UTRmut/+), Bim+/−, and haploinsufficient Bim mice that harbor the germline mutated Bim 3′ UTR in the remaining Bim allele (Bim3′UTRmut/−) (n = 5–7 mice per genotype). (G) Flow cytometry analysis of BIM protein in splenic FO B cells. Samples were processed as a mixture with Ly5.1 wild-type splenocytes. BIM expression was normalized to Ly5.1 FO B cells in the same tube (n = 5–23 mice per genotype). (*) P < 0.05, (**) P < 0.01, (***) P < 0.005. Data are presented as mean ± SD; 7- to 9-wk-old mice were analyzed.
Additional attempts to find evidence for an impact of miR-17∼92:Bim interactions in the B cell compartment also failed. Thus, Bim heterozygosity did not overcome the block at the pro- to pre-B cell transition caused by miR-17∼92 deficiency (Fig. 6E). There were also no differences in the survival of splenic B cells from Mb1-cre and Mb1-cre;Bim3′UTRF/F mice upon in vitro exposure to B cell activating factor (Baff) (Supplemental Fig. S6B) or upon activation by α-IgM antibodies plus interleukin-4 (IL4) or lipopolysaccharide (LPS) (Supplemental Fig. S6C). In all these conditions, loss of BIM had been reported to exert anti-apoptotic activity (Oliver et al. 2006; Woess et al. 2015).
Bim heterozygosity results in lymphoproliferation which is reverted by miR-17∼92 seed match mutation
While lymphopoiesis proceeded normally in the absence of miR-17∼92:Bim interactions, this was not the case in Bim heterozygous mice, which displayed lymphocyte hyperplasia, in accord with earlier work (Fig. 6F; Bouillet et al. 1999; Liu et al. 2018). In this situation, miR-17∼92 seed match mutation in the functional Bim allele corrected the B and T cell hyperplasia (Fig. 6F) and BIM levels in FO B cells. This supports the concept developed by Mukherji and colleagues (Mukherji et al. 2011) that the relative concentrations of miRNAs and their target mRNAs determine thresholds of miRNA control and indicates that, in wild-type mouse lymphocytes, the relative Bim mRNA levels are too high for efficient direct miR-17∼92 control, in contrast to the situation in the heterozygous Bim mutants.
Repression of Bim by miR-17∼92 promotes progenitor B cell fitness during stress responses
In accord with our demonstration that in wild-type mice, miR-17∼92:Bim interactions do not affect B lymphopoiesis in steady-state, they also appeared irrelevant in competitive bone marrow transplantation experiments (Supplemental Fig. S7A), where loss of BIM is advantageous for the fitness of most hematopoietic cell types (Labi et al. 2013). However, a different picture emerged when we FACS-sorted pro-B cells from the bone marrow of Mb1-cre and Mb1-cre;Bim3′UTRF/F mice and cultivated these cells in the presence of IL-7 (Hardy and Hayakawa 2001). Under these conditions, pro-B cells proliferate yet also undergo apoptosis, which ultimately causes the collapse of the cultures. As evidenced by Annexin V staining, Mb1-cre;Bim3′UTRF/F pro-B cells displayed increased fractions of apoptotic cells when compared to Mb1-cre control cells (Fig. 7A; Supplemental Fig. S7B). This corresponded to a substantial reduction of cell numbers over time (Fig. 7B; Supplemental Fig. S7C). Increased apoptosis coincided with elevated BIM protein in Mb1-cre;Bim3′UTRF/F pro-B cells (Fig. 7C), and genotyping confirmed that the Mb1-cre;Bim3′UTRF/F pro-B cells indeed carried the Bim mutant alleles (Supplemental Fig. S7D). Thus, disruption of miR-17∼92:Bim 3′ UTR interactions sensitizes pro-B cells to stress elicited by ex vivo cultivation.
Figure 7.
