

Abstract
Accumulating evidence has revealed that human cancers develop by sequentially mutating pivotal genes, including driver genes, and acquiring cancer hallmarks. For instance, cancer cells are addicted to the transcription factor NRF2 (NFE2L2), which is a driver gene that utilizes the cellular cytoprotection system against oxidative stress and metabolic pathway reprogramming for sustaining high growth. Our group has recently discovered a new addiction to the NRF2‐related factor NRF3 (NFE2L3) in cancer. For many years, the physiological function of NRF3 remained obscure, in part because Nrf3‐deficient mice do not show apparent abnormalities. Nevertheless, human cancer genome databases suggest critical roles of NRF3 in cancer because of high NRF3 mRNA induction in several cancer types, such as colorectal cancer and pancreatic adenocarcinoma, with a poor prognosis. We found that NRF3 promotes tumor growth and malignancy by activating ubiquitin‐independent 20S proteasome assembly through inducing the expression of the proteasome maturation protein (POMP) chaperone and thereby degrading the tumor suppressors p53 and Rb. The NRF3‐POMP‐20S proteasome axis has an entirely different effect on cancer than NRF2. In this review, we describe recent research advances regarding the new cancer effector NRF3, including unclarified ubiquitin‐independent proteolysis by the NRF3‐POMP‐20S proteasome axis. The expected development of cancer therapeutic interventions for this axis is also discussed.
Keywords: 20S proteasome, NRF2, NRF3, p53, ubiquitin‐independent degradation
Cancer cells hijack cellular systems for sustaining high growth by acquiring gene mutations. For example, they are addicted to the transcription factor NRF2 (NFE2L2). We herein summarize recent research advances regarding the new cancer effector NRF3 (NFE2L3), including unclarified ubiquitin‐independent proteolysis by the NRF3‐POMP‐20S proteasome axis.
Abbreviations
- APC
adenomatous polyposis coli
- ARE
antioxidant response element
- bZip
basic‐region leucine zipper
- CNC
cap ‘N’ collar
- CPEB3
cytoplasmic polyadenylation element‐binding protein 3
- DDI1
DNA damage‐inducible protein 1
- DDI2
DDI1 homolog 2
- ER
endoplasmic reticulum
- KEAP1
Kelch‐like ECH‐associated protein 1
- LEF
lymphoid enhancer‐binding factor
- NFE2L3
NFE2‐like 3
- NHB1
N‐terminal homology box 1
- NRF3
NFE2‐related factor 3
- PAAD
pancreatic adenocarcinoma
- POMP
proteasome maturation protein
- Rb
retinoblastoma
- RP
regulatory particle
- TCF4
T‐cell factor 4
1. INTRODUCTION
Within the past half‐century, extensive efforts to overcome cancer, including DNA sequencing of human cancer tissues and bioinformatics analyses, have led directly to the characterization of significant functional mutations in genes and pathways in tumors and the development of new cancer therapies against these targets, in other words, precision medicine.1 These results further highlight that human tumors develop in a multistep process through sequentially acquiring the hallmarks of cancer, comprising at least 8 biological capabilities: sustained proliferative signaling, growth suppressor evasion, cell death resistance, replicative immortality, angiogenesis, invasion and metastasis, energy metabolism reprogramming, and immune destruction evasion.2 These hallmarks are endowed to cells by mutations of pivotal genes.3 Moreover, 2 new concepts of gene mutations related to carcinogenesis have emerged: “driver gene mutations” and “passenger gene mutations”.4, 5 Driver gene mutations confer selective growth advantages to tumor cells, and more than 100 driver genes, such as PIK3CA, SMAD4, and TP53, have been identified. Passenger gene mutations are considered to be just “passengers” and have no effect on the tumorigenic process.
The transcription factor NRF2, which plays crucial roles in cytoprotection against oxidative stress and electrophiles, is one of the cancer driver genes.6, 7, 8 NRF2 mediates the expression of genes involved in the oxidative stress response and metabolic reprogramming, such as glutaminolysis, and its functional activity is regulated by KEAP1, which is a dual functional protein that acts as both an oxidative stress sensor and a ubiquitin E3 ligase.9, 10, 11, 12 Cancer cells depend, namely “addict” these biological functions of NRF2 for their aberrant growth.6, 8 Accordingly, these insights also suggest that the KEAP1‐NRF2 axis provides an attractive target for anticancer drug development, and numerous pharmaceutical companies are struggling to generate both NRF2 inhibitors and activators.13
We recently discovered a new cancer addiction to NFE2L3, which is entirely different from NRF2. In this review, we summarize recent breakthroughs in understanding the physiological function of NRF3 in tumors, especially through ubiquitin‐independent proteolysis by the 20S proteasome.
2. IDENTIFICATION OF NRF3 ADDICTION IN CANCER
2.1. Remarkable upregulation of the NRF3 gene in several cancer tissues
Human cancer databases, such as The Cancer Genome Atlas and Oncomine, strongly suggest the biological relevance of NRF3 in tumors because of the following 4 points:
A substantial number of cancer tissues, namely bladder urothelial carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, colon adenocarcinoma, lymphoid neoplasm diffuse large B‐cell lymphoma, esophageal carcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma, kidney chromophobe, ovarian serous cystadenocarcinoma, PAAD, rectum adenocarcinoma, stomach adenocarcinoma, testicular germ cell tumors, thyroid carcinoma, thymoma, uterine corpus endometrial carcinoma, and uterine carcinosarcoma, show high upregulation of the NRF3 gene compared to adjacent normal tissues (Figure 1).14, 15
Although it is not currently considered as a driver gene, NRF3 is one of 127 significantly mutated genes among 12 cancer types.5
High NRF3 expression is correlated with poor prognosis in PAAD, in terms of both overall survival and disease‐free survival.16
Moreover, recent evidence indicates that NRF3 regulates the growth and metastasis of thyroid, testis, and breast cancer.17, 18, 19, 20
Figure 1.
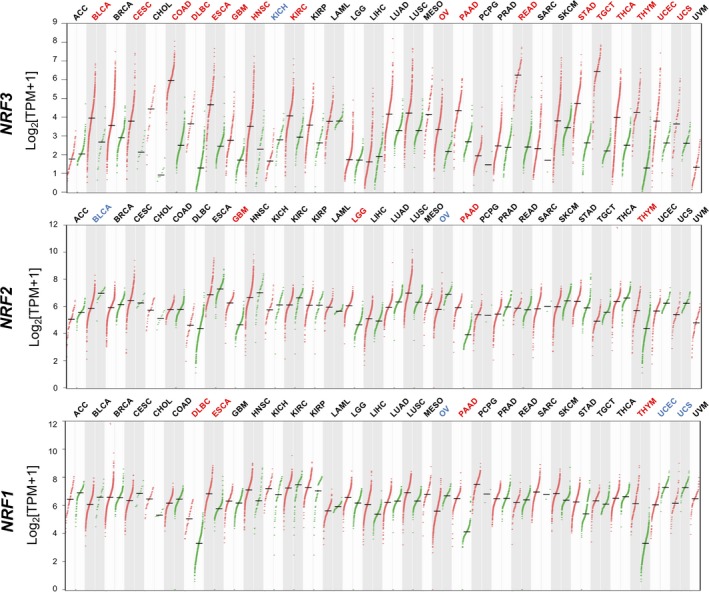
Remarkable upregulation of NFE2‐related factor 3 (NRF3) in various cancer tissues. Dot plots profiling NRF3 (top), NRF2 (middle), and NRF1 (bottom) gene expression across multiple cancer types and paired normal samples from the GEPIA web server.15 Each red or green dot represents a distinct tumor or normal specimen, respectively. Magenta‐ or blue‐colored abbreviations on the upper part of each graph indicate a cancer type with significantly higher or lower NRF gene expression levels, respectively, in cancer tissues than in normal adjacent tissues. ANOVA, q value cut‐off = 0.01. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, diffuse large B‐cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney chromophobe; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemisa; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; TPM, transcripts per million; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma
NRF3, which was initially identified by our group,21 belongs to the CNC‐type bZip transcription factor family, including NRF2 and NRF1 (NFE2L1). As described, NRF2 is well‐known for its cytoprotective effects against oxidative stress, and NRF1 sustains protein homeostasis (eg, proteostasis) by mediating proteasome gene expression following proteasome inhibition.6, 22, 23 These transcription factors augment gene expression by binding to the ARE in genes by heterodimerizing with small Maf proteins. Considering their protein structures and amino acid sequences, it is likely that NRF3 is a close homologue of NRF1 but not NRF2 (as discussed in Section 4). NRF3 shows abundant expression in the cornea, skin, bladder, and placenta, but it shows ubiquitous low‐level expression in other tissues. It has also been reported that ES cells and induced pluripotent stem cells of nonhuman primates show NRF3 upregulation.24 Intriguingly, remarkable NRF3 expression is induced in several types of cancer cells (Figure 1).14, 25 This expression profile of NRF3 is clearly distinct from those of both NRF1 and NRF2, which are expressed ubiquitously and are not significantly upregulated in cancer cells. Regarding the role of NRF2, cancer cells activate their selective growth advantages mainly by somatic mutations of the NRF2 and KEAP1 genes, as well as aberrant posttranslational modifications and protein‐protein interactions.6, 8 These observations imply unique roles of NRF3 in tumors from those of NRF2 and NRF1.
The physiological function of NRF3 remains unclear, in part because Nrf3‐deficient mice, which were generated independently by our and another laboratories, do not show apparent abnormalities under physiological conditions.26, 27 In chemical carcinogenesis experiments, benzo[a]pyrene highly induces T‐cell lymphoblastic lymphoma in Nrf3 KO mice, implying a protective role of Nrf3 in hematopoietic malignancies.28 We herein describe the new mechanisms in tumors in which NRF3 induces tumor growth and malignancy by degrading the tumor suppressor proteins p53 and Rb through activating 20S proteasome assembly.
2.2. Proteasome‐mediated proteostasis in cancer
Before discussing the pathological roles of NRF3 in tumors, we would like to introduce the pivotal roles of the proteasome in cancer. Increasing evidence suggests that cancer cells have a heightened dependence on mechanisms of protein homeostasis (proteostasis) or protein quality control because cancer cells synthesize many WT and mutant proteins (Figure 2).29 To sustain proteostasis, the ubiquitin‐proteasome system is activated for protein degradation mechanisms as well as autophagy. Thus, cancer cells are more susceptible to proteasome inhibition than normal cells, and proteasome inhibitors, such as bortezomib, are consequently utilized to treat multiple myeloma and mantle cell lymphoma.29
Figure 2.
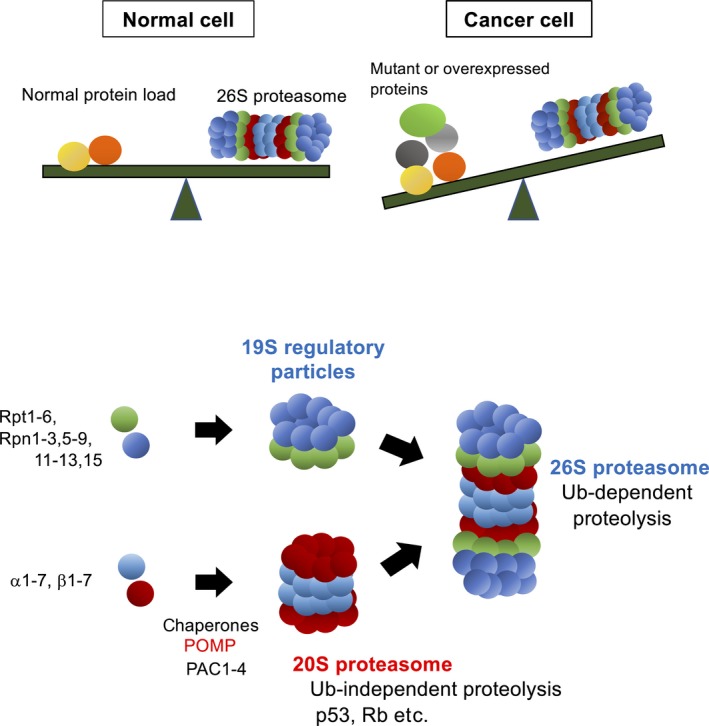
Imbalance of proteostasis in cancer cells. Top panel, In normal cells, the 26S proteasome sustains protein homeostasis (ie, proteostasis) by degrading protein in a ubiquitin‐dependent manner.29 In cancer cells, high levels of normal or mutated proteins are expressed for higher proliferation, consequently causing an imbalance where the degradation load exceeds the ubiquitin‐proteasome system capacity. Bottom panel, Molecular mechanisms of the assembly of the 20S and 26S proteasome complexes. The proteasome maturation protein (POMP) chaperone promotes the assembly of the 20S proteasome along with PAC1‐4 chaperones. The 26S and 20S proteasomes degrade proteins in a ubiquitin (Ub)‐dependent and Ub‐independent manner, respectively.30 PAC, proteasome assembly chaperone; Rpn, regulatory particle non‐ATPase; Rpt, regulatory particle triple‐A protein
The proteasome in the ubiquitin‐proteasome system is the 26S proteasome.29, 30 The 26S proteasome is an unusually large complex that consists of 66 subunits expressed from 33 genes and is composed of 2 portions, the 20S proteasome and the 19S‐RP: the 20S proteasome is a catalytic domain for proteolysis, and 19S‐RP recognizes polyubiquitin chains conjugated to substrate proteins and unfolds their structure by using ATP for proteolysis through the 20S proteasome. The molecular mechanisms underlying proteasome gene expression in mammals remained obscure for a long time. Recently, several transcription factors have been reported to mediate gene expression.31 For example, NRF3‐related NRF1 mediates the gene expression of most 26S proteasome subunits in response to proteasome inhibition32, 33, 34, 35 (it is very confusing that “NRF1” stands for 2 distinct transcription factors, “NFE2L1” and “nuclear respiratory factor 1”). This cellular response is called the “proteasome bounce‐back response” (or proteasome recovery) and sustains proteostasis despite proteasome dysfunction. Similarly, NRF2 directly induces 26S proteasome gene expression under oxidative stress conditions, implying that it supports the degradation of proteins damaged by reactive oxygen species.36
2.3. NRF3‐POMP‐20S proteasome axis attenuates p53/Rb function by degrading in a ubiquitin‐independent fashion
Let us return to the question of the physiological function of NRF3 in cancer cells. Based on the fact that NRF1 is a regulatory factor of 26S proteasome expression, we hypothesized that NRF3 also regulates 26S proteasome gene expression; however, no alteration of proteasome gene expression was observed in NRF3 knockdown cancer cells. Alternatively, NRF3 directly augments the expression of POMP as a chaperone for the assembly of the 20S proteasome complex (Figures 2 and 3)30 (T. Waku, N. Nakamura, M. Koji, H. Watanabe, H. Katoh, C. Tatsumi, N. Tamura, A. Hatanaka, S. Hirose, H. Katayama, M. Tani, Y. Kubo, J. Hamazaki, T. Hamakubo, A. Watanabe, S. Murata, A. Kobayashi, under peer review). Induced POMP expression enhances the assembly and activity of the 20S proteasome, thus degrading the tumor suppressor proteins p53 and Rb in a ubiquitin‐independent manner and consequently increasing cell death resistance and cell cycle arrest in cells. The biological function of the 20S proteasome remains unclear compared with that of the ubiquitin‐dependent 26S proteasome (as discussed below in Section 4). Some in vitro studies revealed that the 20S proteasome degrades unfolded proteins and certain other proteins, including p53 and Rb, in a ubiquitin‐independent fashion because of the lack of 19S‐RP.37 Regarding the relevance of NRF3 to POMP, the related factor NRF2 is also known to mediate POMP gene expression, conferring slight bortezomib resistance in multiple myeloma.38 Additionally, it has been reported that POMP inhibition by microRNA‐101 reduces tumor cell proliferation,39 although this report explores the effects of microRNA‐101 on only the 26S proteasome and not the 20S proteasome.
Figure 3.
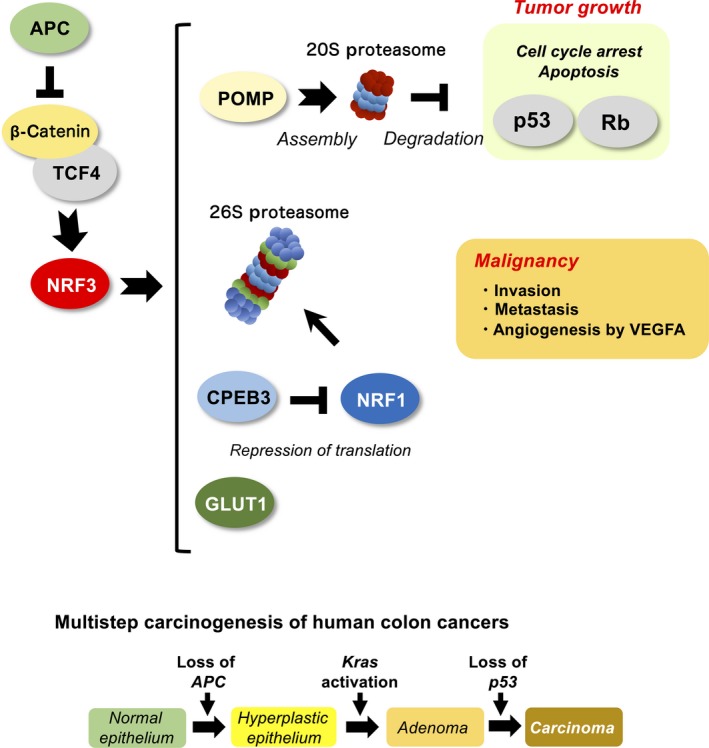
Pathological roles of NFE2‐related factor 3 (NRF3) in cancer. Top panel, NRF3 mRNA is highly induced in various cancer tissues by the β‐catenin/T‐cell factor (TCF)4 complex in the Wnt signaling pathway.42 As a result, NRF3 augments the expression of several genes, such as POMP, 26S proteasome subunits, CPEB3, and GLUT1, in tumors. Bottom panel, Genetic alterations associated with colorectal tumorigenesis. Loss of APC is an initial event of tumorigenesis, thus activating β‐catenin in the Wnt signaling pathway.3 Rb, retinoblastoma; VEGFA, vascular endothelial growth factor A
2.4. NRF3 and NRF1 complementarily regulate basal 26S proteasome activity through a translational mechanism in cancer
Although NRF3 does not likely contribute to proteasome gene expression, we found that both NRF1 and NRF3 complimentarily regulate the basal gene expression of the 26S proteasome in cancer cells (T. Waku, H. Katayama, M. Hiraoka, A. Hatanaka, N. Nakamura, Y. Tanaka, N. Tamura, A. Watanabe, A. Kobayashi, under revision). Intriguingly, NRF3 suppresses NRF1 function at the translational level (Figure 3). The underlying molecular mechanism is due to NRF3‐mediated gene induction of CPEB3, which is known to mediate protein translation both positively and negatively by regulating mRNA polyadenylation.40, 41 NRF3 augments CPEB3 gene expression by binding directly to a species‐conserved ARE site in the gene. Consequently, induced CPEB3 represses NRF1 translation through a cytoplasmic polyadenylation element in the 3′‐UTR of the NRF1 gene. This finding could explain why Nrf3 null mice do not show apparent abnormalities, because Nrf1 could rescue the loss of Nrf3 function in mice.26, 27 Moreover, the existence of a mechanism to switch from NRF1 to NRF3 might suggest that cancer cells favor NRF3 rather than NRF1, or different function of NRF3 from that of NRF1 in transcription is indispensable for sustaining aberrant growth of cancer cells, irrespective that they share similar protein structures.
2.5. Other transcriptional networks of NRF3 in tumors
To comprehensively understand the physiological roles of NRF3 in tumors, genome‐wide identification of NRF3 target genes is indispensable. To this end, we carried out ChIP sequence analyses along with transcriptome analyses using a microarray, and we discovered that the glucose transporter GLUT1 (SLC2A1) is one of the direct NRF3 target genes (Figure 3).42 Consistent with our findings, cancer cells reprogram the metabolic status from oxidative phosphorylation to anaerobic glycolysis by upregulating certain genes, including GLUT1 and GLUT3 (SLC2A3), to increase glucose uptake, which correlates with poor prognosis.43, 44, 45 This metabolic reprogramming of cancer cells is called the Warburg effect. Considering that ARE sites in the GLUT1 gene perfectly match the consensus sequence and are highly conserved among several species, it would be possible that NRF1 and NRF2 also regulate the gene expression. Indeed, we observed NRF1‐mediated regulation under certain conditions. Furthermore, NRF3 has been recently reported to upregulate VEGFA expression, promoting angiogenesis and malignancy in pancreatic cancer.16 Researchers also found NRF3 involvement in the invasion and metastasis of pancreatic cancer. Although we previously reported UHMK1 as an NRF3 target gene,46 it might be regulated indirectly by NRF3 because there are no functional ARE sites in the gene.
