

Abstract
Previous studies utilizing the SSp67phox−/− rat have demonstrated the importance of systemic NADPH oxidase NOX2-derived reactive oxygen species (ROS) production in the pathogenesis of Dahl Salt-Sensitive (SS) hypertension and renal damage. It is established that the immune system contributes to the development of SS hypertension and our laboratory has observed an enrichment of NOX2 subunits in infiltrating T cells. However, the contribution of immune cell-derived ROS in SS hypertension remains unknown. To assess the role of ROS in immune cells, SSp67phox−/− rats underwent total body irradiation and received bone marrow transfer from either SS (+SS) or SSp67phox−/− (+SSp67phox−/−) donor rats. Demonstrated in a respiratory burst assay, response to phorbol 12-myristate 13-acetate stimulus (135 μM) was 10.2-fold greater in peritoneal macrophages isolated from +SS rats compared to nonresponsive +SSp67phox−/− cells, validating that +SS rats were capable of producing NOX2-derived ROS in cells of hematopoietic origin. After 3 weeks of high salt challenge, there was an exacerbated increase in mean arterial pressure in +SS rats compared to +SSp67phox−/− control rats (176.1±4.7 vs 147.9±8.4 mmHg, respectively), which was accompanied by a significant increase in albuminuria (168.3±23.7 vs 107.0±20.4 mg/day) and renal medullary protein cast formation (33.2±4.7 vs 8.1±3.5%). Interestingly, upon analysis of renal immune cells, there was trending increase of CD11b/c+ monocytes and macrophages in the kidney of +SS rats (4.7±0.4 vs 3.5±0.5 ×106 cells/kidney, +SS vs +SSp67phox−/−, p=0.06). These data altogether demonstrate that immune cell production of NOX2-derived ROS is sufficient to exacerbate Dahl SS hypertension, renal damage, and renal inflammation.
Keywords: Hypertension, reactive oxygen species, immune cells, renal damage, NADPH oxidase, p67phox
Graphical Abstract
Introduction
The inappropriate production of reactive oxygen species (ROS) is known to contribute to the development of many pathologies, especially cardiovascular diseases [1], and has been implicated in the progression of hypertension and renal damage in the Dahl salt-sensitive (SS) rat. Specifically, increased production of superoxide and hydrogen peroxide measured via microdialysis has been demonstrated in the renal medulla of SS rats [2, 3]. A major source of ROS comes from the NOX2 isoform of NADPH oxidase, a multi-subunit enzyme comprised of membrane (gp91phox and p22phox) and cytosolic subunits (p47phox, p67phox, p40phox, and Rac 1 or 2) [4]. p67phox was identified as a candidate gene in studies involving congenic Dahl SS rats generated through the substitution of chromosomes from salt-resistant Brown Norway rats into the background of disease-prone SS rats [5, 6]. Furthermore, SS rats have been shown to exhibit higher NADPH oxidase activity in the outer medulla compared to salt-resistant control rats [7] and have increased expression of the p67phox subunit of the enzyme [8]. Null mutation of the p67phox gene in the Dahl SS (SSp67phox−/−) rat by zinc finger nuclease technology resulted in a reduction in salt-induced mean arterial pressure and renal injury compared to SSp67phox+/+, providing the first evidence that ROS production specifically from the NOX2 isoform of NADPH oxidase is crucial for the development of salt-sensitive hypertension in Dahl SS rats [8].
Work from our laboratory as well as others has linked the development of hypertension with immunity and the increased infiltration of immune cells into the kidney [9-12]. It is also known that NOX2, the phagocytic isoform of NADPH oxidase, is highly expressed on immune cells [13]. Our previous studies have shown that compared to whole kidney homogenates, infiltrating T cells isolated from the kidney highly express the p67phox subunit, which becomes exacerbated upon high salt challenge [14]. This is not only true of p67phox, but all other subunits of NOX2 as well. These data altogether suggests that renal infiltrating immune cells may be a significant source of ROS, ultimately contributing to disease progression in the Dahl SS rat.
With previous evidence showing greater levels of renal interstitial ROS in Dahl SS rats, and with role of p67phox already established in their progression of hypertension and renal damage, the current study sought to investigate the effect of oxidative stress inhibition on salt-induced renal immune cell infiltration. Additionally, we utilized a total body irradiation/bone marrow transfer (TBI/BMT) technique to dissociate the contribution of NOX2-derived ROS from immune cells versus the renal parenchyma, and we hypothesize that ROS specifically generated by immune cells is crucial to the development of hypertension and target organ damage in Dahl SS rats.
Methods
Animals.
Experiments were performed on age-matched, inbred, male Dahl SS (SS/JrHSDMcwi) and SSp67phox−/− (SS-Ncf2em1Mcwi) rats maintained on a 0.4% NaCl purified casein-based AIN-76A diet (#113755, Dyets Inc). As previously described [8], SSp67phox−/− rats were generated using zinc finger nuclease (ZFN) technology targeting rat p67phox exon 2. While studies utilizing female Dahl SS rats are currently ongoing, the exclusive focus on male rats is an important limitation of the current study. All experimental protocols were approved by the MCW Institutional Animal Care and Use Committee.
Blood pressure measurement and renal damage phenotyping.
At 7 weeks of age, rats were deeply anesthetized with inhalational anesthesia (isoflurane). Using aseptic technique, rats were instrumented with telemetry transmitters (Data Sciences International) into the carotid artery with antibiotic (25mg/kg cefazolin) and analgesia (0.3mg/kg Buprenorphine-SR) administered after surgery. Following a one-week recovery period, baseline blood pressure was measured continuously while the rats were maintained on the 0.4% NaCl purified casein-based diet. At 9 weeks of age, both groups of rats were switched to a high-salt (HS, 4.0% NaCl) diet for 21 days. Urine was collected during the 0.4% NaCl period, and at 7, 14, and 21 days during HS. Urine electrolytes were measured by flame photometry (Model 410, Corning), urinary protein was measured with an autoanalyzer (ACE, Alfa Wasserman), and urinary albumin was quantified with a fluorescent assay that utilized Albumin Blue 580 dye (Molecular Probes) and a fluorescent plate reader (FL600, Bio-Tek).
Immune cell isolation and flow cytometry.
At the conclusion of the experiment, rats were anesthetized with isoflurane and the kidneys were flushed with heparinized dPBS, minced, and incubated in RPMI 1640 media containing L-glutamine, HEPES, collagenase type IV, and DNase. Mononuclear cells were separated by Percoll density gradient centrifugation, counted on a hemocytometer, and incubated with extracellular markers anti-CD3 (eBioscience), anti-CD4 (BioLegend), and anti-CD8 (BioLegend) for T-cells, anti-CD45R (BD Bioscience) for B-cells, and anti-CD11b/c (eBioscience) for monocytes and macrophages. All cells were analyzed by flow cytometry (LSRII, Becton Dickinson) with FACSDIVA software (Becton Dickinson) and FlowJo software (Tree Star).
Histology.
Kidney tissues were collected for histological analysis and fixed with 10% neutral buffered formalin, paraffin embedded, cut into 4 μm sections, mounted, and stained with Masson’s trichrome. The kidney slices were scanned with a Nikon Super CoolScan 9000 interfaced with VueScan x64 software. The outer medullary cast percentage was determined by color inclusion via MetaMorph Microscopy Automation and Image Analysis software (Molecular Devices).
Total body irradiation and bone marrow transfer (TBI/BMT).
As described previously [15], at 6 weeks of age SSp67phox−/− rats underwent total body irradiation (TBI) at a dose of 11 Gy at a rate of 1.83 Gy/min. The bone marrow cells from either SS or SSp67phox−/− were collected from four donor femora and diluted in 6 ml of dPBS. Within 2 hours of TBI, rats received ~0.3 ml dPBS containing either SS or SSp67phox−/− bone marrow cells via tail vein injection. As described above, two weeks after bone marrow transplantation (BMT), rats were instrumented with telemeters to record blood pressure, switched to HS at 9 weeks of age, and urine collected during the LS period, and at 7, 14, and 21 days during HS. At the conclusion of the experiment, we performed a respiratory burst assay on isolated peritoneal macrophages, followed by tissue harvest, flow cytometric analysis, and histology, as described above.
Respiratory burst assay.
TBI/BMT rats were anesthetized with isoflurane. 10 ml ice-cold 3% FBS in dPBS was injected in to the abdominal cavity followed by a small midline incision to recollect the solution. The collected fluids were centrifuged at 400g for 5 min, followed by incubation with red blood cell lysis buffer (Life Technologies). Similarly performed as previous described [8], Cells were washed, resuspended in Dulbecco’s Modified Eagle Medium (Invitrogen), with 1×106 cells aliquoted into wells in triplicate on a clear-bottom 96-well plate (Bioexpress). The plate was incubated at 37°C for 2 h. Media was then removed and replaced with 0.3 ml of 1 mM luminol derivative L-012 (Wako Pharmaceuticals) dissolved in Hank’s balanced salt solution (Invitrogen). Luminescence of L-012 was used as an index of superoxide production. Luminescence was measured at 37°C on FLUOstar Omega machine (BMG Labtech) for baseline recording. Phorbol 12-myristate 13-acetate (PMA), a PKC activator, was added to each well to yield a final concentration of 135μM. Luminescence was then measured every 5 min for 30 min. To determine the specificity of the assay to detect superoxide, superoxide dismutase (100 U/well) was added as a control.
Statistical Analysis.
Data are expressed as the mean ± one standard error of the mean. Data were assessed for significance using a t-test, a one-way analysis of variance (ANOVA) with a Holm-Sidak post-hoc test, or a two-way repeated measures ANOVA with a Holm-Sidak post-hoc test, as appropriate.
Results
SSp67phox−/− rats demonstrate improved blood pressure, renal damage, and renal immune cell infiltration compared to SS rats.
Even while both groups were maintained on the 0.4% low-salt diet, the SSp67phox−/− rats already demonstrated a lower MAP (~11 mmHg) than the SS (Figure 1A). This protection continued through the entire 3 weeks of high salt challenge, where by HS21 the SSp67phox−/− rats had a 33.2 mmHg lower MAP than the SS rats (140.7±4.8 vs 173.9±8.5 mmHg, SSp67phox−/− vs SS). Shown in Figure 1B, this improved blood pressure phenotype in the SSp67phox−/− was mirrored in their protection from renal damage, where the mutant rats also exhibited less urinary protein (179.3±37.3 vs 323.1±40.7 mg/day, SSp67phox−/− vs SS at HS21) and albumin excretion (98.8±19.4 vs 186.3±25.1 mg/day, SSp67phox−/− vs SS at HS21). SSp67phox−/− rats also demonstrated a significant improvement in renal histological damage, seen by a reduction in medullary tubular protein cast formation (4.9±0.8 vs 19.0±2.1 %, SSp67phox−/− vs SS, Figure 2A). Interestingly, examination of immune cells revealed a specific increase in CD3+ T cells within the circulation (Figure 2B). Upon final examination of the immune cells infiltrating the kidney (Figure 2C), there were fewer total CD45+ leukocytes in the kidneys of the SSp67phox−/− compared to SS (4.8±0.3 vs 6.3±0.5 ×106 cells/kidney, SSp67phox−/− vs SS), which was primarily attributed to a reduction in CD11b/c+ monocytes and macrophages (3.8±0.2 vs 5.2±0.5 ×106 cells/kidney), as well as CD45R+ B cells (1.4±0.3 vs 2.6±0.3 ×105 cells/kidney). Interestingly, there was no difference in the infiltration of CD3+ T cells. These data altogether demonstrated the importance of NOX2-derived ROS in the development of SS hypertension, renal damage, and renal immune cell infiltration.
Figure 1. Attenuation of hypertension and renal damage in SSp67phox−/− rats.

(A) Daily mean arterial pressure (MAP) averages over the course of 3 weeks of 4.0% NaCl challenge. n=7-8. (B) Weekly determination of renal damages indices, urinary protein and albumin excretion. n=8-9. **p<0.005, ***p<0.001 SSp67phox−/− vs SS.
Figure 2. Attenuation of renal histological damage and renal immune cell infiltration in SSp67phox−/− rats.

(A) Percentage of medullary protein cast formation in the kidneys of SS and SSp67phox−/− rats after 3 weeks of 4.0% NaCl challenge. n=7-9. (B) Circulating immune cell profile between SS and SSp67phox−/− rats. (C) Absolute numbers of immune cells infiltrating into the kidneys at the conclusion of the experiment. CD45+: leukocytes, CD11b/c+: monocytes/macrophages, CD3+: T cells, CD4+: T-helper cells, CD8+: cytotoxic T cells, CD45R+: B cells. n=7-9. *p<0.05, **p<0.01 SSp67phox−/− vs SS.
Respiratory burst assay in isolated peritoneal macrophages from TBI/BMT rats demonstrated the ability of SS-derived hematopoetic cells to produce ROS.
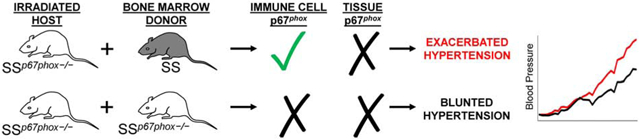
In order to delineate between the contribution of ROS from the renal parenchyma versus ROS from infiltrating immune cells, we utilized a total body irradiation/bone marrow transfer (TBI/BMT) approach ablating the resident bone marrow cells from SSp67phox−/− host rats which then received bone marrow from either an SS or SSp67phox−/− donor rat. With both groups utilizing SSp67phox−/− as the host, neither express p67phox in any of their tissue. However, with the host hematopoietic cells repopulating from the donor, the SSp67phox−/− host receiving bone marrow from an SS donor should have intact p67phox in all cells derived from the hematopoietic cell lineage. We would expect these SS donor cells to produce NOX2-derived ROS in their immune cells, while the cells from the SSp67phox−/− donor would not. As proof of principle, we performed a respiratory burst assay in peritoneal macrophages isolated from rats receiving TBI/BMT to see if these immune cells derived from the reconstituted SS bone marrow were indeed functional. Shown in Figure 3, unstimulated control macrophages from SS donors displayed a higher superoxide production baseline compared to those isolated from SSp67phox−/− donors (1.9-fold higher at 30 min). Upon stimulation with PKC activator and ROS generator phorbol myristate acetate (PMA), there is a striking increase in superoxide detected in the macrophages from SS donors (10.2-fold higher at 30 min), which is not evident in the stimulated macrophages from SSp67phox−/− donors. To assess specificity of the signal for the detection of superoxide, a separate set of stimulated samples were co-incubated with superoxide dismutase which demonstrated levels even lower than baseline, indicating additional sources of O2.− other than NOX2. These results critically demonstrate the functionality and ability of immune cells reconstituted from the SS donor bone marrow to produce ROS.
Figure 3. Respiratory burst assay in isolated peritoneal macrophages from TBI/BMT rats demonstrates the ability of SS-derived hematopoetic cells to produce ROS.

Luminol derivative L-012 was used as an index of superoxide production. Superoxide production was detected in PMA-stimulated macrophages from isolated from rats received bone marrow from SS donors, which was not evident in macrophages from SSp67phox−/− donors. 1×106 peritoneal macrophages/well, PMA = 135 uM, SOD = 100 U/well. n=6-8. pGroup<0.001 SSp67phox−/− vs SS, ***p<0.001 PMA vs Ctrl (SS), †††p<0.001 PMA vs PMA+SOD (SS).
NADPH oxidase-derived ROS production in hematopoietic cells is necessary for the development of salt-sensitive hypertension and renal damage in SS rats.
There was no difference in 0.4% NaCl baseline blood pressure between SSp67phox−/− rats receiving bone marrow from either SS or SSp67phox−/− (Figure 4A). Upon high salt challenge, SS recipients (+SS) demonstrated a clear and significant exacerbation of hypertension compared to SSp67phox−/− recipients (+SSp67phox−/−) (176.1±4.7 vs 147.9±8.4 mmHg, +SS vs +SSp67phox−/− at HS21). This exacerbated blood pressure phenotype in the +SS group was accompanied with greater renal damage evident by more proteinuria (287.2±25.1 vs 179.5±24.6 mg/day, +SS vs +SSp67phox−/− at HS21) and albuminuria (168.3±23.7 vs 107.0±20.4 mg/day, +SS vs +SSp67phox−/− at HS21) (Figure 4B). At the conclusion of the study, there was greater renal histological damage in the +SS rats, evident by the significant percentage of protein casts found in the outer medulla of SS donor rats (33.2±4.7 vs 8.1±3.5 %, +SS vs +SSp67phox−/−, Figure 5A). Inspection of the immune cell subsets found in the circulation revealed no change between +SS and +SSp67phox−/− (Figure 5B), although the total number of circulating immune cells is lower in these TBI/BMT rats compared to non-irradiated rats (Figure 2B), indicative of an incomplete reconstitution of bone marrow cells. Despite this reduced peripheral reconstitution, examination of the infiltrating renal immune cells primarily showed no difference between the two groups, though there was trending increase in CD11b/c+ monocytes and macrophages in the kidneys of +SS rats (4.7±0.4 vs 3.5±0.5 ×106 cells/kidney, +SS vs +SSp67phox−/−, p=0.06, Figure 5C). With the only difference between the +SS and +SSp67phox−/− being the ability to produce NOX2-derived ROS in the hematopoietic cells, this suggests that ROS production in immune cells is important for the development and progression of salt-induced hypertension and renal damage, while not significantly altering the homing of immune cells to the kidney.
Figure 4. NADPH oxidase-derived ROS production in immune cells is necessary for the development of salt-sensitive hypertension and renal damage in SS rats.

(A) Exacerbated rise in mean arterial pressure (MAP) in SSp67phox−/− receiving SS bone marrow (+SS) after 3 weeks of 4.0% NaCl challenge. n=4-5. (B) Renal damage indices, urinary protein and albumin excretion, were also worsened in rats receiving SS bone marrow (+SS) compared to those receiving SSp67phox−/− bone marrow (+SSp67phox−/−). n=9-11. *p<0.05, **p<0.005, ***p<0.001 +SSp67phox−/− vs +SS.
Figure 5. Exacerbation of renal histological damage in SSp67phox−/− receiving SS bone marrow (+SS) rats.

(A) Percentage of medullary protein cast formation in the kidneys of +SS and +SSp67phox−/− rats after 3 weeks of 4.0% NaCl challenge. n=7-9. (B) Circulating immune cell profile between +SS and +SSp67phox−/− rats. (C) Absolute numbers of immune cells infiltrating into the kidneys at the conclusion of the experiment. CD45+: leukocytes, CD11b/c+: monocytes/macrophages, CD3+: T cells, CD4+: T-helper cells, CD8+: cytotoxic T cells, CD45R+: B cells. n=9-10. *p<0.05, **p<0.01 +SSp67phox−/− vs +SS.
Discussion
The present study demonstrated that inhibition of NADPH oxidase via p67phox mutation in Dahl SS rats (SSp67phox−/−) ameliorates salt-induced hypertension, renal injury, and renal immune cell infiltration compared to SS controls. Additionally, transfer of functional, ROS-producing SS bone marrow cells into irradiated SSp67phox−/− worsened disease, indicating that oxidative stress specifically generated by immune cells is sufficient for the exacerbation of hypertension and target organ damage in Dahl SS rats.
Though the importance of NOX2 NADPH oxidase was originally discovered through its association with chronic granulomatous disease, its role in immunity and disease has now been vastly expanded to a whole host of pathologies, including cardiovascular and renal disease [16, 17]. In the Dahl SS rat, high salt challenge resulted in elevated levels of intrarenal ROS (O2.− and H2O2) and hypertension [3, 7]. As evidence for the specific contribution of NOX2, genetic mutation of the p67phox subunit in SS rats via zinc finger nuclease technology reduced salt-sensitive hypertension, reversed reductions in medullary blood flow, maintained GFR, and consequently prevented renal injury [14, 18]. It is worth noting that p67phox has also been demonstrated to also support the activity of both NOX1 and NOX3 [19, 20], though NOX1 is most highly expressed in the colon and NOX3 in the adrenal gland. Aside from increases in NADPH oxidase activity, increased oxidative stress and accelerated disease progression have also been attributed to deficiencies in antioxidants like superoxide dismutase [21, 22]. NADPH oxidase inhibition or antioxidant treatment has been widely demonstrated to have beneficial effects in multiple preclinical hypertension and renal disease settings, either specifically in the Dahl SS rat [2, 3, 7, 23, 24] or in other experimental models [25-29], and is the current target of potential therapeutic agents in ongoing clinical trials.
It is well established that the presence of immune cells in target organs is an important mechanism underlying the development of hypertension and renal injury [30]. While NADPH oxidases in various tissues, including the kidney, have been thoroughly characterized [13, 31], the contribution of ROS derived from either resident or infiltrating immune cells in the kidney remain unknown. NOX2, also known as phagocytic NADPH oxidase, is highly expressed on macrophages/monocytes and granulocytes, and since these CD11b/c+ cells comprise the majority of the total CD45+ leukocytes in the kidney (>80%, Figure 2), it is highly plausible that these renal immune cells can play a pivotal role in determining the oxidant status of the kidney. Furthermore, there is a substantial body of evidence highlighting the integral role of oxidative stress in the activation of the immune system and the progression of hypertensive diseases [32]. This is especially true for NOX2-derived ROS, which has been demonstrated to regulate a wide number of immunological processes related to chemotaxis, monocyte and macrophage infiltration, innate inflammasome activation, antigen cross-presentation, adaptive immune activation, and T cell selection, maturation, and differentiation [33-38]. Dysregulation of these key immune mechanisms may serve as an explanation as to how oxidative imbalance could lead to inappropriate immune activation, thus leading to target organ damage and subsequent hypertension.
The current study revealed through a TBI/BMT approach that the production of NOX2-derived ROS solely in cells of hematopoietic origin (immune cells), but absent in the remainder of the host, was sufficient to drive and exacerbate salt-induced hypertension, renal damage, and immune cell infiltration, highlighting the fundamental role of immune cell ROS in the pathogenesis of Dahl SS disease. More crucially, these results demonstrate the importance of the exact cellular localization and source of ROS production, which becomes critical when determining targets for potential therapeutic applications. Despite extensive, strong pre-clinical data, many clinical trials have failed to provide evidence in support of antioxidant therapy for a number of various diseases [39, 40], for reasons primarily related to short trial duration and poor antioxidant bioavailability [41, 42]. In light of the current study, perhaps taking a more targeted approach to deliver high enough concentrations of antioxidants to the cells specifically responsible for producing injurious ROS would lead to more efficacious therapies and ultimately better outcomes in humans.
Highlights.
Aberrant ROS production has been implicated in the development of salt-sensitive (SS) hypertension and renal damage
Inhibition of NOX2 ROS via p67phox−/− in Dahl SS rats lowered blood pressure and improved renal damage and inflammation
A total body irradiation/bone marrow transfer approach was utilized to assess the specific contribution of ROS derived from immune cells
Immune cell production of NOX2-derived ROS sufficiently exacerbates SS hypertension, renal damage, and renal inflammation
Acknowledgments
We thank Marylou Mader, Tracy Gasperetti, and Brian Fish for their assistance with the TBI/BMT experiments. This work was supported by HL116264, HL137748, DK62803, AHA-15SFRN2391002, AHA-16POST29900004 and 1F32HL136161.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cifuentes ME, Pagano PJ, Targeting reactive oxygen species in hypertension, Curr Opin Nephrol Hypertens 15(2) (2006) 179–86. [DOI] [PubMed] [Google Scholar]
- [2].Makino A, Skelton MM, Zou AP, Cowley AW Jr., Increased renal medullary H2O2 leads to hypertension, Hypertension 42(1) (2003) 25–30. [DOI] [PubMed] [Google Scholar]
- [3].Taylor NE, Cowley AW Jr., Effect of renal medullary H2O2 on salt-induced hypertension and renal injury, Am J Physiol Regul Integr Comp Physiol 289(6) (2005) R1573–9. [DOI] [PubMed] [Google Scholar]
- [4].Paravicini TM, Touyz RM, NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities, Diabetes Care 31 Suppl 2 (2008) S170–80. [DOI] [PubMed] [Google Scholar]
- [5].Moreno C, Kaldunski ML, Wang T, Roman RJ, Greene AS, Lazar J, Jacob HJ, Cowley AW Jr., Multiple blood pressure loci on rat chromosome 13 attenuate development of hypertension in the Dahl S hypertensive rat, Physiol Genomics 31(2) (2007) 228–35. [DOI] [PubMed] [Google Scholar]
- [6].Lu L, Li P, Yang C, Kurth T, Misale M, Skelton M, Moreno C, Roman RJ, Greene AS, Jacob HJ, Lazar J, Liang M, Cowley AW Jr., Dynamic convergence and divergence of renal genomic and biological pathways in protection from Dahl salt-sensitive hypertension, Physiol Genomics 41(1) (2010) 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taylor NE, Glocka P, Liang M, Cowley AW Jr., NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats, Hypertension 47(4) (2006) 692–8. [DOI] [PubMed] [Google Scholar]
- [8].Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM, Cowley AW Jr., Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension, Cell Metab 15(2) (2012) 201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Miguel C, Das S, Lund H, Mattson DL, T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats, Am J Physiol Regul Integr Comp Physiol 298(4) (2010) R1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fehrenbach DJ, Abais-Battad JM, Dasinger JH, Lund H, Mattson DL, Salt-sensitive increase in macrophages in the kidneys of Dahl SS rats, Am J Physiol Renal Physiol 317(2) (2019) F361–F374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Harrison DG, The immune system in hypertension, Trans Am Clin Climatol Assoc 125 (2014) 130–38; discussion 138–40. [PMC free article] [PubMed] [Google Scholar]
- [12].Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL, Immune regulation and vascular inflammation in genetic hypertension, Am J Physiol Heart Circ Physiol 298(3) (2010) H938–44. [DOI] [PubMed] [Google Scholar]
- [13].Bedard K, Krause KH, The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology, Physiol Rev 87(1) (2007) 245–313. [DOI] [PubMed] [Google Scholar]
- [14].De Miguel C, Guo C, Lund H, Feng D, Mattson DL, Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease, Am J Physiol Renal Physiol 300(3) (2011) F734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rudemiller NP, Lund H, Priestley JR, Endres BT, Prokop JW, Jacob HJ, Geurts AM, Cohen EP, Mattson DL, Mutation of SH2B3 (LNK), a genome-wide association study candidate for hypertension, attenuates Dahl salt-sensitive hypertension via inflammatory modulation, Hypertension 65(5) (2015) 1111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schiffrin EL, Antioxidants in hypertension and cardiovascular disease, Mol Interv 10(6) (2010) 354–62. [DOI] [PubMed] [Google Scholar]
- [17].Araujo M, Wilcox CS, Oxidative stress in hypertension: role of the kidney, Antioxid Redox Signal 20(1) (2014) 74–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Evans LC, Ryan RP, Broadway E, Skelton MM, Kurth T, Cowley AW Jr., Null mutation of the nicotinamide adenine dinucleotide phosphate-oxidase subunit p67phox protects the Dahl-S rat from salt-induced reductions in medullary blood flow and glomerular filtration rate, Hypertension 65(3) (2015) 561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ueyama T, Geiszt M, Leto TL, Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases, Mol Cell Biol 26(6) (2006) 2160–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cheng G, Ritsick D, Lambeth JD, Nox3 regulation by NOXO1, p47phox, and p67phox, J Biol Chem 279(33) (2004) 34250–5. [DOI] [PubMed] [Google Scholar]
- [21].Meng S, Roberts LJ 2nd, Cason GW, Curry TS, Manning RD Jr., Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats, Am J Physiol Regul Integr Comp Physiol 283(3) (2002) R732–8. [DOI] [PubMed] [Google Scholar]
- [22].Rodriguez-Iturbe B, Sepassi L, Quiroz Y, Ni Z, Wallace DC, Vaziri ND, Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence, J Appl Physiol (1985) 102(1) (2007) 255–60. [DOI] [PubMed] [Google Scholar]
- [23].Hisaki R, Fujita H, Saito F, Kushiro T, Tempol attenuates the development of hypertensive renal injury in Dahl salt-sensitive rats, Am J Hypertens 18(5 Pt 1) (2005) 707–13. [DOI] [PubMed] [Google Scholar]
- [24].Tian N, Thrasher KD, Gundy PD, Hughson MD, Manning RD Jr., Antioxidant treatment prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension, Hypertension 45(5) (2005) 934–9. [DOI] [PubMed] [Google Scholar]
- [25].Chen X, Touyz RM, Park JB, Schiffrin EL, Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR, Hypertension 38(3 Pt 2) (2001) 606–11. [DOI] [PubMed] [Google Scholar]
- [26].Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG, Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II, Hypertension 40(4) (2002) 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Park JB, Touyz RM, Chen X, Schiffrin EL, Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats, Am J Hypertens 15(1 Pt 1) (2002) 78–84. [DOI] [PubMed] [Google Scholar]
- [28].Schnackenberg CG, Wilcox CS, Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2alpha, Hypertension 33(1 Pt 2) (1999) 424–8. [DOI] [PubMed] [Google Scholar]
- [29].Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL, Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats, Hypertension 36(6) (2000) 957–64. [DOI] [PubMed] [Google Scholar]
- [30].Mattson DL, Immune mechanisms of salt-sensitive hypertension and renal end-organ damage, Nat Rev Nephrol 15(5) (2019) 290–300. [DOI] [PubMed] [Google Scholar]
- [31].Gill PS, Wilcox CS, NADPH oxidases in the kidney, Antioxid Redox Signal 8(9-10) (2006) 1597–607. [DOI] [PubMed] [Google Scholar]
- [32].Loperena R, Harrison DG, Oxidative Stress and Hypertensive Diseases, Med Clin North Am 101(1) (2017) 169–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ, NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy, Arterioscler Thromb Vasc Biol 23(5) (2003) 776–82. [DOI] [PubMed] [Google Scholar]
- [34].Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL, NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia, Antioxid Redox Signal 18(13) (2013) 1537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, Holmdahl R, Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species, J Clin Invest 117(10) (2007) 3020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lam GY, Huang J, Brumell JH, The many roles of NOX2 NADPH oxidase-derived ROS in immunity, Semin Immunopathol 32(4) (2010) 415–30. [DOI] [PubMed] [Google Scholar]
- [37].Singel KL, Segal BH, NOX2-dependent regulation of inflammation, Clin Sci (Lond) 130(7) (2016) 479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, Kuijpers TW, Holmdahl R, van Kooten C, Gelderman KA, Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species, Proc Natl Acad Sci U S A 107(41) (2010) 17686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C, Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases, Cochrane Database Syst Rev (3) (2012) CD007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Steinhubl SR, Why have antioxidants failed in clinical trials?, Am J Cardiol 101(10A) (2008) 14D–19D. [DOI] [PubMed] [Google Scholar]
- [41].Iannitti T, Palmieri B, Antioxidant therapy effectiveness: an up to date, Eur Rev Med Pharmacol Sci 13(4) (2009) 245–78. [PubMed] [Google Scholar]
- [42].A MD, A GH, Why antioxidant therapies have failed in clinical trials, J Theor Biol 457 (2018) 1–5. [DOI] [PubMed] [Google Scholar]