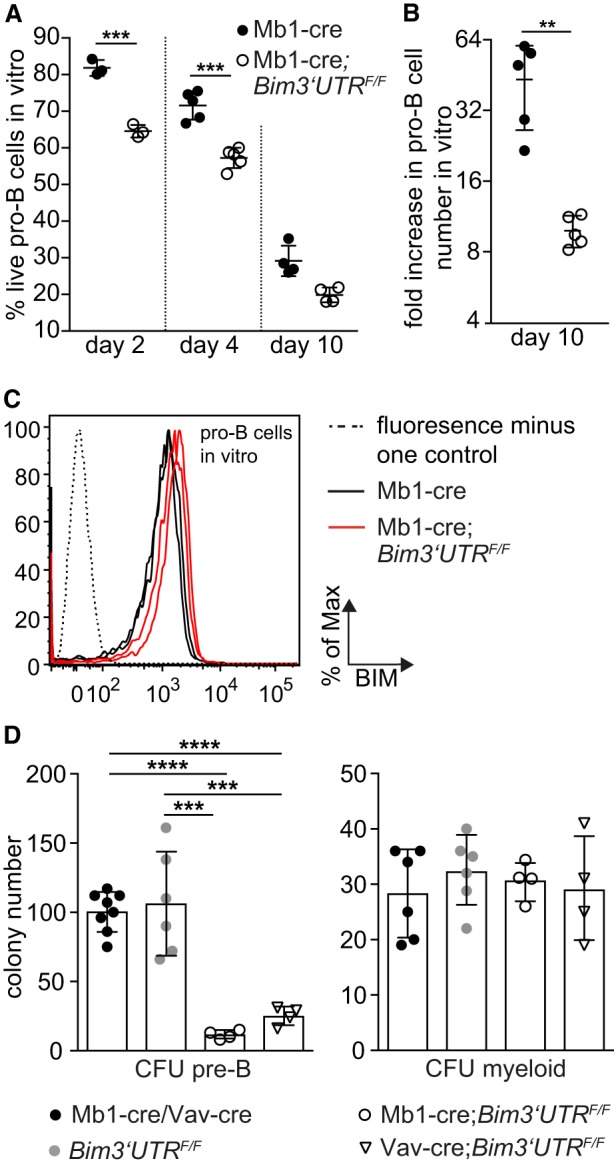
miR-17∼92-mediated repression of BIM promotes the fitness of B cell progenitors during stress responses. (A,B) FACS-sorted pro-B cells (B220hiCD19+AA4.1+IgM−CD25−ckit+) were placed in medium with IL-7. Live cells were defined as AnnexinV−TO-PRO-3− and analyzed by flow cytometry over time (n = 3–5 mice per genotype). (C) Representative flow cytometry analysis of BIM protein expression in pro-B cells from two Mb1-cre and Mb1cre;Bim3′UTRF/F mice, respectively, at culture day 4 (n = 6 mice per genotype). (D) Ex vivo colony formation of bone marrow pro-/pre-B cells (CFU-pre-B) or myeloid cells (CFU-myeloid) (n = 4–8 mice per genotype). (CFU) Colony forming unit. (**) P < 0.01, (***) P < 0.005, (****) P < 0.0001. Data are presented as mean ± SD.
This became most dramatically apparent when we assessed the in vitro colony forming potential of B cell progenitors in semisolid medium. Compared to the controls, Mb1-cre;Bim3′UTRF/F and Vav-cre;Bim3′UTRF/F bone marrow displayed strongly diminished colony formation (Fig. 7D; CFU pre-B), with a fraction of the cells having (partially) escaped 3′ UTR replacement (Supplemental Fig. S7E). Reduced colony formation was restricted to B cells, as under myeloid priming conditions all genotypes produced equal colony numbers (Fig. 7D; CFU myeloid).
Discussion
While miRNAs may fine tune transcriptional and signaling networks (Bartel and Chen 2004), there is also evidence that miRNA control of single key target genes can play a critical functional role in vivo. Apart from more indirect approaches (Xiao and Rajewsky 2009), most convincing in this regard are experiments in which such genes were genetically identified through miRNA seed match mutation (Dorsett et al. 2008; Teng et al. 2008; Kadener et al. 2009; Bassett et al. 2014; Lu et al. 2014, 2015; Ecsedi et al. 2015; McJunkin and Ambros 2017). These experiments primarily addressed miRNA control in specific biological contexts where candidate target genes emerged from the informed inspection of computational predictions. The motivation for the present study arose in a similar situation, namely when we discovered that miRNA control was essential to prevent apoptosis during B cell development (Koralov et al. 2008). From this and parallel work (Ventura et al. 2008), it became clear that one cluster of miRNAs, miR-17∼92, played a prominent role in this process, with the pro-apoptotic BIM protein being a potential key functional target, given the evolutionarily conserved “combinatorial” accumulation of miR-17∼92 seed matches in the Bim gene 3′ UTR.
Considering the close to ubiquitous expression of both the miR-17∼92 miRNAs and BIM, albeit at varying levels, the broad impact of BIM on cellular life-death decisions, the dramatic developmental phenotypes upon miR-17∼92 knock-out, and widespread miR-17∼92 overexpression in human cancers, the problem gained a broader perspective. We therefore set out to explore to what extent miR-17∼92:Bim interactions control apoptosis in and beyond the hematopoietic system by generating a mutant Bim allele in which the 3′ UTR can be conditionally exchanged against a mutant counterpart with all miR-17∼92 seed matches inactivated. PAR-CLIP experiments demonstrated that the selected seed match mutations indeed prevented miRNA binding to the Bim 3′ UTR. This corresponded to increased levels of Bim mRNA and protein in various (mutant) cell types analyzed in later experiments, although averaging over cell populations, the effects varied and rarely exceeded a factor of two.
We note that we have not yet explored the significance of the joint targeting of the Bim 3′ UTR by multiple miR-17∼92 miRNAs. While this feature is expected to enhance the functional efficiency and specificity of the interactions there is also evidence that individual miRNAs of the miR-17∼92 cluster exert distinct regulatory functions. Curiously, we did not detect binding of miR-17-5p to the Bim 3′ UTR in our PAR-CLIP analysis of B cells, despite the abundance of this miRNA and evolutionarily conserved seed matches in the Bim 3′ UTR. Perhaps miR-17-5p binding would be detectable in developing cells of the lung, given that combined knockout of miR-17-5p and miR-92 resulted in embryonic lethality (Han et al. 2015).
Does elimination of miR-17∼92 seed matches in the Bim 3′UTR have physiological consequences and to what extent does it mirror the various effects of miR-17∼92 ablation? Strikingly, Bim seed match mutagenesis in the germ line reproduced the perinatal lethality of miR-17∼92 knockout mice, due in both cases to a failure of the newborns to inflate their lungs. Our experimental evidence indicates that this failure was lung-intrinsic and likely reflects dysregulation at late stages of lung development. This scenario most easily accommodates the rare Bim3′UTRmut/mut long-term survivors, where the lungs did not exhibit detectable histological abnormalities (data not shown). Apart from a lung phenotype with >80% penetrance and reduced body size, Bim3′UTRmut/mut mice displayed only a single additional macroscopically detectable phenotype, namely a kinky tail. Tail deformation has recently been observed in mice upon genetic interference with apoptosis control during development (Ke et al. 2018), and miR-17∼92 deficiency has been linked to skeletal abnormalities in humans and mice (de Pontual et al. 2011; Han et al. 2015). It is therefore quite in order to find a pro-apoptotic miR-17∼92 target critically involved in this context. Note, however, that both in Feingold syndrome patients and in miR-17∼92 heterozygous or homozygous knockout mice, the skeletal abnormalities mainly affected fingers and toes; and that the kinky tail phenotype was absent in the miR-17∼92 knockout embryos (Ventura et al. 2008). While the comparison of the mouse mutants is not straightforward because of major differences of their genetic backgrounds, miR-17∼92 may well affect skeletal development through more than a single target gene. Most importantly, however, mice harbor two miR-17∼92 paralog gene clusters whose concomitant knockout results in early embryonic lethality (Ventura et al. 2008), precluding the identification of tail phenotypes.
In contrast to the lung and skeletal phenotypes, the hematopoietic system was not detectably affected at steady state by ablation of miR-17∼92:Bim interactions. This seemingly contrasts the severe block of B cell development in miR-17∼92 or DICER-deficient animals with massive BIM up-regulation in the affected cells (Koralov et al. 2008; Ventura et al. 2008). However, in the case of DICER deficiency, BIM ablation only moderately rescued B cell development, and the extent of BIM up-regulation in both miR-17∼92 or DICER-ablated B cell progenitors by far exceeded the modest effects we see upon elimination of miR-17∼92:Bim interactions in other cells. We conclude that in the context of B cell development, miR-17∼92 controls cell survival through mechanisms other than direct BIM control. This also applies to the loss of hematopoietic stem/progenitor and B cells in a model of acute miR-17∼92 deletion in adult mice and its partial rescue by BIM deficiency (Brinkmann et al. 2018), given that these cell compartments are unaffected in our mouse model.
Unexpectedly, however, striking deficits in cellular fitness became apparent when mutant B cells that showed no phenotype under physiological conditions were exposed to stress conditions exemplified by in vitro cultivation. Here again, the phenotype was specific for cellular context in that it did not extend to myeloid cells. Decreased stress tolerance and the associated reduced cellular fitness, recurrent themes in the field of miRNA control (Ebert and Sharp 2012), are likely to play a role in vivo where B cells must compete and respond to internal and external stimuli.
Taken together, the present study identifies, through conditional seed match mutagenesis, direct miRNA regulation of BIM expression as a rheostat that controls cell survival in a variety of distinct and diverse cellular and developmental contexts which are hard to predict. We make the case for the mouse miR-17∼92 cluster, members of which collectively target the Bim 3′ UTR in an evolutionarily conserved manner, by showing that the corresponding interactions are critical for life in lung development, control tail morphology, and allow the survival of B cell progenitors under stress conditions. This rheostatic control may depend on BIM concentration: While it plays no role in normal T and B cell development, the T and B cell hyperplasia seen in Bim haploinsufficient animals is corrected upon ablation of miR-17∼92:Bim interactions. Further studies will be needed to test whether the cytoprotective miR-17∼92:Bim relationship operates in other cellular and developmental contexts where BCL-2 family proteins are critical regulators of cell survival. This seems indeed likely as most cells in our body can be subject to mitochondrial apoptosis and the Bim 3′ UTR is a putative target of multiple miRNAs in addition to miR-17∼92 (www.targetscan.com). One of these miRNAs, miR-24, has been specifically invoked in the control of cardiomyocyte survival, with Bim as a potential target gene (Guo et al. 2015).
Finally, in a more general context, the present work reveals the impact and unforeseen complexity of miRNA control at the level of a single, vitally important target gene, demonstrating that the analysis of direct miRNA interactions with individual key targets is essential for the understanding and rational manipulation of miRNA control.
Materials and methods
Mice
Flpe (Dymecki 1996), Mb1-cre (Hobeika et al. 2006), Vav-cre (de Boer et al. 2003), CMV-cre (Schwenk et al. 1995), Bim− (Bouillet et al. 1999), R26hCD2stopF (Calado et al. 2012), and miR-17∼92F (Ventura et al. 2008) mice have been described. Ly5.1 mice were purchased (JAX 002014). Bim3′UTRF mice were generated by us. Genotyping primers for the Bim3′UTR wild-type, floxed, and mutant alleles are detailed in Supplemental Table S3. Cycle conditions: 95°C/5 min; 30 cycles: 95°C/30 sec, 62°C/45 sec, 72°C/45 sec; 72°C/7 min. Animal procedures were approved by the official review boards (Landesamt für Gesundheit und Soziales Berlin, LaGeSo G0374/13; Harvard Medical Area Standing Committee on Animals, #4M0310).
Generation of conditional Bim3′ UTR mutant mice
To collectively disrupt all miR-17∼92:Bim interactions, we mutated three alternate bases in each of the nine PicTar-predicted (pictar.mdc-berlin.de) Bim 3′ UTR miR-17∼92 seed matches by PCR-mediated mutagenesis using primers detailed in Supplemental Table S3. We flanked a 4.9-kb genomic fragment including the entire wild-type Bim exon 4 with loxP sites, which were inserted into nonconserved intronic sequences. To prevent transcription from the wild-type to the mutant exon, we inserted three identical copies of the bovine growth hormone polyadenylation sequence (BGH3XpA) downstream of the wild-type exon. This construct was flanked by two arms of homology, which mediated homologous recombination with the endogenous locus in C57BL/6-derived mouse embryonic stem (ES) cells (Artemis). Identification of neomycin-resistant ES cell clones with a single correct insertion was performed by Southern hybridization. Two independently targeted clones were injected into C57BL/6 albino blastocysts to generate chimeric mice from which germ-line-transmitted animals were obtained by crossing to C57BL/6 mice. Offspring were crossed with FLPe mice to remove the frt-flanked PGK-neomycin sequence and obtain the mice used in this study.
Timed matings and embryos
C-sections were performed to recover E13.5–E18.5 embryos for histology and tissue harvest. Pictures were captured using a binocular microscope (Leica) equipped with a DFC495 digital camera.
Peripheral blood analysis
Blood analysis was performed with a Hemavet HV960FS (Greiner) for E18.5 embryos and a scilVetABC Hematology Analyzer (GML) for adult mice.
Flow cytometry and cell sorting
Unless otherwise stated, staining buffer phosphate buffered saline (PBS) with 3% (v/v) fetal bovine serum (FBS; Gibco 10270-106) was used for preparation, washing, and staining steps.
Single-cell suspensions were prepared by homogenizing spleen through 70-µM filters (Falcon 352350), or flushing both femurs and tibiae using a 23G needle. For erythrocyte lysis, splenocyte suspensions were incubated for 3 min in lysis buffer (pH 7.5) on ice (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). Single-cell suspensions were filtered through a 50-µM cup filcon cell numbers were determined using a hemocytometer (Neubauer) and trypan blue exclusion.
For ex vivo flow cytometry analysis, 3 × 106 cells per staining were incubated with 1 µg/mL αCD16/32 Fc-Block (BioLegend 101320) for 10 min at 4°C and washed and stained for 20 min at 4°C with monoclonal antibody cocktails as detailed in the Supplemental Materials and Methods. Apoptosis was assessed by resuspending cells in Annexin-V-FITC (BioLegend 640945, 1:800) or Annexin-V-eF450 (eBiosciences 48-8006-69, 1:1000) and 10 nM TO-PRO-3 (Invitrogen T3605) in Annexin-V binding buffer (BioLegend 422201) prior to analysis.
For intracellular BIM staining, 3 × 106 cells were cell-surface stained as described above, fixed (BioLegend 420801), and permeabilized (BioLegend 421002) as per the manufacturer's instructions. Cells were incubated with 2 µg/mL Fc-Block in 50 µL permeabilization buffer at 4°C. After 30 min, 50 µL antibody dilution (Abcam ab32158, 1:100) in permeabilization buffer was added and cells were incubated for 30 min. Cells were washed twice with permeabilization buffer and incubated for 30 min with either AlexaFluor-488 goat α-rabbit (Invitrogen A11034, 1:1000) or AlexaFluor-647 goat α-rabbit (Invitrogen A21245, 1:1000) secondary antibody in 50 µL permeabilization buffer. Cells were washed twice with permeabilization buffer and resuspended in staining buffer.
Flow cytometry data were acquired on an LSRII cytometer (BD Biosciences) and analyzed with the FlowJo software (Treestar). Nonsinglet events were excluded based on characteristics of forward and side scatter.
For cell sorting, single-cell suspensions were pre-incubated with 2 µg/mL Fc-Block in 500 µL staining buffer for 10 min at 4°C and washed and stained for 20 min at 4°C with fluorochrome-labeled antibodies in 500 µL staining buffer.
B cell culture
Primary B cells and Abl-B cells were cultured at 37°C in B cell medium: DMEM (Sigma, WHMISDZB) supplemented with 10% (v/v) FBS (Gibco 10270-106), 2 mM L-glutamine (Sigma G7513), 10 mM HEPES (LONZA BE17-737E), 1 mM sodium pyruvate (Gibco 13360-039), nonessential amino acids (Gibco 11140-035), 100 U/mL penicillin/100 µg/mL streptomycin (Sigma 0781), and 50 µM β-mercapto-ethanol (Sigma M3148).
Clonogenicity assay
Colony-forming assays were performed by plating 200,000 bone marrow cells on semisolid medium containing IL-7 (Stem Cell Technologies M3630) for pre-B cells, or by plating 20,000 bone marrow cells on semisolid medium containing 10 ng/mL G-CSF (Peprotech 250-05), 10 ng/mL M-CSF (Peprotech 315-02), 10 ng/mL IL-3 (Peprotech 215-13), and 10 ng/mL IL-6 (Peprotech 216-16) for myeloid progenitors (Stem Cell Technologies M3231) following the manufacturer's instructions. Colony-forming units were enumerated after 7 (preB) or 14 d (myeloid).
Quantitative RT-PCR
RNA of snap-frozen cell pellets or E18.5 lung tissue ground into powder in liquid nitrogen was extracted using the miRNeasy (QIAGEN 217004) or RNeasy mini kit (QIAGEN 74104) as per the manufacturer's instructions. cDNA was generated using the Superscript III RNA reverse transcription kit (Invitrogen 18080044) as per the manufacturer's instructions. cDNA was amplified using a SYBR Green Universal PCR Master Mix (Applied Biosystems 4309155) as per the manufacturer's instructions. qRT-PCR was performed on a StepOnePlus Real-time PCR system (Applied Biosystems) using primer sequences detailed in Supplemental Table S3 with the following cycle conditions: 95°C/10 min; 44 cycles: 95°C/15 sec, 60°C/60 sec. mRNA expression was normalized and expressed relative to the endogenous reference gene Hprt: fold induction = 2(−ΔCt), where ΔCt = Ct(target)–Ct(Hprt).
Immunoblotting
E18.5 lungs were ground into powder in liquid nitrogen and lysed for 30 min in ice-cold extraction buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 100 µg/mL DNase I) completed with protease inhibitors (Roche 11873580001). Lysates were centrifuged at 15,000 g for 20 min, and protein concentration was quantified using Bradford Assay (Bio-Rad 500-0006). Protein extracts were denatured in Laemmli buffer (50 mM Tris-HCL, 2% SDS, 10% glycerol, 0.1% bromphenolblue, 100 mM β-mercaptoethanol), boiled for 5 min, resolved by reducing SDS-polyacrylamide gel electrophoresis (Tris-glycine 14% gel, 20 µg protein/lane), and transferred onto a nitrocellulose membrane (GE Healthcare 10600004). Membranes were blocked with 5% nonfat milk in PBS-T (PBS/0.1% Tween) for 2 h and probed with primary antibodies in blocking buffer overnight at 4°C. Membranes were washed and probed with secondary antibodies in blocking buffer for 2 h at room temperature. The following antibodies were used: Primary: α-BIM (ENZO ALX-804-527, 1:500), α-PTEN (Cell Signaling 9559, 1:500), α-HSP90 (Santa Cruz 13119, 1:1000); Secondary: α-rabbit Ig/HRP (Dako P0448, 1:10,000), α-mouse Ig/HRP (Dako P0161, 1:10,000), α-rat IgG/HRP (Cell Signaling 7077, 1:10,000). Membranes were washed and incubated with an enhanced chemiluminescence reagent (advansta Western Bright ECL Spray, K-12049-D50) before exposure to an X-ray film (GE Healthcare 28906837).
Histology
H&E staining was performed on 7-µm sections from formalin-fixed (4% paraformaldehyde/PBS) paraffin-embedded embryo chests. Images were acquired on a Keyence BZ-9000 microscope and processed with the Keyence BZ-II viewer software.
Skeletal preparations
E18.5 skeletons were prepared as described (Han et al. 2015). Pictures were captured with a binocular microscope (Leica) equipped with a DFC495 digital camera.
nCounter miRNA gene expression profiling
Total RNA was extracted from snap-frozen Abl-B cells or E18.5 lung tissue ground into powder using the miRNeasy mini kit (QIAGEN 217004). Relative miRNA copy number was determined using the nCounter Mouse v1.5 miRNA Expression Assay kit (NanoString Technologies GXA-MMIR15-12) and nCounter SPRINT profiler (NanoString Technologies) as per the manufacturer's instructions.
Sample preparation for PAR-CLIP
Abl-B cells were labeled with 500 µM 4-thiouridine (Carbosynth 13957-31-8) for 5 h and crosslinked at 365 nm with a total dose of 2000 µJ/cm2 (UV Stratalinker 2400). Cell pellets were snap-frozen in liquid nitrogen and stored at −80°C. PAR-CLIP was performed as described in Hafner et al. (2010) with the following modifications. Briefly, cells were lysed in NP-40 lysis buffer (50 mM HEPES-KOH at pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5% [v/v] NP40, 0.5 mM DTT, complete EDTA-free protease inhibitor cocktail). Lysates were incubated with protein G magnetic beads (Invitrogen 100-03D) and precoupled to an α-mouse AGO2 antibody (Wako Chemicals 018-22021) for 1 h at 4°C. Beads were washed with IP wash buffer (50 mM HEPES-KOH, pH 7.5, 150 mM KCl, 0.05% [v/v] NP40, 0.5 mM DTT, complete EDTA-free protease inhibitor cocktail) and subjected to RNase T1 (Fermentas EN0541) treatment (10 U/µL) for 10 min at 37°C. Immunoprecipitations were washed in IP wash buffer and incubated with calf intestinal phosphatase (New England BioLabs M0290), followed by radioactive end-labeling using T4 polynucleotide kinase (New England BioLabs M0201). Protein-RNA complexes were resolved on a 4%–12% NuPAGE gel (Invitrogen WG1402BOX) and transferred onto a nitrocellulose membrane. The protein-RNA complexes at the expected molecular weight were excised. RNA was isolated by Proteinase K (Roche 03115801001) treatment and phenol-chloroform extraction, followed by adapter ligation and cDNA library construction. The amplified cDNA was sequenced on an Illumina HiSeq2000 instrument.
PAR-CLIP data processing
The Illumina CASAVA (v1.8.2) software was used for RNAseq base calling. RNAseq reads had 3′ adaptors trimmed in two passes using (flexbar 2.5) with parameters: –adapter-relaxed –adapter-trim-end RIGHT –max-uncalled 0 –adapter-gap -3 –format i1.8 –pre-trim-phred 30 –min-read-length 12. The reads were collapsed with a custom script to remove PCR amplification biases and consequently trimmed by 2 nt from both 5′ and 3′ ends to remove random nucleotides ligated with the adaptors. A reference based on mm9 was created that contained the corresponding Bim 3′ UTR mutations (mm9_Bim_MUT). Reads that did not map to rRNA (NCBIM37.65 rRNA FASTA downloaded from BioMart) were mapped to mm9 for the wild type and to mm9_Bim_MUT for the Bim3′UTRmut/mut libraries. Only alignments with a mapping quality of at least 13 were retained. For mapping, we used bwa (0.7.12-r1039) in local alignment mode allowing for up to three mismatches in the whole read (bwa aln -n 3 -l 100). Aligned reads that contained RNA T:C conversions were processed using a custom python script. Coverage plots were generated using R and Bioconductor (Lawrence et al. 2013).
Identification of differentially covered miR-17∼92 sites
A list of 3′ UTRs was constructed by taking all transcripts in the UCSC mm9 annotation incorporated by the TxDb.Mmusculus.UCSC.mm9.knownGene Bioconductor package (version 3.2.2) and identifying those that had a UniProt annotation using the org.Mm.eg.db Bioconductor package (version 3.5.0) and a 3′ UTR of at least 50 nt. For each gene, the longest 3′ UTR across all isoforms was selected. The 3′ UTR sequences were constructed using the BSgenome.Mmusculus.UCSC.mm9 Bioconductor genome package (version 1.4.0). A custom python script identified all perfect miR-17∼92 seed matches on them. The 3′ UTR coordinates of a perfect binding site match were converted to genomic coordinates, paying attention to cases of spliced 3′ UTRs. The identified binding sites were then queried using the summarizeOverlaps function of the GenomicAlignments Bioconductor package (version 1.14.2) in the IntersectionNotEmpty mode for the amount of reads overlapping a window of 41 nt centered around them. Only windows covered by at least five reads in at least one sample were kept. Differential coverage was analyzed using the DESeq2 package (Love et al. 2014).
nCounter miRNA gene expression scatterplots
Raw miRNA counts were converted to CPM, counts per million “mapped” (counted). Differential expression analysis was performed using the DESeq2 package, and plots were generated using R.
Statistical analysis
Images were analyzed and processed using Photoshop (Adobe), ImageJ, the Keyence BZ-II viewer software, and Image Access Enterprise 6. Graphs were plotted and statistical analysis was performed with GraphPad Prism 7 software using Student's unpaired two-tailed t test when comparing two groups, one-way ANOVA followed by Tukey post-hoc test when comparing multiple groups and a χ2 test for genotype frequencies. Results are shown as mean and standard deviation (SD). Group and sample sizes were empirically determined and the number of biological repetitions (n) is stated in each figure legend. No randomization or blinding protocol was used for experimental grouping.
Data availability
The PAR-CLIP sequencing, Abl-B cell, and E18.5 lung NanoString data have been deposited in NCBI's Gene Expression Omnibus (GSE123673).
Supplementary Material
Acknowledgments
We thank D. Ghitza, J. Xia, J. Grundy, A. Pellerin, C. Grosse, J. Cernoch, K. Petsch, J. Pempe, B. Unterberger, C. Aydemir, C. Soratroi, I. Gaggl, J. Heppke, A. Koehler, M. Bamberg, and A. Boltengagen for expert technical assistance; J. Kunert, A. Ullmann, X. Richter, K. Rossi, M. Saurwein, N. Heinrich, and N. Schöpf for animal care; H.P. Rahn for support with flow cytometry; M. Ottaviano, M. Bezohra, and R. Lauhkonen-Seitz for administrative assistance; the present and former K.R. lab members, A.Villunger and M. Erlacher for comments and suggestions; M. Reth for Mb1-cre mice; and A. Ventura for miR-17∼92F mice. The pMSCV-v-Abl retroviral vector was a kind gift of F. Alt. This work was supported by grants from the US National Institutes of Health to K.R. (1R01AI064345-01), the European Research Council to K.R. (ERC Advanced Grant 268921), the German Research Foundation to N.R. (RA 838/5-1), the Tyrolean Science Fund to V.L (UNI-0404/1696), the Ingrid Shaker Nessmann Cancer Research Association to V.L. (D-182400-020-012), and the Austrian Cancer Aid to V.L. (KH15017). V.L. acknowledges support from EMBO, F.K. from the German Epigenome Programme, and K.S. from the Austrian Academy of Sciences.
Author contributions: Conceptualization: K.R. and V.L.; methodology: S.P., V.L., and M.M.; formal analysis: F.K. performed the bioinformatics analysis of the PAR-CLIP and nCounter data; investigation: V.L., S.P., T.C., M.M, N.K., K.S., E.D., M.M., G.M., L.R.H.-M, I.L., N.R., and C.K.; resources: K.R., C.B., S.K., L.Q.-M, M.L., N.R., and V.L.; writing: K.R. and V.L.; visualization: V.L., F.K., M.M., and L.R.H.-M.; supervision: K.R. and V.L.; project administration: K.R. and V.L.; funding acquisition: K.R, N.R., and V.L.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.330134.119.