2.6. Upregulation of NRF3 by β‐catenin‐TCF4 in cancer cells
We next addressed the molecular mechanisms underlying NRF3 upregulation in cancer cells and identified that the β‐catenin‐TCF4 complex in the Wnt pathway directly promotes NRF3 expression.42 The Wnt‐β‐catenin pathway, which is essential for normal intestinal growth and development, plays a key role in the initiation of colorectal cancer progression.47, 48, 49 Alterations of the APC gene and the CTNNB1 gene encoding β‐catenin are initial events leading to the development of adenoma in most sporadic cases (Figure 3). As a result, β‐catenin protein stabilization promotes the expression of stemness‐related genes, such as c‐MYC, AXIN2, and LGR5, along with TCF/LEF family proteins, leading to hyperplastic epithelium.45, 50, 51 These insights give rise to the attractive hypothesis that NRF3 cancels the tumor suppressor function of p53 by activating the 20S proteasome following tumorigenesis initiation. Initially, acquiring resistance to apoptosis through gene mutations in tumor suppressors is essential for tumorigenesis, and p53 gene mutations occur at the last step in colon cancers.3
3. MOLECULAR REGULATION OF THE BIOLOGICAL FUNCTION OF NRF3
3.1. Multiple suppression mechanisms of the biological function of NRF3
Our findings regarding the deleterious effects of aberrant NRF3 activation in tumors strongly indicate that NRF3 activity (and probably its expression [Figure 1]) must be tightly regulated to sustain cellular homeostasis. Under physiological conditions (quiescent conditions), NRF3 function is suppressed by sequestration in the ER through the N‐terminal NHB1 domain, and its proteasomal degradation is mediated by the ER‐associated degradation‐related E3 ubiquitin ligase HRD1 (Figure 4).46 In the nucleus, NRF3 is subjected to alternate proteasomal degradation by β‐TRCP, which is an adaptor of Cul1‐based E3 ubiquitin ligase. It has also been reported that NRF3 is degraded in a glycogen synthase kinase 3β‐dependent manner by Fbw7, which is a famous cancer driver and adaptor of Cul1‐based ubiquitin ligase.52 These 2 independent degradation systems should suppress the biological function of NRF3 under quiescent conditions to prevent its detrimental effects.
Figure 4.
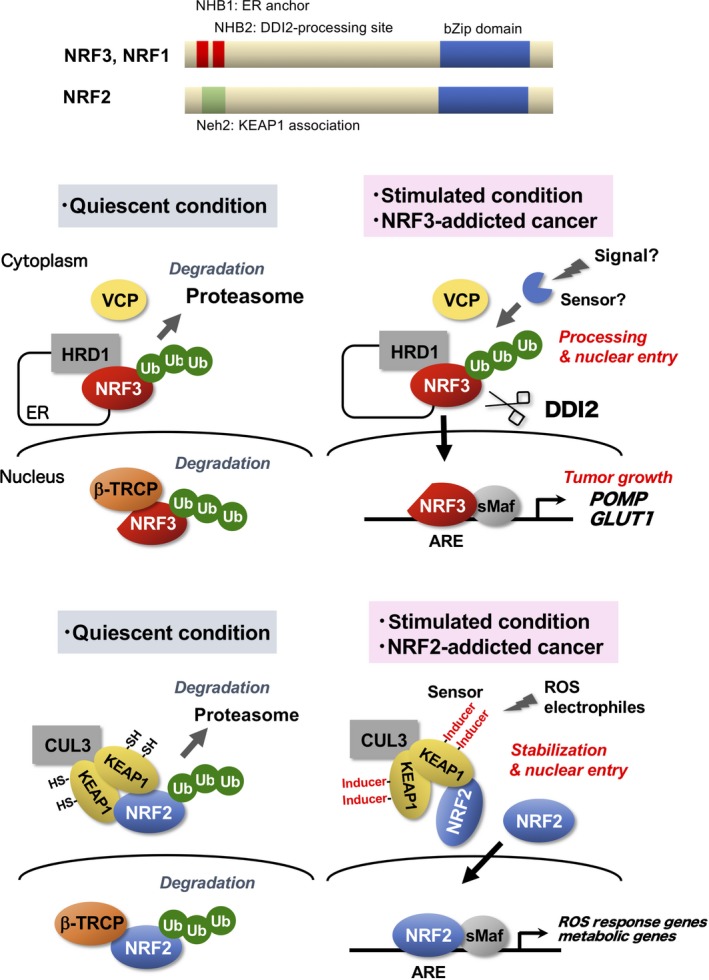
Distinct regulatory mechanisms of NFE2‐related factor 3 (NRF3) and NRF2. Top panel, Schematic structures of NRF3, NRF1 and NRF2. Middle panel, Under quiescent (physiological) conditions, the transcriptional activity of NRF3 is suppressed by sequestration in the endoplasmic reticulum (ER) and HRD1‐mediated proteasome degradation. Under stimulation conditions or in NRF3‐addicted cancer, NRF3 is liberated from multiple suppression mechanisms and processed by the DNA damage‐inducible protein 1 homolog 2 (DDI2) protease, thereby achieving liberation from the ER and activating gene expression in the nucleus.46 Bottom panel, Regulatory mechanisms of the oxidative stress response transcription factor NRF2 are completely different from those of NRF3. Under quiescent conditions, NRF2 function is suppressed by a KEAP1‐mediated proteasomal degradation mechanism.9, 10, 11, 12 The nature of the oxidative stress response by NRF2 is the derepression of this mechanism.62 β‐TRCP, beta‐transducin repeat containing E3 ubiquitin protein ligase; ARE, antioxidant response element; KEAP, Kelch‐like ECH‐associated protein 1; NHB, N‐terminal homology box; sMaf, small Maf; Ub, ubiquitin; VCP, valosin‐containing protein
3.2. Nuclear translocation of NRF3 and NRF1 requires DDI2 protease
The nuclear entry of NRF3 is suppressed by multiple mechanisms in the ER under physiological conditions. Thus, the molecular mechanisms underlying nuclear translocation play a fundamental role in NRF3 functional regulation (Figure 4). Regarding this hypothesis, new insight into the nuclear translocation of NRF1 and Skn1 has led to a major breakthrough. Aspartic proteases DDI2 and DDI1 have been discovered as modulators of their nuclear translocation, respectively.53, 54 Similarly, DDI2 is required for NRF3 nuclear translocation.46 Although there is still no experimental evidence that DDI2 directly cleaves NRF3, an AWLVH motif, which is considered a DDI2 cleavage site in the NHB2 domain, it is highly conserved among several species and in NRF1. This evidence strongly implies that DDI2 could cleave these sites, resulting in their nuclear entry.
