

Abstract
Synthetic biology aims to improve human health and the environment by repurposing biological enzymes for use in practical applications. However, natural enzymes often function with suboptimal activity when engineered into biological pathways or challenged to recognize unnatural substrates. Overcoming this problem requires efficient directed evolution methods for discovering new enzyme variants that function with a desired activity. Here, we describe the construction, validation, and application of a fluorescence-activated droplet sorting (FADS) instrument that was established to evolve enzymes for synthesizing and modifying artificial genetic polymers (XNAs). The microfluidic system enables droplet sorting at ~2–3 kHz using fluorescent sensors that are responsive to enzymatic activity. The ability to evolve nucleic acid enzymes with customized properties will uniquely drive emerging applications in synthetic biology, biotechnology, and healthcare.
Keywords: droplet microfluidics, droplet sorting, DrOPS, enzyme engineering, high throughput screening
Graphical Abstract
Introduction
Synthetic biology is a rapidly growing area of science that aims to reengineer natural biological systems for practical applications that address critical needs in human health and the environment.1, 2 An important aspect of synthetic biology is the ability to quickly and reliably engineer new cellular systems by treating enzymes as interchangeable parts that can be used to design novel biological pathways or act on unnatural substrates that function in completely artificial ways.3, 4 In vitro systems capable of replicating artificial genetic polymers (XNAs) are an example of the latter.5 These systems are currently being pursued as a source of biologically stable affinity reagents (aptamers) and catalysts for biomedicine.6 Unfortunately, natural enzymes often perform with suboptimal activity when introduced into a novel pathway or are challenged to recognize unnatural substrates.7–9 This problem has limited the development of emerging applications in synthetic biology, biotechnology, and healthcare, which rely on carefully tuned enzymes to achieve a desired outcome.10
Directed evolution offers a possible solution to this problem by providing a powerful method for improving the properties of existing enzymes or discovering new enzymes that are not restricted to biological substrates.11, 12 In a typical directed evolution experiment, a protein library is carried through iterative rounds of selection and amplification to enrich for members with a desired characteristic, like ligand binding, stability, or enzymatic activity. For directed evolution to be successful, efficient methods are needed to maintain the genotype-phenotype linkage. This problem has been solved for ligand binding assays, like phage display, where DNA plasmids are transcribed and translated in vivo and proteins are displayed on the surface of a bacteriophage particle or cell.13, 14 More recently, in vitro approaches, like mRNA display, have emerged that link newly translated proteins to their encoding RNA message via a covalent bond or stable ternary complex.15, 16 Using any of these methods, the identity of the selected protein is readily determined by sequencing the gene-coding region of the corresponding plasmid or template.
For protein selection experiments involving catalysis, the genotype-phenotype linkage is generally maintained by spatial segregation using cells, microtiter plates, or artificial man-made compartments that confine the enzyme and encoding plasmid to a defined boundary. Conventional enzymatic screens utilizing agar or microtiter plates are limited to a sample size of ~104 variants and can require several weeks to complete.17 Robotic automation can increase the scale and throughput of library processing by allowing ~106 members to be screened in a few days; however, such systems come with a significant increase in cost and reagent volume.18 A more efficient and economical approach to library screening involves the miniaturization of directed evolution experiments into artificial reaction compartments composed of uniform water-in-oil (w/o) droplets produced by microfluidics.19–22 Compared to robotic systems, microfluidic systems allow researchers to screen ~108 variants per day using 106-fold less sample volume.
Applying microfluidics to directed evolution demands efficient strategies for separating positive droplets away from a large background of negative droplets.23 A straightforward approach is to produce water-in-oil-in-water (w/o/w) double emulsion droplets that can be dispersed in an aqueous phase that is amenable to sorting on a commercial fluorescence-activated cell sorting (FACS) instrument24. FACS-based sorting of double emulsion droplets has been demonstrated for several enzymes,25 including those with alkaline phosphatase and polymerase activity.24, 26 More recently, fluorescence-activated droplet sorting (FADS) devices have been developed that can directly sort single emulsion droplets in a manner analogous to FACS.27 These systems have proven successful in a small but growing number of examples involving directed evolution.19, 28–31
Here, we describe the construction, validation, and application of a FADS instrument (Figure 1) for evolving enzymes that can synthesize and modify XNAs (nucleic acid polymers with unnatural sugar-phosphate backbones).32 We show two different methods for producing single emulsion droplets at 30 kHz that encapsulate individual E. coli cells, which are used as a vehicle for protein expression and delivery. We present thermal, chemical, and enzymatic strategies for releasing recombinant enzyme into the surrounding compartment and demonstrate the use of four different types of fluorescent sensors that are responsive to enzymes that function with polymerase-mediated primer-extension, polymerase-mediated primer-extension with strand displacement, ligase, and restriction endonuclease activities. We show efficient droplet sorting at ~2 kHz with low false-positive and false-negative values. Finally, the power of this approach was demonstrated in a mock screen, which provided an enrichment value of >3000-fold from a doped library containing one active enzyme into a background of 6447 inactive enzyme variants. The FADS system and associated fluorescent sensors make it possible to discover new enzyme variants that can synthesize and manipulate XNA in a manner analogous to natural enzymes to drive new applications in biotechnology and healthcare.
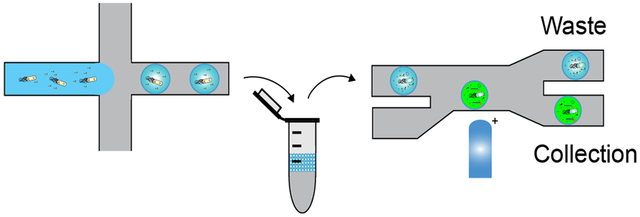
Figure 1. Fluorescence-activated droplet sorting.

