

Abstract
Activity of transcriptional co‐activator with PDZ binding domain (TAZ) protein is strongly implicated in the pathogenesis of human cancer and is influenced by tumor metabolism. High levels of lactate concentration in the tumor microenvironment as a result of metabolic reprogramming are inversely correlated with patient overall survival. Herein, we investigated the role of lactate in the regulation of the activity of TAZ and showed that glycolysis‐derived lactate efficiently increased TAZ expression and activity in lung cancer cells. We showed that the reactive oxygen species (ROS) generated by lactate‐fueled oxidative phosphorylation (OXPHOS) in mitochondria activated AKT and thereby inhibited glycogen synthase kinase 3 beta/beta‐transducin repeat‐containing proteins (GSK‐3β/β‐TrCP)‐mediated ubiquitination and degradation of DNA methyltransferase 1 (DNMT1). Upregulation of DNMT1 by lactate caused hypermethylation of TAZ negative regulator of the LATS2 gene promoter, leading to TAZ activation. Moreover, TAZ binds to the promoter of DNMT1 and is necessary for DNMT1 transcription. Our study showed a molecular mechanism of DNMT1 in linking tumor metabolic reprogramming to the Hippo‐TAZ pathway and functional significance of the DNMT1‐TAZ feedback loop in the migratory and invasive potential of lung cancer cells.
Keywords: DNA methyltransferase 1, lactate, lung cancer, oxidative metabolism, TAZ
Glycolysis-derived lactate efficiently increased TAZ expression and activity in lung cancer cells. ROS generated by lactate-fueled OXPHOS in mitochondria activated AKT and thereby inhibited GSK-3β/β-TrCP-mediated ubiquitination and degradation of DNA methyltransferase 1 (DNMT1). Upregulation of DNMT1 caused hypermethylation of the LATS2 gene promoter. TAZ binds to the promoter of DNMT1 and is necessary for DNMT1 transcription.
1. INTRODUCTION
A fundamental property of cancer cells is to reprogram energy metabolism in order to couple glucose consumption to macromolecular biosynthesis, thus facilitating the survival of rapidly proliferating tumor cells.1, 2 Tumor cells show enhanced glucose uptake and convert pyruvate into lactate under normoxic conditions. This phenomenon of aerobic glycolysis referred to as the Warburg effect not only gives growth advantages to tumor cells, but also has a profound effect on the tumor microenvironment.3 The high levels of lactate accumulation in the tumor microenvironment are correlated with distant metastasis and poor prognosis in a variety of cancers including lung cancer.4, 5 We previously showed that lactate, besides contributing to extracellular acidosis, is emerging as a key signaling molecule that plays a crucial role in senescence suppression and immune escape.6, 7
Aberrant DNA methylation pattern characterized by genome‐wide hypomethylation and localized CpG island promoter hypermethylation has been detected in all types of cancer.8, 9 The hypermethylation of site‐specific CpG island in the promoter of tumor suppressor genes (TSG) catalyzed by DNA methyltransferases (DNMT) can lead to tumorigenesis by silencing transcription of TSG. In mammals, three types of DNMT have been identified.8 It is generally believed that DNMT3a and DNMT3b have de novo methyltransferase activity, whereas DNMT1 has a preference for methylation of hemi‐methylated sites, and is referred to as the maintenance DNA methyltransferase. However, mounting evidence indicates that DNMT1 is also an active de novo methyltransferase.10, 11 Accordingly, there are considerable studies showing upregulation of DNMT1 in cancer. Overexpression of DNMT1 was significantly correlated with hypermethylation of promoters of p16INK4a, fragile histidine triad (FHIT) and retinoic acid receptor β (RARβ) TSG in non‐small cell lung cancer (NSCLC) patients.12, 13 In addition, upregulation of DNMT1 is strongly associated with metastasis and poor prognosis in clinical manifestation.14
A recently identified signaling pathway, the Hippo pathway, has emerged as an important regulator of cancer progression and metastasis.15, 16 The mammalian core components of the Hippo pathway consist of a cascade of kinases that includes mammalian STE20‐like protein kinase 1/2 (MST1/2) kinases, the large tumor suppressor 1/2 (LATS1/2) kinases and their downstream effectors, transcriptional co‐activator with PDZ binding domain (TAZ) and Yes‐associated protein (YAP). The Hippo pathway culminates in phosphorylation of YAP/TAZ by LATS1/2 and subsequent cytoplasmic retention and degradation. Conversely, non‐phosphorylated YAP/TAZ can enter the nucleus to induce gene expression by binding to the TEAD family of transcription factors. Numerous studies showed that LATS1/2 exert tumor‐suppressive effects and TAZ functions as a critical driver of tumor formation and development.17, 18, 19 As TAZ activity is strongly implicated in the pathogenesis of human cancer, a large number of signaling pathways have been identified to modulate TAZ activity and function.20 Our laboratory has contributed to studies that have shown that lactate‐induced TAZ activation plays a critical role in tumor evasion of immune response;7 however, the mechanism of regulation TAZ activity by lactate is not completely established. In the present study, we show a feedback loop between DNMT1 and LATS2/TAZ in lung cancer cells. Upregulation of DNMT1 by extracellular lactate leads to inhibition of TAZ negative regulator of LATS2 kinase to subsequently activate TAZ activity; in turn, TAZ positively activates transcription of DNMT1. Our studies elucidate the mechanism underlying tumor metabolic reprogramming in the regulation of TAZ activity in immune evasion as well as in cell migration.
2. MATERIALS AND METHODS
2.1. Primer and siRNA sequences
All cloning and DNA construction of primer sequences are shown in Table 1. All RT‐PCR primer sequences are shown in Table 2. Sequences of siRNAs are listed in Table 3.
Table 1.
Primers used for PCR amplifications
| Gene | GenBank accession no. | Primer (5' >3') |
|---|---|---|
| DNMT1 |
Forward: GGGGTACCGCATGGGCC TCTATACACTGTGAGATT Reverse: CTAGCTAGCGATGTACCAA ACGGAGAGAGGCGATAC |
|
| DNMT1 | NM_001379.3 |
Forward: GGGGTACCGTACAATCAT GGCTCACTGCAACCTCT Reverse: CTAGCTAGCGATGTACCAA ACGGAGAGAGGCGATAC |
| DNMT1 | NM_001379.3 |
Forward: GGGGTACCTTTGTATATG GTGTAAGGTAAGGGTCC Reverse: CTAGCTAGCGATGTACCAA ACGGAGAGAGGCGATAC |
| DNMT1 | NM_001379.3 |
Forward: GGGGTACCTGCTGTCATA TCCAAGAAATCATTGCC Reverse: CTAGCTAGCGATGTACCAA ACGGAGAGAGGCGATAC |
| DNMT1 |
Forward: GAGTATACACATGCTTT GTAGCTAGTACCAAG Reverse: CTTGGTACTAGCTACAA AGCATGTGTATACTC |
|
| DNMT1 | NM_001379.3 |
Forward: AGTAGTTTGGACTATGGG CACGCGATACAACGAC Reverse: GTCGTTGTATCGCGTGCC CATAGTCCAAACTACT |
| LATS2 | NM_014572.2 |
Forward: GGGGTACCTGATCCACCC GCCTAGACCTCTCAAAG Reverse: CCCAAGCTTTCGGACCTA TTTGACTGGCTGCTATCT |
| DNMT1‐CHIP | NM_001379.3 |
Forward: CTGGGTATAGAAGTGGCATGG Reverse: ATTTGCTGGCTATACGACCTT |
| DNMT1‐CHIP | NM_001379.3 |
Forward: GCCTCTCCGGTTCAAGTGATCT Reverse: AGCCCAGGAGTTTTAGACCAGC |
Abbreviation: DNMT1, DNA methyltransferase 1.
Table 2.