The molecular mechanisms through which DDI2 is activated for processing NRF3 and NRF1 remain obscure. We surmise the presence of certain signals and/or stress promote processing by escape from multiple suppression mechanisms in the ER. This corresponds to oxidative stress in the case of NRF2. Despite artificial stress, proteasome inhibition by MG132 and bortezomib is established as an NRF3 and NRF1 activation signal. Endogenous NRF3 in human colon cancer DLD‐1 and HCT116 cells partly enters into the nucleus, indicating the presence of this signal in these cancer cells.46 Delineating this process, as well as identifying a sensor for the signal, is indispensable for a comprehensive understanding of NRF3 function in cancer.
3.3. Molecular bases underlying functional diversity among NRF1‐3
These observations remind us of the functional diversity among NRF1, NRF2, and NRF3: NRF1 and NRF3 are involved in the regulation of proteasome function, whereas NRF2 is involved in the regulation of oxidative stress response. These transcription factors are thought to have evolved from a common ancestral gene CNC in Drosophila and Skn‐1 in Caenorhabditis elegans.55 For example, they possess similar CNC‐type bZip domains for DNA binding activities, implying that they likely recognize similar DNA sequences and thereby share some target genes, eg, proteasome subunit genes. Nevertheless, the existence of either NHB1/2 domains in NRF1/3 or a Neh2 domain in NRF2 could apparently result in substantial functional differences among these factors because the former is a regulatory interface with the HRD1‐DDI2 axis, whereas the latter is related to KEAP1 (Figure 4). Considering that CncC and Skn‐1a possess both NHB1/2 and Neh2 domains, we surmise that distributing each domain to NRF1‐3 transcription factors in the process of molecular evolution could have led to complexities and diversities in these transcription factors and thereby vertebrates.
3.4. DDI2 inhibitors might be promising anticancer drugs that suppress NRF3
Identification of the DDI2‐mediated NRF3 activation mechanism suggests that DDI2 inhibitors would be promising anticancer drugs to inhibit NRF3 function in tumors. Intriguingly, DDI2 possesses a protease domain similar to that of HIV protease.56 Accordingly, it might be possible to repurpose clinically available HIV protease inhibitors as DDI2 inhibitors. Suppression of DDI2 likely impairs both NRF3 and NRF1 function because DDI2 is required for their nuclear entry.46, 53 Alternatively, DDI2 inhibitors might enhance the treatment effect of the anticancer drug bortezomib, which is utilized for multiple myeloma treatment by targeting proteasomes, because both NRF3 and NRF1 appear to reduce bortezomib efficacy through a proteasome bounce‐back response.
4. CONCLUSIONS: PERSPECTIVE ON FUTURE DIRECTIONS OF NRF3 RESEARCH
Human cancer databases indicate that the biological function and regulatory mechanisms of numerous genes in tumors are still not defined. Moreover, these insights shed light on new issues regarding genetic diversity in each tumor, ie, intratumoral and intertumoral heterogeneity.57, 58 Thus, continuous investigation of unknown mechanisms underlying tumorigenesis remains indispensable for the development of new cancer therapies and precision medicine.
In this regard, we have succeeded in the identification of new roles of the NRF3‐POMP‐20S proteasome axis in cancer cells. This axis eliminates the tumor suppressor function of p53 and Rb in a ubiquitin‐independent manner, thus promoting tumor growth (Figure 3). Conversely, our findings indicate that inhibition of this axis does not likely reduce the proliferation of p53‐deficient tumors, which are approximately half of all human solid tumors.59 Thus, targeting strategies for NRF3 or DDI2 might be suitable for leukemia, in which many blood cells have the intact p53 gene.60 In particular, although the proteasome inhibitor bortezomib is utilized clinically for multiple myeloma therapy, it is assumed that both NRF3 and NRF1 likely confer resistance to this anticancer drug through a “proteasome bounce‐back response” in cells. To increase the therapeutic effects of bortezomib, it might be beneficial to cotreat with a DDI2 inhibitor that likely represses both NRF3‐ and NRF1‐mediated bounce‐back responses. We surmise that the advantage of therapies targeting NRF3 is few side‐effects because Nrf3 KO mice do not show apparent abnormalities.26, 27
We identified that NRF3 promotes 20S proteasome assembly by inducing POMP expression. The biological function of the 20S proteasome itself might be still controversial61 because there are only some in vitro studies that report its function.37 Alternatively, the 20S proteasome is considered to be an intermediate form that ultimately generates 26S or other forms of proteasomes by accompanying accessory factors such as 19S‐RP. Therefore, we do not deny the possibility that our observation reflects the biological effects of the 20S proteasome and other accessory factors. Even so, it remains correct that the NRF3‐POMP‐20S proteasome axis modulates tumor growth and malignancy in a ubiquitin‐independent manner. To establish our findings, we need to undertake the following experiments: compare 20S proteasome activities in many types of cancer cells, comprehensively identify 20S proteasome substrates, and determine the molecular mechanisms of substrate‐specific recognition independent of the ubiquitin chain.
One of the remaining important questions in this research field is the physiological role of NRF3 in normal cells. This question is equivalent to another issue regarding the NRF3 activation signals and/or stress inducers under physiological conditions. To answer the essential question, the identification of whole NRF3 target genes would provide us with much insight. Indeed, we have succeeded in the identification of several NRF3 target genes by combining data from ChIP sequencing and transcriptome analyses (these data will be published in the future). These NRF3 target genes are involved in various biological mechanisms, including the mTORC1 signaling pathway and cholesterol metabolism, consistent with NRF3‐mediated tumor growth. According to these insights, certain nutrients or metabolites might regulate NRF3 function.
CONFLICT OF INTEREST
The authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
We thank all of the members of our laboratory and collaborators for contributions to our research. This work was supported in part by grants‐in‐aid for: Scientific Research (B) (16H03265 to A.K.); Challenging Research (Exploratory) (19K22826 to A.K.); Young Scientists (B) (17K18234 to T.W.); and Scientific Research (C) (19K07650 to T.W.). This work was also supported by The Uehara Memorial Foundation (to T.W.).
Kobayashi A, Waku T. New addiction to the NRF2‐related factor NRF3 in cancer cells: Ubiquitin‐independent proteolysis through the 20S proteasome. Cancer Sci. 2020;111:6–14. 10.1111/cas.14244
REFERENCES
- 1. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell. 2018;173:321‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Review hallmarks of cancer: The next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 3. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto XM, Kensler TW, Motohashi H. The KEAP1‐NRF2 system: A thiol‐based sensor‐effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98:1169‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitamura H, Motohashi H. NRF2 addiction in cancer cells. Cancer Sci. 2018;109:900‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi A, Kang M‐I, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130‐7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1‐BTB protein is an adaptor that bridges Nrf2 to a Cul3‐based E3 ligase: oxidative stress sensing by a Cul3‐Keap1 ligase. Mol Cell Biol. 2004;24:8477‐8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang DD, Lo S‐C, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox‐regulated substrate adaptor protein for a Cul3‐dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941‐10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3‐Roc1 ligase. Mol Cell Biol. 2005;25:162‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cuadrado A, Rojo AI, Wells G, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18:295‐317. [DOI] [PubMed] [Google Scholar]
- 14. Broad Institute TCGA Data . TCGA data version 2016_01_28. Available from http://gepia.cancer-pku.cn/detail.php?gene=NFE2L3. Accessed October 30, 2018.
- 15. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H, Zhan M, Yang R, Shi Y, Liu Q, Wang J. Elevated expression of NFE2L3 predicts the poor prognosis of pancreatic cancer patients. Cell Cycle. 2018;17:2164‐2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C, Saji M, Justiniano SE, et al. RCAN1‐4 is a thyroid cancer growth and metastasis suppressor. J Clin Investig insight. 2017;2:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiu ST, Hsieh FJ, Chen SW, Chen CL, Shu HF, Li H. Clinicopathologic correlation of up‐regulated genes identified using cDNA microarray and real‐time reverse transcription‐PCR in human colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:437‐443. [DOI] [PubMed] [Google Scholar]
- 19. Almstrup K, Ottesen AM, Sonne SB, et al. Genomic and gene expression signature of the pre‐invasive testicular carcinoma in situ. Cell Tissue Res. 2005;322:159‐165. [DOI] [PubMed] [Google Scholar]
- 20. Keun D, Park SH, Jang YK. Molecular signatures associated with transformation and progression to breast cancer in the isogenic MCF10 model. Genomics. 2008;92:419‐428. [DOI] [PubMed] [Google Scholar]
- 21. Kobayashi A, Ito E, Toki T, et al. Molecular cloning and functional characterization of a new cap‘n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443‐6452. [DOI] [PubMed] [Google Scholar]
- 22. Bugno M, Daniel M, Chepelev NL, Willmore WG. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim Biophys Acta. 2015;1849:1260‐1276. [DOI] [PubMed] [Google Scholar]
- 23. Chevillard G, Blank V. NFE2L3 (NRF3): the Cinderella of the Cap‘n’Collar transcription factors. Cell Mol Life Sci. 2011;3:3337‐3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu H, Zhu F, Yong J, et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell. 2008;3:587‐590. [DOI] [PubMed] [Google Scholar]
- 25. BioGPS data . http://biogps.org/#goto=genereport&xml:id=9603. Accessed November 2, 2019.
- 26. Kobayashi A, Ohta T, Yamamoto M, Ohta M, Yamamoto M. Unique function of the Nrf2‐Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273‐286. [DOI] [PubMed] [Google Scholar]
- 27. Derjuga A, Gourley TS, Holm TM, et al. Complexity of CNC transcription factors as revealed by gene targeting of the Nrf3 locus. Mol Cell Biol. 2004;24:3286‐3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chevillard G, Paquet M, Blank V. Nfe2l3 (Nrf3) deficiency predisposes mice to T‐cell lymphoblastic lymphoma. Blood. 2011;117:2005‐2008. [DOI] [PubMed] [Google Scholar]
- 29. Deshaies RJ. Proteotoxic crisis, the ubiquitin‐proteasome system, and cancer therapy. BMC Biol. 2014;12:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009;10:104‐115. [DOI] [PubMed] [Google Scholar]
- 31. Voutsadakis IA. Proteasome expression and activity in cancer and cancer stem cells. Tumor Biol. 2017;39:101042831769224. [DOI] [PubMed] [Google Scholar]
- 32. Steffen J, Seeger M, Koch A, Krüger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD‐dependent feedback loop. Mol Cell. 2010;40:147‐158. [DOI] [PubMed] [Google Scholar]
- 33. Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38:17‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsuchiya Y, Taniguchi H, Ito Y, et al. The casein kinase 2‐Nrf1 axis controls the clearance of ubiquitinated proteins by regulating proteasome gene expression. Mol Cell Biol. 2013;33:3461‐3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sha Z, Goldberg AL. Proteasome‐mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol. 2014;24:1573‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kwak M‐K, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1‐Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786‐8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ben‐nissan G, Sharon M. Regulating the 20S proteasome ubiquitin‐independent degradation pathway. Biomolecules. 2014;4:862‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li B, Fu J, Chen P, et al. The nuclear factor (erythroid‐derived 2)‐like 2 and proteasome maturation protein axis mediate bortezomib resistance in multiple myeloma. J Biol Chem. 2015;290:29854‐29868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X, Schulz R, Edmunds S, et al. MicroRNA‐101 suppresses tumor cell proliferation by acting as an endogenous proteasome inhibitor via targeting the proteasome assembly factor POMP. Mol Cell. 2015;59:243‐258. [DOI] [PubMed] [Google Scholar]
- 40. Charlesworth A, Meijer HA, De Moor CH. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip Rev RNA. 2013;4:437‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y, Tsai Y‐H, Tseng S‐H. Regulation of the expression of cytoplasmic polyadenylation element binding proteins for the treatment of cancer. Anticancer Res. 2016;36:5673‐5680. [DOI] [PubMed] [Google Scholar]
- 42. Aono S, Hatanaka A, Hatanaka A, et al. β‐catenin/TCF4 complex‐mediated induction of the NRF3 (NFE2L3) gene in cancer cells. Int J Mol Sci. 2019;20:3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Labak CM, Wang PY, Arora R, et al. Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am J Cancer Res. 2016;6:1599‐1608. [PMC free article] [PubMed] [Google Scholar]
- 44. Ancey P‐B, Contat C, Meylan E. Glucose transporters in cancer: from tumor cells to the tumor microenvironment. FEBS J. 2018;285:2926‐2943. [DOI] [PubMed] [Google Scholar]
- 45. Satoh K, Yachida S, Sugimoto M, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci USA. 2017;114:7697‐7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chowdhury AMMA, Katoh H, Hatanaka A, et al. Multiple regulatory mechanisms of the biological function of NRF3 (NFE2L3) control cancer cell proliferation. Sci Rep. 2017;7:12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nusse R, Clevers H. Wnt/β‐catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985‐999. [DOI] [PubMed] [Google Scholar]
- 48. Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Symp Quant Biol. 2012;4:1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479‐507. [DOI] [PubMed] [Google Scholar]
- 50. Yochum GS, Cleland R, Goodman RH. A genome‐wide screen for β‐catenin binding sites identifies a downstream enhancer element that controls c‐Myc gene expression. Mol Cell Biol. 2008;28:7368‐7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of β‐catenin binding regions in colon cancer cells using ChIP‐Seq. Nucleic Acids Res. 2010;38:5735‐5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kannan MB, Dodard‐Friedman I, Blank V. Stringent control of NFE2L3 (Nuclear Factor, Erythroid 2‐Like 3; NRF3) protein degradation by FBW7 (F‐box/WD Repeatcontaining Protein 7) and glycogen synthase kinase 3 (GSK3). J Biol Chem. 2015;290:26292‐26302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koizumi S, Irie T, Hirayama S, et al. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife. 2016;5:e18357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lehrbach NJ, Ruvkun G. Proteasome dysfunction triggers activation of SKN‐1A/Nrf1 by the aspartic protease DDI‐1. Elife. 2016;5:e17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fuse Y, Kobayashi M. Conservation of the Keap1‐Nrf2 system: an evolutionary journey through stressful space and time. Molecules. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sirkis R, Gerst JE, Fass D. Ddi1, a eukaryotic protein with the retroviral protease fold. J Mol Biol. 2006;364:376‐387. [DOI] [PubMed] [Google Scholar]
- 57. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Niida A, Nagayama S, Miyano S, Mimori K. Understanding intratumor heterogeneity by combining genome analysis and mathematical modeling. Cancer Sci. 2018;109:884‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood. 2017;130:699‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Demasi M, da Cunha FM. The physiological role of the free 20S proteasome in protein degradation: a critical review. Biochim Biophys Acta – Gen Subj. 2018;1862:2948‐2954. [DOI] [PubMed] [Google Scholar]
- 62. Kobayashi A, Kang M, Watai Y, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]