(a) Overview of microfluidic droplet sorting. A library of genes encoding a specific protein or enzyme are expressed in E. coli and encapsulated in microfluidically generated w/o droplets along with a sensor and corresponding substrates. Expressed proteins are liberated from their E. coli host via chemical, enzymatic, or heat lysis and incubated at the desired reaction temperature off-chip. Following incubation, the droplet population is sorted using the FADS system to enrich for members with a desired functional activity. (b) Overview of the optical train and electronic components of the FADS system. For Cy3 based fluorescence, incident light from a 552 nm laser is focused onto the sorting junction of the microfluidic chip through a 20x objective to excite active droplets. Emitted light is led back through the objective past a series of dichroic mirrors to a PMT with a 563 nm band-pass filter. The signal from the PMT is fed to a FPGA controlled by custom LabView software. Upon detecting a fluorescent signal, the FPGA is instructed to energize a 4M NaCl electrode. A high voltage amplifier amplifies the generated sorting pulse to produce the DEP force necessary to deflect the target drop into the collection channel. Blue light with minimal Cy3 spectral overlap is used to illuminate the sorting chip for monitoring with a high-speed camera. A host PC interfaces with the FPGA to collect data and adjust sorting parameters.
Results
Generating microfluidic droplets for enzyme engineering
We designed and fabricated two different types of fluorocarbon-coated polydimethylsiloxane (PDMS) microfluidic devices based on previous designs that were established for generating uniform water-in-oil (w/o) droplets.33, 34 The fluorocarbon-coated microfluidic devices (Fig. S1–S2) contain similar flow-focusing junctions designed to produce uniform w/o droplets that are stabilized by surfactants in the oil that prevent coalescence at elevated temperatures (as high as 95 °C) and allow for long-term storage at room temperature. The two droplet production devices differ based on the design of the channels carrying the aqueous stream (Fig. S2), which promotes either a single aqueous stream or a double co-flowing aqueous stream upstream of the flow focusing junction. The single-stream device was designed to be compatible with a thermal lysis strategy, while the double flow design allows for the delivery of lysis reagents separate from the population of E. coli cells. Following an iterative design-build-test process, optimal designs produced for both PDMS devices were found to maintain a droplet production rate of 30 kHz for at least 7–8 hours, which is long enough to produce ~109 droplets.
Droplets were formed following a Poisson distribution (λ = 0.1) to ensure that 95% of the occupied droplets contain only one E. coli per droplet. Under these conditions, the droplet population is produced at 10% occupancy (Fig. S3a). This distribution is necessary for maintaining the genotype-phenotype linkage required for directed evolution whereby the genetic information encoded in the DNA plasmids of positively sorted droplets is recovered to determine the sequence of enzyme variants with desired functional properties. Given that E. coli concentration is readily quantified by absorbance (OD600), a conversion factor relating OD600 to the actual cellular concentration of E. coli (σ1.0) can be achieved using the following equation where is the droplet volume.
If we assume that σ1.0 = 5.0 × 108 cells/mL when OD600 = 1.026, then an OD600 of 0.05 will result in λ = 0.1 for 20 μm diameter droplets. This prediction closely agrees with empirical observations obtained using green fluorescent protein (GFP) expressed in E. coli (Fig. S3b).
E. coli lysis inside droplet microcompartments
Using E. coli as a protein expression system for delivering recombinant enzymes to w/o droplets requires efficient methods for lysing the bacteria without disrupting the droplet microcompartment. As described above, droplets are produced under conditions in which the aqueous phase contains all the reagents needed to achieve a desired activity assay once the enzyme is released from the bacteria and allowed to react with the substrate. Since w/o droplets are resistant to coalescence at elevated temperatures, thermophilic enzymes can be released using a thermal lysis strategy, which typically involves heating the droplet population for 5 minutes at 90–95 °C. For thermal lysis, it is advised to use fluorous oils and surfactants that stabilize the compartments at high temperatures.
Mesophilic enzymes are not compatible with heat lysis procedures and necessitate the use of chemical or enzymatic agents that can lyse the E. coli membrane under milder conditions. We therefore chose to explore this process using GFP as a reporter system for natively folded enzymes that escape the cell membrane. Using the co-flow droplet generator design (Fig. S2a), we produced a population of 20 μm diameter droplets in which GFP-expressing E. coli were co-encapsulated with buffer only, buffer and Bug Buster (1x), or buffer and (0.1 mg/mL) lysozyme. Each droplet population was monitored over time at 37 and 55 °C by fluorescence microscopy (Fig. S4). As expected, GFP fluorescence in the buffer only population was confined to the cell. By contrast, droplets encapsulated with Bug Buster became fluorescent after a brief incubation (5 min, 37 °C). Similar images were observed for the lysozyme sample after incubation at a slightly elevated temperature (5 min, 55 °C). Based on these results, we postulate that either condition should be suitable for microfluidic screening of mesophilic enzymes.
Design of a fluorescent-activated droplet soring (FADS) device
Following cell lysis, droplets are incubated for an extended period of time to allow the enzyme to escape the cell membrane and react with the substrate. Since this is a single-cell technique, stochastic factors, such as differences in protein expression levels between individual E. coli cells, differences in the release of the enzyme into the surrounding droplet, and the failure of some enzymes to react with enough substrate molecules to produce a robust signal, may contribute to varying levels of droplet fluorescence. Consequently, it is important that the signal-to-noise ratio (SNR) of the optical sensor used to detect active droplets be as high as possible in order to identify functional enzyme variants with high confidence. Previously, we have shown that the Cy3-Iowa Black fluorophore-quencher pair maintains a higher SNR than other commonly used fluorophore-quencher pairs.26 Based on this observation, we designed a FADS instrument that was compatible with the green-orange fluorescence of a Cy3 organic dye (~550 nm excitation and ~570 nm emission).
An overview of the optical train and associated electronics for signal acquisition and sorting are provided in Figure 1. Focused light from a 552 nm laser is positioned upstream of the sorting junction of a microfluidic chip that is designed for dielectrophoretic sorting using a 4M NaCl salt water electrode that was surrounded by a large ground moat (Fig. 2). This chip has the capacity to screen individual droplets at rates approaching 30 kHz, which is comparable to, if not, slightly faster than commercial FACS instruments. Although faster rates are possible, our experiments were performed with a sorting rate of ~2 kHz using pressure pumps that have a smaller footprint and allow for near instantaneous response times (~100 ms). We found that this rate is more than sufficient to screen modest size libraries (104 members) with high redundancy (~103 copies of each library member) in just a few hours.35
Figure 2. Microfluidic chip design.

