

Abstract
Neurogenic differentiation factor 1 (NeuroD1) is a transcription factor critical for promoting neuronal differentiation and maturation. NeuroD1 is involved in neuroblastoma and medulloblastoma; however, its molecular mechanism in promoting tumorigenesis remains unclear. Furthermore, the role of NeuroD1 in non–neural malignancies has not been widely characterized. Here, we found that NeuroD1 is highly expressed in colorectal cancer. NeuroD1‐silencing induces the expression of p21, a master regulator of the cell cycle, leading to G2‐M phase arrest and suppression of colorectal cancer cell proliferation as well as colony formation potential. Moreover, NeuroD1‐mediated regulation of p21 expression occurs in a p53‐dependent manner. Through chromatin immunoprecipitation and point mutation analysis in the predicted NeuroD1 binding site of the p53 promoter, we found that NeuroD1 directly binds to the p53 promoter and suppresses its transcription, resulting in increased p53 expression in NeuroD1‐silenced colorectal cancer cells. Finally, xenograft experiments demonstrated that NeuroD1‐silencing suppresses colorectal cancer cell tumorigenesis potential by modulating p53 expression. These findings reveal NeuroD1 as a novel regulator of the p53/p21 axis, underscoring its importance in promoting non–neural malignancies. Furthermore, this study provides insight into the transcriptional regulation of p53.
Keywords: cell proliferation, colorectal cancer, neurogenic differentiation factor 1, p21, p53
In this study, we found that NeuroD1 is a novel negative regulator of tumor suppressor p53 and is highly expressed in colorectal cancer. NeuroD1 binds to p53 promoter and suppresses its transcriptional activity, leading to the inhibition of the p53/p21 axis. NeuroD1‐silencing increases p53 and p21 levels, resulting in an inhibition of colorectal cancer cell proliferation and tumorigenesis potential.
1. INTRODUCTION
Colorectal cancer (CRC) shows the third highest incidence among all cancers worldwide.1 In 2018, there were 1 096 601 new cases and 551 269 deaths from CRC, accounting for 6.1% of cancer incidence and 5.8% of total cancer‐related mortality.1 While the 5‐year survival rate of patients with early CRC is approximately 90%, this value is less than 10% in patients with metastases.2 Despite its high incidence and mortality rates, the molecular mechanism of CRC is unclear.
p21 is a negative regulator of the cell cycle and mediates various physiological activities such as cell growth, differentiation, DNA repair, and aging,3, 4 and was identified as a cyclin‐dependent kinase inhibitor.5, 6 It regulates cell cycle progression by affecting multiple cyclins and cyclin‐dependent kinases, whose activities fine‐tune each stage of cell cycle progression.3, 7 p21 is a downstream target of the tumor suppressor p53, as p53 binds to the p21 promoter and activates its transcription. Previous studies revealed that aberrant p21 regulation leads to various diseases, including tumors, Alzheimer’s disease, Huntington’s disease, lupus autoimmunity and Lynch syndrome.7, 8, 9, 10, 11 In several tumors, including melanoma,12 breast cancer,13 gastric cancer,14 colorectal cancer4 and lung adenocarcinoma,15 p21 is downregulated. Unlike p53, whose mutations are detected in approximately 50% of patients with tumors, p21 is rarely mutated in human tumors,3 suggesting that abnormalities in p21 expression regulation may be responsible for its aberrant expression in tumors. Despite its critical role in tumorigenesis, p21 regulation has not been fully elucidated. In an effort to unravel the regulatory mechanism of the p53/p21 axis, we previously screened an shRNA vector library, and identified neurogenic differentiation factor 1 (NeuroD1, also known as ND1) as a potential negative regulator of p21 transcriptional activity.4
Previous studies showed that NeuroD1, a neurogenic basic helix–loop–helix transcription factor, can promote the transformation of human fibroblasts into induced neuronal cells.16 NeuroD1 binds to neuronal genes that are silenced during development, causing them to regain their transcriptional competence and eventually reprogramming other cell types into neurons.17 In mice, NeuroD1 negatively regulates atonal bHLH transcription factor 1 (Atoh1), increasing the transformation of proliferative precursors to differentiating neurons.18 NeuroD1 is also involved in neuronal malignancies. Previous studies have shown that NeuroD1 is highly expressed in neural malignancies, such as neuroblastoma and medulloblastoma, and its silencing suppresses neuroblastoma cell proliferation by regulating the expression of anaplastic lymphoma kinase (ALK) and slit guidance ligand 2 (Slit2).19, 20, 21 NeuroD1 could also function simultaneously with orthodenticle homeobox 2 (OTX2) as regulatory elements and regulate medulloblastoma‐related genes.19 It also promotes tumor cell survival and metastasis in neuroendocrine lung carcinoma.22, 23 Recent studies revealed that NeuroD1 is also involved in non–neural malignancy, as its silencing suppresses the migration and invasion of pancreatic cancer cells.24 However, the roles of NeuroD1 in regulating the tumorigenesis of non–neural cancer are not well‐understood. Furthermore, its molecular mechanism in regulating the tumor cell cycle and proliferation has not been reported.
Here, we found that in CRC cells, NeuroD1 directly binds to the p53 promoter, leading to the suppression of its transcription activity, which, in turn, suppresses the p53 downstream target p21. NeuroD1‐silencing leads to increased p21 expression and increased cyclin B and cyclin‐dependent kinase 1 (CDK1) in CRC cells, resulting in a G2‐M arrest. We showed that the NeuroD1/p53 axis regulates CRC cell proliferation and tumorigenesis potential. These findings reveal not only the role of NeuroD1 as a novel regulator of p53 but also the important role of NeuroD1 in promoting CRC by regulating the p53/p21 axis.
2. MATERIALS AND METHODS
2.1. Plasmids and constructs
According to the algorithm and method previously reported,25, 26 we designed and constructed two shRNA expression vectors with different target sites specifically targeting NeuroD1 (shNeuroD1‐1 [5′‐GCA CAA GCT TGT ATA TAC A‐3′] and shNeuroD1‐2 [5′‐GCT GCA AAG TGC AAA TAC‐3′]), as well as shRNA expression vector targeting p21: 5′‐GAT GGA ACT TCG ACT TTG T‐3′. An shRNA expression vector containing a stretch of 7 thymines terminator sequences exactly downstream of the U6 promoter, namely shCon, was used as a control. An shRNA expression vector targeting p53 was constructed as described previously.27
Reporter vector bringing p21 promoter (p21‐luc), p21 promoter lacking the p53 binding site (p21del‐Luc) and p53 promoter (p53‐luc) were constructed as described previously.4 For reporter vector bringing p53 promoter lacking predicted NeuroD1 binding site (p53del‐luc), the −833 to +17 of the p53 promoter region was cloned into the BamH I and Not I sites of the pGL4.13 (Promega). For reporter vector bringing ALK promoter with NeuroD1 binding site (ALK‐luc), the −670 to +134 of the ALK promoter region was cloned into the EcoR V and Hind III sites of the pGL4.13. Human genome DNA extracted from HCT116WT cells using the TIANamp Genomic DNA Kit (Tiangen Biotech) was used as template for amplifying the promoter regions. p53‐luciferase vector with mutated predicted NeuroD1 binding site (p53mut‐Luc) was constructed based on the site‐specific mutagenesis method using a Site‐directed Gene Mutagenesis Kit (Beyotime).
2.2. Cell lines and cell culture
HCT116WT and HCT116p53null cell lines were provided by Dr Bert Vogelstein at The John Hopkins University Medical School28 and grown in McCoy’s 5A medium (Biological Industries) with 10% FBS (Biological Industries) and 1% penicillin‐streptomycin. Mycoplasma contamination was routinely tested using the Mycoplasma Detection Kit‐QuickTest (Biotool). All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C. Transfection was performed using Lipofectamine 2000 (Invitrogen Life Technologies) according to the manufacturer’s protocol. For gene‐silencing experiments, to eliminate untransfected cells, 24 hours after transfection, transfected cells were selected by using puromycin (final concentration: 1.2 µg/mL) for 36 hours.
For overexpression experiments, cells were transfected with 2 µg of indicated overexpression vector. Twenty‐four hours later, mRNA and protein samples were collected and subjected for further analysis.
For double‐silencing experiments, cells were transfected with 1 µg of indicated shRNA expression vector. Cells were subjected to puromycin selection to eliminate untransfected cells. mRNA and protein were collected 36 hours after puromycin selection. For NeuroD1‐silenced HCT116 cells, NeuroD1, p53‐double silenced HCT116 cells or control stable cell lines, cells were transfected with the indicated vectors (total 2 µg) and selected with puromycin.
2.3. Clinical human colorectal cancer specimens
Human colorectal cancer specimens were obtained from colorectal carcinoma patients undergoing surgery at Chongqing University Cancer Hospital (Chongqing, China), and stored in the Biological Specimen Bank of Chongqing University Cancer Hospital. Patients did not receive chemotherapy, radiotherapy or other adjuvant therapies prior to the surgery. The specimens were snap‐frozen in liquid nitrogen. Written informed consent was obtained from prior patients. The experiments were approved by the Institutional Research Ethics Committee of Chongqing University Cancer Hospital and conducted in accordance with the Declaration of Helsinki.
2.4. Quantitative RT‐PCR and western blotting
Quantitative RT‐PCR (qPCR) and western blotting were performed with methods described in the Supporting Information Material and Methods. The primers and antibodies used are listed in Tables S1 and S2, respectively.
2.5. Statistical analysis
All values of the experimental results are presented as mean ± SD of triplicates. Statistical analysis was performed using Student’s t test, except for clinical samples, which were analyzed by one‐way ANOVA conducted using SPSS Statistics 17.0. A value of P < 0.05 was considered statistically significant; while P < 0.01 was considered highly significant.
3. RESULTS
3.1. Neurogenic differentiation factor 1 regulates cell cycle progression and tumor cell proliferation of colorectal cancer cells by suppressing p21 expression
To examine the effect of NeuroD1 expression modulation on the tumorigenesis potential of solid tumor cells, particularly colon carcinoma cells, we first constructed two shNeuroD1 expression vectors with different target sites to ensure the specificity, and confirmed their silencing effect (Figure S1A). The functional silencing of shNeuroD1 was further verified by confirming its effect on the activity of a firefly luciferase vector containing the promoter sequence of ALK, a previously known target of NeuroD1 (Figure S1B). We next examined the effect of NeuroD1 silencing on the total cell numbers. Knocking down NeuroD1 significantly suppressed the increase in total cell numbers (Figure 1A) and the colony formation potential of HCT116 cells (Figure 1B). Next, we examined the effect of knocking down NeuroD1 on tumor cell cycle progression and apoptosis. We found that while knocking down NeuroD1 enhanced the percentage of apoptotic cells (Figure S1C), it led to significant G2‐M cell cycle arrest (Figure 1C). Concomitantly, NeuroD1 silencing suppressed the expression of cyclin B and CDK1, which are the regulators of G2‐M phase (Figure 1D), while overexpressing NeuroD1 using a NeuroD1 expression vector significantly enhanced their expression (Figure 1E and Figure S1D).
Figure 1.
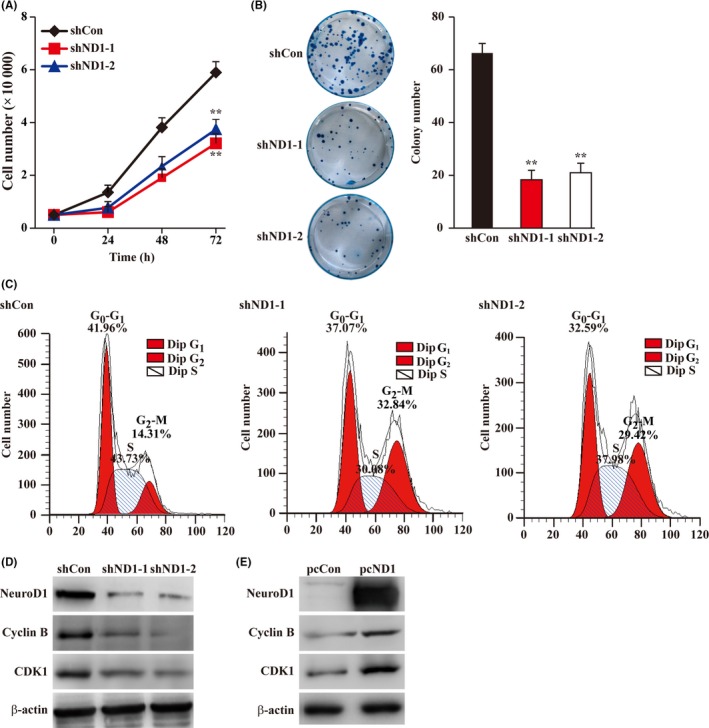
Neurogenic differentiation factor 1 (NeuroD1) regulates colon carcinoma cells proliferation and colony formation potential. A, Cell number of NeuroD1‐silenced HCT116 cells at indicated time points. B, Colony formation potential of NeuroD1‐silenced HCT116 cells. The representative images (left) and quantification results (right) are shown. C, The percentage of NeuroD1‐silenced HCT116 cells at each cell cycle stage, as examined using PI staining and flow‐cytometry. The representative images are shown. D and E, Protein expression levels of cyclin B and cyclin‐dependent kinase 1 in NeuroD1‐silenced (D) and NeuroD1‐overexpressed (E) HCT116 cells, as determined using western blotting. β‐Actin was used as western blotting loading control. Cells transfected with shCon or pcCon were used as controls. Quantitative data were expressed as mean ± SD from three independent experiments. shND1, shRNA expression vector targeting NeuroD1; pcCon, pcDNA3.1(+); pcND1, NeuroD1 overexpression vector. **P < 0.01
p21 is the upstream regulator of cyclins and cyclin‐dependent kinases. Thus, we next examined the transcriptional activity of p21 in NeuroD1‐silenced HCT116 cells. As shown in Figure 2A,B, knocking down NeuroD1 significantly enhanced p21 promoter activity as well as the p21 mRNA level. In agreement with this, NeuroD1 overexpression suppressed the expression of p21 mRNA (Figure 2C). The protein levels of p21 also showed similar expression patterns (Figure 2D,E). These results indicate that NeuroD1 is a novel regulator of p21. Using shp21 expression vector (Figure S2A,B) and shNeuroD1, we next silenced NeuroD1 and p21 simultaneously, and investigated the effect on the number of HCT116 cells. As shown in Figure 2F, knocking down p21 partially restored the decrease in total cell numbers, suggesting that p21 plays a crucial role in NeuroD1 regulation on tumor cell proliferation.
Figure 2.
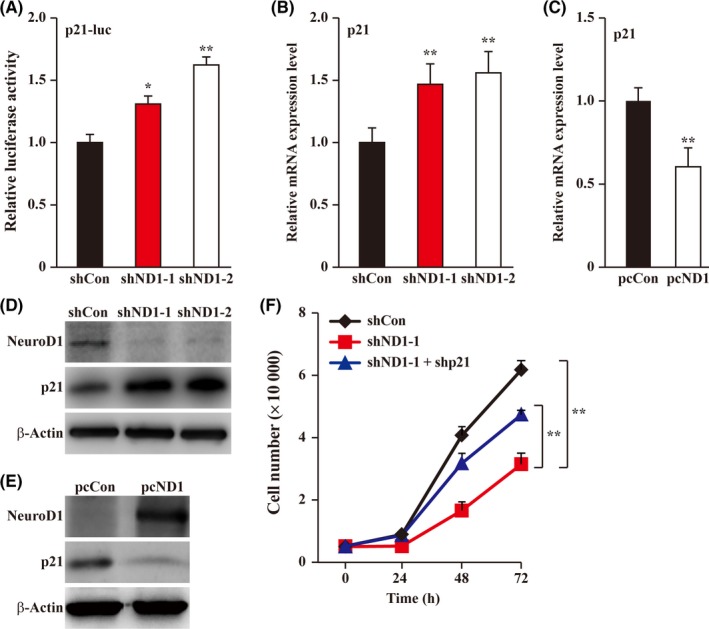
Neurogenic differentiation factor 1 (NeuroD1) regulates the expression level of cell cycle regulator p21. A, Relative luciferase activity of p21‐promoter reporter vector (p21‐luc) in NeuroD1‐silenced HCT116 cells. B and C, p21 mRNA expression level in NeuroD1‐silenced and NeuroD1‐overexpressed HCT116 cells, respectively, as examined using quantitative RT‐PCR (qPCR). D and E, p21 protein expression level in NeuroD1‐silenced and NeuroD1‐overexpressed HCT116 cells, respectively, as determined using western blotting. F, Cell number of NeuroD1, p21‐double silenced HCT116 cells at indicated time points. β‐Actin was used for qPCR normalization and as western blotting loading control. Cells transfected with shCon or pcCon were used as controls. Quantitative data were expressed as mean ± SD from three independent experiments. shND1, shRNA expression vector targeting NeuroD1; pcCon, pcDNA3.1(+); pcND1, NeuroD1 overexpression vector; *P < 0.05; **P < 0.01
3.2. p53 is crucial for NeuroD1 regulation of p21 and tumor cell proliferation
Tumor suppressor p53 is an upstream regulator of p21 that binds to its promoter and activates its transcription. To investigate whether NeuroD1 regulation of p21 occurs via p53, we knocked down NeuroD1 expression in p53‐knocked out HCT116 cells (HCT116p53null cells, Figure S3), and examined the effect of NeuroD1 silencing on p21 transcriptional activity in both wild type HCT116 and HCT116p53null cells. We found that p21 transcriptional activity was not significantly affected in HCT116p53null cells (Figure 3A), indicating that p53 is necessary for NeuroD1 regulation of p21 expression. To further confirm that this regulation occurs through direct regulation of p21 transcription by p53, we next investigated the effect of NeuroD1 silencing in wild‐type HCT116 cells on the activity of a firefly luciferase reporter containing the promoter region of p21 lacking the p53‐binding site (p21del‐luc, Figure S4A). Concomitant with the results observed in HCT116p53null cells, knocking down NeuroD1 in wild‐type HCT116 cells did not significantly affect p21del‐luc (Figure S4B). Furthermore, in contrast to the results in wild‐type HCT116 cells, NeuroD1‐silencing did not affect either p21 mRNA or protein expression in HCT116p53null cells (Figure 3B,C).
Figure 3.
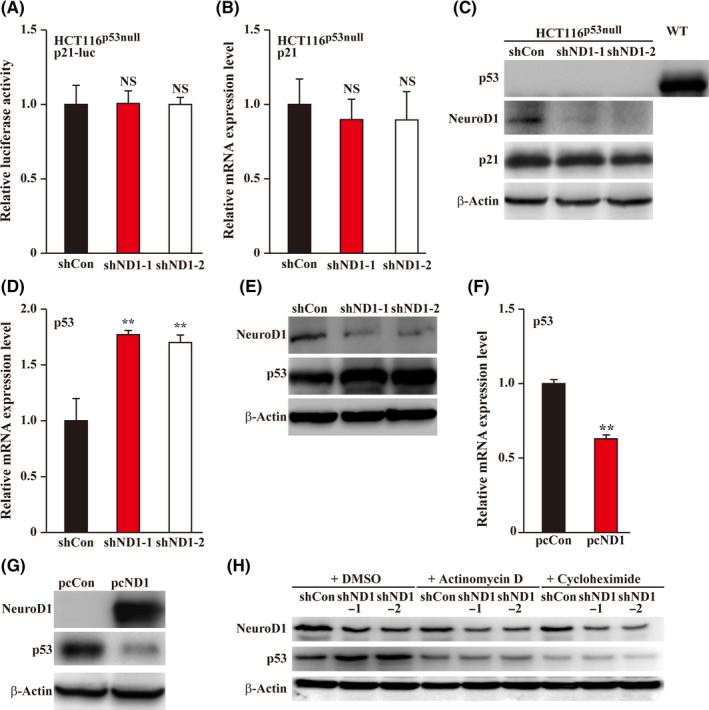
p53 is necessary for Neurogenic differentiation factor 1 (NeuroD1) regulation on p21 expression level. A, Relative luciferase activity of p21‐promoter reporter vector (p21‐luc) in NeuroD1‐silenced HCT116p53null cells. B and C, p21 mRNA (B) and protein (C) expression level in NeuroD1‐silenced HCT116p53null cells as examined using quantitative RT‐PCR (qPCR) and western blotting, respectively. D and E, p53 mRNA (D) and protein (E) expression level in NeuroD1‐silenced wild type HCT116 cells as examined using qPCR and western blotting, respectively. F and G, p53 mRNA (F) and protein (G) expression level in NeuroD1‐overexpressed wild type HCT116 cells as examined using qPCR and western blotting, respectively. H, NeuroD1 and p53 protein expression levels in NeuroD1‐silenced wild type HCT116 cells treated with actinomycin D or cycloheximide, as determined using western blotting. Cells treated with DMSO were used as control. Relative luciferase activity was measured using dual luciferase assay, and the activity of firefly luciferase was normalized with that of Renilla luciferase as inner control. β‐Actin was used for qPCR normalization and as western blotting loading control. Cells transfected with shCon or pcCon were used as controls. Quantitative data were expressed as mean ± SD from three independent experiments. shND1, shRNA expression vector targeting NeuroD1; pcCon, pcDNA3.1(+); pcND1, NeuroD1 overexpression vector; **P < 0.01; NS, not significant
We then investigated the effect of NeuroD1 on p53 expression level. Silencing of NeuroD1 grossly promoted p53 mRNA and protein expression levels (Figure 3D,E), while NeuroD1 overexpression significantly suppressed them (Figure 3F,G), suggesting that NeuroD1 is a negative regulator of p53. Given that NeuroD1 has been reported as a transcription factor,29 we next analyzed its regulatory mechanism of p53 expression. We found that while NeuroD1 silencing greatly enhanced the expression of p53, inhibition of both de novo transcription and protein synthesis abolished this effect (Figure 3H), suggesting that NeuroD1 regulation of p53 expression occurs during the transcription stage.
Next, we examined whether NeuroD1 regulation of HCT116 cell proliferation and colony formation potential also requires the presence of p53. We first investigated the effect of knocking down NeuroD1 and p53 simultaneously on the total cell numbers. The results showed that knocking down p53 partially restored the decrease in total cell numbers and colony formation potential of HCT116 cells (Figure 4A,B) caused by NeuroD1 silencing, and re‐suppressed the caspases activity (Figure S5A). Intriguingly, NeuroD1 silencing also slightly suppressed the colony formation potential of HCT116p53null cells (Figure S5B), indicating that while p53 is critical, NeuroD1 might also regulate tumorigenesis through p53 independent pathway. Furthermore, we found that knocking down p53 re‐suppressed the increase in p21 expression in NeuroD1‐silenced wild‐type HCT116 cells (Figure 4C), indicating that NeuroD1 regulation of p21 occurs through p53. These results strongly suggest that p53 is critical for the NeuroD1‐mediated regulation of the tumorigenesis potential of HCT116 cells.
Figure 4.
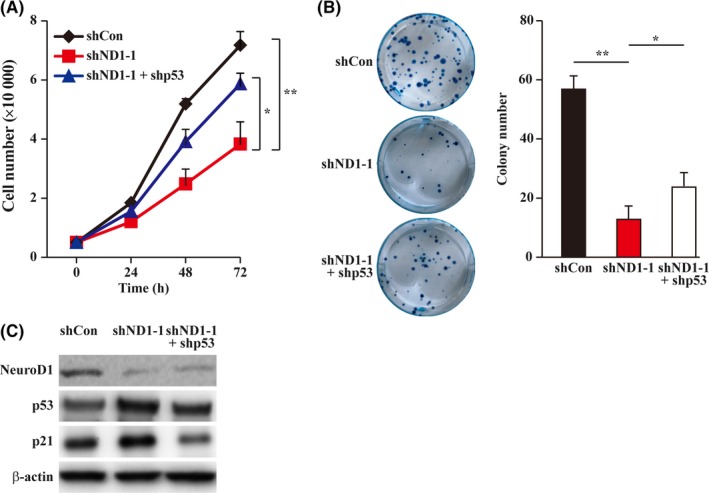
Neurogenic differentiation factor 1 (NeuroD1) regulates cell proliferation through p53. A, Cell numbers in NeuroD1, p53‐double silenced HCT116 cells at indicated time points. B, Colony formation potential of NeuroD1, p53‐double silenced HCT116 cells. Representative images (left) and quantification results (right). C, p21 protein expression level in NeuroD1, p53‐silenced HCT116 cells, as determined using western blotting. β‐Actin was used for western blotting loading control. Cells transfected with shCon were used as controls. Quantitative data were expressed as mean ± SD from three independent experiments. shND1, shRNA expression vector targeting NeuroD1; *P < 0.05; **P < 0.01
3.3. NeuroD1 regulates p53 transcription by directly binding to its promoter region
To reveal the molecular mechanism of NeuroD1 regulation of p53 transcription, we performed a luciferase reporter assay with a firefly luciferase vector containing the p53 promoter sequence (p53‐luc, Figure 5A, upper panel). We found that knocking down NeuroD1 significantly enhanced p53 promoter activity (Figure 5B). Indeed, by using the JASPAR database (http://jaspar.genereg.net/), we predicted a NeuroD1 binding site at −839 to −834 in the p53 promoter and constructed a reporter vector with the p53 promoter but without the predicted NeuroD1 binding site (p53del‐luc, Figure 5A, middle panel). The absence of the predicted NeuroD1 binding site abolished the effect of NeuroD1 silencing on p53 transcriptional activity (Figure 5B). These results indicate that NeuroD1 might directly bind to the p53 promoter and regulates its activity.
Figure 5.
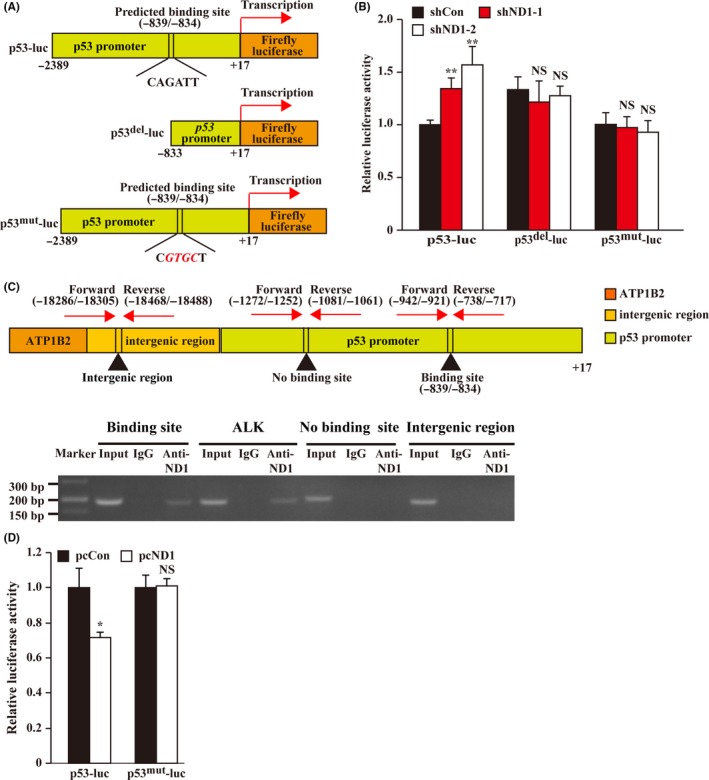
Neurogenic differentiation factor 1 (NeuroD1) directly binds to the p53 promoter and regulates its activity. A, Schematic diagram of p53‐promoter reporter vector (p53‐luc), p53‐promoter reporter vector lacking predicted NeuroD1 binding site (p53del‐luc) and p53 promoter reporter vector with mutated NeuroD1 binding site (p53mut‐luc). The mutated nucleotides are shown in italic red. B, Relative luciferase activities of p53‐luc, p53del‐luc and p53mut‐luc in NeuroD1‐silenced HCT116 cells. C, Binding of NeuroD1 to the p53 promoter region was examined using chromatin immunoprecipitation assay with anti‐NeuroD1 antibody followed by PCR in HCT116WT cells. The predicted NeuroD1 binding site on the p53 promoter and the location of primer sets used for PCR for amplifying p53 promoter region with and without predicted NeuroD1 binding site, as well as for amplifying intergenic region between p53 and ATP1B2 are shown. Primer set used for PCR for amplifying NeuroD1 binding site on the ALK promoter was used as positive control. D, Relative luciferase activity of p53mut‐luc in NeuroD1‐overexpressed HCT116 cells. Relative luciferase activity was measured using dual luciferase assay, and the activity of firefly luciferase was normalized with that of Renilla luciferase as inner control. Cells transfected with shCon or pcCon were used as controls. Quantitative data were expressed as mean ± SD from three independent experiments. shND1, shRNA expression vector targeting NeuroD1; pcCon, pcDNA3.1(+); pcND1, NeuroD1 overexpression vector; * P < 0.05; **P < 0.01; NS, not significant
To further confirm this hypothesis, we examined the binding of NeuroD1 at the predicted site of the p53 promoter region using a chromatin immunoprecipitation assay. As shown in Figure 5C, while NeuroD1 could bind to the −942 to −717 region of the p53 promoter, which includes the predicted NeuroD1 binding site, as well as to ALK promoter at the previously reported binding site,21 it failed to bind to the p53 promoter region outside the predicted binding site and to the intergenic region between p53 and ATP1B2 genes. Moreover, we constructed a p53‐luc reporter vector with mutations in the predicted NeuroD1 binding site (p53mut‐luc, Figure 5A, lower panel) and found that when the “CAGATT” sequences in the −839 to −834 region of the p53 promoter were mutated to “CGTGCT,” NeuroD1 silencing had no significant effect on p53 transcription (Figure 5B). In agreement with this, while NeuroD1 overexpression clearly suppressed p53‐luc reporter activity, it had no significant effect on p53mut‐luc (Figure 5D). These results demonstrate that NeuroD1 binds to the −839 to −834 region of the p53 promoter to directly inhibit p53 transcriptional activity.
3.4. NeuroD1‐silencing suppresses tumorigenesis by enhancing the p53/p21 axis
To determine the role of NeuroD1 in tumorigenesis, we performed in vivo xenograft experiments. To this end, we established NeuroD1‐silenced and NeuroD1, p53‐double silenced HCT116 stable cell lines (Figure S6A), and analyzed their tumorigenesis potential upon being transplanted subcutaneously into Balb/c‐nu/nu mice. As shown in Figure 6A, NeuroD1 silencing significantly suppressed the xenografted tumor growth, while knocking down p53 partially reversed this effect (Figure 6A). The morphological appearance and weight of the tumors showed similar results (Figure 6B,C). We next examined the expression levels of NeuroD1, p53 and p21 in xenografted tumor lesions and found that in the tumors formed by NeuroD1‐silenced HCT116 cells, the expression of p53 and p21 protein were significantly upregulated, while silencing both NeuroD1 and p53 re‐suppressed their expression (Figure 6D). The same results were observed in immunohistochemistry analysis (Figure 6E,F). Furthermore, TUNEL assay and caspase 3 activity assay revealed that knocking down NeuroD1 and p53 simultaneously suppressed the cleaved caspase level and TUNEL positive cells (Figure S6B,C).
Figure 6.
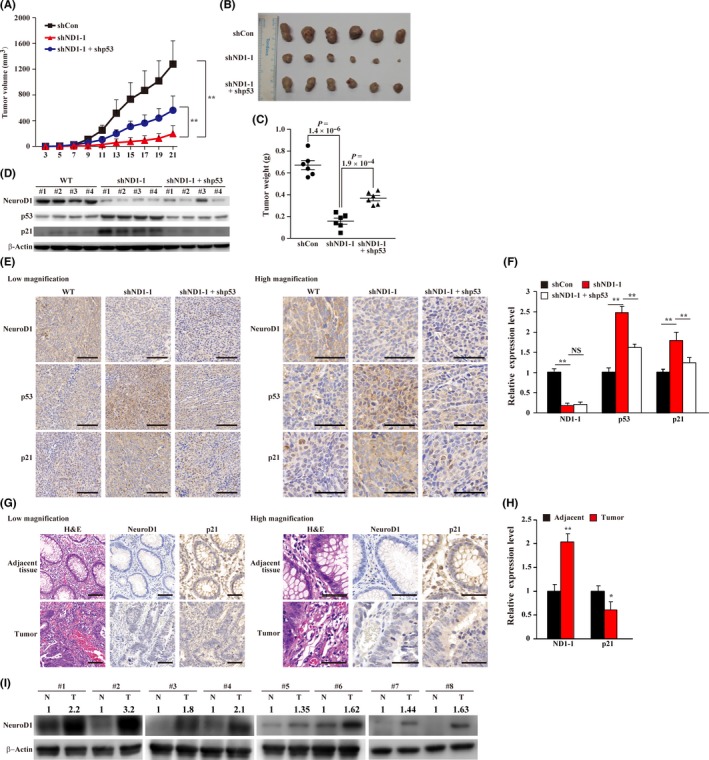
Neurogenic differentiation factor 1 (NeuroD1)‐silencing suppresses tumorigenesis by enhancing the p53/p21 axis. A–C, Tumorigenesis potentials of NeuroD1‐silenced HCT116 cells and NeuroD1, p53‐double silenced HCT116 cells were examined in vivo by subcutaneous injection into Balb/c‐nu/nu mice (n = 6). Volume of the tumor generated was measured at indicated time points (A), and the representative images (B) and the tumor weight (C) at 21 d are shown. D, NeuroD1, p53 and p21 protein expression levels in xenografted tumors in Balb/c‐nu/nu mice injected with indicated cell lines, as determined using western blotting. E and F, Immunohistochemistry staining images against NeuroD1, p53 and p21 in tissue sections of xenografted tumors in Balb/c‐nu/nu mice injected with indicated cell lines. Representative images (E) and quantification of the ratio of the positive cells to the total cell number are shown (F). Quantification results are shown as relative to control. G and H, Immunohistochemistry staining against NeuroD1 and p21 in clinical colorectal cancer patients and normal adjacent tissues. Representative images (G) and quantification of the ratio of the positive cells to total cell number (H). Quantification results are shown as relative to control. I, Protein expression level of NeuroD1 in clinical colon carcinoma and adjacent normal tissues, as determined with western blotting. Cells transfected with shCon were used as controls. Scale bars: 100 µm for low magnification (left panels), and 50 µm for high magnification (right panels). Quantitative data were expressed as mean ± SD. shND1, shRNA expression vector targeting NeuroD1; N, normal adjacent tissue; T, colon carcinoma tissue; *P < 0.05; **P < 0.01
Finally, to reveal the relationship between NeuroD1 regulation of the p53/p21 axis and tumorigenesis, we analyzed NeuroD1 expression level in clinical CRC and normal adjacent tissues. As shown by the results of immunohistochemistry analysis, NeuroD1 was overexpressed while p21 was downregulated in clinical CRC lesions (Figure 6G,H). The results of western blotting further confirmed that the expression level of NeuroD1 was upregulated in CRC tissues (Figure 6I).
Together, our results reveal the critical role of NeuroD1 in regulating CRC tumorigenesis by suppressing the p53/p21 axis. NeuroD1 binds to the −839 to −834 region of the p53 promoter and acts as a transcriptional suppressor, thereby suppressing the p53/p21 axis and promoting tumor cell proliferation and colony formation potential, subsequently leading to the promotion of tumorigenesis (Figure 7).
Figure 7.
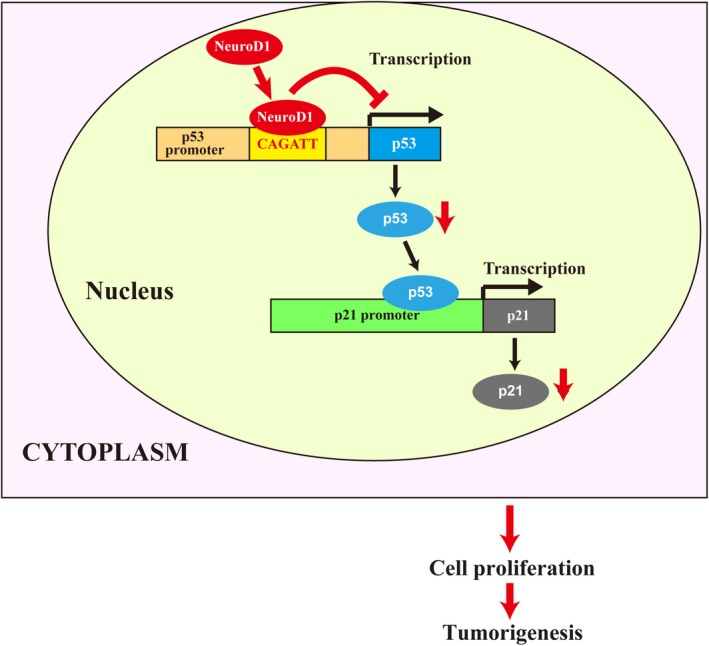
Schematic diagram of Neurogenic differentiation factor 1 (NeuroD1) regulation on tumorigenesis through suppression of the p53/p21 axis
4. DISCUSSION
Neurogenic differentiation factor 1 is a transcription factor first identified as a neuronal determination gene in the Xenopus ectoderm, and is crucial in neuronal differentiation and maturation.16, 17, 18, 29, 30 While it has been reported to be involved in the migration and invasion of pancreatic cancer,24 the role of NeuroD1 in tumorigenesis, particularly in non–neural malignancies, remains unclear. In this study, we found that NeuroD1 is highly expressed in patients with colorectal cancer and that it could directly bind to the p53 promoter, thereby suppressing p53 transcriptional activity. Silencing of NeuroD1 results in the upregulation of p53 and its downstream target gene p21, leading to cell cycle arrest and the suppression of cell proliferation as well as colony formation potential, and, thus, tumorigenesis. To our knowledge, this is the first study to reveal the molecular mechanism of NeuroD1 regulation of non–neuronal malignancy, particularly its function in regulating cell cycle progression and proliferation of CRC cells, suggesting that NeuroD1 is a critical factor in tumorigenesis.
Cell cycle progression is strictly regulated by cyclins and cyclin‐dependent kinases, and disruption of its control is closely related to tumor development. While p21 expression is downregulated in various malignancies,3 p21 mutation is rarely observed in patients with tumors, highlighting the importance of understanding the role of aberrant p21 regulation in tumorigenesis. Our findings showed that NeuroD1 regulates the p53/p21 axis, leading to the negative regulation of p21 transcription, and subsequently affects colorectal cancer cells’ cell cycle progression. Thus, NeuroD1 is associated with aberrant cell cycle regulation and non–neuronal tumorigenesis.
p53, which was discovered in 1979, was the first tumor suppressor gene identified.31 This gene is one of the most important tumor suppressors, and it acts as an upstream regulator of p21. Aberrant p53 expression and activity is an important molecular hallmark of cancer. p53 expression is frequently downregulated in patients with wild‐type p53,32 highlighting the importance of its gene expression regulation. Many previous studies have focused on p53 post–transcriptional regulation.33 Indeed, phosphorylation is critical for p53 protein activation, while ubiquitination‐induced proteasomal degradation is important for maintaining p53 protein homeostasis.4, 34, 35 Meanwhile, acetylation and phosphorylation are crucial for enhancing p53 binding to its target genes, while methylation is critical for regulating p53 acetylation.34 Here, we revealed that NeuroD1 is a novel regulator of p53 transcription and showed that the NeuroD1/p53 axis is crucial for cell cycle regulation, cell proliferation and tumorigenesis. Thus, our results suggest the importance of p53 transcriptional regulation in maintaining the homeostasis of p53 expression.
While the p53/p21 axis is crucial for preventing abnormal cell cycle progression and proliferation, their homeostasis are also critical for maintaining other biological and physiological functions, including DNA repair, stem cells maintenance, differentiation and senescence.3, 36, 37 p53 is also involved in regulating other tumor characteristics, including metabolic reprogramming, motility and invasiveness.38 This indicates that NeuroD1 might also be involved in other biological and physiological pathways. Intriguingly, although with lower extent compared to its effect in p53 wild‐type cells, NeuroD1‐silencing could also suppress colony formation potential in p53 knockout cells. Similarly, xenograft experiments showed that p53‐silencing did not completely restore the effect of NeuroD1‐silencing in suppressing tumor growth. These facts indicate the possibility of the presence of a p53‐independent pathway in NeuroD1 regulation on tumorigenesis.
In conclusion, we identified NeuroD1 as a novel regulator of the p53/p21 axis. This regulation, in turn, regulates cell cycle progression, tumor cell proliferation, and, eventually, tumorigenesis. Our findings provide insight into the regulation of p53 transcriptional activity, as well as demonstrate the potential for targeting NeuroD1 in cancer therapy.
DISCLOSURE
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank Dr Bert Vogelstein (School of Medicine, Johns Hopkins University) for kindly providing wild‐type and p53‐null HCT116 cell lines. This work was supported by grants from the National Natural Science Foundation of China (31871367, 81872273 and 11832008), the Fundamental Research Funds for the Central Universities (2019CDQYSW010) and the Natural Science Foundation of Chongqing (cstc2018jcyjAX0411 and cstc2018jcyjAX0374).
Lei K, Li W, Huang C, et al. Neurogenic differentiation factor 1 promotes colorectal cancer cell proliferation and tumorigenesis by suppressing the p53/p21 axis. Cancer Sci. 2020;111:175–185. 10.1111/cas.14233
Ke Lei, Wenfang Li and Can Huang contributed equally to this work.
Contributor Information
Shourong Wu, Email: shourongwu@cqu.edu.cn.
Vivi Kasim, Email: vivikasim@cqu.edu.cn.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96:1420‐1425. [DOI] [PubMed] [Google Scholar]
- 3. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang C, Wu S, Ji H, et al. Identification of XBP1‐u as a novel regulator of the MDM2/p53 axis using an shRNA library. Sci Adv. 2017;3:e1701383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. el‐Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817‐825. [DOI] [PubMed] [Google Scholar]
- 6. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk‐interacting protein Cip1 is a potent inhibitor of G1 cyclin‐dependent kinases. Cell. 1993;75:805‐816. [DOI] [PubMed] [Google Scholar]
- 7. El‐Deiry WS. p21(WAF1) mediates cell‐cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016;76:5189‐5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yates SC, Zafar A, Rabai EM, et al. The effects of two polymorphisms on p21cip1 function and their association with Alzheimer’s disease in a population of European descent. PLoS ONE. 2015;10:e0114050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Louis Sam Titus ASC, Yusuff T, Cassar M, Thomas E, Kretzschmar D, D’Mello SR. Reduced expression of Foxp1 as a contributing factor in Huntington’s disease. J Neurosci. 2017;37:6575‐6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daszkiewicz L, Vazquez‐Mateo C, Rackov G, et al. Distinct p21 requirements for regulating normal and self‐reactive T cells through IFN‐gamma production. Sci Rep. 2015;5:7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salo‐Mullen EE, Lynn PB, Wang LU, et al. Contiguous gene deletion of chromosome 2p16.3‐p21 as a cause of Lynch syndrome. Fam Cancer. 2018;17:71‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt K, Carroll JS, Yee E, et al. The lncRNA SLNCR recruits the androgen receptor to EGR1‐bound genes in melanoma and inhibits expression of tumor suppressor p21. Cell Rep. 2019;27:2493‐2507 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu RZ, Vo TM, Jain S, et al. NFIB promotes cell survival by directly suppressing p21 transcription in TP53‐mutated triple‐negative breast cancer. J Pathol. 2019;247:186‐198. [DOI] [PubMed] [Google Scholar]
- 14. Xu J, Wang Z, Lu W, et al. EZH2 promotes gastric cancer cells proliferation by repressing p21 expression. Pathol Res Pract. 2019;215:152374. [DOI] [PubMed] [Google Scholar]
- 15. Luo J, Liu K, Yao YU, et al. DMBX1 promotes tumor proliferation and regulates cell cycle progression via repressing OTX2‐mediated transcription of p21 in lung adenocarcinoma cell. Cancer Lett. 2019;453:45‐56. [DOI] [PubMed] [Google Scholar]
- 16. Pang ZP, Yang N, Vierbuchen T, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pataskar A, Jung J, Smialowski P, et al. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. 2016;35:24‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pan N, Jahan I, Lee JE, Fritzsch B. Defects in the cerebella of conditional Neurod1 null mice correlate with effective Tg(Atoh1‐cre) recombination and granule cell requirements for Neurod1 for differentiation. Cell Tissue Res. 2009;337:407‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boulay G, Awad ME, Riggi N, et al. OTX2 activity at distal regulatory elements shapes the chromatin landscape of Group 3 medulloblastoma. Cancer Discov. 2017;7:288‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang P, Kishida S, Cao D, et al. The neuronal differentiation factor NeuroD1 downregulates the neuronal repellent factor Slit2 expression and promotes cell motility and tumor formation of neuroblastoma. Cancer Res. 2011;71:2938‐2948. [DOI] [PubMed] [Google Scholar]
- 21. Lu F, Kishida S, Mu P, et al. NeuroD1 promotes neuroblastoma cell growth by inducing the expression of ALK. Cancer Sci. 2015;106:390‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Osborne JK, Larsen JE, Shields MD, et al. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci USA. 2013;110:6524‐6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Osborne JK, Guerra ML, Gonzales JX, McMillan EA, Minna JD, Cobb MH. NeuroD1 mediates nicotine‐induced migration and invasion via regulation of the nicotinic acetylcholine receptor subunits in a subset of neural and neuroendocrine carcinomas. Mol Biol Cell. 2014;25:1782‐1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Su DW, Gao L, Ding GL, Ni CR, Zhu MH. Effect of NeuroD gene silencing on the migration and invasion of human pancreatic cancer cells PANC‐1. Cell Biochem Biophys. 2014;69:487‐494. [DOI] [PubMed] [Google Scholar]
- 25. Miyagishi M, Taira K. Strategies for generation of an siRNA expression library directed against the human genome. Oligonucleotides. 2003;13:325‐333. [DOI] [PubMed] [Google Scholar]
- 26. Wu S, Kasim V, Kano MR, et al. Transcription factor YY1 contributes to tumor growth by stabilizing hypoxia factor HIF‐1alpha in a p53‐independent manner. Cancer Res. 2013;73:1787‐1799. [DOI] [PubMed] [Google Scholar]
- 27. Wu S, Wang H, Li Y, et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 2018;78:4549‐4562. [DOI] [PubMed] [Google Scholar]
- 28. Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497‐1501. [DOI] [PubMed] [Google Scholar]
- 29. Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix‐loop‐helix protein. Science. 1995;268:836‐844. [DOI] [PubMed] [Google Scholar]
- 30. Gao Z, Ure K, Ables JL, et al. Neurod1 is essential for the survival and maturation of adult‐born neurons. Nat Neurosci. 2009;12:1090‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finlay CA, Hinds PW, Levine AJ. The p53 proto‐oncogene can act as a suppressor of transformation. Cell. 1989;57:1083‐1093. [DOI] [PubMed] [Google Scholar]
- 32. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199‐210. [DOI] [PubMed] [Google Scholar]
- 34. Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang C, Wu S, Li W, et al. Zinc‐finger protein p52‐ZER6 accelerates colorectal cancer cell proliferation and tumour progression through promoting p53 ubiquitination. EBioMedicine. 2019;48:248‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Di Cunto F, Topley G, Calautti E, et al. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science. 1998;280:1069‐1072. [DOI] [PubMed] [Google Scholar]
- 37. Kim H‐S, Heo J‐I, Park S‐H, et al. Transcriptional activation of p21(WAF(1)/CIP(1)) is mediated by increased DNA binding activity and increased interaction between p53 and Sp1 via phosphorylation during replicative senescence of human embryonic fibroblasts. Mol Biol Rep. 2014;41:2397‐2408. [DOI] [PubMed] [Google Scholar]
- 38. Kaiser AM, Attardi LD. Deconstructing networks of p53‐mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials