

In this study, Nutter et al. use rolling circle amplification and CRISPR/Cas9-mediated genome editing to generate Dmpk CTG expansion (CTGexp) mouse knockin models of myotonic dystrophy type 1 (DM1). Their results highlight important interplays between Dmpk and Mbnl expression patterns with CTGexp length and missplicing and also demonstrate that splicing regulation in the choroid plexus, which is responsible for the bulk of cerebral spinal fluid (CSF) production, is adversely affected by CTGexp mutations.
Keywords: MBNL, myotonic dystrophy, neuromuscular disease, RNA splicing, short tandem repeat
Abstract
Short tandem repeats (STRs) are prone to expansion mutations that cause multiple hereditary neurological and neuromuscular diseases. To study pathomechanisms using mouse models that recapitulate the tissue specificity and developmental timing of an STR expansion gene, we used rolling circle amplification and CRISPR/Cas9-mediated genome editing to generate Dmpk CTG expansion (CTGexp) knockin models of myotonic dystrophy type 1 (DM1). We demonstrate that skeletal muscle myoblasts and brain choroid plexus epithelial cells are particularly susceptible to Dmpk CTGexp mutations and RNA missplicing. Our results implicate dysregulation of muscle regeneration and cerebrospinal fluid homeostasis as early pathogenic events in DM1.
Microsatellites, or short tandem repeats (STRs) of ≤10 bp are unstable genetic elements with a propensity to fold into intrastrand DNA structures that compromise replication, recombination, and repair pathways (Usdin et al. 2015; Neil et al. 2017). While STRs are susceptible to both expansions and contractions, expanded STRs cause >40 neurological and neuromuscular hereditary diseases, including C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia (C9-ALS/FTD), Huntington disease (HD) and myotonic dystrophy types 1 (DM1) and 2 (DM2) (Nelson et al. 2013). DM1 is a prominent STR disease as it is the most common adult-onset muscular dystrophy and is caused by CTG expansions (CTGexp) in the 3′ untranslated region (3′UTR) of the DMPK gene. Repeat sizes range from 5 to 37 in unaffected individuals but 50 to >4000 in DM1 patients (Goodwin and Swanson 2014). The DM1 pathomechanism involves transcription of expanded DMPK alleles, which generates toxic CUGexp RNAs that accumulate in nuclear inclusions or RNA foci, where they sequester muscleblind-like (MBNL) proteins. Since MBNL proteins regulate the splicing of their RNA targets during development, loss of MBNL function results in the expression of developmentally inappropriate isoforms in multiple tissues (Thomas et al. 2017).
Attempts at modeling DM1 in mice have relied on heterologous promoters and gene contexts, which disconnects the tissue specificity, developmental timing, and spatial expression of a repeat expansion from its endogenous gene context. This disassociation could mask discoveries that might have important pathomechanistic implications. Here, we use a combination of rolling circle amplification (RCA) and CRISPR/Cas9 genome editing to introduce CTG expansions into the mouse Dmpk 3′UTR. Our results highlight important interplays between Dmpk and Mbnl expression patterns with CTGexp length and missplicing, and also demonstrate that splicing regulation in the choroid plexus, which is responsible for the bulk of cerebral spinal fluid (CSF) production, is adversely affected by CTGexp mutations.
Results and discussion
Dmpk CTGexp knockin mice
Since DMPK exons are well conserved among mammals, with the exception of an uninterrupted CTG repeat tract in the human 3′UTR (Supplemental Fig. S1A), we developed a microsatellite expansion modeling by RCA and genome editing (MERGE) protocol to insert CTGexp mutations into the corresponding region of the mouse Dmpk 3′UTR (see Supplemental Material). Target site selection was validated in C2C12 myoblasts using multiple guide (g)RNAs followed by determination of cleavage efficiencies (Supplemental Fig. S1B; Ran et al. 2013). After establishing the optimal site, adjacent sequences were cloned flanking the CTGexp repeat track as arms of homology (Fig. 1A). The resulting plasmid was amplified by RCA to maintain repeat length, and the resulting DNA served as a template for homology-directed repair (HDR) and for generation of larger repeat templates in vitro using Golden Gate Assembly (Osborne and Thornton 2008). Two rounds of Golden Gate Assembly were performed, with each cycle achieving a repeat content of 2n-2, where n is the starting CTG202 repeat size, resulting in CTG402 and CTG802 HDR templates (Fig. 1B). As proof of concept for this approach, we first generated mouse Dmpk knockin (KI) lines using CRISPR/Cas9 with a CTG202 HDR template. Of 74 newborn pups, four were positive for Dmpk insertion as analyzed by PCR using a forward primer outside the homology arm (Supplemental Fig. S1C). Interestingly, Dmpk CTG202 recombination templates yielded CTG202, but also contracted CTG150 and CTG170, KI lines. Therefore, subsequent injections were performed using the largest CTG802 HDR template (Fig. 1C) with the anticipation that this would result in a greater allelic series of Dmpk CTGexp KI mice. The presence of CTGexp mutations was further confirmed by repeat-primed PCR (Supplemental Fig. S1D). Since off-target insertions could contribute to mutant phenotypes (Nakajima et al. 2016), Southern blotting was performed on the resulting mice from these injections. This analysis identified founders with single and unique integrations of CTG170 and CTG480 into the Dmpk 3′ UTR using both internal and external hybridization probes, respectively. While many additional Dmpk CTGexp mice were generated, these other KI founders showed either concatemeric integration in the Dmpk 3′UTR, off-target insertion events, or excessive mosaicism (Supplemental Fig. S1E), so only CTG170 and CTG480 KI mice were used for subsequent studies. Using genomic DNA from Dmpk+/170 and Dmpk+/480 mice, repeat size was further validated by PCR genotyping (Fig. 1D).
Figure 1.
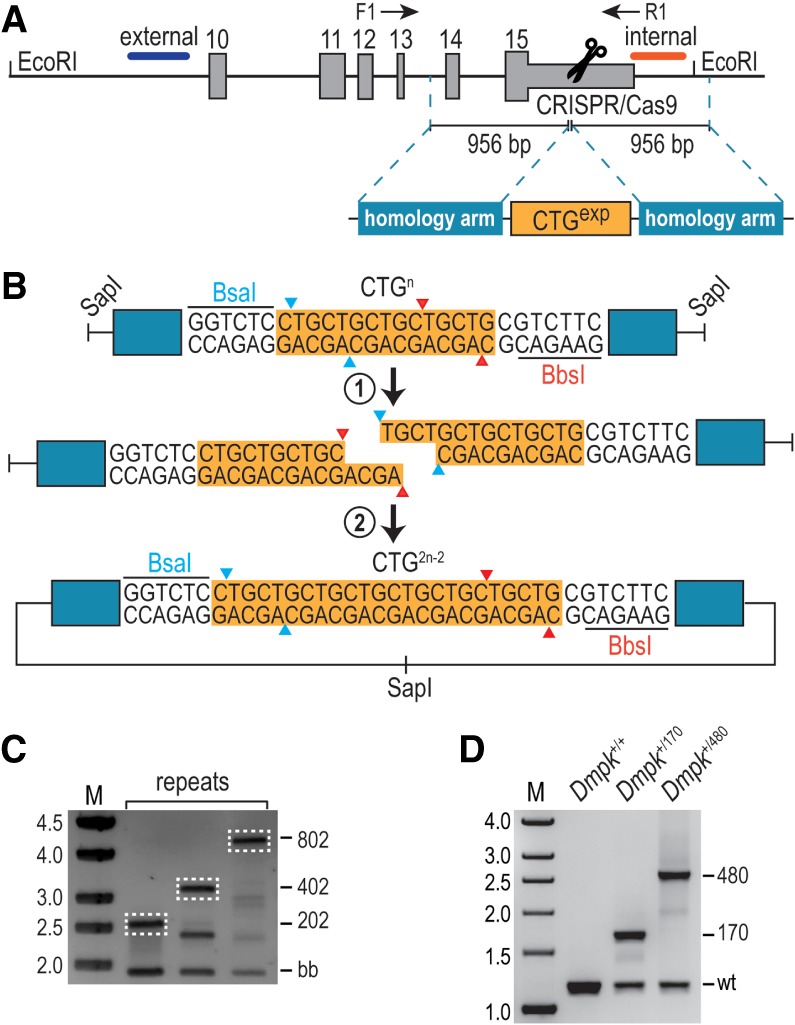
Generation of Dmpk knockin mice carrying CTG 3′UTR expansions. (A) CRISPR/Cas9-mediated recombination of HDR templates containing a CTG expansion flanked by homology arms at the Dmpk 3′UTR. External (blue) and internal (red) hybridization probes are indicated together with forward (F1) and reverse (R1) PCR primers. (B) Repeat dimerization by Golden Gate Assembly followed by RCA. Plasmids containing a repeat expansion flanked by BsaI and BbsI were linearized by SapI, linear constructs were separately digested by BsaI or BbsI, and repeat-containing fragments were gel purified, ligated, and amplified by RCA. This process generates a new construct carrying 2n-2 repeats, where “n” is the initial expansion size. (C) PCR of CTG202, CTG402, and CTG802 HDR templates. Linearized RCA products yielded a 2-kb backbone band (bb) and upper bands corresponding to the recombination template (dashed white lines indicate the isolated HDR templates). (D) PCR genotyping of Dmpk+/+ (wild-type), Dmpk+/170, and Dmpk+/480 mice.
CTGexp mutations induce DMPK protein loss in skeletal muscle and myotubes
Since skeletal muscle weakness/wasting and myotonia are characteristic features of DM1, and Dmpk expression is relatively high in this tissue (Supplemental Fig. S2A), we initially assessed the impact of CTGexp mutations on Dmpk CTGexp KI muscle. CUGexp RNA foci were detectable by RNA fluorescence in situ hybridization (RNA-FISH) in adult skeletal muscles of Dmpkexp mice (Supplemental Fig. S2B), so we tested whether CTGexp insertions altered Dmpk expression in tibialis anterior (TA) muscles from Dmpk+/170 and Dmpk170/170 mice by RT-qPCR. Interestingly, Dmpk RNA levels were affected by these mutant alleles and the total number of CTG repeats (Supplemental Fig. S2C). While this apparent RNA decrease might reflect decreased Dmpk transcription and/or increased Dmpk RNA turnover, previous cell DM1 model studies have shown that the isolation of CUGexp RNAs is impaired using traditional RNA extraction methodologies, possibly due to nuclear retention in RNA foci (Davis et al. 1997). The presence of CUGexp RNA foci and reduced Dmpk RNA levels made us question whether the production of DMPK protein was also altered. Indeed, when assessed by immunoblot analysis we detected a dramatic decrease in DMPK protein levels that was inversely correlated to the number of mutant CTG170 alleles (Supplemental Fig. S2D). DMPK protein levels in the TA from Dmpk+/170 mice decreased by ∼50%, while levels in Dmpk170/170 dropped >90%. These results demonstrated that even a moderate-sized CTG repeat expansion in a mouse Dmpk KI mutant severely compromised protein production from the mutant allele in agreement with early studies that showed decreased expression of DMPK protein in DM1 (Fu et al. 1993). Importantly, we did not detect abnormal muscle phenotypes, including myotonia and centralized myonuclei, in either Dmpk CTG170 or CTG480 heterozygous or homozygous mice (data not shown). This result was consistent with earlier studies on Dmpk knockout mice, which fail to replicate DM-relevant phenotypes and only develop a very mild late-onset myopathy (Jansen et al. 1996; Reddy et al. 1996).
Although RNA-foci were present in Dmpk CTGexp KI mice, the lack of other DM1-associated muscle phenotypes suggested that alternative splicing regulation was not severely altered in this tissue. Indeed, RNAseq followed by RT-PCR validations failed to identify significant changes in DM1-relevant alternative splicing events (Supplemental Table S1; Supplemental Fig. S2E). For example, missplicing of Atp2a1 exon 22 and Clcn1 exon 7a, which are misregulated in DM1 muscle, was not detectable in Dmpk CTG170 or CTG480 KI mice in contrast to Mbnl1−/− knockout TA muscle (Supplemental Fig. S2E). Furthermore, differential gene expression analysis using DESeq2 did not identify major (greater than twofold) changes in gene expression (Supplemental Fig. S2F).
The lack of overt muscle and splicing phenotypes was surprising since our Dmpk KI mutants expressed alleles that are considered to be pathogenic in humans. This result suggested that the temporal and spatial expression of a stable expansion in the Dmpk gene in skeletal muscle did not produce a sufficient CUG repeat load to drive pathology. Based on our earlier finding that human myotubes have higher DMPK expression than adult muscle (Thomas et al. 2017), we tested the possibility that CTGexp mutations might have more severe effects on muscle precursor cells, including primary myoblasts. In agreement with this idea, CUGexp RNA foci were considerably more numerous in myoblasts and differentiated myotubes compared with adult skeletal muscle and were particularly striking in the Dmpk480 heterozygous and homozygous myotubes (Fig. 2A). CUGexp RNA foci number increased upon myogenic differentiation, in agreement with the temporal up-regulation in Dmpk expression during myogenesis. As noted previously for skeletal muscle (Supplemental Fig. S2C), CTGexp mutations led to a decrease in the amount of RNA isolated from both myoblasts and myotubes (Fig. 2B). As suggested above, this was due to an RNA extractability issue since Trizol extraction at an elevated temperature (55°C), a procedure shown to release nuclear-retained RNAs (Chujo et al. 2017), resulted in increased Dmpk RNA levels isolated from KI cells (Fig. 2B; Supplemental Fig. S2G). Myotube DMPK protein levels decreased similarly to Dmpk CTGexp skeletal muscle with no detectable DMPK protein in Dmpk480/480 myotubes (Fig. 2C; Supplemental Fig. S2H).
Figure 2.
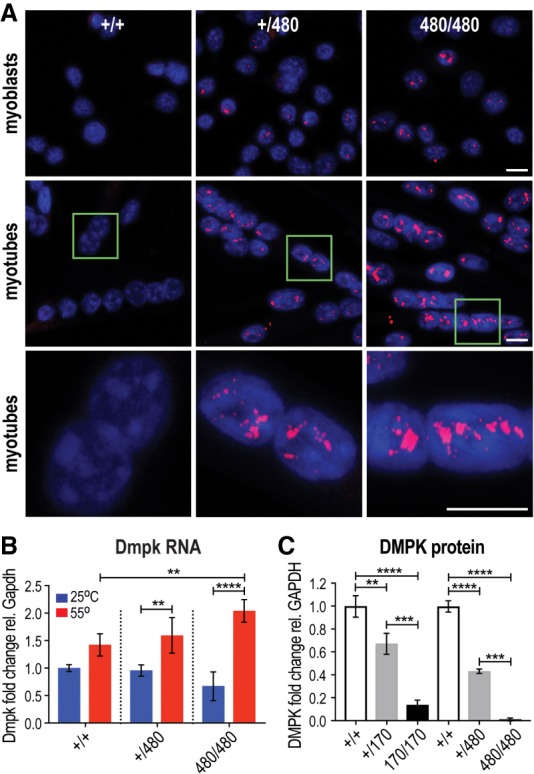
RNA foci and DMPK protein loss in CTG480 KI primary myoblasts and myotubes. (A) RNA-FISH of Dmpk WT (Dmpk+/+), compared with Dmpk CTG+/480 heterozygous and Dmpk CTG480/480 homozygous primary myoblasts and differentiated myotubes, which show numerous nuclear CUG repeat expansion (CUGexp) RNA foci (red) in the nucleus (blue, DAPI). Scale bars = 10 µM. (B) Reduced isolation of myoblast Dmpk CUG480 (heterozygous Dmpk CTG+/480, homozygous CTG480/480) RNAs at 25°C (blue bars) is prevented by high temperature (red, 55°C) Trizol extraction ([*] P < 0.05, [**] P < 0.01, [****] P < 0.0001, one-way ANOVA). (C) Inhibition of Dmpk mRNA nuclear export in cells expressing Dmpk CTGexp genes (Dmpk CTG+/170, Dmpk CTG170/170, Dmpk CTG+/480, Dmpk CTG480/480) leads to decreases in DMPK protein expression that correlate with CTG repeat length. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001; one-way ANOVA.
MBNL sequestration and RNA missplicing in Dmpk CTGexp myotubes
Misregulation of alternative splicing in DM1 results from MBNL sequestration by CUGexp RNA so we next determined whether MBNL proteins were effectively sequestered in nuclear RNA foci of differentiated Dmpk CTGexp KI myotubes. Confocal immunofluorescence microscopy showed both MBNL1 and MBNL2 were localized throughout the nucleoplasm in wild-type (Dmpk+/+) cells, but clearly relocalized to foci in CTGexp myotubes (Supplemental Fig. S3A). In contrast, the localization of another nuclear RNA-binding protein, HNRNPH1, was unaffected. RNA-FISH and IF confirmed that MBNL1 was sequestered in CUGexp RNA foci (Supplemental Fig. S3B), which had a punctate distribution pattern by super-resolution microscopy (Supplemental Fig. S3C). Since MBNL proteins were sequestered in nuclear RNA foci and substantially depleted from the nucleoplasm pool, we next determined whether alternative splicing changes occurred in Dmpk CTGexp myotubes.
RNAs were isolated from Dmpk+/+, Dmpk170/170, Dmpk+/480, and Dmpk480/480 myotubes for RNA-seq analysis. Similar to skeletal muscle, differential gene expression analysis showed only subtle changes due to CTGexp mutations (Supplemental Fig. S2F). In contrast to TA muscle, alternative splicing was significantly dysregulated and the number of missplicing events correlated with CTG repeat load with 243 events in Dmpk480/480, but only 110 in Dmpk+/480 and 19 in Dmpk170/170, myotubes (Fig. 3A; Supplemental Table S2). Many of the top alternative splicing (AS) changes (Cacna1s exon 29, Cacna2d1 exon 19, Ncor2 exon 46 5′ss, Pdlim3 exon 4 5′ss, and MBNL1 exon 5) have been previously characterized in DM1 cells and mouse models (Kimura et al. 2005; Tang et al. 2012; Lee et al. 2013). A prior study proposed 46 AS events as specific biomarkers for DM1 muscle (Wagner et al. 2016) with 26 of these events conserved in mice. Over 40% of these events (11/26) were present in our Dmpk480/480 myotube data set with changes in Cacna1s, Cacna2d1, Clasp1, Dnm1l, Mbnl1, Mxra7, Nfix, Pdlim3, Pdlim3.2, Slain2, and Sorbs1. Selected CUGexp-induced splicing shifts were validated by RT-PCR from separate differentiation experiments using three independent cell lines, and these expansion-induced splicing changes were MBNL dependent since they were similarly dysregulated in Mbnl1−/− knockout myotubes (Fig. 3B; Supplemental Fig. S3D). For example, increased selection of a distal alternative 5′ splice site (ss) in Ncor2 exon 46 occurred with higher CTGexp repeat load, while there was a switch in Ralgapa1 from a more distal to a more proximal 5′ss that resulted in a truncated exon 17 (Supplemental Fig. S3D). Interestingly, we also detected a previously uncharacterized AS change for Dmpk with decreased use of an exon 14 alternative 3′ss in CTG480 myotubes compared with WT (Supplemental Fig. S3E). To clarify why myoblasts and myotubes showed RNA foci and RNA processing changes while skeletal muscle did not, we tested whether Dmpk RNA levels exceeded Mbnl in cells and tissues prone to CUGexp-induced missplicing. Indeed, the Dmpk/Mbnl RNA ratio was much higher in myoblasts and myotubes compared with all skeletal muscle types examined, suggesting that MBNL activity might be more effectively sequestered in myoblasts and myotubes compared with muscle (Supplemental Fig. S3F). Overall, these results suggested that tissue and developmental-stage specific missplicing in DM1 reflects the interplay between repeat load, including repeat expansion length and host gene expression, in combination with MBNL protein levels. To test this possibility, we reduced MBNL1 levels by generating Mbnl1+/−; Dmpk480/480 mice. As expected, significant DM1-associated missplicing events were not detected in Dmpk480/480 or Mbnl1+/−, but were significantly dysregulated in Mbnl1+/−; Dmpk480/480 muscle (Fig. 3C).
Figure 3.
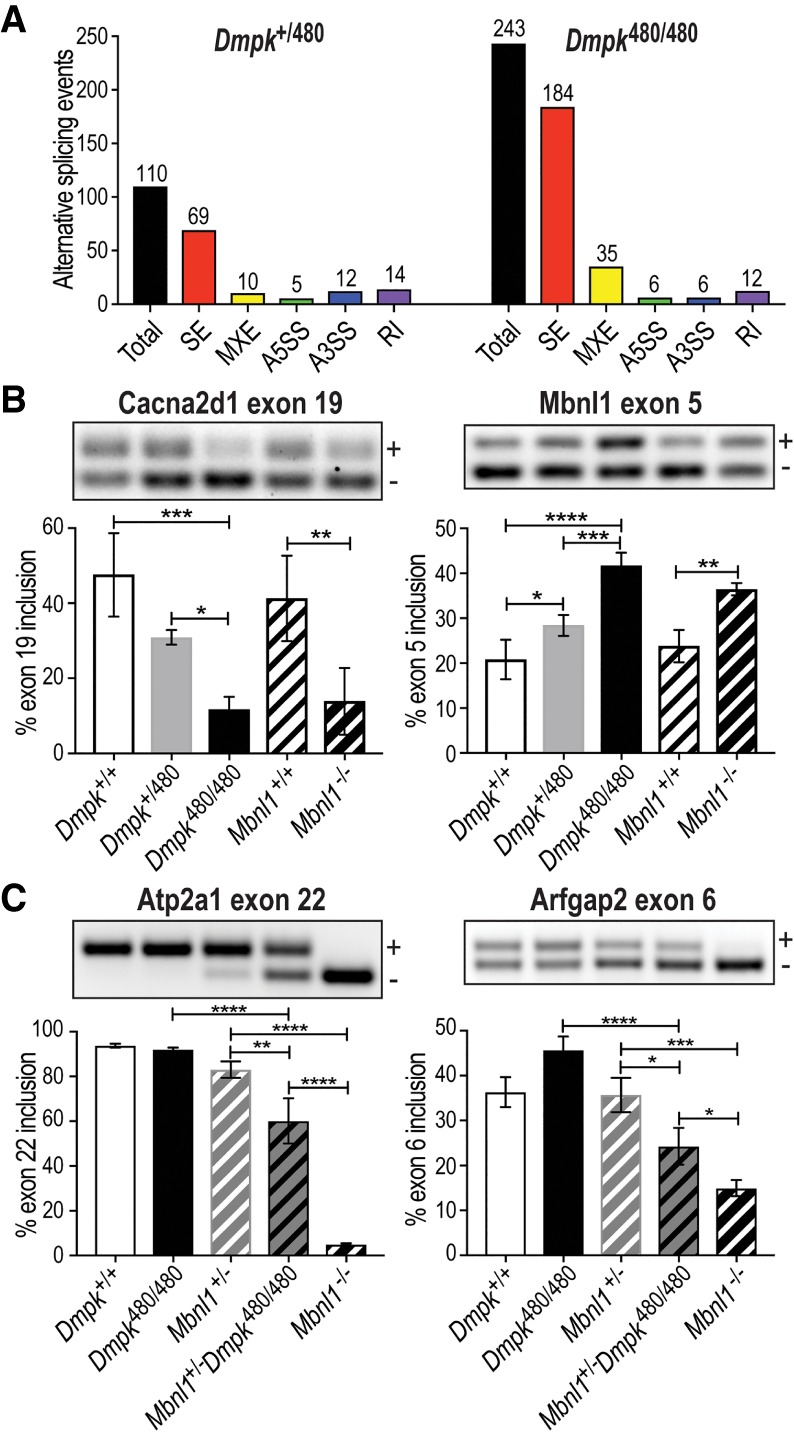
RNA missplicing in Dmpk CTGexp myotubes and diaphragm. (A) Bar graphs illustrating RNA missplicing (Total; SE, skipped exon; MXE, mutually exclusive exons; A5SS, alternative 5′ splice site; A3SS, alternative 3′ splice site; RI, retained intron) in Dmpk CTG+/480 heterozygous (left, total events = 110) and Dmpk CTG480/480 homozygous (right, total events = 243) myotubes compared with WT (Dmpk+/+). (B) RT-PCR validations of Cacna2d1 exon 19 and Mbnl1 exon 5 SE events in Dmpk+/+ versus Dmpk CTG+/480, Dmpk CTG480/480, and Mbnl1−/− myotubes. (C) RT-PCR of Atp2a1 exon 22 and Arfgap2 exon 6 in diaphragm from Dmpk+/+, Dmpk480/480, Mbnl1+/−, Mbnl1+/−; Dmpk480/480, and Mbnl1−/− mice (n = 3, age-matched littermates). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001; one-way ANOVA.
We next tested whether Dmpk CTGexp KI myoblasts were a suitable cell model for testing drugs to correct AS changes in DM1. A recent study reported that furamidine, an analog of the antimicrobial pentamidine, corrected missplicing in both DM1 HeLa cell and HSALR mouse models possibly by promiscuous binding interactions with both DNA CTG · CAG and RNA CUG repeats with nM affinity as well as disrupting MBNL1-CUG complexes at µM concentrations (Jenquin et al. 2018). Myoblasts were differentiated for 24 h prior to treatment with furamidine for 48 h, followed by RNA isolation and RT-PCR to assess the rescue of top AS changes observed in KI myotubes. All furamidine doses showed a shift toward the WT AS pattern and 2 µM was the most effective with ≥50% rescue of two of the top AS events (Supplemental Fig. S3G). This data confirmed that Dmpk CTGexp KI myoblasts provide tractable cell models for screening and assessment of small molecule drugs.
CTGexp mutations induce choroid plexus spliceopathy
While DM1 is classified as a muscular dystrophy, this is a multisystemic disorder in which the CNS is profoundly affected by hypersomnia, cognitive and behavioral abnormalities, progressive memory deficits, cerebral atrophy, and missplicing of MBNL2 target RNAs (Goodwin and Swanson 2014; Meola and Cardani 2015). One of the strengths of our CTGexp KI approach is that repeat expansion expression is driven by the endogenous Dmpk gene, which allows an unbiased screen for cell populations affected in vivo. Since myoblasts were strikingly affected by expression of CTGexp RNAs, we were interested in whether a specific cell type was more susceptible to dysregulation in the brain. Thus, we assembled a panel of multiple CNS regions and performed RT-qPCR to determine Dmpk RNA levels. Although most brain regions had very low expression, Dmpk was highly expressed in the choroid plexus (ChP) with levels exceeding skeletal muscle (Supplemental Figs. S2A, S3F). The ChP is responsible for the majority of cerebral spinal fluid (CSF) production and is composed of epithelial cells surrounding a fenestrated capillary network that protrudes from the brain parenchyma into the ventricular lumen (Lun et al. 2015). Moreover, DMPK protein levels are particularly high in the apical region of these cells (Whiting et al. 1995).
