

Abstract
TP53 alterations are frequent relapse‐acquired mutations in childhood acute lymphoblastic leukemia (ALL). The present study evaluated the clinical significance of relapsed childhood ALL in Taiwan. Diagnostic and/or relapsed bone marrow or peripheral blood was obtained from 111 children with relapsed ALL who were initially treated by using Taiwan Pediatric Oncology Group (TPOG) ALL protocols from January 1997 to May 2018. Mutations were detected by PCR and sequencing, as well as by multiplex ligation‐dependent probe amplification to detect copy number alterations. Copy number and/or sequence alterations of TP53 were detected in 29% (28 of 98) and in 46% (6 of 13) of patients with relapsed B‐cell and T‐cell ALL, respectively. This incidence was much higher than that in several similar studies conducted in Caucasian populations. Seventy percent of all TP53 alterations were gained at relapse in 67 matched samples by back‐tracking matched diagnostic samples. TP53 alterations were associated with lower 5‐year event‐free survival (EFS) and overall survival (OS) rates (P = .013 and P = .0002, respectively). Multivariate analysis confirmed the prognostic significance of TP53 alterations. Forty‐five patients received hematopoietic stem‐cell transplantations post‐relapse. Patients with TP53 alterations (14/45) had inferior 5‐year EFS and OS than patients without TP53 alterations after transplantation (P = .002 and P = .001, respectively). The significance of these TP53 alterations for patients who received transplantations was confirmed by multivariate analysis. In conclusion, TP53 alterations were enriched and useful as prognostic markers in relapsed childhood ALL.
Keywords: MLPA, relapse‐acquired mutations, relapsed childhood ALL, Taiwan, TP53 alterations
Relapsed pediatric ALL patients with TP53 alterations had inferior 5‐year EFS and OS. TP53 alterations were enriched and useful as prognostic markers in relapsed childhood ALL.

1. INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy, accounting for approximately 25% of childhood malignancies.1 Overall 5‐year event‐free survival (EFS) rate for this disease now exceeds 80% in developed countries;2, 3, 4 however, 10%‐20% of patients succumb to recurrences. The prognosis for relapsed ALL is dismal, even with aggressive salvage strategies.5 Thus, relapsed ALL remains a leading cause of cancer‐related deaths in children. Current comprehensive genome sequencing of triplicate diagnostic, remission, and relapsed samples from childhood ALL suggests that most relapsed patients have new, secondary genetic alterations or acquired lesions that likely occurred in a minor clone after the original diagnosis of ALL. One of the pathways enriched in recent relapse pair samples is TP53 mutations.6
TP53 mutations are infrequent (2%‐3%) at initial diagnosis and have not been found to be an important marker for prognosis or in the prediction of treatment response in most childhood ALL clinical trials.7, 8 However, TP53 alterations are frequently found in low‐hypodiploidy ALL, with a somatic mutation rate of up to 90%.9 The incidence of TP53 alterations is higher in relapsed childhood ALL samples than in diagnostic samples.10, 11 Back‐tracking studies showed that most TP53 genetic alterations were relapse‐specific,10 which was confirmed through whole exome sequencing.6
Numerous prognostic markers predict disease outcomes or relapse in newly diagnosed childhood ALL, especially B‐cell ALL.1, 3, 12, 13 TP53 alterations are important genetic events, but poor prognostic markers for relapsed childhood ALL in clinical trials.10, 11 After relapse, minimal residual disease measurements might be the only prognostic marker after reinduction of chemotherapy to predict survival after stem‐cell transplantation.14 TP53 relapse‐specific mutations were observed in approximately 10% of relapsed Caucasian patients.10, 11 However, there are currently few prognostic markers for relapsed patients that indicate a successful return to remission.5
The actual incidence of TP53 alterations in childhood ALL in Taiwan remains unknown. Therefore, we profiled the frequency of genetic alterations and impact of TP53 alterations on survival rate of relapsed childhood ALL patients treated with Taiwan Pediatric Oncology Group (TPOG) ALL protocols in a medical center in Taiwan.
2. MATERIALS AND METHODS
2.1. Patients and protocols
Bone marrow or peripheral blood samples taken at diagnosis, relapse, and/or clinical remission were available for 111 children with relapsed ALL; 98 with B‐cell ALL, and 13 with T‐cell ALL) from January 1997 to December 2015. These patients were enrolled in the TPOG ALL 93, ALL 97 very high risk (VHR), ALL 2002, or ALL 2013 protocols; details of these protocols have been published elsewhere.15 The TPOG ALL 2013 was modified from ALL 2002 by incorporating minimal residual disease (MRD) measurements to stratify treatment. After disease relapse, physicians used different protocols, including the Berlin Frankfurt Münster (BFM) relapse protocol, upgrade to TPOG VHR protocols or FLAG.5, 16, 17 This study was approved by the Institutional Review Board of National Taiwan University Hospital and all participants or their guardians provided written informed consent in accordance with the Declaration of Helsinki.
2.2. Genomic DNA extraction
Lymphoblasts were purified from bone marrow or peripheral blood specimens by the Ficoll‐Paque centrifugation method according to the manufacturer’s instructions (GE Healthcare). Genomic DNA was extracted from leukemic cells using standard phenol/chloroform‐based methods. Briefly, 1 million cells were lysed in 10 mmol/L Tris‐HCl, 10 mmol/L NaCl, 10 mmol/L EDTA, 20 μg proteinase K, and 0.5% SDS by incubating at 37°C for 16 hours. Total RNA was further removed by adding 500 μg PureLink RNase A (Invitrogen) and incubating for 10 minutes at 37°C. An equal volume of phenol‐chloroform‐isopropanol (25:24:1) was added to lysates and mixed by shaking vigorously, followed by centrifugation at 16 100 g at 4°C for 5 minutes. The upper aqueous phase was transferred to a fresh tube; genomic DNA was then precipitated by adding 2× volume −80°C 100% ethanol. The DNA pellet was washed with 75% ethanol and rehydrated with Tris‐EDTA buffer. Concentration of DNA was determined using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific).
2.3. Mutation analysis of TP53
For TP53 (NG_017013) sequence analysis, coding regions were amplified using Phusion Hot Start II High‐Fidelity PCR Master Mix (Thermo Fisher Scientific). PCR was carried out as follows: 98°C for 30 seconds, then 38 cycles of 98°C for 30 seconds, annealing for 30 seconds and 72°C for 30 seconds, followed by a final extension at 72°C for 5 minutes. PCR products were confirmed using 2% agarose gel electrophoresis. We cleaned up the remaining PCR products by incubating with a reaction mixture of FastAP Thermosensitive Alkaline Phosphatase and exonuclease I (both Thermo Fisher Scientific) for 15 minutes at 37°C followed by 85°C for 15 minutes to inactivate the reaction. Sanger sequencing was done using an ABI 3730XL DNA analyzer (Applied Biosystems). The sequencing results aligned to the NCBI GenBank entry NM_000546 using SnapGene 4.1.3 software. Details of PCR, sequencing primers, and annealing temperatures are provided in Table S1.
2.4. Multiplex ligation‐dependent probe amplification
To detect structural alterations of TP53, multiplex ligation‐dependent probe amplification (MLPA) reactions were carried out according to the manufacturer’s instructions. The MRC‐Holland SALSA MLPA probe mix P056‐C1 kit (MRC Holland) was used. PCR fragments were separated by capillary electrophoresis on a Life Technologies 3500 genetic analyzer (Thermo Fisher Scientific). MLPA data were analyzed using Coffalyser.Net v.140721.1958 (MRC Holland). Relative copy numbers were determined after normalization of peaks against controls. Values between 0.75 and 1.3 were considered within the normal range. Values below 0.75 or above 1.3 implied a deletion or gain, respectively. Values below 0.25 indicated biallelic deletion.
2.5. Statistical analyses
Equality between categoric and continuous parameters was tested with the chi‐squared or Fisher’s exact tests. The Kaplan‐Meier method was used to estimate survival curves associated with TP53 alterations for relapsed ALL. The log‐rank test compared different survival curves between TP53 alterations and wild‐type ALL‐relapsed patients. EFS after first relapse was defined as the time from first relapse to any adverse events. OS was defined as time from diagnosis to death. Patients who did not suffer any adverse events within the follow‐up period were censored. EFS of patients with no response to chemotherapy (refractory), death, and second relapse in induction was set to 0. Statistical software SAS 9.4 (SAS Institute) was used for all data analysis. Univariate and multivariate Cox regression analyses were carried out to evaluate hazard ratios (HR) and 95% confidence intervals (CI) of risk factors. We inspected the associations between the presence of alterations and the value of clinical parameters. MRD was not included in the parameters, attributable to inadequate data. All tests were two‐sided, and values of P < .05 were considered significant.
3. RESULTS
3.1. Frequency and type of TP53 alterations in relapsed childhood ALL
Detailed clinical characteristics of the studied cohort are described in Table 1. To obtain the associations of TP53 alterations with different clinical, cytogenetic, and treatment parameters present at the time of relapse, these variables between patients with or without TP53 alterations and between TP53 alteration subgroups were compared statistically (Table 2). Hypodiploidy was enriched in TP53 alterations. However, there were more patients with KMT2A rearrangements and T‐cell ALL in relapsed cases with TP53 alterations than patients with ETV6‐RUNX1, hypodiploidy, and BCR‐ABL1.
Table 1.
TP53 alterations in patients with first relapse of childhood acute lymphoblastic leukemia
| Patient ID | Type | MLPA results TP53 locus | TP53 mutation by Sanger | Exon | Coding DNA position | Base change | Codon number | AA change | Origin of alteration | BMT | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 196 | B | ND | Missense | 5 | 523 | G → A | 175 | R → H | Diagnosis | Yes | Deceased |
| 331 | B | ND | Missense | 8 | 800 | G → C | 267 | R → P | Relapse | No | Deceased |
| 488 | T | ND | Missense | 7 | 733 | G → A | 245 | G → S | Relapse | No | Deceased |
| 549 | B | ND | Missense | 7 | 725 | G → A | 242 | C → Y | Relapse | Yes | Deceased |
| 656 | B | ND | Missense | 7 | 743 | G → A | 248 | R → Q | Relapse | Yes | Deceased |
| 306 | B | NA | Missense | 7 | 743 | G → A | 248 | R → Q | Somatic | No | Deceased |
| 328 | B | NA | Missense | 8 | 817 | C → T | 273 | R → C | Other | No | Deceased |
| 438 | B | NA | Insertion | 8 | 844‐845 | 6nt insertion | 281‐282 | LV insertion | Relapse | Yes | Deceased |
| 463 | B | NA | Missense | 5 | 509 | C → T | 170 | T → M | Relapse | No | Deceased |
| 508 | B | NA | Missense | 6 | 583 | A → T | 195 | I → F | Germline | No | Deceased |
| 9001 | B | NA | Missense | 8 | 844 | C → T | 282 | R → W | Other | No | Deceased |
| 85 | B | ND |
Missense; Missense |
5; 8 |
527; 818 |
G → A; G → A |
176; 273 |
C → Y; R → H |
Relapse; Relapse |
Yes | Deceased |
| 612 | B | ND |
Missense; Missense |
7; 8 |
743; 799 |
G → A; C → G |
248; 267 |
R → Q; R → G |
Relapse; Relapse |
No | Deceased |
| 682 | B | ND |
Missense; Missense |
7; 8 |
743; 817 |
G → A; C → T |
248; 273 |
R → Q; R → C |
Relapse; Relapse |
Yes | Deceased |
| 453 | T | NA | Missense | 7 | 743 | G → C | 248 | R → P | Relapse | Yes | Deceased |
| 522 | B | NA |
Missense; Missense |
5; 7 |
396; 743 |
G → T; G → A |
132; 248 |
K → N; R → Q |
Relapse; Relapse |
No | Deceased |
| 9002 | B | NA | Missense | 7 | 743 | G → A | 248 | R → Q | Other | No | Deceased |
| 915 | T | NA |
Missense; Missense; Missense |
5; 5; 7 |
404; 475; 731 |
G → A; G → C; G → C |
135; 159; 244 |
C → Y; A → P; G → A |
Somatic; Somatic; Somatic |
Yes | Deceased |
| 748 | B | ND |
Missense; Missense; Missense |
5; 8; 8 |
473; 791; 811 |
G → A; T → G; G → C |
158; 264; 271 |
R → H; L → R; E → Q |
Relapse; Relapse; Relapse |
No | Deceased |
| 353 | T | 11 | ND | Relapse | No | Deceased | |||||
| 180 | B | 2‐11 | ND | Other | Yes | Deceased | |||||
| 657 | B | 2‐11 | ND | Somatic | No | Deceased | |||||
| 753 | B | 2‐11 | ND | Other | No | Deceased | |||||
| 863 | B | 2‐11 | ND | Other | No | Deceased | |||||
| 368 | B | 2‐11 | ND | Other | Yes | Deceased | |||||
| 649 | B | 2‐11;10‐11 | ND | Other | Yes | Deceased | |||||
| 50 | B | 2‐11 | Missense | 7 | 742 | C → T | 248 | R → W | Diagnosis | No | Living |
| 342 | T | 2‐11 | Missense | 7 | 742 | C → T | 248 | R → W | Relapse | No | Deceased |
| 630 | B | 2‐11 | Missense | 5 | 524 | G → A | 175 | R → H | Relapse | Yes | Deceased |
| 774 | B | 2‐11 | Frameshift | 10 | 1022 | 1nt deletion | 341 | Frameshift | Diagnosis | No | Living |
| 845 | B | 2‐11 | Nonsense | 5 | 158 | G → A | 53 | W→* | Diagnosis | No | Deceased |
| 641 | B | 2‐11 | Missense | 5 | 482 | C → A | 161 | A → D | Relapse | Yes | Deceased |
| 162 | B | 2‐11 |
Indels; Indels |
8; 8 |
818‐843; 821‐863 |
25nt deletion with 1nt insertion; 41nt deletion with 6nt insertion |
274‐281; 274‐288 |
VCACPGRD deletion; Frameshift |
Somatic; Somatic |
Yes | Deceased |
| 478 | T | 2‐11 |
Missense; Nonsense |
3; 4 |
91; 310 |
G → A; C → T |
31; 104 |
V → I; Q→* |
Relapse; Germline |
No | Deceased |
Abbreviations: AA, amino acid; BMT, bone marrow transplantation; Indels, insertion with deletion; MLPA, multiplex ligation‐dependent probe amplification; NA, data not available; ND, not detected; nt, nucleotide.
Table 2.
Clinical characteristics, genetics, and outcome of acute lymphoblastic leukemia relapsed patients with TP53 alterations
| Characteristics | Analysis of patients with altered vs wild‐type TP53 | Analysis of patients with mutated vs deleted vs mutated and deleted vs wild‐type TP53 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild‐type | Alteration‐type | P‐value | Wild‐type | Exclusive mutation | Exclusive deletion | Mutation and deletion | P‐value | |||||||
| N | % | N | % | N | % | N | % | N | % | N | % | |||
| All patients | 77 | 100 | 34 | 100 | 77 | 100 | 19 | 100 | 7 | 100 | 8 | 100 | ||
| Gender | ||||||||||||||
| Male | 46 | 59.7 | 22 | 64.7 | .677 | 46 | 59.7 | 13 | 68.4 | 3 | 42.9 | 6 | 75.0 | .597 |
| Female | 31 | 40.3 | 12 | 35.3 | 31 | 40.3 | 6 | 31.6 | 4 | 57.1 | 2 | 25.0 | ||
| Age at relapse, years | ||||||||||||||
| <5 | 12 | 15.58 | 9 | 26.47 | .388 | 12 | 15.6 | 4 | 21.1 | 3 | 42.9 | 2 | 25 | .511 |
| ≥5 and <10 | 22 | 28.57 | 9 | 26.47 | 22 | 28.6 | 7 | 36.8 | 1 | 14.2 | 1 | 12.5 | ||
| ≥10 | 43 | 55.84 | 16 | 47.06 | 43 | 55.8 | 8 | 42.1 | 3 | 42.9 | 5 | 62.5 | ||
| Time to relapse | ||||||||||||||
| Very early | 34 | 44.2 | 19 | 55.9 | .022 | 34 | 44.2 | 11 | 57.9 | 4 | 57.1 | 4 | 50.0 | .105 |
| Early | 8 | 10.4 | 8 | 23.5 | 8 | 10.4 | 5 | 26.3 | 2 | 28.6 | 1 | 12.5 | ||
| Late | 35 | 45.5 | 7 | 20.6 | 35 | 45.5 | 3 | 15.8 | 1 | 14.3 | 3 | 37.5 | ||
| ALL subtypes | ||||||||||||||
| T‐ALL | 7 | 9.09 | 6 | 17.6 | .005 | 7 | 9.09 | 3 | 15.8 | 1 | 14.3 | 2 | 25.0 | .089 |
| TCF3‐PBX1 | 1 | 1.3 | 1 | 2.9 | 1 | 1.3 | 1 | 5.3 | 0 | 0.0 | 0 | 0.0 | ||
| 11q23 | 6 | 7.8 | 7 | 20.6 | 6 | 7.8 | 4 | 21.1 | 1 | 14.3 | 2 | 25.0 | ||
| Hyperdiploidy | 5 | 6.5 | 1 | 2.9 | 5 | 6.5 | 1 | 5.3 | 0 | 0.0 | 0 | 0.0 | ||
| BCR‐ABL1 | 5 | 6.5 | 0 | 0.0 | 5 | 6.5 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | ||
| ETV6‐RUNX1 | 6 | 7.8 | 2 | 5.9 | 6 | 7.8 | 2 | 10.5 | 0 | 0.0 | 0 | 0.0 | ||
| Hypodiploidy | 0 | 0.0 | 4 | 11.8 | 0 | 0.0 | 1 | 5.3 | 1 | 14.3 | 2 | 25.0 | ||
| Other | 47 | 61.0 | 13 | 38.2 | 47 | 61.0 | 7 | 36.8 | 4 | 57.1 | 2 | 25.0 | ||
| BMT | ||||||||||||||
| None | 46 | 59.7 | 20 | 58.8 | 1.000 | 46 | 59.7 | 11 | 57.9 | 4 | 57.1 | 5 | 62.5 | 1.000 |
| BMT | 31 | 40.3 | 14 | 41.2 | 31 | 40.3 | 8 | 42.1 | 3 | 42.9 | 3 | 37.5 | ||
Abbreviations: ALL, acute lymphoblastic leukemia; BMT, bone marrow transplantation.
Time to relapse: very early, within 18 months from diagnosis to first relapse of ALL; early, between 18 months and 30 months from diagnosis to first relapse of ALL; late, more than 30 months from diagnosis to first relapse of ALL.
TP53 alterations were identified in 34 patients (31%). Within the B‐cell ALL relapse group, 28 patients (29%) had TP53 gene alterations: six (21%) had deletions in the absence of a mutation (referred to as an “exclusive deletion”), 16 (57%) had mutation(s) in the absence of a deletion (referred to as an “exclusive mutation”), and six (21%) had mutation(s) and deletion(s). In T‐cell ALL patients, TP53 alterations were identified in six patients (46%): three had an exclusive mutation, one had an exclusive deletion, and two had mutation(s) and a deletion (Figure 1A). TP53 P72R, a known polymorphism, exon 1 alterations, and intronic deletions were not evaluated in this study.
Figure 1.
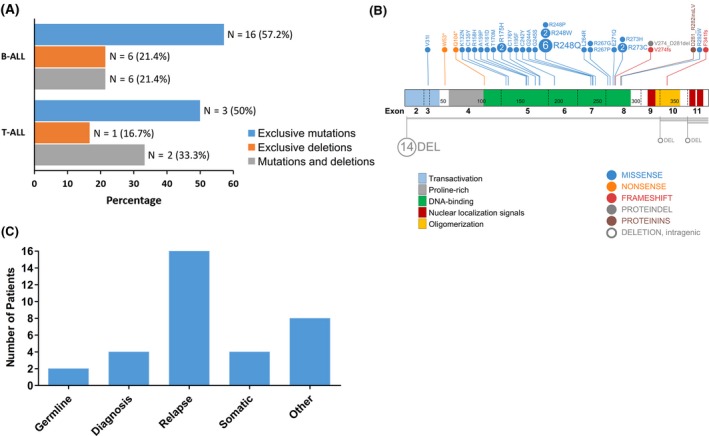
TP53 alterations in first relapse of childhood acute lymphoblastic leukemia (ALL). A, Frequency of TP53 alterations in patients with relapsed B‐cell and T‐cell ALL. B, Distribution of TP53 alterations detected by sequencing in relapsed B‐cell ALL and T‐cell ALL over TP53 coding regions. DNA‐binding domains involved in most TP53 sequence alterations. C, Retrospective copy number and sequence analysis of matched diagnostic and recurrent ALL samples from 67 patients with TP53 alterations at relapse. Bar chart shows the frequency of mutations at both stages of the disease
TP53 alterations are summarized in Table 1. There were 25 missense mutations, four small indels, two exonic deletions, and deletion of the entire TP53 gene among them. No mutations were detected in the remaining coding region in patients with an exclusive TP53 deletion. The most frequent TP53 mutations were detected in the DNA‐binding domain (exons 5‐8) (Figure 1B). In morphological remission samples, we identified missense mutations in two patients (478 and 508). There were eight patients with more than one TP53 alteration. There were six patients with multiple mutations, six with single mutations and single deletions, and two with single deletions and multiple mutations.
3.2. TP53 alterations in matched diagnosis and relapsed patients
Diagnostic and relapse samples were available for 69 patients. Matched‐initial ALL samples were compared with those from relapsed patients with TP53 alterations to trace whether TP53 alterations were persistent or acquired between initial diagnosis and first relapse. Approximately 70% of all mutations and deletions (16 of 24) were relapse‐specific (Figure 1C). One patient acquired an exclusive deletion, seven patients acquired an exclusive mutation, and three patients acquired both a mutation and a deletion at relapse. Five patients had relapse‐specific mutations, but we lacked additional material to check for deletions of TP53. Seven patients had somatic mutations of the TP53 gene at first diagnosis. A single patient had both a mutation and a deletion at initial ALL diagnosis and acquired an additional mutation during relapse.
3.3. Relationships between TP53 alterations and clinical characteristics and survival rate
Five‐year EFS and OS were analyzed for patients with and without TP53 alterations. Five‐year EFS and OS were inferior in patients with TP53 alterations (P = .013 and P = .0002, respectively) after relapse (Figure 2). Of the 69 patients with paired samples, 5‐year EFS and OS were inferior in patients with TP53 alterations (P = .06 and P = .018, respectively) (Figure 3); the significance of TP53 alterations on the inferior survival of relapsed B‐cell ALL was similar (Figure S1). In T‐cell ALL, patients with TP53 alterations had inferior 5‐year EFS and OS, although the number of cases in this subgroup was small (Figure S2). Patients with TP53 alterations tended toward reinduction chemotherapy failure (Fisher’s exact test, P‐values .09 for the ALL cohort and .07 for the paired samples). Forty‐five patients received bone‐marrow transplantation as a rescue procedure; for them, TP53 alterations correlated with lower 5‐year EFS and OS (Figure 4).
Figure 2.
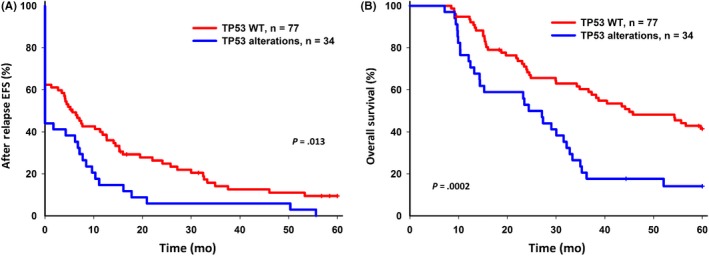
Five‐year event‐free survival (EFS) and overall survival (OS) of relapsed acute lymphoblastic leukemia (ALL) with TP53 alterations. A, 5‐year EFS for relapsed ALL with TP53 wild‐type (WT) and TP53 alterations. B, 5‐year OS for relapsed ALL with TP53 WT and TP53 alterations
Figure 3.
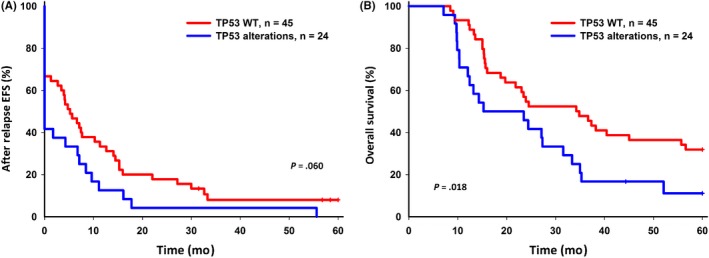
Five‐year event‐free survival (EFS) and overall survival (OS) of relapsed acute lymphoblastic leukemia (ALL) with TP53 alterations in 69 patients with paired samples. A, 5‐year EFS for relapsed ALL patients with TP53 wild‐type (WT) and TP53 alterations. B, 5‐year OS for relapsed ALL patients with TP53 WT and TP53 alterations
Figure 4.
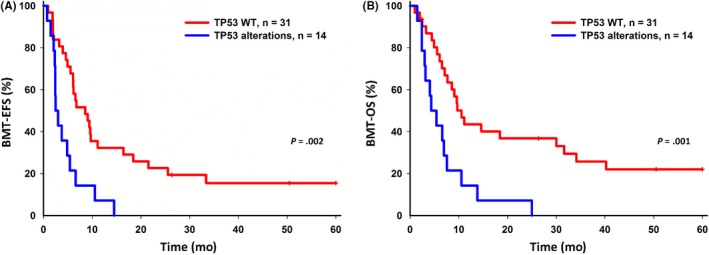
Comparison of bone marrow transplantation (BMT) therapy for 5‐year event‐free survival (EFS) and overall survival (OS) in relapsed acute lymphoblastic leukemia (ALL) patients with TP53 alterations. A, 5‐year EFS for relapsed ALL patients with TP53 wild‐type (WT) and TP53 alterations. B, 5‐year OS for relapsed ALL patients with TP53 WT and TP53 alterations
3.4. Multivariate analysis
Cox univariate regression model identified TP53 alterations (HR = 1.7; 95% CI: 1.12‐2.58, P = .0130) and age at onset of <1 year (HR = 2.34; 95% CI: 1.19‐4.58, P = .014) as the strongest predictive factors for an unfavorable 5‐year‐EFS. Patients who received bone marrow transplantation (BMT) presented with better 5‐year EFS (HR = 0.49; 95% CI: 0.32‐0.73, P = .001) than those who did not (Table 3). Multivariate Cox regression analysis showed that TP53 alterations (HR = 1.52; 95% CI: 0.99‐2.31, P = .054) resulted in marginal statistical significance. However, age at onset of <1 year (HR = 2.74; 95% CI: 1.36‐5.52, P = .005) and BMT (HR = 0.46; 95% CI: 0.30‐0.70, P = .0003) were clinically significant (Table 3).
Table 3.
Five‐year EFS and OS using univariate and multivariate survival analysis in relapsed pediatric ALL patients
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| 5‐year EFS | ||||||
| TP53 alterations | 1.70 | 1.12‐2.58 | .013 | 1.52 | 0.99‐2.31 | .054 |
| Gender | 1.44 | 0.95‐2.17 | .088 | 1.40 | 0.91‐2.14 | .127 |
| Age (<1 y) | 2.34 | 1.19‐4.58 | .014 | 2.74 | 1.36‐5.52 | .005 |
| ALL type | 1.15 | 0.63‐2.10 | .658 | 1.29 | 0.68‐2.44 | .430 |
| BMT | 0.49 | 0.32‐0.73 | .001 | 0.46 | 0.30‐0.70 | .0003 |
| 5‐year OS | ||||||
| TP53 alterations | 2.38 | 1.48‐3.84 | .0004 | 2.13 | 1.30‐3.47 | .003 |
| Gender | 1.41 | 0.87‐2.29 | .162 | 1.22 | 0.74‐2.00 | .438 |
| Age (<1 y) | 4.07 | 2.02‐8.22 | <.0001 | 3.94 | 1.90‐8.15 | .0002 |
| ALL type | 1.83 | 0.96‐3.48 | .065 | 2.24 | 1.13‐4.46 | .021 |
| BMT | 0.81 | 0.51‐1.30 | .389 | 0.71 | 0.44‐1.16 | .173 |
Abbreviations: ALL, acute lymphoblastic leukemia; BMT, bone marrow transplantation; CI, confidence interval; EFS, event‐free survival; HR, hazard ratio; OS, overall survival. Mutation type (reference = TP53 wild‐type); Gender (reference = female); Age (reference = ≥1 y); ALL type (reference = T‐ALL).
TP53 alterations (HR = 2.38; 95% CI: 1.48‐3.84, P = .0004) and age at onset of <1 year (HR = 4.07; 95% CI: 2.02‐8.22, P < .0001) were the strongest predictive factors for an unfavorable 5‐year OS. Clinical significance was noted in patients with TP53 alterations (HR = 2.13; 95% CI: 1.30‐3.47, P = .003) and age at onset of <1 year (HR = 3.94; 95% CI: 1.90‐8.15, P = .0002; Table 3). Patients who received stem‐cell transplantation presented with a better 5‐year OS; however, this trend did not reach statistical significance.
In patients who received BMT, TP53 alterations (HR = 2.93; 95% CI: 1.46‐5.87, P = .003) were identified as the strongest predictive factor for an unfavorable 5‐year EFS. TP53 alterations (HR = 2.88; 95% CI: 1.37‐6.07, P = .005) and age at onset of <1 year (HR = 3.05; 95% CI: 1.19‐7.79, P = .02) were clinically significant (Table S2).
4. DISCUSSION
Approximately 31% of relapsed childhood ALL cases harbor genetic alterations of TP53, and the TP53‐acquired alteration rate was 35% in sixty‐nine patients with diagnostic samples to backtrack mutations, which is higher than that observed in Caucasian populations. These alterations were associated with a higher rate of reinduction failure; most were relapse‐specific.
The incidence of TP53 alterations has increased in relapsed childhood ALL over the incidence in diagnostic samples, which has been documented in several large clinical trials. Hof et al10 sequenced TP53 hot‐spot exons 5‐8 (DNA‐binding domain) and deletion status in 265 first‐relapsed childhood ALL patients, whereas Irving et al11 analyzed a larger cohort of relapsed childhood B‐cell ALL patients. These large international trials reported similar frequencies of TP53 alterations for relapsed B‐cell ALL, 11%‐12%. Richter‐Pechańska et al18 conducted another cohort study of relapsed T‐cell ALL patients, reporting that 13% of samples from relapsed T‐cell ALL patients harbored TP53 alterations; only 1.4% of the initial diagnostic samples had TP53 alterations. Our case number is approximately half of those of these recent studies; however, the results are similar to those of previous studies using small case numbers, irrespective of B‐cell or T‐cell ALL.19, 20, 21 The reason for the higher incidence of TP53 alterations in relapsed samples remains unknown, although we did not use next‐generation sequencing to target the sequences. TP53 alterations might be identified in other pediatric cancers, such as medulloblastoma, osteosarcoma, and rhabdomyosarcoma. However, this information is not available from Taiwanese patients; there are no reports of the incidence of somatic, acquired TP53 alterations or germline variants in childhood ALL or other pediatric malignancies. This is the first report of acquired somatic mutations in relapsed ALL patients in Taiwan. Given the much higher incidence of acquired TP53 alterations in childhood ALL, a larger clinical trial to detail the true somatic and germline variants of TP53 in other pediatric malignancies, particularly in relapsed samples, is worthy of investigation in Taiwan.
The prognostic values of TP53 alterations have been documented in several large‐scale international clinical trials for relapsed childhood ALL. TP53 alterations are associated with nonresponse to chemotherapeutic treatment and poor EFS and OS.10 Most groups used time to relapse, relapse site, and cytogenetic alterations to define risk groups after recurrence.10 Patients with a TP53 alteration tended to have a very early relapse and should be classified and treated as high‐risk patients.11 Irrespective of the initial chemotherapy response, patients with TP53 alterations tend to have refractory disease, second relapses, and dismal outcomes. For relapsed T‐cell ALL, TP53 mutations were the best predictor of a second event in the cohort of Richter‐Pechańska.18 Almost all patients with TP53 alterations died of disease very soon after relapse, whereas patients without TP53 alterations survived. In this cohort, 47 patients received BMT; infant ALL patients apparently did not benefit from stem‐cell transplantation. Other treatment strategies might be indicated for this aggressive ALL.22, 23, 24 Our analyses suggest that somatic alteration of TP53 at relapse is a good predictor of transplantation outcomes.
We identified two patients with TP53 mutations from remission samples, suggesting that these mutations were not somatic, but of germline origin. TP53 germline mutations are enriched in low‐hypodiploidy patients and are common in Li‐Fraumeni syndrome.9 TP53 germline mutations result in a poorer outcome than other common subtypes of childhood ALL, such as ETV6‐RUNX1 and hyperdiploidy. A recent study showed TP53 germline variants (not common mutations) also contribute to significant clinical outcomes.25 Excluding the low hypodiploidy subtype, which occurred in close relationship with TP53 germline mutations, children with TP53 pathogenic variants still experienced significantly worse outcomes than those without.
There have also been studies investigating the significance of TP53 alterations in diagnostic ALL samples. Such alterations were profiled in B‐cell ALL by Forero‐Castro et al26 They reported that patients with TP53 alterations had lower 5‐year OS and EFS and higher relapse rates after multivariate analysis. In a study of 625 ALL patients by Stengel et al,27 overall TP53 mutation incidence was 16%, increasing with age. In addition to the low hypodiploidy subtype, TP53 mutations were also enriched in ALL with MYC rearrangements, recently classified as a subtype of B‐cell ALL.28 Stengel et al used next‐generation sequencing to profile the TP53 mutations. In addition to low hypodiploidy, high TP53 mutation loads also correlated with high hyperdiploidy and complex karyotypes. B‐cell ALL had a higher mutation load than T‐cell ALL. Median OS was significantly shorter for patients with TP53 mutations than for patients with wild‐type TP53.27 Although the clinical significance of diagnostic TP53 alterations requires validation in other childhood ALL clinical trials, TP53 alterations, including diagnostic, germline, and relapse alterations, might have clinical significance in childhood ALL and, therefore, warrant identification in future ALL clinical trials.
Although TP53 alterations have clinical significance, it remains difficult to ablate poor clinical outcomes. Just as in other cancers, in childhood ALL, the most common TP53 missense mutations are detected in the DNA‐binding domain. This might result in accumulation of mutant p53 and poor therapy response, with inferior outcomes. Restoration of the wild‐type p53 protein or ablation of the mutant p53 protein might be a fruitful approach for targeting these TP53 alterations. In a preclinical patient‐derived TP53‐mutant ALL xenograft model by Demir et al, a small molecule, APR‐246, had a therapeutic effect and resulted in excellent leukemia‐free survival. Rebuilding tumor‐suppressive functions by targeting mutant p53 proteins seems to be a reasonable therapeutic strategy and is likely worth future clinical trials.29, 30
In conclusion, our findings indicate that TP53 alterations at relapse may be used as a molecular marker for therapeutic failure in relapsed pediatric ALL patients. The frequency of TP53 alterations at relapse in the present study was much higher than that reported in Caucasians. This finding shows the need for a larger prospective clinical trial in Taiwan. TP53 alterations may aid in future risk classification of children with relapsed ALL. Targeting of mutant p53 proteins might warrant further clinical trials as a treatment strategy.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
The authors express their gratitude to all patients who participated in this study and their parents. The authors also acknowledge the efforts of the TPOG and the Childhood Cancer Foundation in Taiwan. This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST‐106‐2314‐B‐002‐199‐ and MOST‐107‐2314‐B‐002‐173‐MY2 to YLY).
Yu C‐H, Chang W‐T, Jou S‐T, et al. TP53 alterations in relapsed childhood acute lymphoblastic leukemia. Cancer Sci. 2020;111:229–238. 10.1111/cas.14238
Chih‐Hsiang Yu and Wan‐Ting Chang contributed equally to this article.
Contributor Information
Hsuan‐Yu Chen, Email: hychen@stat.sinica.edu.tw.
Yung‐Li Yang, Email: yangyl92@ntu.edu.tw.
REFERENCES
- 1. Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019;16:227‐240. [DOI] [PubMed] [Google Scholar]
- 2. Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730‐2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541‐1552. [DOI] [PubMed] [Google Scholar]
- 4. Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33:2938‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14:e205‐e217. [DOI] [PubMed] [Google Scholar]
- 6. Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B‐acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high‐risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:3080‐3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wada M, Bartram C, Nakamura H, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82:3163‐3169. [PubMed] [Google Scholar]
- 9. Holmfeldt L, Wei L, Diaz‐Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hof J, Krentz S, Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185‐3193. [DOI] [PubMed] [Google Scholar]
- 11. Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B‐cell precursor acute lymphoblastic leukemia. Blood. 2016;128:911‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pui CH, Mullighan CG, Evans WE, et al. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high‐risk ALL and implementing precision medicine. Blood. 2015;125:3977‐3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bader P, Kreyenberg H, Henze GHR, et al. Prognostic value of minimal residual disease quantification before allogeneic stem‐cell transplantation in relapsed childhood acute lymphoblastic leukemia: The ALL‐REZ BFM Study Group. J Clin Oncol. 2009;27:377‐384. [DOI] [PubMed] [Google Scholar]
- 15. Liang DC, Yang CP, Lin DT, et al. Long‐term results of Taiwan Pediatric Oncology Group studies 1997 and 2002 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:397‐405. [DOI] [PubMed] [Google Scholar]
- 16. Einsiedel HG, Av S, Hartmann R, et al. Long‐term outcome in children with relapsed ALL by risk‐stratified salvage therapy: results of trial acute lymphoblastic leukemia‐relapse study of the Berlin‐Frankfurt‐Münster Group 87. J Clin Oncol. 2005;23:7942‐7950. [DOI] [PubMed] [Google Scholar]
- 17. Tallen G, Ratei R, Mann G, et al. Long‐term outcome in children with relapsed acute lymphoblastic leukemia after time‐point and site‐of‐relapse stratification and intensified short‐course multidrug chemotherapy: results of trial ALL‐REZ BFM 90. J Clin Oncol. 2010;28:2339‐2347. [DOI] [PubMed] [Google Scholar]
- 18. Richter‐Pechańska P, Kunz JB, Hof J, et al. Identification of a genetically defined ultra‐high‐risk group in relapsed pediatric T‐lymphoblastic leukemia. Blood Cancer J. 2017;7:e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gump J, McGavran L, Wei Q, et al. Analysis of TP53 mutations in relapsed childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2001;23:416‐419. [DOI] [PubMed] [Google Scholar]
- 20. Diccianni M, Yu J, Hsiao M, et al. Clinical significance of p53 mutations in relapsed T‐cell acute lymphoblastic leukemia. Blood. 1994;84:3105‐3112. [PubMed] [Google Scholar]
- 21. Felix CA, Nau MM, Takahashi T, et al. Hereditary and acquired p53 gene mutations in childhood acute lymphoblastic leukemia. J Clin Invest. 1992;89:640‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tomizawa D. Recent progress in the treatment of infant acute lymphoblastic leukemia. Pediatr Int. 2015;57:811‐819. [DOI] [PubMed] [Google Scholar]
- 23. Tomizawa D, Koh K, Sato T, et al. Outcome of risk‐based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia. 2007;21: 2258‐2263. [DOI] [PubMed] [Google Scholar]
- 24. Andersson AK, Ma J, Wang J, et al. The landscape of somatic mutations in infant MLL‐rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qian M, Cao X, Devidas M, et al. TP53 germline variations influence the predisposition and prognosis of B‐cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018;36:591‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forero‐Castro M, Robledo C, Benito R, et al. Mutations in TP53 and JAK2 are independent prognostic biomarkers in B‐cell precursor acute lymphoblastic leukaemia. Br J Cancer. 2017;117:256‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stengel A, Schnittger S, Weissmann S, et al. TP53 mutations occur in 15.7% of ALL and are associated with MYC‐rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014;124:251‐258. [DOI] [PubMed] [Google Scholar]
- 28. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Demir S, Boldrin E, Sun Q, et al. Therapeutic targeting of mutant p53 in pediatric acute lymphoblastic leukemia. Haematologica. 2019. 10.3324/haematol.2018.199364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bykov VJN, Eriksson SE, Bianchi J, et al. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18:89‐102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials