

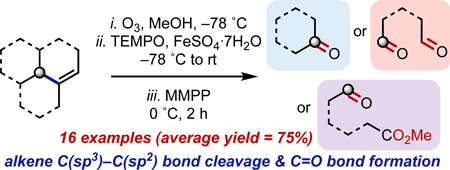
Abstract
Herein we report a one-pot protocol for the oxodealkenylative introduction of carbonyl functionalities into terpenes and terpene-derived compounds. This transformation proceeds via Criegee ozonolysis of an alkene, reductive cleavage of the resulting α-alkoxy hydroperoxide, trapping of the generated alkyl radical with TEMPO, and subsequent oxidative fragmentation with MMPP. Using readily available starting materials from chiral pool, a variety of carbonyl-containing products have been accessed rapidly in good yields.
Keywords: ozonolysis, alkene, ferrous, radical, terpene, redox
Graphical Abstract
Natural products have had a longstanding influence on both the natural world and society.[1] In particular, they have played an important role in drug development, with approximately 65% of FDA-approved small-molecule drugs being, in some way, dependent on natural products.[2] Nature’s chiral pool is also frequently exploited in enantiospecific synthesis, especially in the total synthesis of small and complex molecules of biological relevance.[3] One interesting facet relevant to these context is the prevalence of alkenes (39.9%) relative to that of ketones (15.9%), enones (6.0%), and aldehydes (2.4%) found in natural products (Scheme 1).[4] With the chiral pool materials being useful in many types of chemical processes, a simple method for the conversion of natural alkenes into carbonyl-containing compounds would be highly valuable.
Scheme 1.

Prevalence of functional groups in natural products, halide-to-carbonyl conversion, and the oxodealkenylative process. Magnesium bis(monoperoxyphthalate) hexahydrate, MMPP.
In 2004, Inokuchi and Kawafuchi reported a simple protocol for the conversion of alkyl halides to carbonyl-containing compounds (Scheme 1).[5] This process involves SN2 displacement of an alkyl halide with the anion of 2,2,6,6-tetramethylpiperidin-1-yl (TEMPO) and subsequent m-chloroperoxybenzoic acid (mCPBA)-mediated oxidation of the O-alkyl TEMPO intermediate to give the corresponding carbonyl compound. While only a few natural products contain halide functionalities (chloride, 1.8%; bromide, 1.5%), terpenes and terpenoids are abundant.[6] Because these natural alkenes can be converted into O-alkyl TEMPO intermediates through redox-based radical processes, we envisioned that it is possible to prepare carbonyl compounds from terpene-derived starting materials.
In this paper, we report a simple one-pot protocol for the cleavage of an alkene C(sp3)–C(sp2) bond followed by formation of a C=O bond. The reaction involves several steps: ozonolysis of an alkene 1, Fe(II)-mediated single-electron-transfer (SET)-based reduction of the intermediate α-alkoxy hydroperoxide, alkoxy radical-induced β-fragmentation, trapping of the resultant alkyl radical with a persistent radical TEMPO,[7] and subsequent oxidation, ultimately providing the carbonyl-containing product 2 (Scheme 1). This transformation proceeds under mild reaction conditions and open to the air, employs common terpenes and terpene derivatives as starting materials, and is tolerant of functional groups that are typically prone to degradation and/or reaction in acidic, basic, and/or oxidative conditions (e.g., β-hydroxy ketones, acetals, enones, ketones, alcohols). Furthermore, the initial oxidant (ozone) is renewable, the ferrous salt (FeSO4·7H2O) is plentiful,[8] and the terminal oxidant (MMPP) is less expensive and more stable than similar peracids.[9]
Our strategy relies on the decomposition of organic peroxides using ferrous salts, a process that has been known for over 100 years.[10] Pertinent to this study, the first explicit mention of radical intermediates in the Fe(II)-mediated degradation of α-alkoxy hydroperoxides (and the formation of alkyl radical dimers) came from Hawkins in 1955 (Scheme 2).[11a] Later, Kumamoto, De La Mare, Rust, and Kochi described the trapping of these alkyl radicals with cupric species to form alkyl halides or alkenes.[11b,c] Although Criegee had in 1949 reported the formation of α-alkoxy hydroperoxides through ozonolysis of alkenes in alcoholic solvents,[12] it was not until 1964 that Murai, Sonoda, and Tsutsumi combined this method of hydroperoxide generation with the established iron/copper couple to produce dimer and halide products from alkenes.[11d] In 1980, Schreiber applied these findings in the synthesis of the natural product (±)-recifeiolide and in the conversion of (–)-dihydrocarvone to (+)-6-methylcyclohex-2-enone.[11e] Since then, many groups have employed this strategy in total synthesis and in the preparation of various biologically relevant molecules.[13] Furthermore, several recent reports have expanded upon these pioneering studies to establish a number of other useful functionalization protocols involving radical-based fragmentations of α-alkoxy hydroperoxides.[14]
Scheme 2.

Pioneering examples of Fe(II)-mediated radical fragmentations of α-alkoxy hydroperoxides.
Initially, we used the hydroxy ketone 1a to examine the conditions necessary for the conversion of an alkene to a ketone (Table 1). The optimal temperature for the addition of TEMPO and ferrous sulfate (added as a 5% wt/vol aqueous solution) was –78 ˚C, followed by warming to room temperature (entries 1–4). The reaction itself was extremely rapid and typically complete within a minute of the addition of the iron salt. Using 1.0 equivalent of TEMPO gave a slightly lower yield (versus 1.5 equiv, entry 5), while no benefit was gained when using 2.0 equivalents (entry 6). For the conversion of the O-alkyl TEMPO adduct to ketone, it was 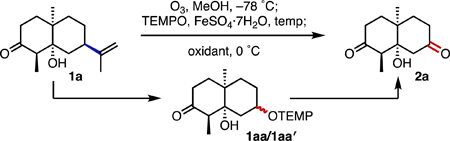 also found that MMPP performed better than mCPBA (the oxidant reported by Inokuchi and Kawafuchi) and other commonly used oxidizing agents such as hydrogen peroxide, urea hydrogen peroxide (UHP), and potassium peroxymonosulfate (Oxone™) (entries 7–11). Furthermore, the water-solubility of MMPP allowed simple work-up conditions and facile removal of by-products. The highest yield of 2a was achieved when employing 2.5 equivalents of MMPP (entries 12–15). One reason for the use of excess MMPP was to ensure the oxidation of TEMPO hydroxyl to TEMPO free radical, a process supported by the observation that MMPP-mediated oxidation of pure 1aa to give 2a was accompanied by the regeneration of TEMPO. An attempt to convert the intermediate α-alkoxy hydroperoxide directly to oxygenated products by trapping the alkyl radical with O2 in the presence of PhSiH3 produced the ketone 2a in 24% yield (entry 16).[14f,15]
also found that MMPP performed better than mCPBA (the oxidant reported by Inokuchi and Kawafuchi) and other commonly used oxidizing agents such as hydrogen peroxide, urea hydrogen peroxide (UHP), and potassium peroxymonosulfate (Oxone™) (entries 7–11). Furthermore, the water-solubility of MMPP allowed simple work-up conditions and facile removal of by-products. The highest yield of 2a was achieved when employing 2.5 equivalents of MMPP (entries 12–15). One reason for the use of excess MMPP was to ensure the oxidation of TEMPO hydroxyl to TEMPO free radical, a process supported by the observation that MMPP-mediated oxidation of pure 1aa to give 2a was accompanied by the regeneration of TEMPO. An attempt to convert the intermediate α-alkoxy hydroperoxide directly to oxygenated products by trapping the alkyl radical with O2 in the presence of PhSiH3 produced the ketone 2a in 24% yield (entry 16).[14f,15]
Table 1.
Optimization of the conditions for the oxodealkenylation.[a]
| entry | TEMPO (equiv) |
Oxidant (equiv) |
Temp (°C) |
Yield[b] 1aa+1aa’ |
Yield[c] 2a |
|---|---|---|---|---|---|
| 1 | 1.5 | – | –78 to rt | 91 | – |
| 2[d] | 1.5 | – | −78 to rt | 94 | – |
| 3[d] | 1.5 | – | 0 | 85 | – |
| 4[d] | 1.5 | – | rt | 92 | – |
| 5[d] | 1.0 | – | −78 to rt | 79 | – |
| 6[d] | 2.0 | – | −78 to rt | 93 | – |
| 7[d] | 1.5 | mCPBA (1.2) | −78 to rt | – | 33 |
| 8[d] | 1.5 | H2O2 (1.2) | −78 to rt | – | Trace |
| 9[d] | 1.5 | UHP (1.2) | −78 to rt | – | 0 |
| 10[d] | 1.5 | MMPP (1.2) | −78 to rt | – | 53 |
| 11[d] | 1.5 | Oxone™ (1.2) | −78 to rt | – | Trace |
| 12[d] | 1.5 | MMPP (1.5) | −78 to rt | – | 62 |
| 13[d] | 1.5 | MMPP (2.0) | −78 to rt | – | 78 |
| 14[d] | 1.5 | MMPP (2.5) | −78 to rt | – | 84 |
| 15[d] | 1.5 | MMPP (3.0) | −78 to rt | – | 81 |
| 16[e] | – | O2 | 0 to rt | – | 24 |
Reaction conditions: alkene 1a (0.100 mmol, 1.0 equiv) in MeOH (0.025 M), FeSO4·7H2O (1.2 equiv). See the Supporting Information (SI) for further experimental details.
Yield based on NMR spectral analysis, using 1-chloro-2,4-dinitrobenzene as the internal standard.
Isolated yield.
aq. FeSO4·7H2O (5% wt/vol)
2.5 equiv FeSO4·7H2O and 2.5 equiv PhSiH3.
Under the optimized conditions, a number of terpenes and terpene derivatives were subjected to the oxodealkenylation (Scheme 3). The β-hydroxy-decalinones 1a and 1b gave their products in yields of 87 and 75%, respectively, while the dimethyl ketal 1c gave the corresponding ketone in 95% yield. (+)-Nootkatone (1d) also cleanly provided the expected enedione 2d in 82% yield. Although cis-(–)-limonene oxide (1e) initially generated the desired epoxy-ketone product, this species was unstable during work-up and purification and underwent conversion to the γ-hydroxy enone 2e in 81% yield. Subsequently, we found that treatment of the crude mixture with triethylamine promoted clean conversion to the γ-hydroxy enone product 2e. (–)-Dihydrocarveol (1f) and the ethylene glycol acetal of trans-(+)-dihydrocarvone (1g) produced their corresponding ketone products in yields of 80 and 75%, respectively. Markedly, the hydroxy ketone 2f, which is used in the synthesis of JNK1 and JNK2 inhibitors, has previously been accessible only through a resolution-based method.[16] After basic workup (similar to 2e), the carvone-derived β-hydroxy epoxide 1h supplied the dihydroxyenone 2h in 61% yield. The diol 1i smoothly furnished the β-hydroxy ketone 2i in 83% yield, while the carvone-derived hydroxy ketone 1j gave its corresponding hydroxydione product 2j in 50% yield. Notably, one of the OH groups in the diol 1i was converted into the acetate during the ozonolysis/reductive fragmentation; this reaction amounts to selective mono-oxidation of one OH group of a putative triol, accompanied by chemoselective protection of another, demonstrating the potential power of the current reaction. The carvone-derived phosphine oxide (1k) and the limonene oxide-derived phosphine oxide (1l) both gave their respective ketone products in yields of 60 and 82%, respectively. Applying this transformation to exo-cyclic methylene and endo-cyclic alkenes generated some unique scaffolds. (±)-Sabinene (1m) fragmented to form its supposed primary radical, with subsequent trapping and oxidation providing the cyclopropane aldehyde 2m in 58% yield. Methyleneadamantane (1n) was also able to generated its keto ester 2n in 87% yield. Applying the oxodealkenylation to a more complex terpene, we found that (+)-aromadendrene (1o) underwent less discriminative radical scission to give the ketone 2o and the aldehyde 2o′ (1.3:1 r.r.) in a combined yield of 82%. Finally, oxodealkenylative cleavage of (+)-α-pinene (1p) produced the cyclobutanone 2p and the ketoaldehyde 2p′ in 67% yield. Notably, with the exception of 2d,[17] 2e,[18] 2f,[16] (±)-2g,[19] and 2n,[20] all of these ketone, α,β-enone, and aldehyde products are new, yet they contain multiple stereocenters, a breadth of complexity, and arise from readily accessible starting materials.
Scheme 3.

Examples of oxodealkenylation. Reaction conditions: alkene 1 (0.5–2.0 mmol, 1.0 equiv) in MeOH (0.025 M), TEMPO (1.5 equiv), aq. FeSO4·7H2O (5% wt/vol, 1.2 equiv), MMPP (2.5 equiv). See the SI for further experimental details. [a] Isolated yields after SiO2 chromatography. [b] The crude reaction mixture was treated with Et3N during workup.
To demonstrate the utility of this methodology, several studies were performed (Scheme 4). First, a gram-scale reaction employing the ethylene glycol-protected trans-(+)-dihydrocarvone (1g) was shown to be efficient, supplying the mono-protected cyclohexane-1,3-dione 2g in an isolated yield of 75% (714 mg). Next, it was shown that reductive N–O bond cleavage of the O-alkyl TEMPO adduct 1fa derived from (–)-dihydrocarveol (1f) furnished the cis-diol 3 in 92% yield. This sequence appears to be a convenient method for the introduction of OH groups into terpenes and terpenoids. We also performed several mechanistic studies. Subjecting (1S)-(+)-3-carene (1q) to the oxodealkenylation reaction conditions resulted in the generation of the transient cyclopropyl carbinyl radical, with subsequent ring opening and then trapping with TEMPO, supplying the aldehyde 1qa in a yield of 50%.
Scheme 4.

Synthetic utility and mechanistic studies of the oxodealkenylation. See the SI for experimental details.
Typically, we found that the combination of Criegee ozonolysis and SET-based fragmentation, and subsequent trapping of the alkyl radical intermediates converted the terpenoid starting materials cleanly to their desired products. Nevertheless, in some cases (primarily when employing cycloalkenes), we also observed side products. To investigate the pathways leading to these side products, we isolated all of the detectable products from the reaction of (+)-α-pinene (1p). Through NMR spectroscopic and mass spectrometric analyses, we identified the products as the O-alkyl TEMPO adducts 1pa and 1pa′, the ketoester 4, and the O-alkyl TEMPO adduct 5 (1pa+1pa′/4/5, 5.4:1.6:1). These products arose through two possible molozonide (A) fragmentation pathways.[21] In the major pathway, the tertiary α-alkoxy hydroperoxide B is generated. When treated with a ferrous species, the resulting alkoxy radical can undergo β-scission smoothly to give the desired O-alkyl TEMPO adducts 1pa and 1pa′ (which, upon oxidation, provides the cyclobutanone 2p). In the minor pathway, the secondary α-alkoxy hydroperoxide C is generated. Secondary alkyl hydroperoxides are prone to Fe(II)-catalyzed dehydration to produce carboxylic esters 4.[22] This transformation occurs through SET from Fe(II), cleavage of the O–O bond to form the alkoxy radical intermediate, and α-hydrogen abstraction by Fe(III) hydroxide. Alternatively, β-fragmentation of the alkoxy radical D and subsequent trapping of the alkyl radical E gives the O-alkyl TEMPO adduct 5 (which, upon oxidation, gives the product 2p′).
In summary, we have developed a practical one-pot method for converting the C(sp3)–C(sp2) bonds of alkenes into C=O groups in a variety of terpenes, terpenoids, and terpenoid-derived compounds from the chiral pool. The ease of operation enables the rapid generation of complex molecules suitable for further functionalization from abundant plant-based natural products. We have also found that this oxodealkenylation reaction is scalable, and that the intermediate O-alkyl TEMPO adduct can be converted into the corresponding alcohol through reductive cleavage of the N–O bond. Furthermore, our mechanistic studies have demonstrated the various types of byproducts that can arise during ozonolysis of terpenes and Fe(II)-mediated radical fragmentations of α-alkoxy hydroperoxides.
Supplementary Material
Acknowledgements
Financial support for this study was provided by the NIH (R01GM071779). A.J.S. thanks the Majeti–Alapati fellowship and the UCLA Dissertation Year Fellowship for funding. We thank the UCLA Molecular Instrumentation Center for providing the instrumentation for NMR spectroscopy and mass spectrometry.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Khan RA, Saudi Pharm. J. 2018, 26, 739–753; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gershenzon J, Dudareva N, Nat. Chem. Bio. 2007, 3, 408–414. [DOI] [PubMed] [Google Scholar]
- [2].Newman DJ, Cragg GM, J. Nat. Prod. 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- [3].a) Brill ZG, Condakes ML, Ting CP, Maimone TJ, Chem. Rev. 2017, 117, 11753–11795. For selected examples, reported this year, of total syntheses starting from chiral pool materials, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pfaffenbach M, Bakanas I, O’Conner NR, Herrick JL, Sarpong R, Angew. Chem. Int. Ed. 2019, 58, 1–6; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Johnson RE, Ree H, Hartmann M, Lang L, Sawano S, Sarpong R, J. Am. Chem. Soc. 2019, 141, 2233–2237; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Boyko TD, Huck CJ, Sarlah D, J. Am. Chem. Soc. 2019, 141, 14131–14135; [DOI] [PubMed] [Google Scholar]; e) Sittihan S, Jamison TF, J. Am. Chem. Soc. 2019, 141, 11239–11244; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xu J, Rawal VH, J. Am. Chem. Soc. 2019, 141, 4820–4823; [DOI] [PubMed] [Google Scholar]; g) Yu X, Xiao L, Wang Z, Luo T, J. Am. Chem. Soc. 2019, 141, 3440–3443; [DOI] [PubMed] [Google Scholar]; h) Nakamura H, Yasui K, Kanda Y, Baran PS, J. Am. Chem. Soc. 2019, 141, 1494–1497; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kim JH, Kim I, Song Y, Kim MJ, Kim S, Angew. Chem. Int. Ed. 2019, 58, 11018–11022; [Google Scholar]; j) Takao K-I, Kai H, Yamada A, Fukushima Y, Komatsu D, Ogura A, Yoshida K, Angew. Chem. Int. Ed. 2019, 58, 9851–9855; [DOI] [PubMed] [Google Scholar]; k) Xu B, Wang B, Xun W, Qiu FG, Angew. Chem. Int. Ed. 2019, 58, 5754–5757; [DOI] [PubMed] [Google Scholar]; l) He C, Xuan J, Rao P, Xie P-P, Hong X, Lin X, Ding H, Angew. Chem. Int. Ed. 2019, 58, 5100–5104; [DOI] [PubMed] [Google Scholar]; m) Hu N, Dong C, Zhang C, Liang G, Angew. Chem. Int. Ed. 2019, 58, 6659–6662; [DOI] [PubMed] [Google Scholar]; n) Tao Y, Reisenauer K, Taube JH, Romo D, Angew. Chem. Int. Ed. 2019, 58, 2734–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ertl P, Schuhmann T, J. Nat. Prod. 2019, 82, 1258–1263. [DOI] [PubMed] [Google Scholar]
- [5].Inokuchi T, Kawafuchi H, Tetrahedron 2004, 60, 11969–11975. [Google Scholar]
- [6].Firn R, Nature’s Chemicals: The Natural Products that shaped our world, Oxford, New York, 2010. [Google Scholar]
- [7].Studer A, Chem. Eur. J. 2001, 7, 1159–1164. [DOI] [PubMed] [Google Scholar]
- [8].Caille S, Science 2019, 364, 635. [DOI] [PubMed] [Google Scholar]
- [9].a) Brougham P, Cooper MS, Cummerson DA, Heaney H, Thompson N, Synthesis 1987, 11, 1015–1017; [Google Scholar]; b) Heaney H, Aldrichimica Acta 1993, 26, 35–45; [Google Scholar]; c) Carvalho JFS, Silva MMC, Sá e Melo ML, Tetrahedron 2009, 65, 2773–2781. [Google Scholar]
- [10].a) Brodie C, J. Chem. Soc. 1864, 17, 266–281; [Google Scholar]; b) Fenton HJH, J. Chem. Soc., Trans. 1894, 65, 899–911; [Google Scholar]; c) Mummery CS, J. Soc. Chem. Ind. 1913, 32, 889–893; [Google Scholar]; d) Haber F, Weiss J, Naturwissenschaften. 1932, 20, 948–950. [Google Scholar]
- [11].a) Hawkins EGE, J. Chem. Soc. 1955, 3463–3467; [Google Scholar]; b) Kumamoto J, De La Mare HE, Rust FF, J. Am. Chem. Soc. 1960, 82, 1935–1939; [Google Scholar]; c) De Le Mare HE, Kochi JK, Rust FF, J. Am. Chem. Soc. 1961, 83, 2013; [Google Scholar]; d) Murai S, Sonoda N, Tsutsumi S, Bull. Chem. Soc. Jpn. 1964, 37, 1187–1190; [Google Scholar]; e) Schreiber SL, J. Am. Chem. Soc. 1980, 102, 6163–6165. [Google Scholar]
- [12].Criegee R, Wenner G, Justus Liebig Ann. Chem. 1949, 564, 9–15. [Google Scholar]
- [13].a) Jansen BJM, Kreuger JA, de Groot A, Tetrahedron 1989, 45, 1447–1452; [Google Scholar]; b) Meulemans TM, Stork GA, Macaev FZ, Jansen BJM, de Groot A, J. Org. Chem. 1999, 64, 9178–9188; [Google Scholar]; c) Jiang C-H, Bhattacharyya A, Sha C-K, Org. Lett. 2007, 9, 3241–3243; [DOI] [PubMed] [Google Scholar]; d) Velthuisen EJ, Danishefsky SJ, J. Am. Chem. Soc. 2007, 129, 10640–10641; [DOI] [PubMed] [Google Scholar]; e) Beaulieu M-A, Sabot C, Achache N, Guérard KC, Canesi S, Chem. Eur. J. 2010, 16, 11224–11228; [DOI] [PubMed] [Google Scholar]; f) Liffert R, Linden A, Gademann K, J. Am. Chem. Soc. 2017, 139, 16096–16099; [DOI] [PubMed] [Google Scholar]; g) Schuppe AW, Newhouse TR, J. Am. Chem. Soc. 2017, 139, 631–634; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Manoni F, Rumo C, Li L, Harran PG, J. Am. Chem. Soc. 2018, 140, 1280–1284. [DOI] [PubMed] [Google Scholar]
- [14].a) Gui J, Wang D, Tian W, Angew. Chem. Int. Ed. 2011, 50, 7093–7096; [DOI] [PubMed] [Google Scholar]; b) Huang D, Schuppe AW, Liang MZ, Newhouse TR, Org. Biomol. Chem. 2016, 14, 6197–6200; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smaligo AJ, Vardhineedi S, Kwon O, ACS Catal. 2018, 9, 1867–1872; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Smaligo AJ, Swain M, Quintana JC, Tan MF, Kim DA, Kwon O, Science 2019, 364, 681–685; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Smaligo AJ, Kwon O, Org. Lett. 2019, DOI: 10.1021/acs.orglett.9b03186; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bao J, Tian H, Yang P, Deng J, Gui J, ChemRxiv 2019, DOI: 10.26434/chemrxiv.88774455. [DOI] [Google Scholar]
- [15].Crossley SWM, Obradors C, Martinez RM, Shenvi RA, Chem. Rev. 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ferretti AC, Man H-W, Muslehiddinoglu J, Xu J, Yong KH-Y, Beauchamps MG, Kothare MA, Zhou N, Boersen NA, Li Y, Hilgraf R, Nagy MA, Zou D, Huang L, Solid forms of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use. WO 2015116755 A2, January 30, 2014. [Google Scholar]
- [17].McGuire HM, Odom HC, Pinder ARJ, J. Chem. Soc., Perkin Trans. 1974, 1, 1879–1883. [Google Scholar]
- [18].a) Bueno AB, Carreño MC, Ruano JLG, Tetrahedron Lett. 1995, 36, 3737–3740; [Google Scholar]; b) Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS, Nature 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vankar YD, Chaudhuri NC, Rao CT, Tetrahedron Lett. 1987, 28, 551–554 [Google Scholar]
- [20].Renzoni GE, Borden WT, J. Org. Chem. 1983, 48, 5231–5236. [Google Scholar]
- [21].a) Kamens R, Jang M, Chien C-J, Leach K, Environ. Sci. Technol. 1999, 33, 1430–1438; [Google Scholar]; b) Zhang D, Zhang R, J. Chem. Phys. 2005, 122, 1–12; [Google Scholar]; c) Zhang X, Chen Z, Wang H, He S, Huang D, Atmos. Environ. 2009, 43, 4465–4471. [Google Scholar]
- [22].a) Hartmann M, Seiberth M, Helv. Chim. Acta. 1932, 15, 1390–1392; [Google Scholar]; b) Robertson A, Nature 1948, 162, 153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.