Primers used for RT‐PCR
| Gene | GenBank accession no. | Primer (5' >3') |
|---|---|---|
| LATS2 | NM_014572.2 |
Forward: GGGACAAAGGCGGAAAGGA Reverse: GCTTAGCCTCTCCATCGGAC |
| DNMT1 | NM_001379.3 |
Forward: AGGAGGGCTACCTGGCTAAA Reverse: GCTTAGCCTCTCCATCGGAC |
| OCT4 | NM_002701.6 |
Forward: GAGAACCGAGTGAGAGGCAACC Reverse: CATAGTCGCTGCTTGATCGCTTG |
| Nanog | NM_024865.4 |
Forward: AATACCTCAGCCTCCAGCAGATG Reverse: TGCGTCACACCATTGCTATTCTTC |
| Notch1 | NM_017617.5 |
Forward: GTGACTGCTCCCTCAACTTCAAT Reverse: CTGTCACAGTGGCCGTCACT |
| SOX2 | NM_003106.4 |
Forward: TACAGCATGTCCTACTCGCAG Reverse: GAGGAAGAGGTAACCACAGGG |
| CD133 | NM_001145847.2 |
Forward: AGAGCTTGCACCAACAAAGTACAC Reverse: AAGCACAGAGGGTCATTGAGAGA |
| CD44 | NM_000610.4 |
Forward: TGGCACCCGCTATGTCCAG Reverse: GTAGCAGGGATTCTGTCTG |
|
CD117 |
NM_000222.2 |
Forward: GGCGACGAGATTAGGCTGTT Reverse: CATTCGTTTCATCCAGGATCTCA |
|
CD24 |
NM_013230.3 |
Forward: ACTGGAACTTCAAGTAACTCCTCCC Reverse: AGACTGGCTGTTGACTGCAGG |
| β‐actin | NM_001101.5 |
Forward: CCTTCCTGGGCATGGAGTCCT Reverse: GGAGCAATGATCTTGATCTTC |
Abbreviation: DNMT1, DNA methyltransferase 1.
Table 3.
Sequences of siRNA
| Gene | GenBank accession no. | Target sequence (5′–3′) |
|---|---|---|
| siDNMT1 | NM_001379.3 | GCAATGAGACTGACATCAAA |
| siLDHA | NM_001165414.1 | GCCAUCAGUAUCUUAAUGATT |
| siLDHB | NM_002300.7 | AAGAUUGUAGUGGUAACUGCATT |
| siGPR81 | NM_032554.3 | CTGCTAGACTCTATTTCCT |
| siYAP | NM_001130145.3 | GACATCTTCTGGTCAGAGA |
| siTEAD1 | NM_021961.6 | GCCCUGUUUCUAAUUGUGGTT |
| siTEAD4 | NM_003213.4 | CCCATGATGTGAAGCCTTT |
Abbreviations: DNMT1, DNA methyltransferase 1; LDHA, lactate dehydrogenase A; LDHB, lactate dehydrogenase B.
2.2. Supporting materials and methods
Cells, transfection, antibodies, plasmids, reagents, cloning and DNA construction and extended experimental procedures can be found in Data S1.
3. RESULTS
3.1. Glycolysis‐derived lactate is a key driver of TAZ upregulation in lung cancer cells
Publicly available data from two lung cancer patient cohorts (Gene Expression Omnibus [GEO]: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101929) and (GEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19804) showed that the expression of lactate dehydrogenase A (LDHA) correlated inversely with levels of LATS2, raising a possibility that downregulation of LATS2 may contribute to lactate‐induced activation of TAZ. To address this hypothesis, we examined the effect of lactate on expression of several key components of the Hippo pathway in H1299 and A549 lung carcinoma cell lines (Figure 1A; Figure S1A). Cells were starved and subsequently incubated with various concentrations of lactate for 3 hours. Concentrations of lactate used in this study are physiologically relevant because lactate levels in tumor can reach up to 40 mmol/L.21 Exogeneous lactate induced a dramatic dose‐dependent increase in TAZ expression and activity in both cell lines. Lactate‐induced TAZ activation is supported by the parallel increase in TAZ target gene CTGF expression. In contrast, lactate had no overt effect on both levels of steady‐state and phosphorylated form YAP. Importantly, lactate triggered a robust induction of synthetic TEAD reporter (8xGTIIC‐Luc), a readout of TAZ/YAP activity in vivo (Figure 1B). This induction was dependent on TAZ activity, as depletion of TAZ by siRNA abolished the effect of lactate. Crucially, suppression of YAP failed to affect the lactate‐induced activity of 8xGTIIC‐Luc (Figure 1B), again indicating that TAZ rather than YAP is the target of lactate signaling. A hallmark of TAZ regulation is its phosphorylation by LATS1/2 to drive cytoplasmic sequestration and degradation. Consistently, we found that lactate exposure caused a reduction in both TAZ phosphorylation and the levels of LATS2, whereas the amount of LATS1 remained unchanged (Figure 1A), suggesting lactate‐induced TAZ activation might involve a LATS2‐dependent arm. Therefore, transfections were done to generate cells with increased levels of LATS2; overexpression of LATS2 markedly reduced lactate‐induced TAZ expression compared with control‐vector‐transfected cells (Figure 1C; Figure S1B). These data together suggest that LATS2 inhibition is required for the induction of TAZ activity by lactate in lung cancer cells.
Figure 1.
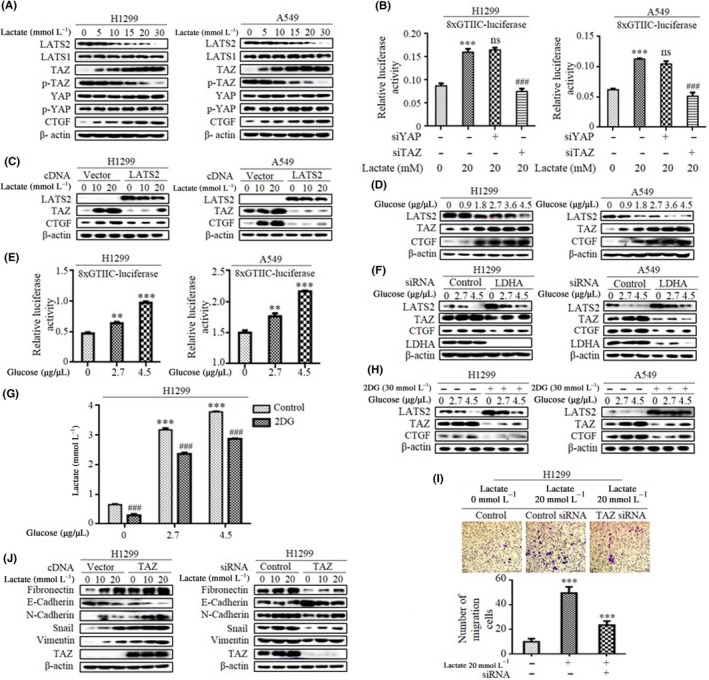
Glycolysis‐derived lactate is a key driving transcriptional co‐activator with PDZ binding domain (TAZ) upregulation in lung cancer cells. A, Expression levels of several key components of Hippo pathway in cells were analyzed by western blotting. B, Relative luciferase activities were measured when cells were cotransfected with synthetic TEAD reporter 8xGTIIC‐luc in combination with siRNA against either YAP or TAZ. ***Difference from control cells, ###difference from control siRNA‐transfected cells, ns, no statistical difference. C, Western blot shows TAZ and CTGF expression in cells after transfection with LATS2 cDNA. D, Western blot examines LATS2, TAZ and CTGF expression in cell lines after treatment with glucose. E, Glucose stimulation dose‐dependently increased synthetic TEAD reporter 8xGTIIC‐luc activity. F, Western blot shows LATS2 expression in both cell lines after transfection with lactate dehydrogenase A (LDHA) siRNA. G, Cells were treated with 2‐deoxy‐d‐glucose (2‐DG) and then extracellular lactate concentration was measured by Lactate Colorimetric/Fluorometric Assay Kit (Biovision). ***Difference from control cells, ###difference from 2‐DG‐treated cells. H, Western blot shows LATS2, TAZ and CTGF expression in cells treated with 2‐DG. I, Transwell chamber images (upper panel) and quantitative analysis (lower panel) show that the increased potential for the invasion of lactate‐treated cells is lost after TAZ silencing. J, Western blot shows expression levels of epithelial‐mesenchymal transition markers and Snail after transfection with TAZ cDNA and siRNA. All, *P < .05; **P < .01; ***P < .001; ### P < .001. (Western blotting quantitative analysis of Figure 1 is shown in Figure S1.)