References
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, Chen C-Z. 2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5: 396–400. 10.1038/nrg1328 [DOI] [PubMed] [Google Scholar]
- Bassett AR, Azzam G, Wheatley L, Tibbit C, Rajakumar T, McGowan S, Stanger N, Ewels PA, Taylor S, Ponting CP, et al. 2014. Understanding functional miRNA-target interactions in vivo by site-specific genome engineering. Nat Commun 5: 4640 10.1038/ncomms5640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Köntgen F, Adams JM, Strasser A. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738. 10.1126/science.286.5445.1735 [DOI] [PubMed] [Google Scholar]
- Brinkmann K, Ng AP, de Graaf CA, Di Rago L, Hyland CD, Morelli E, Rautela J, Huntington ND, Strasser A, Alexander WS, et al. 2019. miR17∼92 restrains pro-apoptotic BIM to ensure survival of haematopoietic stem and progenitor cells. Cell Death Differ 116: 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, de Alborán IM, Janz M, Rodig S, Rajewsky K. 2012. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol 13: 1092–1100. 10.1038/ni.2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, et al. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33: 314–325. 10.1002/immu.200310005 [DOI] [PubMed] [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, Van Haeringen A, Geneviève D, Goldenberg A, Oufadem M, et al. 2011. Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat Genet 43: 1026–1030. 10.1038/ng.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai T-H, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, et al. 2008. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 28: 630–638. 10.1016/j.immuni.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki SM. 1996. Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc Natl Acad Sci 93: 6191–6196. 10.1073/pnas.93.12.6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524. 10.1016/j.cell.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, Großhans H. 2015. The let-7 microRNA directs vulval development through a single target. Dev Cell 32: 335–344. 10.1016/j.devcel.2014.12.018 [DOI] [PubMed] [Google Scholar]
- Feild JA, Zhang L, Brun KA, Brooks DP, Edwards RM. 1999. Cloning and functional characterization of a sodium-dependent phosphate transporter expressed in human lung and small intestine. Biochem Biophys Res Commun 258: 578–582. 10.1006/bbrc.1999.0666 [DOI] [PubMed] [Google Scholar]
- Grabow S, Kueh AJ, Ke F, Vanyai HK, Sheikh BN, Dengler MA, Chiang W, Eccles S, Smyth IM, Jones LK, et al. 2018. Subtle changes in the levels of BCL-2 proteins cause severe craniofacial abnormalities. Cell Rep 24: 3285–3295.e4. 10.1016/j.celrep.2018.08.048 [DOI] [PubMed] [Google Scholar]
- Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, Rajewsky N. 2014. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell 54: 1042–1054. 10.1016/j.molcel.2014.03.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Deng Y, Liu J, Qian L. 2015. Cardiomyocyte-specific role of miR-24 in promoting cell survival. J Cell Mol Med 19: 103–112. 10.1111/jcmm.12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. 10.1016/j.cell.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y-C, Vidigal JA, Mu P, Yao E, Singh I, González AJ, Concepcion CP, Bonetti C, Ogrodowski P, Carver B, et al. 2015. An allelic series of miR-17∼92-mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nat Genet 47: 766–775. 10.1038/ng.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K. 2001. B cell development pathways. Annu Rev Immunol 19: 595–621. 10.1146/annurev.immunol.19.1.595 [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. 2005. A microRNA polycistron as a potential human oncogene. Nature 435: 828–833. 10.1038/nature03552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, Tollervey D. 2013. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153: 654–665. 10.1016/j.cell.2013.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. 2006. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci 103: 13789–13794. 10.1073/pnas.0605944103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J-P, Lu Y, Loeb GB, Leslie CS, Rudensky AY. 2018. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat Immunol 19: 1137–1145. 10.1038/s41590-018-0208-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, Kang SG, Liu W-H, Sabouri-Ghomi M, Cullen BR, et al. 2013. MicroRNA-17∼92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J 32: 2377–2391. 10.1038/emboj.2013.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadener S, Rodriguez J, Abruzzi KC, Khodor YL, Sugino K, Marr MT, Nelson S, Rosbash M. 2009. Genome-wide identification of targets of the drosha-pasha/DGCR8 complex. RNA 15: 537–545. 10.1261/rna.1319309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, Strasser A. 2018. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173: 1217–1230.e17. 10.1016/j.cell.2018.04.036 [DOI] [PubMed] [Google Scholar]
- Kollek M, Voigt G, Molnar C, Murad F, Bertele D, Krombholz CF, Bohler S, Labi V, Schiller S, Kunze M, et al. 2017. Transient apoptosis inhibition in donor stem cells improves hematopoietic stem cell transplantation. J Exp Med 214: 2967–2983. 10.1084/jem.20161721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, et al. 2008. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 132: 860–874. 10.1016/j.cell.2008.02.020 [DOI] [PubMed] [Google Scholar]
- Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, Wert SE, Stripp BR, Morris RE, Glasser SW, et al. 1996. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci 93: 9594–9599. 10.1073/pnas.93.18.9594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500. 10.1038/ng1536 [DOI] [PubMed] [Google Scholar]
- Labi V, Erlacher M, Kiessling S, Villunger A. 2006. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ 13: 1325–1338. 10.1038/sj.cdd.4401940 [DOI] [PubMed] [Google Scholar]
- Labi V, Bertele D, Woess C, Tischner D, Bock FJ, Schwemmers S, Pahl HL, Geley S, Kunze M, Niemeyer CM, et al. 2013. Haematopoietic stem cell survival and transplantation efficacy is limited by the BH3-only proteins Bim and Bmf. EMBO Mol Med 5: 122–136. 10.1002/emmm.201201235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V, Schoeler K, Melamed D. 2019. miR-17∼92 in lymphocyte development and lymphomagenesis. Cancer Lett 446: 73–80. 10.1016/j.canlet.2018.12.020 [DOI] [PubMed] [Google Scholar]
- Lai M, Gonzalez-Martin A, Cooper AB, Oda H, Jin HY, Shepherd J, He L, Zhu J, Nemazee D, Xiao C. 2016. Regulation of B-cell development and tolerance by different members of the miR-17∼92 family microRNAs. Nat Commun 7: 12207 10.1038/ncomms12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. 2014. MYC through miR-17-92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell 26: 262–272. 10.1016/j.ccr.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773. 10.1038/nature03315 [DOI] [PubMed] [Google Scholar]
- Liu R, King A, Bouillet P, Tarlinton DM, Strasser A, Heierhorst J. 2018. Proapoptotic BIM impacts B lymphoid homeostasis by limiting the survival of mature B cells in a cell-autonomous manner. Front Immunol 9: 592 10.3389/fimmu.2018.00592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, Boiani S, Leyland R, Galloway A, Andrews S, et al. 2014. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. J Exp Med 211: 2183–2198. 10.1084/jem.20140338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L-F, Gasteiger G, Yu I-S, Chaudhry A, Hsin J-P, Lu Y, Bos PD, Lin L-L, Zawislak CL, Cho S, et al. 2015. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity 43: 52–64. 10.1016/j.immuni.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, Ambros V. 2017. A microRNA family exerts maternal control on sex determination in C. elegans. Genes Dev 31: 422–437. 10.1101/gad.290155.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilyansky E, Rigoutsos I. 2013. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 20: 1603–1614. 10.1038/cdd.2013.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris JK, McColl KS, Distelhorst CW. 2011. Glucocorticoid-mediated repression of the oncogenic microRNA cluster miR-17∼92 contributes to the induction of Bim and initiation of apoptosis. Mol Endocrinol 25: 409–420. 10.1210/me.2010-0402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, van Oudenaarden A. 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859. 10.1038/ng.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordigården A, Kraft M, Eliasson P, Labi V, Lam EW-F, Villunger A, Jönsson J-I. 2009. BH3-only protein Bim more critical than Puma in tyrosine kinase inhibitor-induced apoptosis of human leukemic cells and transduced hematopoietic progenitors carrying oncogenic FLT3. Blood 113: 2302–2311. 10.1182/blood-2008-07-167023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PM, Vass T, Kappler J, Marrack P. 2006. Loss of the proapoptotic protein, Bim, breaks B cell anergy. J Exp Med 203: 731–741. 10.1084/jem.20051407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. 2004. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 64: 3087–3095. 10.1158/0008-5472.CAN-03-3773 [DOI] [PubMed] [Google Scholar]
- Rajewsky N. 2006. microRNA target predictions in animals. Nat Genet 38: S8–S13. 10.1038/ng1798 [DOI] [PubMed] [Google Scholar]
- Rosenberg N, Baltimore D, Scher CD. 1975. In vitro transformation of lymphoid cells by Abelson murine leukemia virus. Proc Natl Acad Sci 72: 1932–1936. 10.1073/pnas.72.5.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. 1995. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23: 5080–5081. 10.1093/nar/23.24.5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrard R, Luehr S, Holzkamp H, McJunkin K, Memar N, Conradt B. 2017. miRNAs cooperate in apoptosis regulation during C. elegans development. Genes Dev 31: 209–222. 10.1101/gad.288555.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stripp BR, Sawaya PL, Luse DS, Wikenheiser KA, Wert SE, Huffman JA, Lattier DL, Singh G, Katyal SL, Whitsett JA. 1992. cis-acting elements that confer lung epithelial cell expression of the CC10 gene. J Biol Chem 267: 14703–14712. [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. 2008. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 28: 621–629. 10.1016/j.immuni.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzlak S, Kaufmann T, Villunger A. 2016. Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis. Genes Dev 30: 2133–2151. 10.1101/gad.289298.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, et al. 2008. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132: 875–886. 10.1016/j.cell.2008.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woess C, Tuzlak S, Labi V, Drach M, Bertele D, Schneider P, Villunger A. 2015. Combined loss of the BH3-only proteins Bim and Bmf restores B-cell development and function in TACI-Ig transgenic mice. Cell Death Differ 22: 1477–1488. 10.1038/cdd.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Rajewsky K. 2009. MicroRNA control in the immune system: basic principles. Cell 136: 26–36. 10.1016/j.cell.2008.12.027 [DOI] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. 2008. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 9: 405–414. 10.1038/ni1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PAR-CLIP sequencing, Abl-B cell, and E18.5 lung NanoString data have been deposited in NCBI's Gene Expression Omnibus (GSE123673).