RNA-FISH of Dmpk CTGexp KI brains revealed numerous CUGexp RNA foci in ChP (Fig. 4A) but less in other brain regions including hippocampus (Supplemental Fig. S4A). RNA-seq analysis of isolated ChP (Fig. 4B) identified many missplicing events (Fig. 4C; Supplemental Table S3) in the absence of significant gene expression changes (Supplemental Fig. S4B). Many of the top AS events were validated by RT-PCR (Fig. 4D; Supplemental Fig. S4C) and these AS changes in Dmpk CTGexp KI mice ChP were MBNL dependent since they were similarly dysregulated in Mbnl2−/− knockout ChP. These observations demonstrate that a key cell population important for CSF production accumulates RNA foci that compromise MBNL-mediated splicing activity, which could negatively impact CSF homeostasis and composition.
Figure 4.
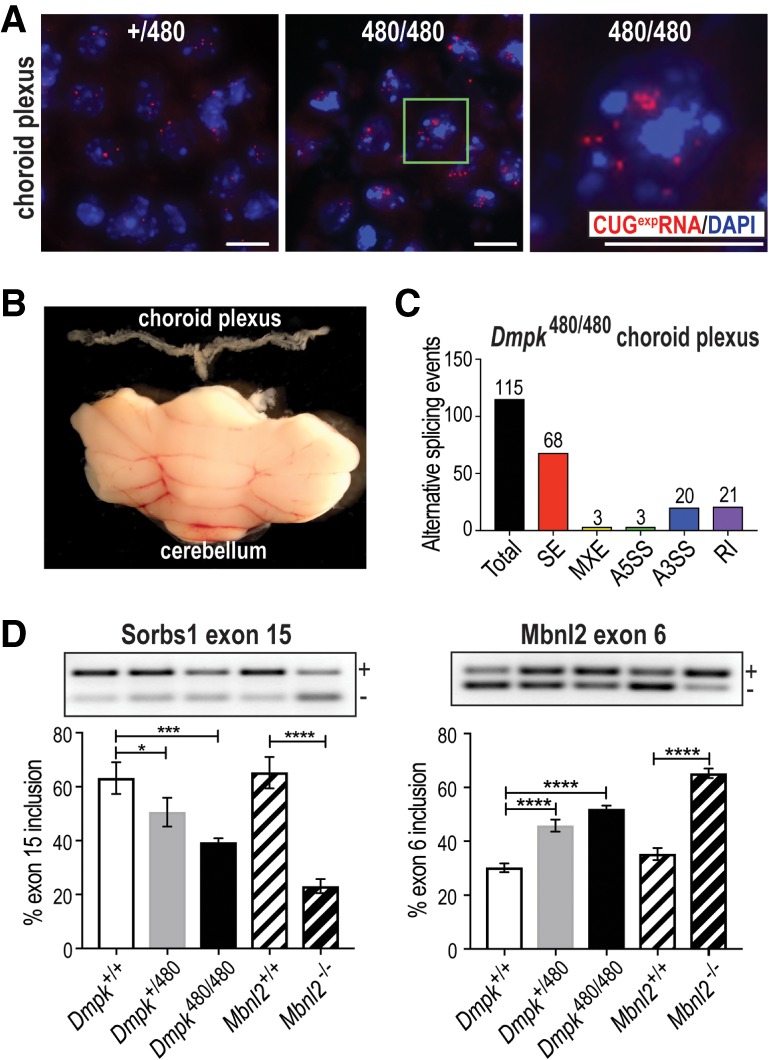
Dmpk CTGexp mutations disrupt alternative splicing regulation in the brain choroid plexus. (A) RNA-FISH of Dmpk CUG480 RNAs (red) retained in the nucleus (blue, DAPI). The green box in the middle panel corresponds to the magnified region in the right panel (scale bars = 10 µM). (B) Size comparison of the isolated fourth ventricle choroid plexus (top) to the cerebellum (bottom). (C) Bar graph illustrating RNA missplicing (total events = 115) in Dmpk CTG480/480 homozygous choroid plexus compared with WT. (D) RT-PCR validations of Sorbs1 exon 15 and Mbnl2 exon 6 missplicing in WT (Dmpk+/+, Mbnl2+/+) versus Dmpk CTG+/480, Dmpk CTG480/480 KI and Mbnl2−/− KO ChP. (*) P < 0.05; (***) P < 0.001; (****) P < 0.0001; one-way ANOVA.
Dmpk CTGexp knockins reveal cell-specific susceptibility to STR expansions
While somatic mosaicism due to the expansion of CTG repeats is a characteristic feature of DM1, it is unclear whether these increases in repeat length determine disease onset and progression or whether other genetic and environmental factors are involved (Nakamori et al. 2013). Here, we attempted to test whether discrete CTGexp lengths are determinative for specific pathological features of DM1 by inserting the largest expansions to date in the Dmpk 3′UTR. We demonstrate that zygote injections of an HDR template with a specific CTGexp length resulted in a range of repeat sizes in adult mice, possibly due to repeat instability during homologous recombination. Thus, microinjection of the largest expansion obtained by RCA is an effective strategy to generate an allelic series of STR expansion knock-ins. Furthermore, CTGexp mutations were stable with no germline contractions or expansions observed to date (<12 months of age), and although it is possible that somatic mosaicism will occur in older Dmpk CTGexp mice, this lack of CTG repeat instability may explain the absence of a severe muscle phenotype in vivo. Nevertheless, this repeat stability allows a determination of the CTGexp thresholds that produce specific molecular and physiological pathological effects. Since Dmpk CTG480 heterozygous and homozygous mice did not develop adult muscle DM1 disease phenotypes, the pathological threshold for this disorder has not yet been achieved. Thus, CUGexp RNA loads of >1000 repeats may be required to produce DM1-relevant muscle pathology, a result that agrees with previous human studies that detect CTG repeats up to 13-fold greater in DM1 muscle compared with repeat numbers in blood leukocytes (Thornton et al. 1994).
Another finding reported here with potential ramifications for our understanding of DM1 pathomechanisms is that the ChP is affected by alternative splicing deficits prior to skeletal muscle. The role of the ChP in CSF production involves selective transport so it is significant that several alternative splicing changes in Dmpk CTGexp KI mice ChP are involved in calcium and glutamate transport. Although the physiological outcome of these splicing changes in ChP is currently unclear, DM1 patients are affected by increases in ventricular volume and hypersomnia, which could be influenced by CSF homeostasis (Krogias et al. 2015; Meola and Cardani 2015). Future studies are required to determine whether Dmpk CTGexp KI mice have abnormal sleep patterns similar to Mbnl2−/− knockout mice and whether DM patients have altered CSF content attributable to missplicing of ChP epithelial cell transport genes.
Materials and methods
DMPK CTGexp knockin mice
Optimal CRISPR/Cas9 Dmpk 3′UTR targeting sites were identified (http://crispr.mit.edu) and crRNA, tracrRNA, and SpCas9 protein (IDT) were transfected into C2C12 cells as an RNP complex using RNAiMAX (ThermoFisher) (Supplemental Table S4). Genomic DNA was isolated and T7E1 assays (Alt-R Genome Editing Detection kit, IDT) were performed following the manufacturer's instructions. Homology arms flanking the Dmpk cleavage site were PCR amplified and cloned into a CTG202 plasmid (Dr. Charles Thornton, University of Rochester) by Gibson Assembly (New England Biolabs, NEB). RCA was performed using 100 ng of CTG202 annealed to 100 pmols of random hexamers (ThermoFisher) in 0.45× T4 DNA ligase buffer in a total volume of 22 µL. Next, 3 µL of 10× phi29 buffer, 3 µL of 10 mM dNTP solution mix, 0.6 µL of 10 mg/mL BSA, 0.4 µL of yeast inorganic pyrophosphatase, and 1 µL of phi29 DNA polymerase (NEB) were added, followed by incubation at 37°C for 18 h followed by polymerase inactivation (65°C, 10 min).
To expand repeats, RCA products were separately digested by SapI-BsaI or SapI-BbsI, and the repeat-containing fragments were gel extracted, purified using AMPure XP beads (Beckman Coulter), and ligated overnight at 16°C using T4 DNA ligase (NEB). Ligation products containing 2n-2 CTG repeats were amplified by RCA. Prior to zygote microinjections, RCA products carrying 202 or 802 repeats were linearized by AfeI, gel extracted, and purified using AMPure XP beads. Linear recombination templates, together with sgRNA and Cas9 protein (System Biosciences), were microinjected into C57BL/6J zygotes (Harvard University Genome Modification Facility). Tail snips were genotyped by PCR and repeat-primed PCR (ABI3730 DNA Analyzer, Applied Biosystems) was performed to test for Dmpk CTG insertions. Results were processed using Gene Marker software (SoftGenetics). Southern blot analysis included internal and external probes to ensure single integrations at the expected site. Dmpk CTG170 C57BL/6J mice were backcrossed to minimize potential unlinked Cas9-induced mutations, whereas CTG480 BL6 males showed impaired mutant allele transmission so these mutants were outcrossed onto FVB/NJ. All animal procedures were reviewed and approved by the University of Florida IACUC. All primers and oligonucleotides are listed in Supplemental Table S4.
Myoblast isolation, differentiation, and furamidine treatment
Primary myoblasts were isolated and treated as described (see Supplemental Material).
RNA-seq
Total RNA was quality checked using a Fragment Analyzer, depleted of rRNA and cDNA libraries prepared (UltraII Directional RNA Library Prep kit, NEB). Tibialis anterior and myotube libraries used 500 ng, whereas choroid plexus libraries used 200 ng of total RNA. Sequencing was performed on the NextSeq500 Illumina platform. Fastq files were inspected using Fastqc. Differential gene expression studies used DESeq2 (Love et al. 2014). For splicing analysis, reads were aligned to the mouse genome (mm10) using STAR(v2.6.0a) (Dobin et al. 2013) and splicing was quantified using rMATS(v4.0.2) (Shen et al. 2012). The rMATS output tables were filtered with the R package maser, with cutoff criteria of average reads ≥5, FDR <0.05, and minimum change in splicing of 10% (|ΔΨ| ≥0.1). RNA-seq data sets are deposited in GEO under accession no. GSE137494.
Statistical analysis
Statistical significance was determined in GraphPad Prism by ordinary one-way ANOVA with Tukey's multiple comparisons test and all statistical analyses were based on at least three biologically independent samples. A detailed description of the Materials and Methods is available in Supplemental Material.
Supplementary Material
Acknowledgments
We thank J. Thomas, J. Cleary, and M. Scotti for technical assistance and D. Borchelt for comments on the manuscript. M.S.S. acknowledges support from the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS058901; NS98819) and the Muscular Dystrophy Association (MDA; RG480539), and C.A.N. and Ł.J.S. (MDA546770) received postdoctoral support from the Muscular Dystrophy Foundation and the MDA, respectively.
Author contributions: C.A.N., J.L.B., R.O., and M.S.S. designed the project; R.O. generated Dmpk CTGexp lines, C.A.N., B.M.K., R.O., and B.S.P. performed RNA-FISH and IF; C.A.N. and E.A.V. conducted super-resolution microscopy; C.A.N., J.L.B., B.A.O., Ł.J.S., H.A.C., and E.T.W. generated RNA-seq and RT-PCR results; R.O. and F.I. performed Southern blot analysis; C.A.N., J.L.B., R.O., and M.S.S. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.328963.119.
References
- Chujo T, Yamazaki T, Kawaguchi T, Kurosaka S, Takumi T, Nakagawa S, Hirose T. 2017. Unusual semi-extractability as a hallmark of nuclear body-associated architectural noncoding RNAs. EMBO J 36: 1447–1462. 10.15252/embj.201695848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. 1997. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci 94: 7388–7393. 10.1073/pnas.94.14.7388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A, Ashizawa T, Perryman MB, Scarlato G, Fenwick RG, et al. 1993. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science 260: 235–238. 10.1126/science.8469976 [DOI] [PubMed] [Google Scholar]
- Goodwin M, Swanson MS. 2014. RNA-binding protein misregulation in microsatellite expansion disorders. Adv Exp Med Biol 825: 353–388. 10.1007/978-1-4939-1221-6_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Groenen PJ, Bächner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, et al. 1996. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet 13: 316–324. 10.1038/ng0796-316 [DOI] [PubMed] [Google Scholar]
- Jenquin JR, Coonrod LA, Silverglate QA, Pellitier NA, Hale MA, Xia G, Nakamori M, Berglund JA. 2018. Furamidine rescues myotonic dystrophy type I associated mis-splicing through multiple mechanisms. ACS Chem Biol 13: 2708–2718. 10.1021/acschembio.8b00646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S. 2005. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet 14: 2189–2200. 10.1093/hmg/ddi223 [DOI] [PubMed] [Google Scholar]
- Krogias C, Bellenberg B, Prehn C, Schneider R, Meves SH, Gold R, Lukas C, Schneider-Gold C. 2015. Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J Neurol 262: 365–374. 10.1007/s00415-014-7566-6 [DOI] [PubMed] [Google Scholar]
- Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, et al. 2013. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med 5: 1887–1900. 10.1002/emmm.201303275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Monuki ES, Lehtinen MK. 2015. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci 16: 445–457. 10.1038/nrn3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola G, Cardani R. 2015. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 1852: 594–606. 10.1016/j.bbadis.2014.05.019 [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kazuno AA, Kelsoe J, Nakanishi M, Takumi T, Kato T. 2016. Exome sequencing in the knockin mice generated using the CRISPR/Cas system. Sci Rep 6: 34703 10.1038/srep34703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, et al. 2013. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74: 862–872. 10.1002/ana.23992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil AJ, Kim JC, Mirkin SM. 2017. Precarious maintenance of simple DNA repeats in eukaryotes. Bioessays 39 10.1002/bies.201700077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, Orr HT, Warren ST. 2013. The unstable repeats–three evolving faces of neurological disease. Neuron 77: 825–843. 10.1016/j.neuron.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ, Thornton CA. 2008. Cell-free cloning of highly expanded CTG repeats by amplification of dimerized expanded repeats. Nucleic Acids Res 36: e24 10.1093/nar/gkn025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, et al. 1996. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet 13: 325–335. 10.1038/ng0796-325 [DOI] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. 2012. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res 40: e61 10.1093/nar/gkr1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA. 2012. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet 21: 1312–1324. 10.1093/hmg/ddr568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Sznajder LJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, Nishino I, Nakamori M, Wang ET, Swanson MS. 2017. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev 31: 1122–1133. 10.1101/gad.300590.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA, Johnson K, Moxley RT, 3rd 1994. Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol 35: 104–107. 10.1002/ana.410350116 [DOI] [PubMed] [Google Scholar]
- Usdin K, House NC, Freudenreich CH. 2015. Repeat instability during DNA repair: insights from model systems. Crit Rev Biochem Mol Biol 50: 142–167. 10.3109/10409238.2014.999192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, Thornton CA, Wang ET, Berglund JA. 2016. Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet 12: e1006316 10.1371/journal.pgen.1006316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting EJ, Waring JD, Tamai K, Somerville MJ, Hincke M, Staines WA, Ikeda JE, Korneluk RG. 1995. Characterization of myotonic dystrophy kinase (DMK) protein in human and rodent muscle and central nervous tissue. Hum Mol Genet 4: 1063–1072. 10.1093/hmg/4.6.1063 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.