Metabolic features of the Warburg effect are a hallmark of cancer, where most tumors drive glucose toward lactate for rapid energy production. To show the biological relevance of TAZ activation induced by lactate, we first evaluated whether the metabolic status of lung cancer cells influences TAZ expression. As shown in Figure 1D, glucose stimulation dose‐dependently increased the expression of TAZ and the target gene CTGF (Figure S1C). In contrast, the levels of LATS2 protein were significantly decreased by glucose addition. Accordingly, glucose treatment also induced the 8xGTIIC‐Luc reporter in a dose‐dependent method in both H1299 and A549 cells (Figure 1E). To validate the effect of glycolysis on TAZ activation in a lactate‐dependent method, we used an RNA interference approach to knock down LDHA, which directs the conversion of pyruvate into lactate and sustains aerobic glycolysis. Depletion of LDHA dramatically elevated LATS2 expression, and diminished glucose‐induced TAZ activation (Figure 1F and Figure S1D). To further confirm the involvement of glycolysis in TAZ activation, we treated cells with glycolysis blocker 2‐deoxy‐d‐glucose (2‐DG), a competitive inhibitor of hexokinase. As expected, extracellular lactate level, an indicator of glycolysis, was markedly decreased in the presence of 2‐DG (Figure 1G), notably, 2‐DG treatment significantly attenuated glucose‐stimulated TAZ and CTGF expression (Figure 1H; Figure S1E), reinforcing the key role of glycolysis‐derived lactate for induction of TAZ activity.
Elevated lactate has been associated with the acquisition of an aggressive and invasive phenotype. To confirm the importance of lactate‐induced TAZ activation, we next assessed the effect of manipulation of TAZ levels on lactate‐induced migration and invasion in lung cancer cells. We first used overexpression approaches to determine whether TAZ is sufficient to induce cellular mobility. Indeed, introduction of TAZ into H1299 cells led to an increase in cell motility in wound‐closure assays as well as invasiveness judged by the cell numbers that invaded the Matrigel‐coated chamber (Figure S1F). The increased migratory and invasive potential of lactate‐treated cells was lost after TAZ silencing (Figure 1I). In concert with the observed increases in motile and invasive responses induced by lactate, a similar upregulation of epithelial marker E‐cadherin accompanied by a decrease in mesenchymal markers fibronectin, Vimentin and N‐cadherin expression was induced in a TAZ siRNA‐sensitive method (Figure 1J; Figure S1G). In addition to enhanced migration and invasion, TAZ activation also enables other key attributes of cancer cells including aberrant cell proliferation and acquisition of cancer stem cell traits. Consistently, we found that overexpression of TAZ in H1299 cells significantly increased colony formation (Figure S1H), and cell proliferation (Figure S1I). Previous studies reported that expression of Oct4, Nanog, CD133,22 CD44,23 CD117,24 Notch1,25 SOX226 and CD2427 correlate with stemness potential in lung cancer cells. Strikingly, qRT‐PCR analysis confirmed upregulation of these lung cancer stem cell signature genes in TAZ‐overexpressing cells (Figure S1J). Furthermore, we evaluated the potential correlations among LDHA, TAZ and their associations with patient survival. We found that LDHA and TAZ expression were negatively associated with overall survival and first progress survival in patients with NSCLC in the combined data sets (http://www.kmplot.com; Figure S1K). Together, these data support the proposition that TAZ activation regulated by the Warburg effect controls lactate‐mediated migratory and invasive phenotype and are clinically associated with patient outcomes.
3.2. DNA methyltransferase 1‐dependent methylation is required for lactate‐induced LATS2 suppression
As the ability of lactate to activate TAZ requires LATS2 inhibition, we next dissected the molecular mechanism by which lactate regulates LATS2 expression in lung cancer cells. We first measured the mRNA levels of the LATS2 gene; qRT‐PCR results showed that lactate decreased mRNA levels in a dose‐dependent method in both H1299 and A549 cells (Figure 2A). The promoter activity of LATS2 was also measured using dual luciferase report assay. Consistent with mRNA expression, lactate significantly decreased LATS2 promoter activity in both cell lines (Figure 2B). A feature closely linked to transcriptional inhibition is focal DNA methylation of the gene promoter, and hypermethylation of the LATS2 gene promoter has been reported in breast cancer and leukemia.28, 29 To address the possibility that diminished expression of LATS2 arose from its promoter methylation, we then examined the relationship between the methylation profile of the LATS2 promoter and its expression in human lung cancer cases from The Cancer Genome Atlas (TCGA) databases. We found that expression levels of LATS2 inversely correlated with the methylation status of LATS2 promoter (R = −.3854, P < .0001) (Figure 2C). Intriguingly, no correlation was found between LATS1 levels and its promoter methylation.
Figure 2.
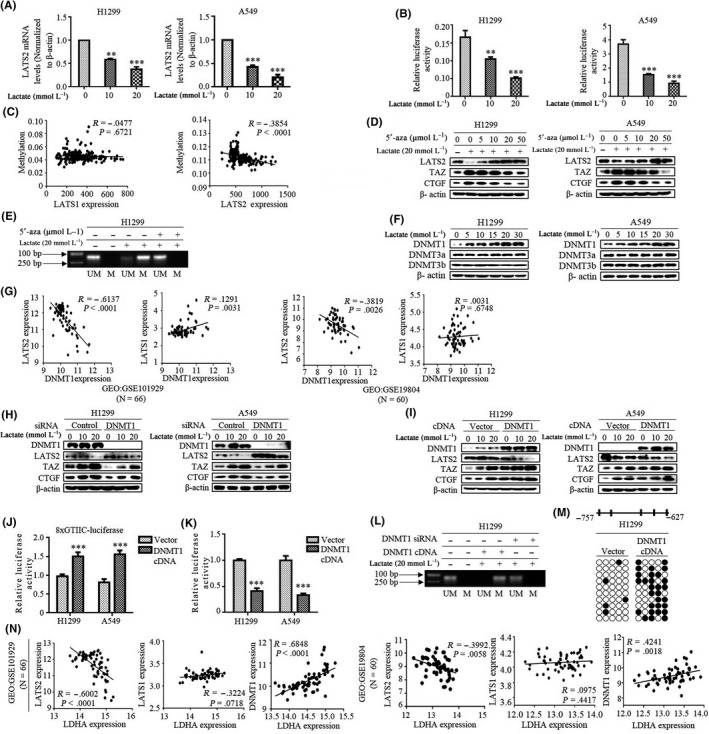
DNA methyltransferase 1 (DNMT1)‐dependent methylation is required for lactate‐induced LATS2 suppression. A, LATS2 mRNA was measured by qRT‐PCR in cells with the indicated lactate treatment. B, LATS2 promoter luciferase activities were measured after treatment with lactate. C, Expression levels of LATS2 inversely correlated with the methylation status of LATS2 promoter. D, In cells treated with demethylating agent 5′‐aza in the presence of lactate, western blot shows LATS2, transcriptional co‐activator with PDZ binding domain (TAZ) and CTGF expression. E, methylation‐specific PCR (MSP) analysis around the area of LATS2 CpG island in H1299 cells treated with lactate and 5′‐aza. M, methylated; UM, unmethylated. F, Western blot examines DNMT1, DNMT3A/3B expression in cells treated with lactate. G, Gene Expression Omnibus (GEO) DataSet analysis with lung cancer patients shows that DNMT1 expression was inversely correlated with LATS2 expression, but there was no obvious correlation with LATS1 expression. H and I, Western blot analysis of LATS2, TAZ and CTGF after transfection with DNMT1 siRNA (H) and DNMT1 cDNA (I). J and K, Either 8xGTIIC‐Luc (J) or LATS2 promoter (K) relative luciferase activities were measured after overexpression with DNMT1 cDNA. L, H1299 was transfected with DNMT1 cDNA or siRNA, and MSP was used to analyze the methylation of the LATS2 promoter. M, Sequencing of the amplified bisulfite‐treated DNA (BSP) analysis of DNMT1‐induced methylation of CpG islands in the LATS2 promoter (from −648 to −748) compared with control vector. Open circles indicate unmethylated CpG sites, whereas solid circles indicate methylated CpG sites. Data are shown as results of 10 clones. N, GEO DataSet analysis of the correlation of LDHA expression with LATS2, LATS1 and DNMT1 expression in lung cancer patients. All **P < 0.01, ***P < .001. (Western blotting quantitative analysis of Figure 2 is shown in Figure S2.)
To determine the direct involvement of methylation in the decreased expression of LATS2 induced by lactate, cells were treated with demethylating agent 5′‐aza‐2′deoxycytidine (5′‐aza) in the presence of lactate. 5′‐Aza resulted in a considerable increase in LATS2 expression in a dose‐dependent way, which was paralleled by the reduction of TAZ and CTGF (Figure 2D; Figure S2A). To further address this issue, we carried out methylation‐specific PCR (MSP) analysis around the area of CpG islands from −648 to −748 in the promoter of LATS2 in H1299 cells. We detected lactate‐induced hypermethylation at this location (Figure 2E); importantly, hypermethylation status was lost after the addition of cells with 5′‐aza, indicating that methylation is clearly responsible for downregulation of LATS2 expression induced by lactate.
DNA methylation is typically mediated by DNMT. To explore the molecular basis of lactate‐induced hypermethylation of the LATS2 gene, we investigated the possible implication of DNMT in LATS2 promoter methylation. Figure 2F shows that lactate dose‐dependently increased DNMT1 levels, but did not cause discernable change in the levels of DNMT3a and DNMT3b in both cell lines (Figure S2B). In agreement with this, DNMT1 level was found to be inversely correlated with LATS2 expression in the GEO database, as no obvious correlation was observed between the levels of DNMT1 and LATS1 (Figure 2G). To further confirm the key role of DNMT1 in LATS2 methylation, we analyzed LATS2 expression following DNMT1 knockdown. Validated DNMT1 siRNA effectively downregulated DNMT1 and also caused a dramatic increase in the levels of LATS2 in tandem with a decrease in TAZ and CTGF expression (Figure 2H; Figure S2C). Surprisingly, there was still a lactate effect on LATS2 expression because even DNMT1 knockdown was efficient, suggesting that DNMT1 is unlikely to be the only driving factor responsible for reduction of LATS2 levels by lactate. Further studies using transient transfection with DNMT1 cDNA showed that overexpression of DNMT1 strongly inhibited LATS2 expression, which also led to TAZ activation as shown by elevated levels of TAZ and CTGF (Figure 2I; Figure S2D) and activity of TEAD reporter (8xGTIIC‐Luc) (Figure 2J). In addition, cells with DNMT1 overexpression showed significant reduction in the promoter activity of the LATS2 gene (Figure 2K), suggesting that DNMT1 is a key methyltransferase that silences LATS2 expression through selective methylation of the LATS2 promoter. To further validate these findings, MSP was carried out in H1299 cells to determine the methylation status of the LATS2 promoter after DNMT1 silencing. Levels of methylation were significantly attenuated by knockdown of endogenous DNMT1, whereas DNMT1 overexpression resulted in a large induction of LATS2 promoter methylation (Figure 2L). Similarly, bisulfite sequencing analysis confirmed transfection of DNMT expression vector induced hypermethylation of the LATS2 promoter at the same CpG islands compared with control vector (Figure 2M). Interestingly, analysis of GEO data indicated that LDHA expression was positively correlated with DNMT1 and negatively correlated with LATS2 expression (Figure 2N). In summary, these results indicate that lactate‐induced LATS2 suppression is due to transcription silencing by DNMT1‐dependent methylation.
3.3. Lactate oxidation facilitated by monocarboxylate transport 1 contributes to lactate‐induced DNMT1 upregulation
To further explore the mechanism of DNMT1 regulation by lactate, we first investigated whether GPR81, a G‐protein‐coupled receptor for lactate,7, 30 is required for induction of DNMT1 by lactate. Surprisingly, lactate‐induced DNMT1 levels were significantly decreased when H1299 cells were transfected with GPR81 expression vector (Figure 3A; Figure S3A). In contrast, knockdown of endogenous GPR81 dramatically enhanced the induction of DNMT1 by lactate (Figure 3B; Figure S3B), suggesting that the GPR81 receptor functions as sink for lactate and the levels of extracellular lactate might play a key role in the regulation of DNMT1.
Figure 3.
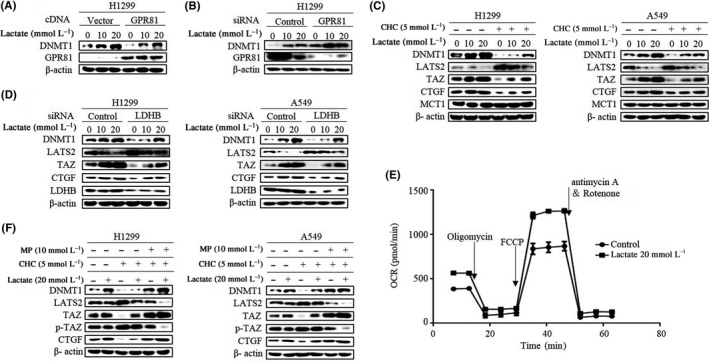
Lactate oxidation facilitated by monocarboxylate transport 1 (MCT1) contributes to lactate‐induced DNA methyltransferase 1 (DNMT1) upregulation. A and B, After transfection with GPR81 cDNA (A) or GPR81 siRNA (B), western blot was carried out for analysis of DNMT1 levels. C, Western blot analysis of DNMT1, LATS2, transcriptional co‐activator with PDZ binding domain (TAZ), CTGF and MCT1 in cells treated with lactate in the presence of α‐cyano‐4‐hydroxycinnamate (CHC). D, Western blot analysis of DNMT1, LATS2, TAZ and CTGF in cells transfected with lactate dehydrogenase B (LDHB) siRNA following lactate treatment. E, Cells were treated with lactate, then oxygen consumption rate (OCR) was analyzed using Agilent Seahorse instrument (Agilent). FCCP, p‐trifluoromethoxy carbonyl cyanide phenylhydrazone. F, Cells were either pretreated with CHC or left untreated, and then western blot analysis of DNMT1, LATS2, TAZ, p‐TAZ and CTGF in cells in the presence of lactate or methyl pyruvate (MP) was carried out. (Western blotting quantitative analysis of Figure 3 is shown in Figure S3.)
As cancer cells use exogenous lactate for metabolic purposes, and monocarboxylate transport 1 (MCT1) is a key transport protein for the uptake of lactate into cells,31 we used MCT1 inhibitor α‐cyano‐4‐hydroxycinnamate (CHC) to manipulate lactate transport. No difference of MCT1 expression level was observed in control and CHC‐treated cells. In contrast, CHC (5 mmol/L) diminished lactate‐induced DNMT1 expression and suppressed TAZ activation as shown by increased LATS2 levels as well as decreased TAZ and its downstream target CTGF expression in both cell lines (Figure 3C; Figure S3C), suggesting that lactate‐induced DNMT1 required MCT1‐facilitated uptake of lactate into the cell. Inside cells, lactate dehydrogenase B (LDHB) catalyzes lactate into pyruvate, which then fuels the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS).4 We therefore aimed to characterize the contribution of LDHB and OXPHOS in the regulation of DNMT1. Silencing of LDHB caused significant reduction of DNMT1 protein levels induced by lactate (Figure 3D; Figure S3D). To further confirm the potential involvement of OXPHOS, we analyzed the oxygen consumption rate (OCR), an indicator of cellular OXPHOS, in lactate‐treated cells. Lactate treatment resulted in higher OCR measured in H1299 cells after serial addition of oligomycin, FCCP (p‐trifluoromethoxy carbonyl cyanide phenylhydrazone), antimycin A and rotenone (Figure 3E). Consistent with the above findings, methylpyruvate, a membrane permeable form of pyruvate, also increased DNMT1 expression and TAZ activation even in the presence of MCT1 inhibitor CHC (Figure 3F; Figure S3E). Together, these data showed the significance of lactate oxidation and OXPHOS in the regulation of DNMT1 expression as well as TAZ activation.