(a) Schematic illustration of the microfluidic sorting chip. Shallow channels are shown in grey and deeper channels appear in green. Red and black channels indicate the electrode and ground moat, respectively. Boxed regions represent portions of the chip that are magnified in subsequent images. (b) Regular spacing of re-injected droplets prior to the sorting junction. (c) Still image from a 2 kHz sorting video showing positive fluorescent drops (transparent) being separated from negative drops containing an opaque green dye. (d) Pressure equilibration channels immediately prior to the outlets offset differences in hydraulic resistance between the waste and collection channels. Scale bars are 50 μm.
The FADS chip was designed with a second flow focusing junction which allows droplets entering the device to become evenly spaced inside a microfluidic channel (Fig. 2b). Flowing droplets pass through a focused laser line that is located immediately upstream of a Y-junction that leads to waste and collection channels. Droplets passing through the laser produce a fluorescent signal with a ~570 nm emission wavelength that is detected by a photomultiplier tube (PMT) in the optical train. If the optical signal for a fluorescent droplet exceeds a user-defined value set for photon counting intensity and occupancy time, three 600 Vpp (50 kHz, 50% duty cycle) square wave pulses are applied across the salt water electrode within 5 μs of detecting the droplet. Based on the principle of dielectrophoresis, the non-uniform electric field polarizes the droplet causing it to deflect into the collection line located at the Y–junction (Fig. 2c). Dim droplets that are either empty or contain inactive enzyme maintain their trajectory and flow into the waste line. As a precaution, an oil bias flow acts to prevent the migration of inactive droplets into the collection line. As needed, the device may be illuminated with blue light to visualize droplet sorting and capture video data with a high-speed camera (35,000 fps), as blue light does not overlap with the spectral properties of Cy3.
Sorting fluorescent droplets
The efficiency of droplet sorting is a critical parameter for directed evolution experiments aimed at producing enzymes with tailormade properties as a high false negative rate causes active variants to be removed from the pool, while a high false positive rate leads to reduced selection efficiency. To evaluate the sorting efficiency of our home-built FADS instrument, we calculated the efficiency of droplet sorting using a defined mixture of fluorescent and non-fluorescent droplets. The fluorescent and non-fluorescent droplets were generated at full occupancy by encapsulating either a Cy3-labeled DNA (ST.1G.HP.44.Cy3, Table S1) or a green dye inside w/o droplets, respectively. The population of non-fluorescent droplets appears opaque when illuminated with blue light (Fig. 2b–d), making them distinguishable from the population of fluorescent droplets on still and video images.
The two droplet samples were mixed together to create a pre-sorted population of droplets that contained ~25% fluorescent droplets. The mixed sample was introduced into the FACS device and sorted with a photon counting threshold of 60 and a temporal residence time of 50 – 75 μs. These parameters were selected based on a series of flow experiments where we observed the distribution of peak heights and widths for homogeneous populations of fluorescent drops flowing at a rate of ~2–3 kHz. A sample of the droplet traces from a 1-hour sorting run (~10 million droplets) performed at screening rate of 3 kHz (image acquisition rate of 200 kHz) is provided in Figure 3a. A movie made to illustrate the droplet sorting process is provided in the supplementary information. Fluorescence microscopy images taken of the pre- and post-sorted droplets (Fig. 3b–d) indicate that the positively sorted droplet population was composed of ~99.75% fluorescent drops, while the negatively sorted population contained only ~ 0.18% fluorescent drops.
Figure 3. FADS-based sorting of a fluorescent droplet population.

(a) Sample of data acquired during a sorting run (~1.4 kHz) showing the PMT-detected fluorescence signal (red, left axis) produced by positive and negative drops along with the voltage pulse applied to the electrode (blue, right axis) when a positive drop is detected. Insets show a magnified image of the shaded region (left) illustrating a positive drop along with the generated sorting pulse, and the histogram (right) showing the distribution of peak droplet fluorescence values for an active population. Fluorescence microscope images showing the (b) pre-sorted droplet population, (c) positively sorted droplets, and (d) negative droplets. Negative droplets contained an opaque green dye and positive droplets contained 1 μM Cy3-labeled DNA. Scale bars are 100 μm.
Videos were taken at regular intervals to monitor the deflection of fluorescent droplets into the collection line. Close inspection of the droplet sorting process reveals that the small number of non-fluorescent droplets present in the collection line was mainly due to irregular droplet spacing, which causes smaller size droplets (produced by droplet splitting at the sorting junction) to pack behind regular size droplets. When the regular size droplets are fluorescent, the smaller size droplets are deflected into the collection line by dielectrophoresis. A second source of background was detected in the equilibrium channels located downstream of the sorting junction. In this case, large equilibrium channels were allowing some non-fluorescent droplets to pass from the waste line into the collection line. To overcome both problems, a new FADS device was produced that increased droplet spacing in the sorting channel and modified the equilibrium channel between the waste and collection lines. The new device -reduced the frequency of non-fluorescent droplets in the sorted pool to <0.01%.
Establishing optical sensors for a diverse set of nucleic acid enzymes
DNA modifying enzymes play a critical role in biotechnology and medicine by allowing genetic information to be amplified, ligated, and sequenced.36 Consequently, these enzymes represent a toolkit of reagents that are routinely used for basic and applied research involving genes, gene families, and entire genomes. However, in many cases, natural enzymes do not perform as well as expected because they are being used in a non-natural context and would benefit from optimization by directed evolution.37 As a first step in this direction, optical sensors are needed that can report on the functional activity of DNA modifying enzymes in w/o droplets. Toward the broader goal of establishing a set of nucleic acid modifying enzymes that function on nucleic acid polymers with non-natural backbone structures, we investigated four different types of enzymatic activity that are central to molecular biology. These include DNA synthesis by primer-extension, DNA synthesis with strand displacement, restriction enzyme digestion, and DNA ligation (Fig. 4).
Figure 4. Optical sensing modalities tested for droplet sorting based on DNA modifying enzymes.

Droplet based detection of four different classes of nucleic acid modifying enzymes were performed using recombinant enzymes that catalyze (a) polymerase extension, (b) strand displacement, (c) restriction enzyme digestion, and (d) DNA ligation activities. Data is presented as the distribution of peak droplet fluorescence for each population. The peak value fluorescence describes the maximum fluorescence reading of a droplet as it passes in front of the laser line. The max standard is a chemically synthesized version of the product obtained from the enzymatic reaction.
Polymerase extension.
DNA polymerases are one of the most important enzymes found in nature, and as such, have become the cornerstone of biotechnology applications that involve DNA synthesis, amplification, and sequencing.38 Polymerases with modified activities are continually being developed to support new applications in healthcare, which has created a demand for engineered polymerases with properties that exceed their natural counterparts. To support this process, we have established an optical polymerase activity assay that allows high-throughput screening in uniform w/o/w droplets that are sorted by FACS.26 The sensor consists of a 3’-Cy3 labeled self-priming template that is quenched at room temperature by a short DNA strand carrying a 5’-quencher. At elevated temperatures, the quencher strand dissociates from the template and if the polymerase is able to extend the primer to full-length product, the droplets fluoresce because the quencher is no longer able to reanneal with the template.
To evaluate the polymerase sensor in the context of a FADS device, three different commercial thermophilic DNA polymerases (Bst, Taq, and Q5) were encapsulated in droplets, incubated for 1 hour at 55 °C, and sorted based on their fluorescence (activity). A histogram analysis of ~106 droplet-sorting events from each polymerase population (Fig. 4a) confirms that each sample exhibits robust polymerase activity that is 95% of the max standard, a chemically synthesized version of the product obtained from the enzymatically driven reaction (Fig. S5a). These results are in close agreement with bulk solution experiments that were assayed by denaturing polyacrylamide gel electrophoresis (PAGE) (Fig. S6a). Based on the signal intensity of our no polymerase control, the maximum SNR for this sensor is ~5.4 (Z’ = 0.79), which is more than sufficient to distinguish the fluorescent droplets from non-fluorescent droplets.
Strand displacement.
DNA polymerases with strand displacement activity have found practical utility in numerous DNA amplification techniques, including loop-mediated isothermal amplification (LAMP), strand displacement amplification (SDA), helicase-dependent amplification (HDA), and the nicking enzyme amplification reaction (NEAR). These techniques are critical components of several point-of-care diagnostic tools that rely on a simple workflow, quick turnaround time, and minimal analytical instrumentation. Given the strong interest in polymerases with strand displacement activity, we sought to create an assay to promote the directed evolution of these enzymes in w/o droplets. For this application, we used a modified version of our polymerase sensor that contained a longer, more thermally stable version of the quencher strand whose displacement is needed to create a fluorescent signal. The sensor was evaluated in droplets using commercial DNA polymerases that exhibit (Bst) and lack (Q5) strand displacement activity. Analysis of ~106 droplet-sorting events from each polymerase (Fig. 4b) confirms that Bst DNA polymerase exhibits robust strand displacement activity with a cumulative droplet count that is ~85% of the max standard, while Q5 DNA polymerase is similar to the no polymerase control. These results are in close agreement with bulk solution experiments assayed by traditional PAGE (Fig. S6b). The sensor yields an SNR of ~18 (Z’ = 0.86) (Fig. S5b), which is 3-fold better than the polymerase sensor developed for primer-extension activity. The increased sensitivity of the strand displacement sensor is due to the higher thermal stability of the quencher strand, which is less likely to dissociate from the template at room temperature than the shorter strand developed for standard primer-extension activity (ΔΔG = −42.7 kcal/mole).
Restriction digestion.
Restriction endonucleases catalyze the sequence-specific double-stranded cleavage of DNA to produce cut DNA products with blunt or sticky-ends. These enzymes are commonly used for cloning and plasmid linearization. Because the evolution of enzymes with custom restriction endonuclease activity is an attractive area of research, we adapted an optical sensor that was previously developed for use in bulk solution.39 Accordingly, the sensor consists of a nicked duplex in which enzymatic activity disrupts a fluorophore-quencher pair adjacent to the digestion site (Fig. 4c). Using a sensor that was engineered to contain a Pst I nuclease digestion site, we measured the strand cleavage activity of two commercial DNA endonucleases (Pst I and Not I) in droplets. A histogram produced from ~106 droplet-sorting events (Fig. 4c) indicates that Pst I is specific for the Pst I nuclease digestion site, with Not I indistinguishable from the no enzyme control. The sensor produced a modest SNR of 2.6 (Z’ = 0.29), which is consistent with 50% cleavage in bulk solution (Fig. S6c) and agrees with manufacturer specifications.
DNA ligation.
Ligases are critical for a variety of biotechnology applications, including cloning and next-generation DNA sequencing (NGS). We therefore adapted a previous DNA ligase sensor to function in microfluidic droplets.40 The sensor is based on a molecular beacon design with two short substrates annealed to the loop region of a DNA stem-loop structure. Enzymes that ligate the two DNA strands together produce a fluorescent signal by converting the stem-loop into a linear duplex that separates the donor-quencher pair. We evaluated the activity of the sensor in droplets using T4 DNA ligase (Fig. S5d). Analysis of ~106 droplet-sorting events demonstrates that the enzyme functions with high activity, producing fluorescent values equivalent to the max standard (SNR of ~12, Z’ = 0.84, Fig. S5d). Notably, the sensor fluorescence in droplets lacking the enzyme or lacking a phosphorylated substrate remained quenched. Interestingly, we found that the ligase sensor is highly sensitive to the length of the acceptor and donor strands (Fig. S7), which required careful optimization to create a high activity sensor.
