

Abstract
Increased expression of protein arginine methyl transferase 6 (PRMT6) correlates with worse prognosis in lung cancer cases. To interrogate the in vivo functions of PRMT6 in lung cancer, we developed a tamoxifen-inducible lung targeted PRMT6 gain-of-function (GOF) mouse model, which mimics PRMT6 amplification events in human lung tumors. Lung targeted overexpression of PRMT6 accelerated cell proliferation de novo and potentiated chemical carcinogen (urethane)-induced lung tumor growth. To explore the molecular mechanism/s by which PRMT6 promotes lung tumor growth, we employed proteomics-based approaches and identified Interleukin enhancer binding protein 2 (ILF2) as a novel PRMT6-associated protein. Furthermore, by using a series of in vitro gain- and loss-of-function experiments we defined a new role for PRMT6-ILF2 signaling axis in alternate activation of tumor-associated macrophages (TAMs). Interestingly, we have also identified macrophage migration inhibitory factor (MIF), which has been recently shown to regulate alternate activation of TAMs, as an important downstream target of PRMT6-ILF2 signaling. Collectively, our findings reveal a previously unidentified non-catalytic role for PRMT6 in potentiating lung tumor progression via the alternate activation of TAMs.
Keywords: PRMT6, ILF2, MIF, protein-protein interaction, arginine methylation
Introduction
Lung cancer is the leading cause of cancer-related deaths in the world, and non-small cell lung cancer (NSCLC) accounts for ~85% of all lung cancers with a 5-year survival of only ~16% (1, 2). Lung cancer, a genetically heterogeneous disease, is most often diagnosed at an advanced inoperable stage where systemic therapies show only a modest benefit. However, the molecular mechanisms in the initiation and the progression of lung cancer remain poorly understood. Hence, identification of novel gene/s, which drive tumor initiation and progression, is required for future development of more effective therapies to treat advanced lung cancers. Post-translational modifications (PTM) on proteins expand the functional diversity of the proteome, favoring robust dynamic modulation of protein behavior for precise temporal and spatial control of gene expression and/or signaling. One such PTM is protein arginine methylation, which is de-regulated in cancers (3).
Protein arginine methylation is catalyzed by a class of enzymes known as protein arginine methyl transferases (PRMTs). Depending on the position of the guanidino nitrogens being methylated, PRMTs are classified as monomethylated or di-methylated and the latter can be either symmetric or asymmetric (4, 5). Increased expression of PRMT isoforms e.g., PRMT1, CARM1, PRMT5, PRMT6, PRMT9 in several tumor types have been correlated with poor overall survival (6-11). In recent years several small molecule compounds and peptide inhibitors that target the catalytic/substrate binding domains of PRMTs have been developed and tested (12-14). However, since some PRMT isoforms display very limited substrate specificity, development of catalytic inhibitors of PRMTs should not be the sole approach to treat cancers. Therefore, the identification of catalytic independent functions of PRMTs may help in the development of new therapies with increased specificity and efficacy. Interestingly, among the members of arginine methyl transferase family that are upregulated in several cancers including lung cancer, PRMT6 is known to display a narrow substrate specificity (15). However, studies investigating PRMT6 function in vivo, and in lung cancer in particular, have lagged behind other genes, leaving several open questions about their role.
In this study, to closely mimic PRMT6 amplification events in human lung tumors, we developed a lung targeted PRMT6 gain-of-function (GOF) mouse model. Overexpression of PRMT6 was sufficient to drive de novo hyperproliferation in the lungs. Furthermore, PRMT6 overexpression potentiated chemical carcinogen (urethane)-induced lung tumor growth. We also demonstrate that PRMT6 overexpression promotes lung tumor progression via the alternate activation of tumor-associated macrophages (TAMs). To explore the molecular mechanism/s by which PRMT6 promotes alternate activation of TAMs and lung tumor growth, we employed proteomics-based approaches and identified a protein-protein interaction (PPI) between PRMT6 and Interleukin enhancer binding protein 2 (ILF2), which is critical for the regulation of macrophage migration inhibitor factor (MIF). Collectively, our findings reveal a unique role for PRMT6 in potentiating lung tumor progression via the alternate activation of TAMs. Therefore, targeting the newly identified PRMT6/ILF2/MIF axis may open new possibilities for the therapeutic intervention of lung cancer.
Materials and Methods
Animal studies
1. Ethics statement:
Animal experiments were conducted in a strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). The animals were housed in Biologics Research laboratory vivarium, UIC. All the animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC).
2. PRMT6Tg founder mice generation:
The full-length cDNA sequences of open reading frame (ORF) for human PRMT6 was inserted into the multiple cloning site (MCS) of pCAG-floxed STOP-3XFlag-MCS plasmid backbone. The constructed plasmid encoding the PRMT6 ORF were sequenced to confirm that is was mutation-free, and PRMT6 protein overexpression was verified through transient transfection into 293T cells with the PRMT6 STOP plasmid and Cre plasmid, followed by Western blot analysis using a Flag antibody. To generate the PRMT6 overexpression transgenic mice, the STOP plasmid was linearized and introduced into the pronucleus of day 1 fertilized embryos (FVB/N), by microinjection. Injected embryos were then transferred into day 1 plugged pseudo-pregnant female mice. Founder pups were genotyped by PCR, using the tail clip to examine the germline transmission.
PRMT6Tg founder mice were backcrossed with Sftpc-CreERT2 mouse line to generate PRMT6Tg; Sftpc-Cretm mice. Parental stocks of Sftpc-CreERT2 were a generous gift from Dr. Brigid Hogan [(Duke University, (17)]. The genetic background of the mice was determined using PCR of DNA from tail biopsies.
3. TLA analysis:
Viable frozen splenocytes from PRMT6Tg mouse were used and processed according to CerGentis’ TLA protocol (44). Briefly, two primer sets were designed based on the hPRMT6 transgene and were used in individual TLA amplifications. PCR products were purified and library was prepared using the Illumina Nextera flex protocol, followed by sequencing on an Illumina sequencer. Sequence reads were mapped using Burrows-Wheeler Aligner’s Smith-Waterman Alignment [BWA-SW,(45)] and NGS reads were aligned to the transgene sequence and host genome (mouse). Integration sites were detected based on the coverage peaks in the genome and the identification of fusion reads between transgene sequence and host genome.
4. Urethane treatment:
Tumors were initiated in tamoxifen treated PRMT6Tg; Sftpc-Cretm mice and PRMT6Tg mice via four weekly intraperitoneal injections (IP) of either 0.9% saline or Urethane 1 g/Kg body weight. The mice were euthanized and dissected after 20 weeks to assess the formation of lung tumors. Lung tumors were counted and measured using a digital calipers (Fisher Scientific, Waltham, MA, USA).
5. Subcutaneous xenografts:
H2122 parental cells and H2122 PRMT6 sgRNA cells (2 ×106 viable cells/flank) were injected subcutaneously (s.c.) into the lower flanks of 6-8 week old athymic nude mice (Jackson laboratories). Tumor growth was assessed by weekly caliper measurements. Four weeks after the implantation of the cells, mice were sacrificed; flank tumors were removed and weighed.
Cell culture
Human bronchial epithelial (Beas2B) cells and the NSCLC cell lines (H2122 and H1299) were obtained from ATCC (Manassas, VA, USA). All cell lines were cultured in RPMI 1640 medium (10-040-CV, Cellgro, Mediatech Inc., Manassas, VA) supplemented with 10% fetal bovine serum (FBS) in a humidified 5% CO2 incubator at 37°C. Cell lines were cultured bi-weekly and stocks of cell lines were passaged no more than ten times for use in experiments. The cell lines were routinely tested for Mycoplasma contamination.
Gene editing of PRMT6
Guide RNAs (sgRNAs) targeting the first exon of PRMT6 (GAAAAGAAAGCTTGAGTCGG) were cloned into the BbsI site of px330 plasmid (Addgene plasmid # 42230). H2122 cells were transfected with the guide plasmids using Lipofectamine reagent (Invitrogen) according to the vendor’s recommendations. A day after transfection, cells were trypsinized and were diluted to a concentration of 1 cell/100 μl, seeded into 96-well plates, and were grown for 3 weeks. Thirty single clones were isolated and assayed by western blot analysis for PRMT6 expression. Multiple clones that showed complete loss of PRMT6 expression were selected for downstream analysis.
Knock-down protocol
Double-stranded RNAs (siRNAs) targeting the 3’UTR of human ILF2 (AGCUGCCUGCUACUGGGCUUUACAU) were purchased from Invitrogen (Invitrogen, Carlsbad, CA), while siRNAs targeting MIF were purchased from Santa Cruz Biotechnology (sc-37137). Control siRNAs were purchased from Santa Cruz Biotechnology. NSCLC cells were treated with 100 nM siRNAs by using Lipofectamine 2000 reagent according to the manufacturer’s protocol.
Immunoblot Analysis
Lung tissue protein extracts were obtained from mouse lungs homogenized in tissue protein extraction lysis buffer containing a cocktail of 1 m HEPES, 5 m NaCl, 10% Triton X-100, 1 m DTT, 0.5% EDTA, 20 mm NaVO3, 10 mm PMSF, 0.5 m Na-β-glycerophosphate and protease inhibitors using Qiagen TissueLyser LT (Qiagen), while NSCLC cells were lysed in a lysis buffer (0.5% Triton X-100, 50 mM β-glycerophosphate, pH 7.2, 0.1 mM sodium vanadate, 2 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 2 μg/ml leupeptin, and 4 μg/ml aprotinin), and the western blotting analysis was carried out as previously described (6, 46). Briefly, aliquots of various protein extracts were resolved on SDS-PAGE gels and transferred to nitrocellulose. The filters were blocked in Tris-buffered saline (10 mM Tris-HCl, pH 7.4, 140 mM NaCl, containing 0.1% Tween-20 (TTBS) and 3% non-fat dry milk and then incubated with the same blocking solution containing the indicated antibodies at 0.5 μg/ml for 16 h. Filters were extensively washed in TTBS, and bound antibodies were visualized with horseradish peroxidase (HRP)-coupled secondary antibodies. The following antibodies were used for immunoblotting: PRMT6 (14641), and GAPDH (5174) antibodies were from Cell Signaling Technology, FLAG (F3165) and actin (A3853) were from Sigma-Aldrich, ILF2 (ab28772) antibodies were from Abcam, and MIF (FL-115) antibodies were from Santa Cruz Biotechnology.
Cell Proliferation Studies
For five-day cell growth assay, 25,000 cells were seeded in duplicates in a 24-well plate. Cell proliferation was measured for five days by trypsinization of cells, followed by cell counting using a hemocytometer. Cell numbers were represented in the graphs. For clonogenic cell proliferation assays, 1000 cells per well were seeded in a 12-well culture plate followed by incubation at 37°C in a 5% CO2 incubator. After 5-7 days colonies were stained using a staining solution (0.5% Crystal Violet, 12% Glutaraldehyde, 87.5% H2O) for 1 hr at room temperature. After de-staining in water and drying, colonies were quantified using Biorad Chemidoc Imaging System and Quantity One Software. Cloning efficiency represents the mean number of colonies formed per well.
Cell migration assays
For assessing cell migration, 30,000 cells in serum free media were seeded into the transwell inserts (Corning) containing 8 μm permeable pores and allowed to migrate towards 10% FBS containing medium. Later, the cells in the transwell inserts were removed and the inserts were washed in PBS for three times. The migrated cells on the bottom of the insert were fixed with 2% glutaraldehyde solution followed by crystal violet (1%) staining. After washing the inserts three times with PBS, the inserts were allowed to air dry and pictures were taken using an inverted microscope. Ten independent fields were counted for each transwell and the average number of cells/field were represented in the graphs.
Anchorage-independent growth
Soft agar assays were performed as described previously (47). Briefly, 5,000 cells were plated in duplicates in a six-well plate in growth medium containing 0.3% noble agar. After 14 days, colonies were stained and visualized with nitroblue tetrazolium chloride.
In-gel tryptic digestion and mass spectrometry
To identify PRMT6-associated proteins, lung lysates from FVB/NJ mice were incubated with GST or GST-PRMT6. Following SDS-PAGE analysis of PRMT6 immunocomplexes, the gel bands corresponding to the molecular range of 50–80 kDa was excised, destained, reduced, aklyated, and digested with trypsin. The resulting concentrated peptide extract was analyzed by automated microcapillary liquid chromatography tandem mass spectrometry. Full mass (MS) spectra were recorded on the peptides over a 400–2000 m/z range, Only peptides with a p value of at least 0.02 were analyzed further.
Cytokine array
Cell lysates derived from Beas2B cells expressing empty vector of pCMV-HA-hILF2 were subjected to immunoblotting based cytokine profiling using proteome profiler human XL cytokine array kit (ARY022B, R&D systems).
Isolation of conditioned medium from mouse alveolar epithelial cells
Lungs from the PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice 10 days after last tamoxifen treatment were harvested, and cells were isolated using mouse tumor cell isolation kit (130-110-187, Miltenyi Biotec) according to the manufacturer’s recommendations. The cell suspension was incubated in a 100 mm cell culture dish pre-coated with mouse IgG for 2h. Later, the non-adherent cells were collected, centrifuged, resuspended in DMEM complete medium (10% FBS, 1% Penicillin/Streptomycin, 25 mM HEPES, and 10 ng/ml keratinocyte growth factor). The cells were plated on culture dishes coated with fibronectin, collagen, and BSA, and the medium was changed every 2 days. On day 5, the medium was collected, centrifuged to remove the cells, aliquoted, and stored at −80 °C.
Bone marrow-derived macrophage culture
Bone marrow-derived monocytes were isolated from femurs and tibias of wildtype FVB/NJ mice and cultured in the presence of macrophage-colony stimulating factor (M-CSF, R&D systems) to promote macrophage maturation as previously described (48). After 3-7 days, these cells display the morphology of mature (M0) macrophages. M0 macrophages were later treated with the conditioned medium isolated from PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice for 24 h. Later total RNA was isolated from the macrophages and markers of M1 and M2 polarization were assayed by qPCR.
Isolation of lung tumor-associated macrophages (TAMs)
Lungs from PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice 20 weeks after tamoxifen and urethane treatment were enzymatically digested and CD11b+ cells were positively selected using autoMACs ProSeparator (Miltenyi Biotec). Purity of the isolated cells was later confirmed by flow cytometry using CD11b and F4/80 macrophage markers. RNA from the isolated cells was isolated and the expression of macrophage M1 and M2 markers were determined by quantitative PCR (qPCR).
RNA isolation and quantitative PCR
Total RNA from the lung tissues, NSCLC cells, CD11b+ cells, and macrophages were obtained using Trizol reagent (Invitrogen) as per the manufacturer’s recommendations. For quantitative RT-PCR, 3 μg of total RNA was reverse transcribed using random primers and real-time PCRs were performed using the QuantiTect SYBR Green PCR kit (204050, Qiagen, Venlo, Limburg) and the Bio-Rad CFX qPCR detection system. The primers utilized in the PCR experiments were shown in Supplementary table 2.
Indirect immunofluorescence
Lung tumor sections from PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice 20 weeks after tamoxifen and urethane treatment were employed in immunostaining procedure using the following antibodies: CD68 (FA-11, Bio-Rad) for identifying macrophages, iNOS (ab15323, Abcam) to detect M1 polarization, Arginase I (sc-18351, Santa Cruz Biotechnology) to detect M2 polarization, EPCAM (VU1D9, Cell signaling technology), alpha smooth muscle actin (ab5694, Abcam), and CD31 (550274, BD Biosciences) using procedures described previously (49). Briefly, lung sections were fixed with 3% paraformaldehyde and permeabilized with 0.01% Triton-X-100 followed by blocking with 3% normal horse serum. The samples were then exposed to primary antibodies and detected with fluorescent dye-conjugated secondary antibodies. Image acquisition was performed using an Evos digital inverted microscope (Thermofisher) with a 40x objective.
In vitro methylation assays
In vitro methylation assays were performed as described previously (12,13,20). Briefly, GST-ILF2, or Histone 3 (1 μg) was incubated with HA-affinity matrix containing bound PRMT6 and 1 μCi of S-adenosyl-L-[methyl-3H] methionine (NEN radiochemicals, 250 μCi, 9.25 MBq), at 30 °C for 1 h. After 1 h, the reactions were stopped and separated on an SDS-PAGE gel. The separated proteins were later transferred to nitrocellulose membrane, amplified (Autofluor, National Diagnostics, 2 h), dried and fluorography was performed.
Data analysis
Data were compiled from at least three independent, replicate experiments, each performed on separate cultures and on separate occasions. The responses were displayed as “fold-changes.” Comparisons of data among experimental groups were performed using student’s t-test to assess variance. Increase in statistical significance (p value of <0.05) is denoted with an “asterisk” symbol, while a decrease in statistical significance (p value of <0.05) is denoted with a “hash” symbol.
Results
PRMT6 is upregulated in lung cancer
PRMT6 was shown to be upregulated in lung tumors (11). Consistent with the publication by Yoshimatsu et al., (11), mining the cancer genome atlas (TCGA) lung adenocarcinoma (LUAD) datasets revealed a significant upregulation of PRMT6 in lung tumors when compared to normal lung tissues (Supplementary Fig. 1A). To further assess the prognostic significance of high PRMT6 expression in lung cancer patients, we analyzed TCGA LUAD datasets by using Kaplan-Meier plotter (www.kmplot.com). Our analyses indicated that high PRMT6 expression is significantly correlated with poor overall survival (Supplementary Fig. 1B). To determine whether PRMT6 expression is also elevated at the protein level, we employed formalin-fixed paraffin embedded (FFPE) human tissue samples and fresh frozen lung tumor tissues (Supplementary Fig. 1C, 1D). IHC staining of FFPE sections with PRMT6 antibody showed a strong nuclear staining of PRMT6, while no such staining was observed in secondary antibody alone control (Fig. 1C). The slides were later scanned using Aperio imagescope scanner (Leica biosystems), followed by the scoring of the tissue slides by an H-score method that combines the staining intensity with the percentage of cells stained (16). A significant increase in PRMT6 staining was observed in the tumor tissues when compared to adjacent uninvolved lung tissues (N=8, Supplementary Fig. 1C). Similarly, immunoblotting of fresh frozen lung tumor tissues also revealed a robust increase in PRMT6 expression in the tumors when compared to uninvolved lung tissues of the same patient (N=5, Supplementary Fig. 1D). Together, these results indicate that PRMT6 might be a significant contributor to LUAD and provide evidence that PRMT6 may represent a novel prognostic indicator for this disease.
Figure 1. Lung targeted PRMT6 expression accelerates cell proliferation.
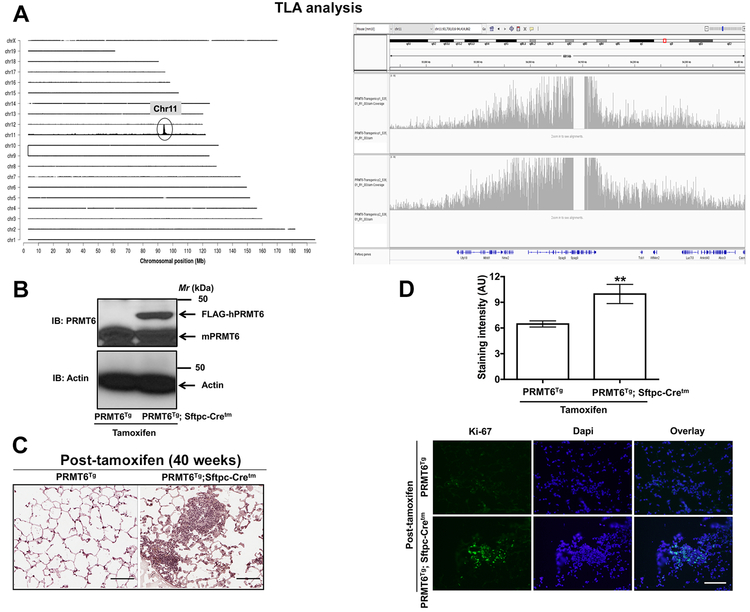
A. TLA coverage and analysis plots. Left panel represents the sequence coverage: mouse chromosomes 1 through X are arranged on the Y-axis and X-axis represents chromosomal position. While, right panel displays the graphical representation of transgene integration site and structural changes. B. Tissue lysates from the lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen treatment were subjected to immunoblotting with anti-PRMT6 antibodies that detects both human and mouse PRMT6 isoforms. C, D. 40 weeks after tamoxifen administration to induce PRMT6 expression, lung sections were subjected to H&E staining (C, scale bar: 100 μM) and Indirect immunofluorescence with anti-ki67 antibodies (D, scale bar: 200 μM). Upper panel represents the quantification of ki67 staining, while representative images were displayed in the lower panel. **, p< 0.01; versus PRMT6Tg control.
Overexpression of PRMT6 drives de novo hyperproliferation in the lungs
PRMT6 was shown to be upregulated in lung tumors (11). However, mouse models to investigate PRMT6 function in vivo, and in lung cancer in particular, have lagged behind other genes, leaving several open questions about their role. To closely mimic PRMT6 amplification events in human lung tumors, we created a PRMT6 gain-of-function (GOF) mouse model by cloning the open reading frame (ORF) for human PRMT6 isoform into the multiple cloning site (MCS) of pCAG-Lox-STOP-Lox-3XFlag-MCS plasmid backbone (LSL-hPRMT6) that keeps the LSL-hPRMT6 allele silent until the ‘stop’ cassette is deleted by a Cre recombinase. The LSL-hPRMT6 plasmid was later linearized and introduced into the pronucleus of day 1 fertilized embryos (FVB/N), by microinjection. Injected embryos were then transferred into day 1 plugged pseudo-pregnant female mice. Founder pups were genotyped by PCR, using the tail clip to examine the germline transmission. In order to identify the exact transgene integration sites, we performed targeted locus amplification analysis (TLA analysis) on splenocytes from LSL-hPRMT6 mice (hereafter referred to as PRMT6Tg). TLA analysis mapped the transgene insertion site to mouse chromosome 11 at intronic region between exon 10 and 11 and ending in the intronic region between exon 24 and 25, which results in a 21kb deletion of Spag9 (Fig. 1A). The copy number is estimated to be around 5-8 copies. To avoid possible phenotypes arising due to random insertion of PRMT6 transgene, transgenic mice were maintained in heterozygous genotype.
For lung-targeted expression of PRMT6, we generated an inducible hPRMT6 GOF transgenic by crossing PRMT6Tg to a tamoxifen-regulated surfactant protein C-driven Cre-recombinase mouse line [(Sftpc-Cretm) (17)]. Daily injections of tamoxifen (100 mg/Kg body weight) for five days led to a robust induction of hPRMT6 expression in the lungs of PRMT6Tg; Sftpc-Cretm, but not PRMT6Tg control mice, within a week after the last tamoxifen administration (Fig. 1B). PRMT6 overexpression caused hyperproliferation of the lung epithelium as demonstrated by an increase in ki-67 immunostaining (Fig. 1C, 1D). Consistent with our observations, a recent report demonstrated hyper-branching of the mammary glands and increased ki-67 staining in the breast epithelium of PRMT6 GOF transgenic mice (18).
Overexpression of PRMT6 potentiates urethane-induced lung tumor growth
Since PRMT6 and KRas mutations are co-occurring in 24% (272 patients) of queried patients/samples [cBioPortal (www.cbioportal.org)], we determined whether PRMT6 overexpression can potentiate mutant KRas-driven lung tumor formation. For these studies, we interrogated the effects of urethane (ethyl carbamate) on PRMT6Tg and PRMT6Tg; Sftpc-Cretm mice, a prototypical model to study lung tumorigenesis (19-21). Urethane is a chemical carcinogen, which causes activating mutations in KRas (19-21), leading to the formation of lung tumors in mice. Several studies have highlighted a strain specific response to Urethane; FVB/NJ showed lung tumorigenesis within 20 weeks after 1 dose (1 g/Kg body weight) of Urethane administration, while C57Bl/6J, being resistant, require 6 weekly doses (1 g/Kg body weight) of Urethane and develop lung tumors within 40 weeks (19-21). Since the PRMT6Tg; Sftpc-Cretm mice were in mixed background, we determined the urethane administration regimen empirically that results in 5-10 lung tumors in control mice after intraperitoneal administration. Strikingly, 4 weekly injections (intraperitoneal, 1 g/Kg body weight) of urethane to PRMT6Tg; Sftpc-Cretm mice after tamoxifen administration (for PRMT6 induction, Fig. 2A) resulted in a more severe phenotype in terms of tumor size (Fig. 2B) and number (Fig. 2C) when compared to PRMT6Tg control mice treated with tamoxifen and urethane. Masked histological examination by a lung pathologist revealed that while control mice mostly developed microscopic adenomas, the PRMT6Tg; Sftpc-Cretm mice on the other hand developed macroscopic aggressive lung adenocarcinomas. Furthermore, indirect immunofluorescence staining of the tumor sections with CD31 antibodies consistently revealed more mature blood vessels in the tumors of PRMT6Tg; Sftpc-Cretm mice, when compared to the tumors of PRMT6Tg control mice (Fig. 2D). Taken together, we show that PRMT6 is an important contributor to tumor progression and metastasis in mutant KRas-driven lung tumors.
Figure 2. Lung targeted PRMT6 expression augments chemical carcinogen (urethane)-induced lung tumor growth.

A. Tissue lysates from the lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatments were subjected to immunoblotting with anti-PRMT6 antibodies that detects both human and mouse PRMT6 isoforms. B. 20 weeks after tamoxifen and urethane administration lung sections were subjected to H&E staining (scale bar: 100 μM). C. Lung tumor numbers from PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatments were counted and were represented in the graph. D. Lung tumor sections of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatment (20 weeks) were subjected to indirect immunofluorescence staining with anti-EPCAM, anti-SMA, and anti-CD31 antibodies as described in the methods (scale bar: 10 μM).
Depletion of PRMT6 reduces cell proliferation, cell migration, and anchorage-independent growth of NSCLC cells
To investigate the function of PRMT6 in NSCLC, we employed CRISPR-Cas9 mediated gene editing of PRMT6 in NSCLC cell lines: H2122 (3A-3E) and H1299 (3F-3J). For generating PRMT6 knockout cell lines, we transfected H2122 and H1299 cells with single guide RNAs (sgRNAs) targeting PRMT6 followed by clonal isolation via limiting dilutions. H2122 and H1299 PRMT6 knockout clones displayed a consistent reduction in cell proliferation when compared to their parental cells, as determined by cell growth curves (Fig. 3B, 3G) and clonogenic cell proliferation assays (Fig. 3C, 3H). We next investigated the effects of PRMT6 knockout on cell migration by using trans-well assays. H2122 and H1299 PRMT6 knockout clones displayed reduced cell motility when compared to their respective parental cells (Fig. 3D, 3I). To further explore the functional consequence of PRMT6 gene knockout, we evaluated anchorage-independent growth capabilities of H2122 and H1299 PRMT6 knockout clones. Clearly, depletion of PRMT6 in H2122 and H1299 cells resulted in a reduced number of colonies in soft agar when compared to their corresponding parental cells (Fig. 3E, 3J).
Figure 3: PRMT6 is required for NSCLC cell proliferation, migration, and anchorage-independent growth.

H2122 (A-E) and H1299 (F-J) NSCLC cells were transfected with PRMT6 single guide RNAs (sgRNA), followed by limited dilutions to develop PRMT6 knockout H2122 and H1299 clones. A, F. Lysates of H2122 (A) and H1299 (F) PRMT6 knockout clones and their parental cells were immunoblotted with anti-PRMT6 antibodies. The proliferation rates of H2122 and H1299 PRMT6 knockout clones were determined by hematocytometer cell count (B, G) and clonogenic (C, H) cell proliferation assays as described in the methods. Data represents mean ± SEM from 3 independent highly reproducible experiments. *, p< 0.05; versus parental control. Cell migration rates of H2122 (D) and H1299 (I) PRMT6 knockout clones were assayed in transwell inserts as described in the methods. *, p< 0.05; versus parental control. Anchorage-independent growth of H2122 (E) and H1299 (J) PRMT6 knockout clones were assayed in soft-agars as described in the methods. *, p< 0.05; versus parental control. K-M. H2122 parental cells and PRMT6 knockout H2122 cells were subcutaneously injected into athymic nude mice. Tumor bearing mice (K), extracted tumors (L), and tumor weights (M) were displayed in the figure. **, p< 0.01; versus control.
We also tested the role of PRMT6 in lung tumor growth in vivo by performing xenograft studies in athymic nude mice (Fig. 3K-M). For these studies, H2122 parental cells and H2122 PRMT6 sgRNA cells were injected subcutaneously (s.c.) into the lower flanks of athymic nude mice. Tumor growth was assessed by weekly caliper measurements. It was striking to observe that PRMT6 depletion resulted in a dramatic reduction in tumor growth within 4 weeks of implantation of cells (Fig. 3K-M). Collectively, these data support an important role for PRMT6 in promoting cell proliferation, cell migration, anchorage-independent growth, and in vivo tumor growth of NSCLC cells.
Identification of PRMT6-associated proteins that mediate lung tumor progression
Since the molecular functions of PRMT6 are governed by the proteins it interacts with and/or its substrates, we sought to identify PRMT6-associated proteins of the lung. For these studies, mouse lung lysates were employed in pulldowns by using GST-PRMT6 as a “bait” followed by proteomics. Affinity pulldown reactions resulted in the identification of several proteins that are specific to GST-PRMT6, but not GST pulldowns. Of the identified proteins, only the proteins with four or more unique peptides were considered as potential PRMT6-associated proteins (Fig. 4A, Supplementary table 1). STRING database provides critical assessment and integration of protein-protein interactions that are both direct and indirect (22). We created a STRING-curated protein interaction network (Fig. 4B) for the proteins that met our selection criteria (Supplementary table 1). With the exception of 3 proteins, the remaining proteins had at least one interaction with another protein (either predicted or experimentally determined), indicating an interconnectivity among the identified proteins (Fig. 4B). We elected to focus on Interleukin-enhancer binding factor 2 (ILF2) as it is identified as an arginine methylated protein in a proteomic screen (23), and given its previous association with malignant tumors of diverse origins (24-29).
Figure 4: ILF2 is a PRMT6 interacting protein.

A. Mouse lung lysates were employed in affinity pulldowns with either GST- or GST-PRMT6 agarose followed by SDS-PAGE separation. Later, PRMT6-associated proteins were identified by in gel tryptic digestion followed by mass spectrometry. B. STRING protein-protein interaction network of PRMT6 interacting proteins. C. Beas2B cells were transiently transfected with HA-PRMT6 for 48 hours followed by cell lysis and affinity pulldowns with anti-HA antibodies. Interaction of PRMT6 and ILF2 was visualized by probing the immunoblots with anti-ILF2 antibodies. D. His-tagged PRMT6 and GST-tagged ILF2 were purified from E.Coli. Later, equal amounts of His-PRMT6 were mixed with either GST or GST-ILF2 followed by affinity pulldowns with GST-agarose beads. Direct interaction of PRMT6 and ILF2 were visualized by probing the blots with anti-His antibodies. E. In vitro methylation assays were performed by employing HA-PRMT6 purified from Beas2B cell lysates, GST-ILF2 or histone 3 as substrates and 3H-SAMe as a methyl donor. Representative fluorographs were shown in the figure.
To validate the interaction of ILF2 with PRMT6, affinity pulldowns were performed. For these studies, human bronchial epithelial cells (Beas2B) were engineered to express HA-PRMT6. Later, the cell lysates were employed in pulldown assays with anti-HA antibodies. The presence of endogenous ILF2 in the HA-PRMT6 immunocomplex was made possible by probing the immunoblots with anti-ILF2 antibodies. A strong PRMT6-ILF2 interaction was detected (Fig. 4C). Furthermore, to determine whether the PRMT6-ILF2 interactions were direct or indirect, we purified recombinant His-PRMT6 and GST-ILF2 in E.Coli. Later, the two proteins were mixed, and pulldowns were performed with GST-agarose beads. A strong PRMT6-ILF2 interaction was detected in GST-ILF2 pulldowns, but not in GST pulldowns (Fig. 4D). Collectively, these data provide evidence for a direct interaction between PRMT6 and ILF2.
To evaluate if ILF2 is a PRMT6 substrate, we performed in vitro methylation assays (Fig. 4E). For these studies, HA-PRMT6 was expressed and isolated from Beas2B cells via HA-affinity pulldowns. While, recombinant GST-ILF2 was purified from E. Coli. Later, the abilities of HA-PRMT6 to methylate GST-ILF2 were evaluated in the presence of radiolabeled (3H) S-adenosyl L-methionine (SAMe), a methyl group donor. To our surprise, PRMT6 failed to catalyze ILF2 methylation (Fig. 4E). The fact that PRMT6 could methylate Histone 3, a known PRMT6 substrate (30), indicates that the PRMT6 employed in in vitro methylation reactions was catalytically active (Fig. 4E). Taken together, these data indicate that ILF2 is a PRMT6 interacting protein, but not a catalytic substrate of PRMT6.
Pro-proliferative effects of PRMT6 are ILF2-dependent
It was shown earlier that increased expression of ILF2 is associated with poor overall survival in tumors of diverse origin (25, 26, 28). However, the upstream regulators of ILF2 expression remain to be identified. In this study, we have identified ILF2 as a novel PRMT6 interacting protein (Fig. 4). Interestingly, spearman rank correlation analysis of the TCGA lung cancer datasets revealed a significant positive correlation between PRMT6 and ILF2 (Fig. 5A). Furthermore, mutually-dependent expression of PRMT6 and ILF2 proteins in the lung tumors of the patient samples was also detected (Fig. 5B). These observations suggest that PRMT6 is a novel regulator of a pro-tumorigenic protein ILF2 in the lung. To explore this exciting possibility, we performed over expression of HA-tagged hPRMT6 in non-transformed bronchial epithelial cells (Beas2B). Overexpression of PRMT6 resulted in a dramatic increase in ILF2 protein levels, as determined by western blotting with anti-ILF2 antibodies (Fig. 5C). To fully complement our overexpression studies, we also evaluated ILF2 levels in PRMT6 knockout H2122 and H1299 clones. It was striking to observe a complete loss of ILF2 protein levels, but not mRNA levels (data not shown), upon PRMT6 depletion in both H2122 (Fig. 5D) and H1299 (Fig. 5E). Consistent with the effects of PRMT6 modulation on ILF2 protein levels in vitro, probing mouse lung lysates of PRMT6Tg; Sftpc-Cretm mice (post-tamoxifen and urethane administration) also resulted in a robust induction of ILF2 levels when compared to the PRMT6Tg control mice (Fig. 5F). In vitro methylation assays demonstrate that ILF2 is not a catalytic substrate of PRMT6 (Fig. 4E). To evaluate whether PRMT6 catalytic activity is required for ILF2 regulation, we performed over expression of HA-hPRMT6 and HA-hPRMT6-KLD (methyl transferase dead mutant) in non-transformed bronchial epithelial cells (Beas2B). Overexpression of either PRMT6 or PRMT6-KLD mutant resulted in a dramatic increase in ILF2 protein levels (Fig. 5G). Taken together, these observations establish that PRMT6 regulates ILF2 protein levels in a catalytic-independent manner.
Figure 5: PRMT6 is a novel regulator of ILF2 expression.

A. Spearman rank correlation analysis of TCGA lung cancer datasets revealed a positive correlation between PRMT6 and ILF2. B. Protein lysates from human lung tumor and adjacent uninvolved lung tissue from the same patient were probed for the expression of PRMT6 and ILF2 via immunoblotting. C. Human non-transformed bronchial epithelial cells (Beas2B) were transfected with either control or HA-PRMT6. The cell lysates were later probed for the expression of PRMT6 (HA-tag) and ILF2 via immunoblotting. D, E. Lysates of H2122 (D) and H1299 (E) PRMT6 knockout clones and their parental cells were immunoblotted with anti-PRMT6 and anti-ILF2 antibodies. F. Tissue lysates from the lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatments were subjected to immunoblotting with anti-PRMT6 and anti-ILF2 antibodies. G. Human non-transformed bronchial epithelial cells (Beas2B) were transfected with either HA-PRMT6 or HA-PRMT6-KLD mutant. The cell lysates were later probed for the expression of PRMT6 (HA-tag) and ILF2 via immunoblotting. H. The proliferation rates of H1299 cells co-transfected with siRNA-resistant HA-ILF2 plasmid with either control or ILF2 siRNAs were determined by clonogenic cell proliferation assays as described in the methods. The cell lysates were later probed for the expression of ILF2 via immunoblotting. **, p< 0.01; *, p< 0.05. I. The proliferation rates of Beas2B cells co-transfected with HA-PRMT6 plasmid with either control or ILF2 siRNAs were determined by hematocytometer cell count as described in the methods. The cell lysates were later probed for the expression of HA-PRMT6 and ILF2 via immunoblotting. **, p< 0.01.
To determine if ILF2 mediates the pro-proliferative effects of PRMT6, we employed RNA interference by designing siRNAs targeting the 3’ untranslated region (UTR) of ILF2 (Fig. 5H). Treatment of H1299 cells with ILF2 siRNAs resulted in a reduction in cell proliferation as determined by clonogenic cell proliferation assays (Fig. 5H). Furthermore, the effects of ILF2 depletion on cell proliferation could be rescued by the expression of siRNA-resistant HA-ILF2 plasmid (Fig. 5H), indicating the specificity of the siRNAs to ILF2. To determine the biological significance of PRMT6-mediated ILF2 regulation, we performed overexpression of PRMT6 together with ILF2 siRNAs in Beas2B cells (Fig. 5I). As anticipated, overexpression of PRMT6 resulted in a robust increase in cell growth, as determined by hemocytometer cell count (Fig. 5I). Depletion of ILF2 in PRMT6 overexpressing cells resulted in a reduction in PRMT6-induced cell proliferation (Fig. 5I), indicating that the biological effects of PRMT6 were indeed ILF2 dependent.
PRMT6-ILF2 signaling axis is a novel regulator of pro-inflammatory cytokine MIF
Since ILF2 is an important regulator of the T-cell expression of IL2 (31), we hypothesized that PRMT6/ILF2 signaling potentiates lung tumor growth by modulating cytokine levels. To identify the cytokines regulated by ILF2, we employed whole cell lysates of bronchial epithelial cells (Beas2B) expressing either empty vector or HA-ILF2 on a 105-cytokine antibody array (Proteome profiler Human XL Cytokine Array kit, R&D systems). Interestingly, forced expression of ILF2 resulted in a robust induction of four cytokines e.g., ICAM1, IL-8, IL-32, and MIF (Fig. 6A). Of note, these cytokines were not previously associated with either ILF2 or PRMT6. Macrophage migration inhibitory factor (MIF) was of interest due to its pro-tumorigenic role in a number of cancer types (32-35), and due to its most prominent induction by ILF2 (Fig. 6B).
Figure 6: PRMT6/ILF2 signaling axis regulates the expression of MIF.

A. Lysates of Beas2B cells expressing either control or HA-ILF2 were screened for differentially expressing cytokines by using a human cytokine array as described in the methods. B. Expression of ICAM1, IL8, IL32, and MIF transcripts in Beas2B cells expressing either control or HA-ILF2 using qPCR. *, p< 0.05; **, p< 0.01; versus control. C. Beas2B cells were transfected with either control or HA-ILF2. The cell lysates were later probed for the expression of ILF2 and MIF via immunoblotting. D, E. Lysates of H2122 (D) and H1299 (E) transfected with ILF2-specific siRNAs were immunoblotted with anti-ILF2 and anti-MIF antibodies. F, G. Lysates of H2122 (F) and H1299 (G) PRMT6 knockout clones and their parental cells were immunoblotted with anti-PRMT6, anti-ILF2, and anti-MIF antibodies. H. Tissue lysates from the lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatments were subjected to immunoblotting with anti-PRMT6, anti-ILF2, and anti-MIF antibodies. I. Beas2B cell lysates co-transfected with HA-PRMT6 plasmid with either control or ILF2 siRNAs were probed for the expression of HA-PRMT6, ILF2, and MIF via immunoblotting. J-L. The proliferation rates of Beas2B cells co-transfected with HA-ILF2 plasmid with either control or MIF siRNAs were determined by hematocytometer cell count (K) and clonogenic (L) cell proliferation assays as described in the methods. **, p< 0.01. The cell lysates were later probed for the expression of HA-ILF2, and MIF via immunoblotting (J).
To validate the regulation of MIF by ILF2, we performed overexpression of HA-tagged ILF2 in Beas2B cells (Fig. 6C). Overexpression of ILF2 resulted in a robust increase in MIF expression (Fig. 6C). Complementary studies involving siRNA-mediated knockdown of ILF2 in H2122 (Fig. 6D) and H1299 (Fig. 6E) cells resulted in a dramatic reduction in MIF expression. We also determined if PRMT6, an upstream regulator of ILF2 (Fig. 5), could also regulate MIF. For these studies, we probed the cell lysates of H2122 and H1299 PRMT6 knockout clones. Depletion of PRMT6 in H2122 and H1299 cells resulted in a dramatic reduction in MIF expression (Fig. 6F, 6G). In agreement with the effects of PRMT6 depletion on MIF expression in vitro, probing mouse lung lysates of PRMT6Tg; Sftpc-Cretm mice (post-tamoxifen and urethane administration) also resulted in a robust induction of MIF expression when compared to the PRMT6Tg control mice (Fig. 6H). Taken together, these observations indicate that PRMT6 and ILF2 are novel regulators of MIF. To further investigate whether PRMT6 stimulated MIF expression was ILF2-dependent, we performed overexpression of PRMT6 in Beas2B cells (Fig. 6I). As anticipated, overexpression of PRMT6 in Beas2B cells resulted in a robust increase in MIF expression (Fig. 6I), which was blocked by depletion of ILF2 (Fig. 5I). Taken together, these data indicate that the PRMT6/ILF2 signaling axis is a novel regulator of MIF expression in NSCLC.
To determine the biological significance of ILF2-mediated MIF expression, we performed overexpression of ILF2 together with MIF siRNAs in Beas2B cells (Fig. 6J). Overexpression of ILF2 resulted in a significant increase in cell growth, as determined by hemocytometer cell count (Fig. 6K), and clonogenic assays (Fig. 6L), which was blocked by the depletion of MIF (Fig. 6K, 6L), indicating that the biological effects of ILF2 are MIF-dependent.
PRMT6 potentiates lung tumor progression via alternate activation of macrophages
Prior studies highlighted an important role for MIF in the alternate (M2) activation of macrophages within the tumor microenvironment (36). Since PRMT6/ILF2 signaling axis regulates MIF (Fig. 6), we hypothesized that increased expression of PRMT6 potentiates lung tumor growth through the alternate activation of macrophages. To explore this possibility, we probed the expression of classical and alternate activation markers within the macrophages via indirect immunofluorescence staining of lung tumor sections of PRMT6Tg; Sftpc-Cretm mice and PRMT6Tg control mice post-tamoxifen and urethane treatment (Fig. 7A). Tumor-associated macrophages (TAMs) of PRMT6Tg control mice displayed classical activation of macrophages with a pro-inflammatory phenotype [high M1 marker (iNOS) and low M2 (Arginase I) marker expression (Fig. 7A)], while the TAMs of PRMT6Tg; Sftpc-Cretm mice were skewed towards an M2-like tumor promoting phenotype [low M1 marker (iNOS) and high M2 marker (Arginase I) expression (Fig. 7A)]. To gain more insights into PRMT6-mediated macrophage polarization, CD11b+ cells were isolated from total lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice treated with tamoxifen and urethane. Eighty three percent (83%) of CD11b+ cells were positive for F4/80, an alveolar macrophage-specific marker, displaying the purity of the TAM population (Fig. 7B). Later RNAs from the CD11b+ cells were isolated and the expression of M1 and M2 macrophage markers were determined by quantitative PCR (qPCR). Consistent with the indirect immunofluorescence data (Fig. 7A), CD11b+ cells isolated from PRMT6Tg; Sftpc-Cretm lung tumors showed increased expression of M2 markers (Arg I and IL10) and decreased expression of M1 markers [TNFα and iNOS, (Fig. 7B)]. In contrast, CD11b+ cells isolated from PRMT6Tg control lung tumors displayed increased expression of M1 markers (Fig. 7C). These studies demonstrate for the first time that PRMT6 is an important modulator of lung tumor microenvironment via the alternate activation of TAMs.
Figure 7: PRMT6 overexpression promotes alternate activation of tumor-associated macrophages.
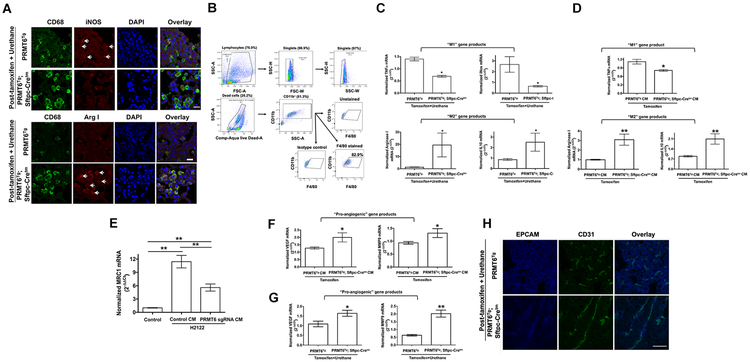
A. Lung tumor sections of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatment (20 weeks) were subjected to indirect immunofluorescence staining with anti-CD68, anti-iNOS, and anti-arginase I antibodies as described in the methods. Scale bar: 20 μM. B. Gating strategy for the purification of TAMs using CD11b antibodies. Sixty% of live cells are CD11b+, of which 83% are F4/80+. C. Total RNA from CD11b+ cells from the lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatment (20 weeks) were employed in qPCR analysis for the indicated M1 and M2 macrophage markers. *, p< 0.05; versus control. D. Bone marrow-derived macrophages were treated with the conditioned medium isolated from AECs isolated from PRMT6Tg; Sftpc-Cretm or PRMT6Tg control mice after tamoxifen treatment. Later, total RNA from the macrophages were isolated and qPCRs were performed for M1 and M2 macrophage markers as described in the methods. **, p< 0.01; *, p< 0.05; versus control. E. THP1 cells were incubated with 150 mM phorbol myristate acetate (PMA) for 24h. Later, the differentiated macrophages were incubated with conditioned medium from H2122 PRMT6 knockout clone or parental cells for additional 24 h. Total RNA from the macrophages were isolated and qPCRs were performed for M2 macrophage marker as described in the methods. **, p< 0.01. F. Total RNA isolated from bone marrow-derived macrophages treated with the conditioned medium isolated from AECs isolated from PRMT6Tg; Sftpc-Cretm or PRMT6Tg control mice after tamoxifen treatment were employed in qPCRs for pro-angiogenic gene markers as described in the methods. *, p< 0.05; versus control. G. Total RNA isolated from lungs of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatment (20 weeks) were subjected to qPCR analysis of pro-angiogenic gene markers as described in the methods. **, p< 0.01 *, p< 0.05; versus control. H. Lung tumor sections of PRMT6Tg; Sftpc-Cretm and PRMT6Tg control mice after tamoxifen and urethane treatment (20 weeks) were subjected to indirect immunofluorescence staining with anti-CD31 antibodies as described in the methods. Scale bar: 10 μM.
To determine if the secreted factors from PRMT6 overexpressing alveolar epithelial cells (AECs) induce macrophage polarization towards the M2 phenotype, we isolated and cultured alveolar epithelial cells (AECs) from PRMT6Tg; Sftpc-Cretm and PRMT6Tg mice treated with tamoxifen only (Fig. 7D). Later, conditioned medium from AECs were collected. We also cultured bone marrow-derived monocytes in the presence of macrophage colony stimulating factor (mCSF) to promote macrophage maturation (M0). Treatment of mouse bone marrow-derived macrophages (M0) with the conditioned medium from PRMT6 overexpressing AECs resulted in an increase in M2 markers (Arginase 1 and IL10), and a decrease in M1 marker (TNFα), when compared to control conditioned medium treated macrophages (Fig. 7D). As a complementary strategy, we employed conditioned medium collected from H2122 PRMT6 knockout clones (Fig. 3) on THP1 monocytic cells-derived macrophages (5). Macrophages treated with the conditioned medium collected from PRMT6 knockout 2122 clone showed a significant reduction in M2 macrophage polarization as detected by Mannose receptor 1 (MRC1) expression when compared to macrophages treated with the conditioned medium collected from parental H2122 cells (Fig. 7E). These observations indicate that PRMT6 induced paracrine signaling products promote the alternate activation of macrophages.
Alternate activation of TAMs is known to promote tumor progression by supporting tumor neo-angiogenesis (36). To determine whether PRMT6-stimulated M2 macrophage polarization promotes macrophage angiogenic potential, we measured the levels of vascular endothelial growth factor (VEGF) and matrix metalloproteinase 9 (MMP9), which are the markers of angiogenesis. Mouse bone marrow-derived macrophages treated with the conditioned medium from PRMT6 overexpressing AECs, which were M2 polarized (Fig. 7C), showed a significant increase in the expression of VEGF and MMP9 transcripts (Fig. 7F) when compared to the control conditioned medium treated macrophages (Fig. 7E). Furthermore, we also detected increased VEGF and MMP9 expression in the lung tumors of PRMT6 overexpressing mice post-tamoxifen and urethane treatment (Fig. 7G), suggesting increased angiogenesis in the tumors of PRMT6 overexpressing mice. In agreement with the increased expression of VEGF, there were more mature blood vessels in the tumors of PRMT6Tg; Sftpc-Cretm mice when compared to the tumors of control (PRMT6Tg), as determined by CD31 immunostaining (Fig. 7H). Collectively, these findings indicate a new role for PRMT6 in potentiating lung tumor progression and tumor angiogenesis via the alternate activation of TAMs.
Discussion
Despite several studies highlighting the importance of PRMTs in multiple tumor types (6-11), pharmacologic inhibitors of PRMTs have not reached the clinic. This is in part due to the current strategies aimed at targeting the catalytic domains of PRMTs, which may not work for PRMTs that display narrow substrate specificities. Therefore, the identification of new roles, apart from their classical catalytic roles, of PRMTs in tumor development would be beneficial to develop new therapeutics with increased specificity. To our knowledge, PRMT6, which is overexpressed in lung cancer, is the only member of arginine methyl transferase family that displays narrow substrate specificity (15). These observations hint at previously undisclosed non-catalytic functions of PRMT6 in lung tumor development.
In the current study, we made several observations that will further our knowledge of lung tumor progression. First, to closely mimic PRMT6 amplification events in human lung adenocarcinoma, we created a lung targeted PRMT6 gain-of-function (GOF) mouse model. Induction of PRMT6 expression resulted in hyper-proliferation of the alveolar epithelium, as determined by ki-67 staining (Fig. 1B, 1C). Consistent with our observations, a recent report demonstrated hyper-branching of the mammary glands and increased ki-67 staining in the breast epithelium of PRMT6 GOF transgenic mice (18). These observations are indicative of a genetic predisposition of PRMT6 GOF mice to increased carcinogen-induced lung tumor growth. Indeed, when we interrogated the effects of a common oncogenic driver in lung cancer e.g., mutant KRas in PRMT6 GOF mice we observed a striking increase in lung tumor number in mouse tumor models (Fig. 2). We also observed differences in tumor histology; while control mice mostly developed microscopic adenomas, the PRMT6Tg; Sftpc-Cretm mice on the other hand developed macroscopic aggressive lung adenocarcinomas (Fig. 2B). Furthermore, the tumors of PRMT6Tg; Sftpc-Cretm mice (experimental) also showed more mature blood vessels, an indicative of neo-angiogenesis (Fig. 2D). Taken together, we show that PRMT6 is an important contributor to tumor progression and metastasis in mutant KRas-driven lung tumors. Of note this mouse model can also serve as an important pre-clinical tool to evaluate the efficacy of PRMT6 inhibitors.
Second, we have also identified a PPI between PRMT6 and ILF2, which contributes to lung tumor development (Fig. 4B, 4C). We also demonstrate that ILF2 is not a catalytic substrate of PRMT6 (Fig. 4D). Similar role for PRMT6-mediated regulation of proteins independent of arginine methylation has been reported earlier (15). These findings suggest that PRMT6 displays non-catalytic functions, probably as a scaffold that contributes to lung tumor growth. However, how PRMT6 recruits, organizes, and regulates their binding partners in molecular terms awaits further study.
Third, ILF2 is a known regulator of T-cell expression of cytokines (31, 37-39). By employing cytokine arrays, we have identified macrophage migration inhibitory factor (MIF), a pro-inflammatory cytokine, as a downstream target of ILF2. We also demonstrate that the pro-proliferative effects of PRMT6-ILF2 signaling axis were MIF-dependent (Fig. 6J-L). However, the other known functions of ILF2 in DNA metabolism (40), transcription (41), RNA export (42), and microRNA biogenesis (24) in lung cancer development remains to be discerned.
Fourth, we have identified that PRMT6 promotes lung tumor growth by reprogramming macrophages to a tumor promoting (M2) phenotype. Earlier studies highlighted an important role for MIF in alternate activation of macrophages to a tumor-promoting phenotype (36). In this study, we have identified that TAMs in PRMT6Tg; Sftpc-Cretm mice (post-tamoxifen + urethane) were observed to be alternatively activated to M2 phenotype (Fig. 7). We also show that the PRMT6-ILF2 axis as a novel regulator of MIF expression (Fig. 6). Taken together, these observations suggest that PRMT6-ILF2-MIF signaling axis contributes to the alternate activation of TAMs during lung adenocarcinoma development. However, more detailed studies employing MIF neutralizing antibodies and/or inhibitors are needed to fully demonstrate the new paradigm, which is clearly a major contributing factor for immunosuppression and neo-angiogenesis (Fig. 7H) during lung tumor progression. Furthermore, the PRMT6 GOF lung tumor model can also serve as a pre-clinical tool for the investigation of immunotherapy efficacy and function.
In conclusion, we have identified a unique role for PRMT6/ILF2/MIF signaling axis in promoting lung tumor growth. We also demonstrate that PRMT6 promotes lung tumor growth via its scaffold functions, but not its catalytic function (Fig. 4), offering new therapeutic opportunities for the development of inhibitors of PRMT6 and ILF2 heterodimerization as anti-neoplastic agents. Furthermore, PRMT6 is the only methyl transferase that displays automethylation, which is critical for its stability and regulation (43). Another therapeutic approach would be to target the PRMT6 automethylation. Therapeutic targeting based on these unique features e.g., scaffold and/or automethylation would not only provide us novel drugs to treat lung cancers, but also provide us new tools to study the role of PRMT6 in alternate activation of macrophages.
Supplementary Material
Implications:
This is the first study to demonstrate an in vivo role for protein arginine methyl transferase 6 in lung tumor progression via the alternate activation of tumor-associated macrophages.
Acknowledgements
We thank UIC genome editing core (GEC) for designing PRMT6 guide RNAs, Research Histology and Tissue Imaging core at UIC Research Resources Center for IHC staining of lung tumor FFPE sections with PRMT6 antibody, and UIC Biorepository for providing human lung tumor biopsy tissues. We thank Biological Mass Spectrometry Core, University of Colorado, Denver for their help in in-gel tryptic digestion and protein identification. We also would like to thank CerGentis for TLA analysis. We thank Dr. Juel Chowdhury for spearman rank correlation analysis of lung cancer TCGA datasets. This study was supported in part by A Breath of Hope Lung Foundation (ABOHLF) fellowship to RKB, by a Merit Award from the U.S. Department of Veterans Affairs, and NIH grant R01CA138528 to RAW. MTB is supported by NIH grant R01GM126421. VG is supported from a grant from Bears Care.
Footnotes
Conflict of Interest: M.T.B is a co-founder of EpiCypher and V.G is the founder of Adhaere Pharmaceuticals. The remaining authors declare that there is no conflict of interest.
References:
- 1.Herbst RS, Heymach JV, Lippman SM. Lung cancer. The New England journal of medicine. 2008;359:1367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA: a cancer journal for clinicians. 2008;58:71–96. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nature reviews Cancer. 2013;13:37–50. [DOI] [PubMed] [Google Scholar]
- 4.Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007;120:4243–6. [DOI] [PubMed] [Google Scholar]
- 5.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avasarala S, Van Scoyk M, Karuppusamy Rathinam MK, Zerayesus S, Zhao X, Zhang W, et al. PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J Biol Chem. 2015;290:13479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin RM, Morettin A, Cote J. Role of PRMTs in cancer: Could minor isoforms be leaving a mark? World journal of biological chemistry. 2014;5:115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–6. [DOI] [PubMed] [Google Scholar]
- 9.Le Romancer M, Treilleux I, Leconte N, Robin-Lespinasse Y, Sentis S, Bouchekioua-Bouzaghou K, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–21. [DOI] [PubMed] [Google Scholar]
- 10.Mathioudaki K, Papadokostopoulou A, Scorilas A, Xynopoulos D, Agnanti N, Talieri M. The PRMT1 gene expression pattern in colon cancer. Br J Cancer. 2008;99:2094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011;128:562–73. [DOI] [PubMed] [Google Scholar]
- 12.Halby L, Marechal N, Pechalrieu D, Cura V, Franchini DM, Faux C, et al. Hijacking DNA methyltransferase transition state analogues to produce chemical scaffolds for PRMT inhibitors. Philos Trans R Soc Lond B Biol Sci. 2018;373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha SH, Owens EA, Feng Y, Yang Y, Xie Y, Tu Y, et al. Synthesis and evaluation of carbocyanine dyes as PRMT inhibitors and imaging agents. European journal of medicinal chemistry. 2012;54:647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Jiang H, Jin J, Xie Y, Chen Z, Zhang H, et al. Development of Potent Type I Protein Arginine Methyltransferase (PRMT) Inhibitors of Leukemia Cell Proliferation. Journal of medicinal chemistry. 2017;60:8888–905. [DOI] [PubMed] [Google Scholar]
- 15.Fisk JC, Zurita-Lopez C, Sayegh J, Tomasello DL, Clarke SG, Read LK. TbPRMT6 is a type I protein arginine methyltransferase that contributes to cytokinesis in Trypanosoma brucei. Eukaryotic cell. 2010;9:866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao J, Di Lorenzo A, Lin K, Lu Y, Zhong Y, Sebastian MM, et al. Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res. 2019;79:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keith RL, Karoor V, Mozer AB, Hudish TM, Le M, Miller YE. Chemoprevention of murine lung cancer by gefitinib in combination with prostacyclin synthase overexpression. Lung Cancer. 2010;70:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.To MD, Rosario RD, Westcott PM, Banta KL, Balmain A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene. 2013;32:4028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boisvert FM, Cote J, Boulanger MC, Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol Cell Proteomics. 2003;2:1319–30. [DOI] [PubMed] [Google Scholar]
- 24.Higuchi T, Todaka H, Sugiyama Y, Ono M, Tamaki N, Hatano E, et al. Suppression of MicroRNA-7 (miR-7) Biogenesis by Nuclear Factor 90-Nuclear Factor 45 Complex (NF90-NF45) Controls Cell Proliferation in Hepatocellular Carcinoma. J Biol Chem. 2016;291:21074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Q, He X, Qiu X, Liu X, Sun G, Guo J, et al. Expression of NF45 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:10149–57. [DOI] [PubMed] [Google Scholar]
- 26.Ni S, Zhu J, Zhang J, Zhang S, Li M, Ni R, et al. Expression and clinical role of NF45 as a novel cell cycle protein in esophageal squamous cell carcinoma (ESCC). Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:747–56. [DOI] [PubMed] [Google Scholar]
- 27.Shamanna RA, Hoque M, Pe’ery T, Mathews MB. Induction of p53, p21 and apoptosis by silencing the NF90/NF45 complex in human papilloma virus-transformed cervical carcinoma cells. Oncogene. 2013;32:5176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wan C, Gong C, Ji L, Liu X, Wang Y, Wang L, et al. NF45 overexpression is associated with poor prognosis and enhanced cell proliferation of pancreatic ductal adenocarcinoma. Mol Cell Biochem. 2015;410:25–35. [DOI] [PubMed] [Google Scholar]
- 29.Wen X, Liu X, Mao YP, Yang XJ, Wang YQ, Zhang PP, et al. Long non-coding RNA DANCR stabilizes HIF-1alpha and promotes metastasis by interacting with NF90/NF45 complex in nasopharyngeal carcinoma. Theranostics. 2018;8:5676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iberg AN, Espejo A, Cheng D, Kim D, Michaud-Levesque J, Richard S, et al. Arginine methylation of the histone H3 tail impedes effector binding. J Biol Chem. 2008;283:3006–10. [DOI] [PubMed] [Google Scholar]
- 31.Zhao G, Shi L, Qiu D, Hu H, Kao PN. NF45/ILF2 tissue expression, promoter analysis, and interleukin-2 transactivating function. Exp Cell Res. 2005;305:312–23. [DOI] [PubMed] [Google Scholar]
- 32.Lin S, Wang M, Liu X, Zhu W, Guo Y, Dai Z, et al. Association of genetic polymorphisms in MIF with breast cancer risk in Chinese women. Clinical and experimental medicine. 2017;17:395–401. [DOI] [PubMed] [Google Scholar]
- 33.Nobre CC, de Araujo JM, Fernandes TA, Cobucci RN, Lanza DC, Andrade VS, et al. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathology oncology research : POR. 2017;23:235–44. [DOI] [PubMed] [Google Scholar]
- 34.Rafiei S, Gui B, Wu J, Liu XS, Kibel AS, Jia L. Targeting the MIF/CXCR7/AKT Signaling Pathway in Castration-Resistant Prostate Cancer. Mol Cancer Res. 2019;17:263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36:2095–104. [DOI] [PubMed] [Google Scholar]
- 36.Yaddanapudi K, Putty K, Rendon BE, Lamont GJ, Faughn JD, Satoskar A, et al. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol. 2013;190:2984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi H, Hu YH, Xiao ZZ, Sun L. Nuclear factor 45 of tongue sole (Cynoglossus semilaevis): evidence for functional differentiation between two isoforms in immune defense against viral and bacterial pathogens. Developmental and comparative immunology. 2014;42:125–31. [DOI] [PubMed] [Google Scholar]
- 38.Kiesler P, Haynes PA, Shi L, Kao PN, Wysocki VH, Vercelli D. NF45 and NF90 regulate HS4-dependent interleukin-13 transcription in T cells. J Biol Chem. 2010;285:8256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi L, Qiu D, Zhao G, Corthesy B, Lees-Miller S, Reeves WH, et al. Dynamic binding of Ku80, Ku70 and NF90 to the IL-2 promoter in vivo in activated T-cells. Nucleic Acids Res. 2007;35:2302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchesini M, Ogoti Y, Fiorini E, Aktas Samur A, Nezi L, D’Anca M, et al. ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell. 2017;32:88–100 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Belshan M. NF45 and NF90 Bind HIV-1 RNA and Modulate HIV Gene Expression. Viruses. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nie Y, Ding L, Kao PN, Braun R, Yang JH. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol. 2005;25:6956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singhroy DN, Mesplede T, Sabbah A, Quashie PK, Falgueyret JP, Wainberg MA. Automethylation of protein arginine methyltransferase 6 (PRMT6) regulates its stability and its anti-HIV-1 activity. Retrovirology. 2013;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Vree PJ, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJ, et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nature biotechnology. 2014;32:1019–25. [DOI] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avasarala S, Bikkavilli RK, Van Scoyk M, Zhang W, Lapite A, Hostetter L, et al. Heterotrimeric G-protein, Galpha16, is a critical downstream effector of non-canonical Wnt signaling and a potent inhibitor of transformed cell growth in non small cell lung cancer. PLoS One. 2013;8:e76895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borowicz S, Van Scoyk M, Avasarala S, Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK, et al. The soft agar colony formation assay. Journal of visualized experiments : JoVE. 2014:e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiser-Evans MC, Wang XQ, Amin J, Van Putten V, Choudhary R, Winn RA, et al. Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res. 2009;69:1733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, et al. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am J Pathol. 2010;176:2972–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.