3.4. Lactate inhibits GSK‐3β/β‐TrCP‐mediated ubiquitination and degradation leading to DNMT1 upregulation
It has been established that DNMT1 protein levels are post‐transcriptionally regulated by the AKT/GSK‐3β/β‐TrCP ubiquitylation pathway.13 We questioned whether the ubiquitin‐proteasome system also plays a role in lactate‐induced DNMT1. We first measured DNMT1 protein half‐life upon lactate treatment. Indeed, protein synthesis inhibitor cycloheximide (CHX) chase assay confirmed lactate treatment prolonged DNMT1 protein half‐life compared with untreated H1299 cells (Figure 4A; Figure S4A). Moreover, graded overexpression of β‐TrCP diminished endogenous DNMT1 levels and TAZ activation in a dose‐dependent method (Figure 4B; Figure S4B). Strikingly, immunoprecipitation assay (IP) confirmed that DNMT1 ubiquitylation was reduced after lactate treatment (Figure 4C). This finding of reduction in ubiquitylation of DNMT1 by lactate promoted us to ask whether lactate also regulates the activity of the AKT‐GSK‐3β pathway. We found that lactate was able to stimulate AKT activity as indicated by enhanced phosphorylation of AKT and GSK‐3β in a dose‐dependent way (Figure 4D; Figure S4C). AKT phosphorylates and inactivates GSK‐3β which attenuates DNMT1 phosphorylation and recruitment of β‐TrCP. In line, ectopic expression of wild‐type GSK‐3β markedly decreased DNMT1 expression and increased LATS2 expression accordingly (Figure 4E; Figure S4D). In contrast, treatment with GSK‐3β inhibitor elevated lactate‐induced DNMT1 levels (Figure 4F; Figure S4E), suggesting that the AKT‐GSK‐3β pathway is required for induction of DNMT1 protein levels induced by lactate. This conclusion was further potentiated by ectopic AKT expression; it showed that overexpression of AKT dose‐dependently increased DNMT1 levels (Figure 4G; Figure S4F), and a consistent result was obtained with AKT inhibitor LY294002. LY294002 significantly diminished DNMT1 levels accompanied by increased LATS2 levels (Figure 4H; Figure S4G). IP assay also showed that ubiquitination of DNMT1 was attenuated in LY294002‐treated H1299 cells (Figure 4I). In addition, levels of phosphorylation of AKT and GSK‐3β induced by lactate were markedly decreased in the presence of CHC (Figure 4J; Figure S4H), firmly supporting the involvement of the MCT1‐facilitated transport of lactate in the regulation of the AKT/GSK‐3β/β‐TrCP pathway and DNMT1 accumulation.
Figure 4.
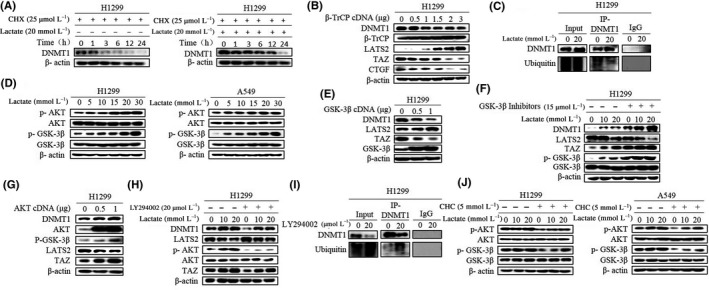
Lactate inhibits glycogen synthase kinase 3 beta/beta‐transducin repeat‐containing proteins (GSK‐3β/β‐TrCP)‐mediated ubiquitination and degradation leading to DNA methyltransferase 1 (DNMT1) upregulation. A, Cells were treated with cycloheximide (CHX) for the indicated time in the presence or absence of lactate. Western blot was used to determine DNMT1 protein levels. B, After cells were transfected with β‐TrCP cDNA, western blot showed DNMT1 protein decreased in a dose‐dependent way. C, Cells were treated with lactate after which cell lysates were immunoprecipitated with anti‐DNMT1 antibody and then western blotted with anti‐ubiquitin. Normal IgG was used as a negative control. D, Expression levels of p‐AKT and p‐GSK‐3β were analyzed by western blotting. E and F, Western blotting shows the levels of DNMT1, transcriptional co‐activator with PDZ binding domain (TAZ) and LATS2 after overexpression with GSK‐3β cDNA (E) and treatment with GSK‐3β inhibitor (F). G and H, Western blotting shows the levels of DNMT1, TAZ and LATS2 after overexpression with AKT cDNA (G) and treatment with LY294002 (H). I, Cells were treated with LY294002, after which cell lysates were immunoprecipitated with anti‐DNMT1 antibody and then western blotted. Normal IgG was used as a negative control. J, Levels of phosphorylation of AKT and GSK‐3β induced by lactate were markedly decreased in the presence of CHC. (Western blotting quantitative analysis of Figure 4 is shown in Figure S4.)
3.5. Lactate‐induced ROS regulates AKT and TAZ activity
We further explored the mechanism by which lactate regulates AKT activity. Once exogenous lactate enters cells, it must be converted to pyruvate and go through OXPHOS in the mitochondria. Given that mitochondria ROS activated the AKT pathway in our previous study,32 we then explored whether extracellular lactate might increase intracellular ROS levels. The fluorescent probe DCFH‐DA was used to detect endogenous ROS. Immunofluorescence showed that lactate exposure caused a marked increase in fluorescent signal in dose‐dependent way (Figure 5A). To explore whether the increased ROS levels during lactate‐fueled OXPHOS were responsible for AKT activation, we then used the antioxidant N‐acetyl‐l‐cysteine (NAC) to inhibit the lactate‐induced increase in ROS, and it showed that both p‐AKT and p‐GSK‐3β levels were attenuated and both DNMT1 levels and TAZ activation were inhibited in a dose‐dependent way (Figure 5B; Figure S5A). In line with the role of ROS in activating AKT, treatment of cells with hydrogen peroxide (H2O2) induced AKT and GSK‐3β phosphorylation and increased DNMT1 and TAZ expression levels (Figure 5C; Figure S5B). Furthermore, analyses with MitoTracker Green FM (Invitrogen), which measures mitochondrial mass, indicated that lactate treatment did not alter mitochondrial mass. In contrast, staining cells with mitochondrial potentiometric dye MitoTracker Red CMXRos (Invitrogen), which measures actively respiring mitochondria and therefore mitochondrial ROS,33 showed a significant increase in fluorescent intensity in the presence of lactate (Figure 5D).
Figure 5.
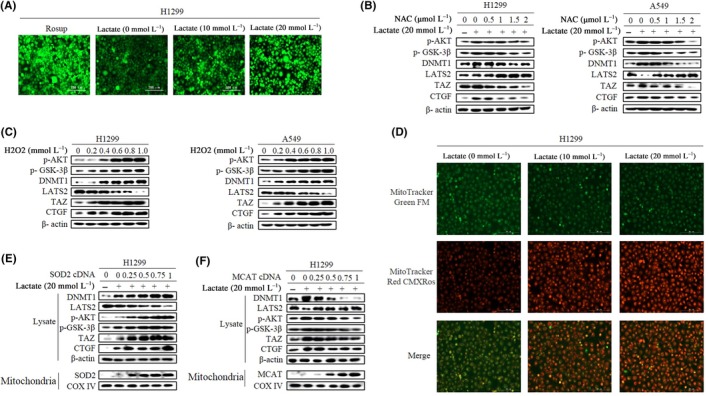
Lactate‐induced reactive oxygen species (ROS) regulates AKT and transcriptional co‐activator with PDZ binding domain (TAZ) activity. A, H1299 cells were treated with lactate and positive control (Rosup, a compound mixture for oxidative stress). ROS levels were analyzed by fluorescent probe DCFH‐DA. (Original magnification × 100. Scale bar, 200 μm). B, Western blotting shows expression levels after treatment with N‐acetyl‐l‐cysteine (NAC) in the presence of lactate. C, Cells were treated with H2O2 and protein expression levels determined by western blot analysis. D, H1299 cells were treated with lactate, mitochondria were labeled with MitoTracker Green FM (Invitrogen), and ROS in mitochondria were labeled with MitoTracker Red CMXRos (Invitrogen) and photographed under a fluorescence microscope (original magnification × 100. Scale bar, 200 μm). E and F, H1299 cells were transfected with either superoxide dismutase 2 (SOD2) expression plasmid (E) or plasmid mitochondrial targeted catalase (MCAT) (F). Protein levels were prepared and analyzed by western blot with antibodies against indicated proteins. (Western blotting quantitative analysis of Figure 5 is shown in Figure S5.)
To further determine whether lactate‐induced AKT activation was dependent on mitochondrial ROS, we took advantage of routinely used ROS enzyme superoxide dismutase 2 (SOD2), which converts superoxide into hydrogen peroxide (H2O2) in mitochondria, and mitochondrial targeted catalase (MCAT), which is targeted to mitochondria for decomposing H2O2. Increasing concentration of SOD2 levels as shown by blotting of mitochondrial fraction significantly augmented lactate‐induced AKT and TAZ activation as indicated by increasing levels of p‐AKT, TAZ and CTGF (Figure 5E; Figure S5C). Both AKT and TAZ activation was dramatically mitigated when cells were transfected with MCAT in a dose‐dependent method (Figure 5F; Figure S5D). Taken together, our results provide strong evidence for a central role of lactate‐induced H2O2 in the activation of AKT and TAZ.
3.6. DNA methyltransferase 1 is a direct transcriptional target of the TAZ‐TEAD complex
DNA methyltransferase 1 is essentially involved in the initiation and progression of many types of cancer including lung cancer. We showed that glycolysis‐derived lactate post‐transcriptionally increased DNMT1 stability and activity. Interestingly, we also noted strong induction of DNMT1 mRNA levels by lactate (Figure 6A). Therefore, a luciferase reporter harboring WT promoter DNMT1 was generated. Strikingly, the promoter activity of DNMT1 was markedly induced by lactate treatment in both H1299 and A549 cells (Figure 6B), indicating that lactate also transcriptionally increases DNMT1 expression.
Figure 6.
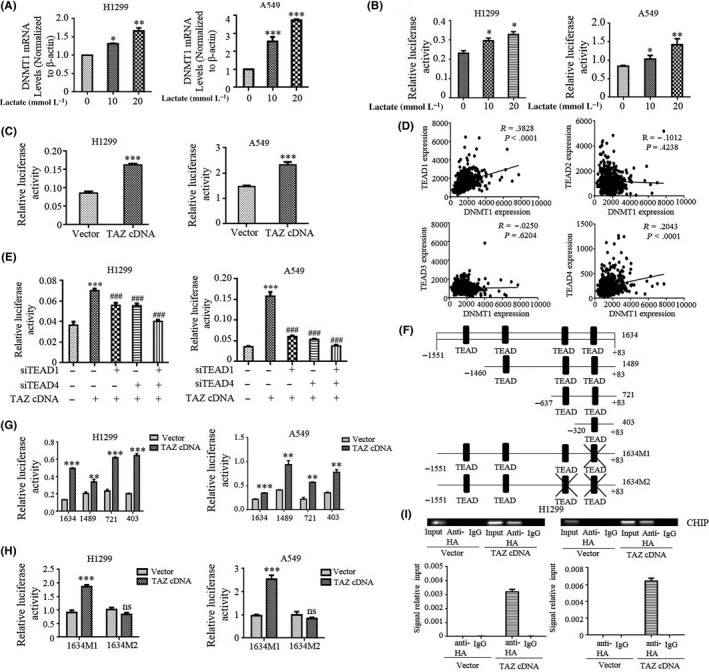
DNA methyltransferase 1 (DNMT1) is a direct transcriptional target of the transcriptional co‐activator with PDZ binding domain (TAZ)‐TEAD4 complex. A, DNMT1 mRNA was measured by qRT‐PCR in cells with indicated lactate treatment. B, DNMT1 promoter luciferase activities were measured after treatment with lactate. C, DNMT1 full‐length promoter luciferase activities were measured after transfection with TAZ cDNA in cells. D, LUAD cohort of The Cancer Genome Atlas (TCGA) analyzed the correlation of TEAD expression with DNMT1 expression. E, Cells were cotransfected with DNMT1 promoter and TAZ cDNA and siTEAD1 or siTEAD4 (***P < .001 for difference from control cells; ### P < .001 for difference from control siRNA‐transfected cells). F, Schematic representation of four different lengths of the DNMT1 promoter normal construct and two TEAD1/4 mutant constructs. G and H, Cells were transiently cotransfected with TAZ cDNA, four different lengths of the DNMT1 promoter luciferase reporter (G) and two mutant TEAD binding site constructs (H), and luciferase activity was determined. I, H1299 cells were subjected to ChIP assays by using anti‐HA antibodies or control IgG after transfection with HA‐TAZ. (Upper panel) Standard PCR products were run and scanned. (Lower panel) qRT‐PCR results were quantified and are indicated. All *P < .05, **P < .01, ***P < .001
To address the mechanism by which lactate promotes DNMT1 transcription, we carried out bioinformatic analysis to examine the promoter sequence of DNMT1 for potential binding sites of lactate‐induced transcriptional factors and we found four putative TEAD binding sites. As TAZ does not bind DNA directly, transcriptional responses to TAZ activity are mediated by members of the transcriptional factor TEAD family, and we questioned whether TAZ regulates the transcription of DNMT1. To examine this, DNMT1 promoter activity was tested with the dual luciferase reporter assay in TAZ‐transfected cells. Ectopic expression of TAZ efficiently induced DNMT1‐luciferase reporter in both cell lines (Figure 6C), consistent with direct regulation by TAZ.
To more generally determine the contribution of TAZ/TEAD to DNMT1 expression, we first analyzed the LUAD cohort of TCGA and found that TEAD1 and TEAD4 were positively correlated with DNMT1 expression (Figure 6D). Accordingly, TEAD1 and TEAD4 knockdown markedly reduced the activity of TAZ‐stimulated DNMT1 promoter reporter in both H1299 and A549 cells (Figure 6E), suggesting that induction of DNMT1 by TAZ was mediated at least by TEAD1/4. To specifically address whether TAZ/TEAD directly regulates DNMT1 transcription, set deletion constructs of DNMT1 promoter reporter containing various predicted TEAD binding sites were generated (Figure 6F). Constructs that contained all four or even one predicted TEAD binding site all responded to TAZ overexpression (Figure 6G). Surprisingly, the promoter activity of DNMT1 (1634M1) was still responsive to TAZ when the last TEAD putative site was mutated (Figure 6H). However, mutation of the last two TEAD binding sites (1634M2) abrogated TAZ‐induced expression of the DNMT1 promoter reporter in both H1299 and A549 cells (Figure 6H). Moreover, ChIP assays in the lysates prepared from the HA‐TAZ transfected cells showed that TAZ is specifically associated with the region containing the last two TEAD binding sites in the DNMT1 promoter (Figure 6I). Collectively, these observations support a model in which DNMT1 induces TAZ activation to constitute a feedback mechanism in the lactate‐induced processes of tumorigenesis.