Polymerase-mediated DNA, RNA, and XNA synthesis from E. coli generated enzymes
Engineering DNA polymerases to synthesize nucleic acid polymers with backbone structures that are distinct from those found in nature has enabled the evolution of affinity reagents (aptamers) and catalysts that are resistant to nuclease digestion.32, 41–44 However, substantially more work is needed to establish new examples of engineered polymerases that recognize different XNA polymers. To demonstrate how researchers could evolve new examples of XNA polymerases, we encapsulated populations of E. coli cells expressing three different types of thermophilic polymerases in w/o droplets. The set of polymerases included a natural DNA polymerase isolated from the bacterial species Thermococcus gorgonarius (Tgo) that synthesizes DNA and coincidentally a close structural analog of DNA known as 2’-fluoroarabino nucleic acid (FANA),42 a DNA polymerase (DV-QGLK) that was engineered to synthesize RNA,45 and a DNA polymerase (Kod-RS) that was engineered to synthesize threose nucleic acid (TNA).26 In each case, E. coli cells expressing these enzymes were encapsulated in w/o droplets with the polymerase sensor responsive to primer-extension activity and the correct set of nucleoside triphosphates (dNTPs, NTPs, FANA-NTPs, and tNTPs), heat lysed, and incubated for 18 hours at 55 °C. Fluorescent microscope images collected afterwards reveal a strong fluorescence dependence on the presence of nucleoside triphosphates that are supplied ex vivo, as droplets lacking nucleoside triphosphates remain dim. Importantly, this result demonstrates that endogenous nucleoside triphosphates (dNTPs and NTPs) from the E. coli host are present at insufficient quantities to produce a false positive signal, confirming that polymerase engineering in microfluidic droplets can proceed without interference from endogenous substrates.
Mock enrichment assay for DNA synthesis activity
We evaluated the performance of our FADS system by performing a mock selection designed to enrich for droplets with recombinant DNA polymerase activity. E. coli cells expressing wild-type Kod DNA polymerase (KOD-wt) and a null mutant (KOD-null) containing the D542G mutation in the enzyme active site were mixed at a molar ratio of 1:1000 and 1:10000 (active to inactive variants). The E. coli populations were separately encapsulated in microfluidic droplets with the polymerase sensor and substrates necessary for primer-extension activity, heat lysed, incubated en masse (1 h, 55 °C), and individual droplets were sorted for DNA synthesis activity using the FADS device. Plasmids from positively sorted droplets were isolated, the gene-coding region was amplified by PCR and the expression vector was reconstructed by Gibson assembly, transformed into fresh E. coli, grown to confluency, and induced to express a new population of E. coli cells that had been enriched in polymerases with DNA synthesis activity. The regenerated population of E. coli were encapsulated in microfluidic droplets along with the sensor, buffer, and dNTP substrates, and the number of fluorescent droplets were counted by flowing the enriched droplet population through the FADS device.
We carefully measured the enrichment values for both doped library populations using flow data obtained from the droplet sorting instrument (~106 droplets per experiment). These experiments were performed separately from the actual droplet sorting experiments, which involved ~107 droplets for each doped library. Analysis of homogeneous populations of wild-type and null mutant polymerase reveal that the false negative rate is ~2% and false positive rate is ~0.005% (Fig. 6a). Evaluation of the starting populations before selection reveals that the two doped libraries contain 1 in 722 and 1 in 6447 active to inactive members, which approximates the anticipated doping levels of 1:1000 and 1:10000 and provides the upper limit for an absolute enrichment value (Fig. 6b). However, one should keep in mind that the true maximum enrichment value is slightly less due to Poisson loading of the E. coli into the droplets, which allows for a small number of co-encapsulation events where a positively sorted droplet could contain more than one E. coli cell. Analysis of both libraries indicates that after 1 round of selection, ~50–55% of the E. coli containing droplets have functional enzymes. This corresponds to enrichment values of 407- and 3227-fold for the 1:1000 and 1:10000 doped libraries, respectively, which is close to the theoretic maximum enrichment values of 656- and 5861-fold, respectively, calculated using Poison statistics of experimentally determined encapsulation values (Fig. 6b and Fig. S8–S9). These levels are sufficient to allow for both purity and yield sorting depending on the needs of the experiment.
Figure 6. Enrichment assay based on DNA polymerase activity.

a) Images and distributions of peak fluorescence for clonal populations of E. coli expressing wild-type KOD DNA polymerase (left) and a null mutant KOD-D542G (right) encapsulated in droplets. Scale bars are 50 μm. b) Tabulated results and theoretical enrichment values. E. coli expressing wild-type KOD DNA polymerase were spiked at cellular ratios of 1:1000 and 1:10000 into a population of E. coli expressing the null mutant KOD-D542G. ε0 and ε1 represent the fraction of KOD-wt expressing cells in the starting and sorted populations, respectively. Raw ε0 and ε1 values were normalized using experimentally determined false negative and false positive rates obtained from the clonal populations. Experimental enrichment (ηexp) is a ratio of ε1,norm/ε0,norm. The absolute enrichment (ηabs) is 1/ε0,norm, while the maximum theoretical enrichment (ηmax) is mathematically derived based on Poisson statistics.
Discussion
We describe a microfluidic-based droplet sorting platform that enables the directed evolution of DNA processing enzymes. In contrast to previous droplet sorting approaches, our methodology is specifically tailored for applications that involve the development of artificial genetic polymers for synthetic biology.5 A key aspect of our approach was the design and validation of four different fluorescent sensors that transduce various polymerase activities (normal primer extension and primer-extension with downstream strand displacement), as well as ligase and restriction endonuclease activity inside w/o droplets.
Quantitative analysis of various nucleic acid processing assays suggested extremely high statistical robustness. Apart from the restriction endonuclease sensor, each sensor functions with a Z’ of 0.79 – 0.86, indicating exceptional statistical discrimination as assays of biological activity.46 Sorting and enrichment experiments here were comparisons of no activity and full activity for the purposes of obtaining Z’ values, however, a directed evolution experiment attempts to enrich mutants with modestly increased activity above some initial level of activity. In a power analysis of the droplet-scale polymerase activity assay (Z’ = 0.79), the separation between background and wild-type polymerase activity is 28σ (σ, standard deviation, ~ 2 RFU). Only a 1.5-fold increase in signal over background corresponds to 4σ (32 ppm false discovery rate). In our companion piece,35 a KOD polymerase mutant library screen was conducted using a 60-RFU threshold (4-fold signal over background, ~22σ), demonstrating the feasibility and high statistical power of the approach to identify polymerase mutants with novel activity. Although the lower quality of the endonuclease activity assay would normally preclude its use in a standard-scale high-throughput protein engineering workflow, the ultra-miniaturized scale of the FADS system makes implementation of the assay relatively risk-free. Collectively, these enzymatic activities represent the core functional units of the molecular biology toolkit, which impacts nearly every area of healthcare research from drug discovery to personalized medicine.
The platform facilitates the production of a monodisperse population of small (20 μm diameter) droplets at a frequency of 30 kHz (>100 million droplets per hour) using a highly stable dripping regime that can be maintained for a standard workday. However, in practice, most droplet production runs are complete after just 1 hour, as a population of 108 droplets is more than sufficient for typical directed evolution experiments. Although we use E. coli as a protein expression system for delivering recombinant enzymes to the microfluidic compartment, the technology platform is compatible with coupled cell-free transcription and translation (TNT) systems.47, 48 The one exception is polymerase engineering where dNTP and NTP substrates interfere with the selection.26 Relative to commercial TNT systems, E. coli benefits from lower reagent costs, ease of production, and user-friendly methods for storing engineered cell lines as glycerol stocks. An additional benefit is the fact that DNA plasmids provide a convenient format that allows for the immediate expression, purification, and characterization of selected variants.
Once the enzymes have been released from the E. coli and allowed to react with the fluorescent sensor, the sorting device is used to screen fluorescent droplets at a rate of 3 kHz (10 million droplets per hour). This frequency compares favorably with automated liquid handling robots that screen 106 samples in 2–3 days but require far greater cost and sample volume.18 Custom software allows for user-defined parameters in which droplet threshold is set based on photon counts and residence time. The ability to adjust the fluorescent droplet parameters allows for high instrument sensitivity and makes it possible to establish droplet thresholds that are specific for different optical sensors and enzymatic applications. Through iterative design-build-test cycles, we were able to show that the optimal droplet sorting device reduces the frequency of non-fluorescent droplets in the sorted pool to values that are less than 1 in 10,000 positively sorted droplets. This number is more than sufficient to meet the needs of most droplet sorting experiments involving enzymes that are used to replicate and modify XNAs.
Single cell directed evolution experiments that utilize E. coli as a protein expression system face stochastic problems that can limit the enrichment of functional enzymes during the first few rounds of selection. These include differences in protein expression levels between individual E. coli cells, differences in the release of the enzyme into the surrounding droplet, and the failure of some enzymes to react with enough substrate molecules to produce a strong fluorescent signal. It should be noted that recombinant enzymes expressed in E. coli are diluted ~1000-fold when they enter the droplet compartment. This is generally not a problem for natural enzymes which have been evolved by natural selection to recognize their substrates with high catalytic efficiency. However, the same is not true for enzyme libraries which typically function with reduced activity and protein stability. For this reason, it is important to ensure that the optical sensor functions with a high SNR and to adjust the droplet sorting parameters accordingly to ensure that enough droplets are selected to enable efficient recovery and amplification of the positively sorted library.
Although FACS-based sorting of double-emulsion droplets provides a user-friendly approach for performing directed evolution experiments in microfluidic droplets,24 custom FADS-instruments offer a number of unique advantages.49 First, FADS instruments utilize single-emulsion droplets, which are easier to produce and significantly more stable than double-emulsion droplets. Second, the higher stability of single emulsion droplets reduces the level of background contamination in positively sorted droplets, as single emulsion droplets are less prone to self-lysing. Third, FADS-based sorting is specifically designed for droplet sorting applications while FACS-based sorting was designed for eukaryotic cells, and later adapted for droplet sorting applications.24 In the case of small w/o droplets, it is unlikely that FACS instruments will be able to sort individual droplets. Fourth, FADS provides a higher partitioning efficiency than w/o/w sorting in FACS, as microfluidic devices are specifically engineered for the size and charge of single emulsion droplets. Fifth, high speed cameras enable researchers to visualize each droplet sorting event, which is not possible with FACS. Last, FADS devices offer a cheaper alternative to conventional FACS instruments by accelerating the pace of research with a dedicated instrument for directed evolution.
We anticipate that directed evolution using FADS will make it possible to address real-world applications that currently limit the fields of synthetic biology, biotechnology, and molecular medicine. In the area of polymerase engineering, we expect FADS-based instruments to increase the speed at which new polymerases are developed that can faithfully copy genetic information back and forth between DNA and XNA. Current XNA polymerases, by contrast, function with reduced catalytic activity and fidelity as compared to their natural counterparts.50 By analogy, the same methodology could also be used to evolve enhanced DNA and RNA polymerases for NGS applications that involve modified substrates or require long read lengths. Similarly, polymerases with reverse-transcriptase activity could be developed that are better suited for structured RNA molecules that cause existing polymerases to stall. In the area of ligases, we expect to see an emergence of activities that allow engineered ligases to synthesize XNA strands on lengths and scales that are currently not possible by solid-phase synthesis. These same techniques could be used to improve the quality of DNA ligases that are currently used to barcode NGS libraries, which suffer from sequence-bias. Last, the ability to rapidly search vast regions of sequence space makes FADS perfectly suited for discovering polymerases that can replicate XNA independent of DNA. This last application would have a dramatic impact on synthetic biology projects that utilize XNA polymers by providing a convenient method for amplifying XNA directly without conversion to DNA.
In summary, this work presents a single-droplet sorting instrument and fluorescent sensors that enable the directed evolution of enzymes that can synthesize and modify artificial genetic polymers. Because of the flexibility of the technology, the same methodology developed for XNA could also be used to optimize natural enzymes that synthesize and modify DNA and RNA. Such projects open the door to custom enzymes for synthetic biology, biotechnology, and molecular medicine.
Supplementary Material
Figure 5. Polymerase-mediated synthesis of natural, non-cognate, and xeno-nucleic acid polymers in uniform droplet microcompartments.

E. coli expressing natural and engineered polymerases were encapsulated in droplet microcompartments, lysed, and assayed for polymerase activity in the presence (left) and absence (right) of the appropriate nucleoside triphosphate. Enzymatic activities include (a) TGO-mediated DNA synthesis; (b) DV-QGLK-mediated RNA synthesis, (c) TGO-WT-mediated FANA synthesis, and (d) KOD-RS-mediated TNA synthesis. The absence of florescence in the no nucleoside triphosphate controls demonstrates that endogenous E. coli nucleotides do not interfere with polymerase activity.
Acknowledgements
We would like to thank members of the Chaput laboratory for helpful discussions and critical reading of the manuscript. This work was supported by new laboratory start-up funds from the University of California, Irvine. B.M.P. was supported by grants from the National Institutes of Health (GM120491) and the National Science Foundation (1255250).
Footnotes
Competing Financial Interests
The authors declare no competing interests.
Additional Information
Supplementary information contains materials and methods, supplementary tables S1, supplementary figures S1–S9, and mathematical model.
References
- 1.Leonard E, Nielsen D, Solomon K, and Prather K (2008) Engineering microbes with synthetic biology frameworks, Trends Biotechnol 26, 674–681. [DOI] [PubMed] [Google Scholar]
- 2.Weber W, and Fussenegger M (2009) Engineering of synthetic mammalian gene networks, Chem. Biol 16, 287–297. [DOI] [PubMed] [Google Scholar]
- 3.Canton B, Labno A, and Endy D (2008) Refinement and standardization of synthetic biological parts and devices, Nat. Biotechnol 26, 787–793. [DOI] [PubMed] [Google Scholar]
- 4.Benner SA, and Sismour AM (2005) Synthetic biology, Nat. Rev. Genet 6, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaput JC, Yu H, and Zhang S (2012) The emerging world of synthetic genetics, Chem. Biol 19, 1360–1371. [DOI] [PubMed] [Google Scholar]
- 6.Dunn MR, Jimenez RM, and Chaput JC (2017) Analysis of aptamer discovery and technology, Nat. Rev. Chem 1, 0076. [Google Scholar]
- 7.Paddon CJ, and Keasling JD (2014) Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development, Nat. Rev. Microbiol 12, 355–367. [DOI] [PubMed] [Google Scholar]
- 8.Chaput JC, Ichida JK, and Szostak JW (2003) DNA polymerase-mediated DNA synthesis on a TNA template, J. Am. Chem. Soc 125, 856–857. [DOI] [PubMed] [Google Scholar]
- 9.Kempeneers V, Vastmans K, Rozenski J, and Herdewijn P (2003) Recognition of threosyl nucleotides by DNA and RNA polymerases, Nucleic Acids Res 31, 6221–6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner NJ (2009) Directed evolution drives the next generation of biocatalysts, Nat. Chem. Biol 5, 567–573. [DOI] [PubMed] [Google Scholar]
- 11.Packer MS, and Liu DR (2015) Methods for the directed evolution of proteins, Nat. Rev. Genet 16, 379–394. [DOI] [PubMed] [Google Scholar]
- 12.Zeymer C, and Hilvert D (2018) Directed Evolution of Protein Catalysts, Annu. Rev. Biochem 87, 131–157. [DOI] [PubMed] [Google Scholar]
- 13.Smith G, and Petrenko V (1997) Phage Display, Chem. Rev 97, 391–410. [DOI] [PubMed] [Google Scholar]
- 14.Cherf GM, and Cochran JR (2015) Applications of Yeast Surface Display for Protein Engineering, Methods Mol. Biol 1319, 155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts RW, and Szostak JW (1997) RNA-peptide fusions for the in vitro selection of peptides and proteins, Proc. Natl. Acad. Sci. U.S.A 94, 12297–12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanes J, and Pluckthun A (1997) In vitro selection and evolution of functional proteins by using ribosome display, Proc. Natl. Acad. Sci. U.S.A 94, 4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, and Arnold FH (1997) Combinatorial protein design: strategies for screening protein libraries, Curr. Opin. Struct. Biol 7, 480–485. [DOI] [PubMed] [Google Scholar]
- 18.Martis E, Radhakrishnan R, and Badve R (2011) High-throughput screening: the hits and leads of drug discovery, J. Appl. Pharm. Sci 1, 2–10. [Google Scholar]
- 19.Agresti JJ, Antipov E, Abate AR, Ahn K, Rowat AC, Baret JC, Marquez M, Klibanov AM, Griffiths AD, and Weitz DA (2010) Ultrahigh-throughput screening in drop-based microfluidics for directed evolution, Proc. Natl. Acad. Sci. U.S.A 107, 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price AK, and Paegel BM (2016) Discovery in Droplets, Anal. Chem 88, 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaerli Y, Kintses B, and Hollfelder F (2012) Protein engineering in microdroplets, In Protein Engineering Handbook (Lutz S, and Bornscheuer UT, Eds.), pp 73–85, Wiley VCH, Weinheim. [Google Scholar]
- 22.Aharoni A, Griffiths AD, and Tawfik DS (2005) High-throughput screens and selections of enzyme-encoding genes, Curr. Opin. Chem. Biol 9, 210–216. [DOI] [PubMed] [Google Scholar]
- 23.Duncombe TA, Tentori AM, and Herr AE (2015) Microfluidics: reframing biological enquiry, Nat. Rev. Mol. Cell Biol 16, 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zinchenko A, Devenish SRA, Kintses B, Colin P-Y, Fischlechner M, and Hollfelder F (2014) One in a million: flow cytometric sorting of single cell-lysate assays in monodisperse picoliter double emulsion droplets for directed evolution, Anal. Chem 86, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang G, and Withers SG (2009) Ultrahigh-throughput FACS based screening for directed enzyme evolution, ChemBioChem 10, 2704–2715. [DOI] [PubMed] [Google Scholar]
- 26.Larsen AC, Dunn MR, Hatch A, Sau SP, Youngbull C, and Chaput JC (2016) A general strategy for expanding polymerase function by droplet microfluidics, Nat. Commun 7, 11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xi HD, Zheng H, Guo W, Ganan-Calvo AM, Ai Y, Tsao CW, Zhou J, Li W, Huang Y, Nguyen NT, and Tan SH (2017) Active droplet sorting in microfluidics: a review, Lab Chip 17, 751–771. [DOI] [PubMed] [Google Scholar]
- 28.Tran DT, Cavett VJ, Dang VQ, Torres HL, and Paegel BM (2016) Evolution of a mass spectrometry-grade protease with PTM-directed specificity, Proc. Natl. Acad. Sci. U.S.A 113, 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero PA, Tran TM, and Abate AR (2015) Dissecting enzyme function with microfluidic-based deep mutational scanning, Proc. Natl. Acad. Sci. U.S.A 112, 7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kintses B, and al., e. (2012) Picoliter cell lysate assays in microfluidic droplet compartments for directed evolution, Chem. Biol 19, 1001–1009. [DOI] [PubMed] [Google Scholar]
- 31.Colin PY, Kintses B, Gielen F, Miton CM, Fischer G, Mohamed MF, Hyvonen M, Morgavi DP, Janssen DB, and Hollfelder F (2015) Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics, Nat. Commun 6, 10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinheiro VB, Taylor AI, Cozens C, Abramov M, Renders M, Zhang S, Chaput JC, Wengel J, Peak-Chew SY, McLaughlin SH, Herdewijn P, and Holliger P (2012) Synthetic genetic polymers capable of heredity and evolution, Science 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tawfik DS, and Griffiths AD (1998) Man-made cell-like compartments for molecular evolution, Nat. Biotechnol 16, 652–656. [DOI] [PubMed] [Google Scholar]
- 34.Tan YC, Cristini V, and Lee AP (2006) Monodispersed microfluidic droplet generation by shear focusing microfluidic device, Sens. Actuators B Chem 114, 350–356. [Google Scholar]
- 35.Nikoomanzar A, Vallejo D, and Chaput JC (2019) Elucidating the determinants of polymerase specificity by microfluidic-bsaed deep mutational scanning, ACS Synth. Biol, revise and resubmitted. [DOI] [PubMed] [Google Scholar]
- 36.Cohen R, and Hoerner CL (1996) Recombinant DNA technology: a 20-year occupational health retrospective, Rev. Environ. Health 11, 149–165. [DOI] [PubMed] [Google Scholar]
- 37.Buchholz F (2009) Engineering DNA processing enzymes for the postgenomic era, Curr. Opin. Biotechnol 20, 383–389. [DOI] [PubMed] [Google Scholar]
- 38.Aschenbrenner J, and Marx A (2017) DNA polymerases and biotechnological applications, Curr. Opin. Biotechnol 48, 187–195. [DOI] [PubMed] [Google Scholar]
- 39.Zhao G, Li J, Tong Z, Zhao B, Mu R, and Guan Y (2013) Enzymatic cleavage of type II restriction endonucleases on the 2’-O-methyl nucleotide and phosphorothioate substituted DNA, PLoS One 8, e79415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B, Tong Z, Zhao G, Mu R, Shang H, and Guan Y (2014) Effects of 2’-O-methyl nucleotide on ligation capability of T4 DNA ligase, Acta Biochim. Biophys. Sin. (Shanghai) 46, 727–737. [DOI] [PubMed] [Google Scholar]
- 41.Mei H, Liao JY, Jimenez RM, Wang Y, Bala S, McCloskey C, Switzer C, and Chaput JC (2018) Synthesis and Evolution of a Threose Nucleic Acid Aptamer Bearing 7-Deaza-7-Substituted Guanosine Residues, J. Am. Chem. Soc 140, 5706–5713. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Ngor AK, Nikoomanzar A, and Chaput JC (2018) Evolution of a General RNA-Cleaving FANA Enzyme, Nat. Commun 9, 5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor AI, Pinheiro VB, Smola MJ, Morgunov AS, Peak-Chew S, Cozens C, Weeks KM, Herdewijn P, and Holliger P (2015) Catalysts from synthetic genetic polymers, Nature 518, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Zhang S, and Chaput JC (2012) Darwinian evolution of an alternative genetic system provides support for TNA as an RNA progenitor, Nat. Chem 4, 183–187. [DOI] [PubMed] [Google Scholar]
- 45.Dunn MR, Otto C, Fenton KE, and Chaput JC (2016) Improving Polymerase Activity with Unnatural Substrates by Sampling Mutations in Homologous Protein Architectures, ACS Chem. Biol 11, 1210–1219. [DOI] [PubMed] [Google Scholar]
- 46.Zhang JH, Chung TD, and Oldenburg KR (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J. Biomol. Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- 47.Murray CJ, and Baliga R (2013) Cell-free translation of peptides and proteins: from high throughput screening to clinical production, Curr. Opin. Chem. Biol 17, 420–426. [DOI] [PubMed] [Google Scholar]
- 48.Hartsough EM, Shah P, Larsen AC, and Chaput JC (2015) Comparative analysis of eukaryotic cell-free expression systems, Biotechniques 59, 149–151. [DOI] [PubMed] [Google Scholar]
- 49.Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD, and Heyman JA (2013) Single-cell analysis and sorting using droplet-based microfluidics, Nat. Protoc 8, 870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikoomanzar A, Dunn MR, and Chaput JC (2017) Evaluating the rate and substrate specificity of laboratory evolved XNA polymerases, Anal. Chem 89, 12622–12625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.