4. DISCUSSION
As a consequence of metabolic reprogramming, tumor cells rely on aerobic glycolysis and secrete a large amount of lactate into the tumor microenvironment.34 High levels of lactate concentration in the microenvironment are now recognized as a hallmark of cancer cells and are correlated with poor clinical outcome. Many studies have suggested that lactate has a critical immunosuppressive role.5, 35 We previously identified that TAZ mediates lactate‐induced programmed death‐ligand 1 (PD‐L1) expression.7 However, the mechanism of how TAZ expression and activity are regulated by lactate has been relatively unexplored. The present study showed that lactate accumulation induced hyperactivity of TAZ through the interplay between glycolysis and oxidative metabolism. Induction of TAZ involves metabolism of lactate in the mitochondria facilitated by MCT1‐mediated transport of lactate into cells. Mitochondrial ROS generated by lactate‐fueled OXPHOS was able to stimulate AKT activity, thereby attenuating the GSK‐3β/β‐TrCP ubiquitination pathway which, in turn, led to DNMT1 accumulation and activation. DNMT1 methylated the LATS2 promoter, thus overcoming its inhibitory effect on TAZ activity. Moreover, TAZ in complex with TEAD directly stimulated DNMT1 transcription, constituting a DNMT1‐TAZ positive feedback loop. Together, our findings show that the DNMT1‐mediated feedback mechanism enables robust regulation of TAZ induced by tumor metabolic reprogramming.
Tumor cells profoundly rely on aerobic glycolysis to meet their bioenergetic and biosynthetic demands. The enhanced glycolysis, in turn, leads to production of high levels of lactate in the tumor microenvironment. However, many studies have suggested that rather than acidifying the tumor environment, lactate functions as an oxidative metabolite to fuel OXPHOS. Transport of lactate into or out of cells is mediated by monocarboxylate transporters (MCT), among which MCT1 and MCT4 have been identified to transport lactate across cell membranes. MCT4 operates almost exclusively as a lactate‐exporting protein, whereas MCT1 is primarily involved in lactate uptake.4 After being transported into cells, lactate is converted into pyruvate by LDHB, pyruvate then fuels the TCA cycle and OXPHOS. Activity of oxidative metabolism in tumor cells has been the subject of intensive discussion. This has originated in part from Warburg’s claim that the mitochondria in cancer cells are permanently impaired. However, strong evidence has indicated that the TCA cycle and oxidative metabolism in tumor cells are intact, and that global ATP concentration changes only marginally.36, 37, 38 Our results showing that lactate can be substituted for glucose to fuel oxidative metabolism might provide several advantages to tumor cells. Oxidation of lactate yields 18 ATP per lactate molecule, thereby maintaining adequate energy production and also facilitating the cell’s use of TCA cycle intermediates as biosynthetic precursors. Moreover, the increased ROS levels generated from lactate‐fueled OXPHOS can also activate pro‐survival pathways such as AKT as shown by the present study. In line with our results, MCT1 and LDHB were significantly associated with poor patient outcome, and an inhibitor of MCT1, AZD3965, has entered clinical trials.1
Over the past decade, TAZ and its related protein YAP have emerged as important drivers of tumor growth and metastasis. YAP/TAZ function as transcriptional co‐activators complexed mainly with the transcription factor TEAD to promote cancer development through the simultaneous induction of cell proliferation, stem cell amplification, and promotion of immunosuppression and metastasis. In the present study, we presented evidence showing that TAZ is located downstream of lactate signaling and plays a crucial role in some lactate‐induced biological functions. Consistent with our previous study,7 we again failed to detect changes in YAP protein steady‐state levels and activity upon lactate treatment. TAZ is negatively regulated by two closely related kinase LATS1/2 in the canonical Hippo pathway. On phosphorylation by LATS1/LATS2, TAZ are transcriptionally inactivated through cytoplasmic translocation. Although LATS1 and LATS2, which are located on chromosomes 6q25.1 and 13q12.11, respectively, show extensive sequence similarity, many of their functions are nevertheless distinct.18 For example, LATS2 plays a crucial role in embryonic stem cells and differentiation, which is not shared with LATS1. LATS2 was also reported to increase the invasive potential of breast cancer cell lines harboring mutant p53, suggesting that LATS1 and LATS2 may play different roles in tumorigenesis. Classically, tumor suppressors may undergo loss‐of‐function through mutations; however, mutations in LATS1/2 are rarely found in human cancers. Promoter methylation is the main mechanism by which LATS1/2 are often inactivated, and they are also differentially regulated at the transcriptional level.18 In this study, we found that lactate treatment exclusively repressed LATS2 but not LATS1 expression through promoter methylation. These results also shed new light on the modalities by which the Hippo pathway controls TAZ/YAP activity. Prior to this study, we considered that TAZ/YAP activity was equitably regulated by LATS1/2. Herein, we show that lactate regulates TAZ expression in a LATS2‐dependent method. We speculated that under certain cellular conditions, LATS2 might interact with TAZ only. Complementary to this work, the Luo group has identified that LATS2 rather than LATS1 is critical for Ski‐mediated inhibition of TAZ.39 Consistently, SnoN, a Ski homolog, inhibits binding of LATS2 to TAZ and subsequent phosphorylation of TAZ.40 However, we reason that the specific relevance of LATS2/TAZ interaction will likely depend on the cellular context as, in many cell types or experimental conditions, TAZ was controlled by both LATS1/2.
Epigenetic disorder has an essential role in tumors, many of which are mediated by altered expression and activity of DNMT1. DNMT1 is generally referred to as a maintenance methyltransferase and is responsible for maintaining the methylation pattern after DNA replication. However, mounting evidence indicates that DNMT1 also functions as a de novo methyltransferase in both stem cells and differentiated cells. Accordingly, prevalent upregulation of DNMT1 correlates well with aberrant DNA methylation in various cancers including lung cancer, resulting in lymph node metastasis and shorter survival in patients. The mechanism underlying DNMT1 overexpression has been extensively studied. Herein, we provide compelling evidence that TAZ directly regulates DNMT1 transcription mediated by the TEAD4 transcriptional factor.
Notably, post‐transcriptional regulation has been reported to mediate protein stability and activity of DNMT1. Acetyltransferase Tip60 and GSK‐3β have been shown to acetylate and phosphorylate DNMT1, respectively, thus triggering ubiquitination‐mediated degradation of DNMT1. Consistent with this, we showed that lactate activates AKT through the generation of ROS in the mitochondria, then inhibits GSK‐3β/β‐TrCP‐mediated protein degradation, leading to accumulation of DNMT1. Taken together, our study describing the lactate‐induced feedback loop between DNMT1 and TAZ enhances our understanding of the complex and dynamic relationship between glycolysis and oxidative metabolism in tumor cells (Figure 7). In addition, rigorous dissection of the role of lactate in lung cancer could provide novel mechanistic insights into how metabolism reprogramming functions in lung cancer formation and progression.
Figure 7.
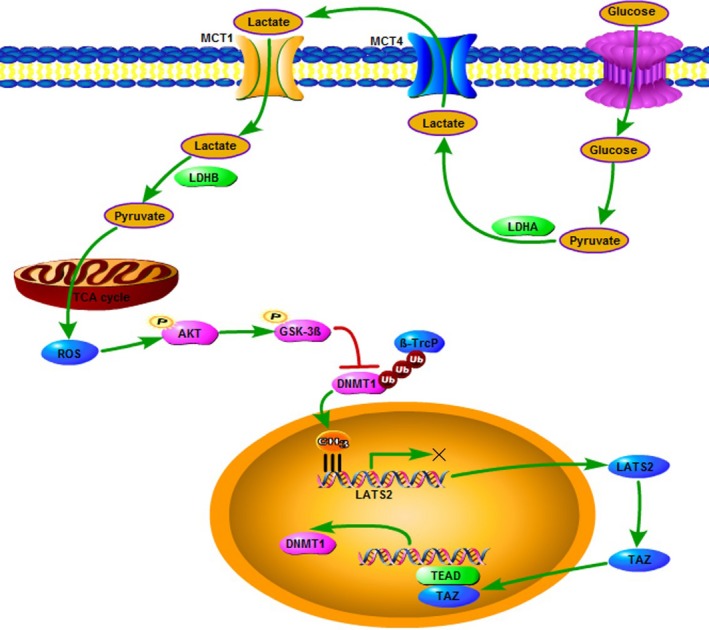
Schematic representation of lactate signaling in the regulation of DNA methyltransferase 1‐ transcriptional co‐activator with PDZ binding domain (DNMT1‐TAZ) feedback loop. GSK‐3β, glycogen synthase kinase 3 beta; LDH, lactate dehydrogenase; MCT1, monocarboxylate transport 1; ROS, reactive oxygen species; TCA, tricarboxylic acid; β‐TrCP, beta‐transducin repeat‐containing proteins
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (81872371 to Zhihao Wu), Anhui Provincial Natural Science Foundation (1708085MH203 to Zhihao Wu), Open Project Program of State Key Laboratory of Molecular Oncology (SKL‐KF‐2019‐11 to Zhihao Wu) and Natural Science Foundation of the Higher Education Institutions of Anhui Province (KJ2018A0248 Huijun Wei). Tao Huang, Xinglu Zhou, Xike Mao, Chenxi Yu, Tianyu Su and Chenchen Chen are team members of Precision Medicine Interesting Group.
Huang T, Zhou X, Mao X, et al. Lactate‐fueled oxidative metabolism drives DNA methyltransferase 1‐mediated transcriptional co‐activator with PDZ binding domain protein activation. Cancer Sci. 2020;111:186–199. 10.1111/cas.14246
Tao Huang and Xinglu Zhou contributed equally to this work.
Xike Mao and Chenxi Yu contributed equally to this work.
REFERENCES
- 1. Martinez‐Outschoorn UE, Peiris‐Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11‐31. [DOI] [PubMed] [Google Scholar]
- 2. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruocco MR, Avagliano A, Granato G, et al. Metabolic flexibility in melanoma: a potential therapeutic target. Semin Cancer Biol. 2019;59:187‐207. [DOI] [PubMed] [Google Scholar]
- 4. Gallo M, Sapio L, Spina A, Naviglio D, Calogero A, Naviglio S. Lactic dehydrogenase and cancer: an overview. Front Biosci (Landmark Ed). 2015;20:1234‐1249. [DOI] [PubMed] [Google Scholar]
- 5. Gottfried E, Kreutz M, Mackensen A. Tumor metabolism as modulator of immune response and tumor progression. Semin Cancer Biol. 2012;22:335‐341. [DOI] [PubMed] [Google Scholar]
- 6. Li X, Zhang Z, Zhang Y, Cao Y, Wei H, Wu Z. Upregulation of lactate‐inducible snail protein suppresses oncogene‐mediated senescence through p16(INK4a) inactivation. J Exp Clin Cancer Res. 2018;37:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng J, Yang H, Zhang Y, et al. Tumor cell‐derived lactate induces TAZ‐dependent upregulation of PD‐L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36:5829‐5839. [DOI] [PubMed] [Google Scholar]
- 8. Zhang W, Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630‐641. [DOI] [PubMed] [Google Scholar]
- 10. Benetatos L, Vartholomatos G. On the potential role of DNMT1 in acute myeloid leukemia and myelodysplastic syndromes: not another mutated epigenetic driver. Ann Hematol. 2016;95:1571‐1582. [DOI] [PubMed] [Google Scholar]
- 11. Gowher H, Stockdale CJ, Goyal R, Ferreira H, Owen‐Hughes T, Jeltsch A. De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry. 2005;44:9899‐9904. [DOI] [PubMed] [Google Scholar]
- 12. Kim JS, Kim H, Shim YM, Han J, Park J, Kim DH. Aberrant methylation of the FHIT gene in chronic smokers with early stage squamous cell carcinoma of the lung. Carcinogenesis. 2004;25:2165‐2171. [DOI] [PubMed] [Google Scholar]
- 13. Lin RK, Hsieh YS, Lin P, et al. The tobacco‐specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 2010;120:521‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piperi C, Vlastos F, Farmaki E, Martinet N, Papavassiliou AG. Epigenetic effects of lung cancer predisposing factors impact on clinical diagnosis and prognosis. J Cell Mol Med. 2008;12:1495‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246‐257. [DOI] [PubMed] [Google Scholar]
- 16. Lin JI, Poon CL, Harvey KF. The Hippo size control pathway–ever expanding. Sci Signal. 2013;6:pe4. [DOI] [PubMed] [Google Scholar]
- 17. Yu T, Bachman J, Lai ZC. Mutation analysis of large tumor suppressor genes LATS1 and LATS2 supports a tumor suppressor role in human cancer. Protein Cell. 2015;6:6‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24:1488‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cordenonsi M, Zanconato F, Azzolin L, et al. The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell. 2011;147:759‐772. [DOI] [PubMed] [Google Scholar]
- 20. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walenta S, Mueller‐Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol. 2004;14:267‐274. [DOI] [PubMed] [Google Scholar]
- 22. Petpiroon N, Bhummaphan N, Soonnarong R, et al. Ti0.8O2 nanosheets inhibit lung cancer stem cells by inducing production of superoxide anion. Mol Pharmacol. 2019;95:418‐432. [DOI] [PubMed] [Google Scholar]
- 23. Leung EL, Fiscus RR, Tung JW, et al. Non‐small cell lung cancer cells expressing CD44 are enriched for stem cell‐like properties. PLoS ONE. 2010;5:e14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levina V, Marrangoni A, Wang T, et al. Elimination of human lung cancer stem cells through targeting of the stem cell factor‐c‐kit autocrine signaling loop. Cancer Res. 2010;70:338‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Xu W, Guo H, et al. NOTCH1 signaling regulates self‐renewal and platinum chemoresistance of cancer stem‐like cells in human non‐small cell lung cancer. Cancer Res. 2017;77:3082‐3091. [DOI] [PubMed] [Google Scholar]
- 26. Ooki A, Dinalankara W, Marchionni L, et al. Epigenetically regulated PAX6 drives cancer cells toward a stem‐like state via GLI‐SOX2 signaling axis in lung adenocarcinoma. Oncogene. 2018;37:5967‐5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu H, Mu J, Xiao J, et al. CD24 negative lung cancer cells, possessing partial cancer stem cell properties, cannot be considered as cancer stem cells. Am J Cancer Res. 2016;6:51‐60. [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi Y, Miyoshi Y, Takahata C, et al. Down‐regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res. 2005;11:1380‐1385. [DOI] [PubMed] [Google Scholar]
- 29. Jimenez‐Velasco A, Roman‐Gomez J, Agirre X, et al. Downregulation of the large tumor suppressor 2 (LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19:2347‐2350. [DOI] [PubMed] [Google Scholar]
- 30. Liu C, Wu J, Zhu J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G‐protein‐coupled receptor, GPR81. J Biol Chem. 2009;284:2811‐2822. [DOI] [PubMed] [Google Scholar]
- 31. Romero‐Garcia S, Moreno‐Altamirano MM, Prado‐Garcia H, Sanchez‐Garcia FJ. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol. 2016;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo L, Li L, Wang W, Pan Z, Zhou Q, Wu Z. Mitochondrial reactive oxygen species mediates nicotine‐induced hypoxia‐inducible factor‐1α expression in human non‐small cell lung cancer cells. Biochim Biophys Acta. 2012;1822:852‐861. [DOI] [PubMed] [Google Scholar]
- 33. Basu S, Rajakaruna S, Dickinson BC, Chang CJ, Menko AS. Endogenous hydrogen peroxide production in the epithelium of the developing embryonic lens. Mol Vis. 2014;20:458‐467. [PMC free article] [PubMed] [Google Scholar]
- 34. Avagliano A, Granato G, Ruocco MR, et al. Metabolic reprogramming of cancer associated fibroblasts: the slavery of stromal fibroblasts. Biomed Res Int. 2018;2018:6075403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seth P, Csizmadia E, Hedblom A, et al. Deletion of lactate dehydrogenase‐A in myeloid cells triggers antitumor immunity. Cancer Res. 2017;77:3632‐3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate‐fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930‐3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345‐19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015;79:253‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rashidian J, Le Scolan E, Ji X, et al. Ski regulates Hippo and TAZ signaling to suppress breast cancer progression. Sci Signal. 2015;8:ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu Q, Le Scolan E, Jahchan N, Ji X, Xu A, Luo K. SnoN antagonizes the hippo kinase complex to promote TAZ signaling during breast carcinogenesis. Dev Cell. 2016;37:399‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials