

Abstract
Breast cancer is the most prevalent malignancy among women. Although endocrine therapy is effective, the development of endocrine resistance is a major clinical challenge. The tumor microenvironment (TME) promotes tumor malignancy, and tumor‐associated macrophages (TAM) within the TME play a crucial role in endocrine resistance. Herein, we aimed to elucidate the relationship between TAM and the endocrine‐resistant phenotype of breast cancer. Macrophages were cultured with conditioned medium (CM) from tamoxifen‐sensitive (MCF7‐S) or ‐resistant (MCF7‐R) MCF7 breast cancer cells. M2 polarization was detected by CD163 immunofluorescence. To determine the effect on endocrine resistance, MCF7 cells were cultured in the supernatant of different TAM, and then treated with tamoxifen. CC‐chemokine ligand 2 (CCL2) immunohistochemistry was carried out on pathological sections from 100 patients with invasive estrogen receptor‐positive breast cancer. We found that macrophages cultured in the CM of MCF7‐S and MCF7‐R cells were induced into TAM, with a more obvious M2 polarization in the latter. Tamoxifen resistance was increased by culture in TAM medium. TAM secreted CCL2, which increased endocrine resistance in breast cancer cells through activation of the PI3K/Akt/mTOR signaling pathway. High expression of CCL2 was correlated with infiltration of CD163+macrophages (r = 0.548, P < .001), and patients with high CCL2 expression presented shorter progression‐free survival than those with low CCL2 expression (P < .05). We conclude that CCL2 secreted by TAM activates PI3K/Akt/mTOR signaling and promotes an endocrine resistance feedback loop in the TME, suggesting that CCL2 and TAM may be novel therapeutic targets for patients with endocrine‐resistant breast cancer.
Keywords: breast cancer, CCL2, tamoxifen resistance, tumor microenvironment, tumor‐associated macrophage
This study investigated the mechanisms underlying tumor‐associated macrophage (TAM)‐mediated endocrine resistance in breast cancer cells. We found that endocrine‐resistant breast cancer cells can induce M2 polarization of TAM, and M2‐polarized TAM in turn further promote endocrine resistance in breast cancer cells. We believe that this article will be of interest to the readership of this journal because we uncovered the underlying mechanism of TAM‐induced endocrine resistance: TAM secrete the cytokine CCL2, which activates the PI3K/Akt/mTOR signaling pathway.

Abbreviations
- CCL2
CC‐chemokine ligand 2
- CM
conditioned medium
- ER
estrogen receptor
- MCP‐1
monocyte chemoattractant protein 1
- MR
tumor‐associated macrophage from a tamoxifen‐resistant tumor microenvironment
- MS
tumor‐associated macrophage from a tamoxifen‐sensitive tumor microenvironment
- PFS
progression‐free survival
- TAM
tumor‐associated macrophage
- TME
tumor microenvironment
1. INTRODUCTION
Breast cancer is the most common cause of cancer‐related mortality in women.1 Approximately 70% of breast cancers are hormone receptor‐positive.2 Although endocrine therapy remains the most effective treatment, its efficacy is limited by intrinsic or acquired endocrine resistance.3, 4, 5 Tamoxifen, a selective ER modulator, is the most common endocrine therapy worldwide 6; hence, resistance to tamoxifen is a major clinical challenge.
Solid tumors such as breast cancer usually contain heterogeneous populations of neoplastic cells, immune cells, and collections of tissue‐specific resident and recruited stromal cell types, which together form the TME.7, 8 The relevance of this tumor heterogeneity is linked to various hallmarks of cancer including drug resistance.9, 10, 11, 12, 13, 14 Macrophages are diverse cells that show distinct phenotypes depending on their anatomical location and activating stimuli.15, 16 In breast cancer, macrophages can comprise up to 50% of the TME.17 Among them, M2‐type macrophages, commonly referred to as TAM, are associated with tumor malignancy.18, 19 CD163, a scavenger receptor, is regarded as a highly specific monocyte/macrophage marker for M2 polarization.20 Escamilla et al reported that TAM are involved in the resistance of patients with prostate cancer to androgen blockade therapy.21 In our previous study, we found a correlation between TAM and tamoxifen resistance in patients with breast cancer, but the underlying mechanism has not been elucidated.22
Tumor‐associated macrophages secrete a variety of chemokines into the TME including CCL2, which is secreted mainly by macrophages when cocultured with MCF7 cells.23 CCL2 is also known as MCP‐1 and small inducible cytokine A2. CCL2 belongs to the CC chemokine family that recruits monocytes, memory T cells, and dendritic cells to sites of inflammation.24, 25 Several studies have reported that CCL2 and the inflammatory environment promote the malignant behavior of different tumors.26, 27, 28 Xu et al found that CCL2 recruits monocytes to the TME and polarizes macrophages into the M2 type.25 Secretion of CCL2 is also an important manifestation of TAM polarization in macrophages.29 Ueno et al showed that a high level of CCL2 expression is a significant indicator of earlier relapse.30 However, there have been only a few studies on the role of CCL2 in endocrine resistance of breast cancer.
The molecular mechanism underlying endocrine resistance of breast cancer is highly complicated and involves many molecules and pathways such as overexpression of epidermal growth factor receptor and/or the HER‐2/neu/ErbB2 oncogene,31 mutations in ER or loss of ER‐alpha expression,32, 33 activation of the MAPK 32 and PI3K/Akt/mTOR signaling pathways,34 and overexpression of cytochrome P450 (CYP450) enzymes.35 Among these factors, the activation of the PI3K/Akt/mTOR signaling pathway is dominant. Thota et al observed induction of endocrine resistance by tumor macrophages, although the intrinsic molecular mechanism remains unclear.36
The aim of the present study was to explore the interaction between TAM and the endocrine‐resistant phenotype in breast cancer. CM from tamoxifen‐sensitive and ‐resistant breast cancer cells was tested for its ability to induce macrophage M2 polarization. We then examined the role of TAM in promoting tamoxifen resistance. We also identified a specific cytokine, CCL2, secreted by TAM to promote endocrine resistance and investigated its underlying mechanism of action. CCL2 immunohistochemistry was carried out on pathological sections from patients with invasive ER‐positive breast cancer to examine the correlation between CCL2 expression and disease prognosis. Our findings will further the understanding of how TAM promote tamoxifen resistance in breast cancer cells and may identify novel therapeutic targets for patients with endocrine‐resistant breast cancer.
2. MATERIALS AND METHODS
2.1. Reagents and cell culture
4‐Hydroxytamoxifen (4‐OH Tam), recombinant human MCP‐1 (CCL2), the CCL2 synthesis inhibitor Bindarit, and PMA were purchased from Sigma‐Aldrich. Antibodies against CCL2, CD163, nuclear factor kappa B (NF‐κB), and components of the PI3K/Akt/mTOR pathway were purchased from Boster. P‐FOXK1 was purchased from Invitrogen. Breast cancer cell lines MCF7 and T47D and human monocyte cell line THP‐1 were purchased from the Type Culture Collection of the Chinese Academy of Sciences and maintained in RPMI‐1640 medium supplemented with FBS. Tamoxifen‐resistant MCF7 (MCF7‐R) cells were obtained by treating MCF7 cells with 1000 nmol/L 4‐OH Tam for 3 months. MCF7‐R cells were grown in RPMI‐1640 with 5% FBS and 1000 nmol/L 4‐OH Tam to maintain resistance against tamoxifen.
2.2. Cell viability assay
Cell proliferation was assessed using CCK‐8 (Vazyme) assay according to the manufacturer’s instructions. Briefly, the cells were seeded in 96‐well plates at a density of 1 × 104 cells/well and incubated with 5 μmol/L tamoxifen for 0, 12, 24, 36, 48, 60, and 72 hours. At the end of the respective incubation times, the cells were incubated in complete medium containing 10% CCK‐8 reagent for 2 hours. Subsequently, absorbance of the sample was measured at 450 nm in a microplate reader (Tecan). The experiments were repeated three times.
2.3. Immunofluorescence staining
MΦ, MS, and MR human macrophages grown in small culture dishes were fixed with 4% paraformaldehyde for 30 minutes, followed by washing with PBS thrice. Cells were blocked with 5% BSA for 30 minutes, and then incubated with the anti‐CD163 antibody (diluted 1:1000) at 4°C overnight. Signal was detected using Alexa Fluor 488 and 594‐conjugated secondary antibodies (Invitrogen), and the samples were imaged using the Axioskop 2 mot plus fluorescence microscope equipped with Plan Apochromat 203/0.8 NA and 403/0.95 NA objectives and the AxioCam MRc camera with AxioVision software 4.7.1 (Carl Zeiss).
2.4. Real‐time quantitative PCR
Total RNA was extracted from MΦ, MS, and MR macrophages using TRIzol reagent and 1 μg RNA was reverse transcribed to cDNA using reverse transcriptase (Vazyme). Real‐time quantitative PCR (RT‐PCR) was done using the SYBR Green I Real‐Time Detection kit (CWBio) on the CFX96 Detection System (Bio‐Rad). Relative gene expression was normalized to macrophage expression. Primers for the target genes are listed in Table S1.
2.5. ELISA
CCL2 and tumor necrosis factor (TNF)α present in culture supernatants from MCF7‐S, MCF7‐R, MΦ, MS, and MR macrophages were quantified using specific ELISA kits (Quantikine Human CCL2 Immunoassay and Quantikine Human TNFα Immunoassay; R&D Systems) following the manufacturer’s instructions. Concentration of the samples was estimated from the standard curve. CCL2 and TNFα levels below the detection limit of the assay were recorded as zero.
2.6. Flow cytometry analysis
Breast cancer cells were suspended in RPMI‐1640 with 10% FBS at a concentration of 1 × 106 cells/mL. For annexin V‐FITC/propidium iodide (PI) staining, 100 μL cells were transferred to a flow cytometer tube, diluted with PBS, and centrifuged at 300 g for 5 minutes. The supernatant was then removed by decanting and blotting. For each sample, the pellet was suspended in 100 μL of 1× binding buffer, and 5 μL Annexin V‐FITC was added. The samples were incubated in dark for 15 minutes. Before flow cytometer analysis, 400 μL of 1× binding buffer and 5 μL of 50 μg/mL PI were added to each sample. For flow cytometry analysis, PI green fluorescence was dot plotted on the y‐axis and Annexin V‐FITC orange fluorescence on the x‐axis.
Macrophages were washed with PBS. PE mouse anti‐human CD163 (BD Pharmingen) was used to analyze M2 polarization of macrophages. The cells were filtered and resuspended in buffer before sorting. All analyses were conducted on FACSDiva (Version 6.1.3).
2.7. Western blotting
Cells were lysed in RIPA buffer. Protein samples were sonicated followed by centrifugation at 17 153 g for 15 minutes. Supernatants were collected and protein concentrations were determined using the Bradford Assay (Bio‐Rad). Protein lysates were then subjected to 4%‐20% Tris‐glycine SDS PAGE, and then transferred onto PVDF membranes according to the manufacturer’s instructions (Invitrogen). The membranes were blocked in 5% milk‐Tris‐buffered saline with 0.1% Tween (TBS‐T) at 23°C for 1 hour, followed by incubation with primary antibodies at 4°C overnight. On the following day, the membranes were washed with TBS‐T thrice before incubation with HRP‐conjugated secondary antibodies (Cell Signaling) at 23°C for 1 hour. Protein expression was visualized by ECL chemiluminescence (Promega) and quantified using Image J software (National Cancer Institute).
2.8. Chemotaxis assay
Microporous membrane (8‐μm pore size) Transwell inserts (Costar) were used in the chemotaxis assay. Briefly, 2 × 105 cells in 200 μL RPMI‐1640 were added to the upper chamber, and 500 μL CM from MΦ, MS, MR, and MR + Bindarit was added to the lower chamber. THP‐1 cells were allowed to migrate at 37°C for 1 hour in an atmosphere of 5% CO2/95% air, and then the inserts were fixed and stained with 0.1% crystal staining solution. Non‐migratory cells were removed before the membrane was mounted and migratory cells were observed and counted under a microscope.
2.9. Immunohistochemistry
The protocol for this study was approved by the Ethics Committee of Harbin Medical University Cancer Hospital and conforms to the provisions of the Declaration of Helsinki. Breast cancer specimens were obtained from Harbin Medical University Cancer Hospital. All patients signed informed consent forms for medical record review and tissue sample donation. Between December 2003 and June 2014, 100 patients with ER‐positive breast cancer were enrolled in this retrospective study. Slides were incubated with anti‐CCL2 antibody (1:400 dilution; BioSource International) and anti‐CD163 antibody (1:250 dilution; 10D6; Novocastra). Yellow granules indicated CCL2 positivity. CCL2 immunoreactivity was scored by staining intensity (negative, weak, moderate, or strong staining) and the percentage of positive tumor cells per core (≤25%, >25%‐50%, >50%‐75%, and >75%). Tissues were considered positive for CCL2 expression with ≥moderate staining intensity in >25% of the cells examined. Yellow granules indicated CD163 positivity. CD163 immunoreactivity was scored as the infiltration density of CD163+ macrophages ranging from 0 (absent) to 3 (dense). A score equal to or greater than 1 was regarded as positive.
2.10. Statistical analysis
Data are expressed as mean ± SD. Statistical analyses were carried out using GraphPad Prism software (version 6.0) and SPSS Statistics software (Version 19.0). Multiple comparisons were evaluated using the one‐way ANOVA. Correlation between CD163 and CCL2 was determined using Spearman rank‐order correlation. Survival curves were analyzed using Kaplan‐Meier method and curves were compared using the log‐rank test. Multivariate survival analysis using the Cox regression proportional hazards model was carried out to adjust for clinical variables that may have statistical significance for prognosis in a univariate analysis. Results with P values <.05 were considered to be statistically significant (*P < .05, **P < .01).
3. RESULTS
3.1. Conditioned medium from breast cancer cells induces macrophage M2 polarization
THP‐1 cells were first differentiated into macrophage (MΦ) by treatment with 320 nm PMA for 24 hours. Then, CM from tamoxifen‐sensitive MCF7 (MCF7‐S) and tamoxifen‐resistant MCF7 (MCF7‐R) cells was added to MΦ for another 24 hours of culture (Figure 1A). Finally, we obtained two types of TAM: TAM from a tamoxifen‐sensitive TME (MS) and TAM from tamoxifen‐resistant TME (MR).
Figure 1.
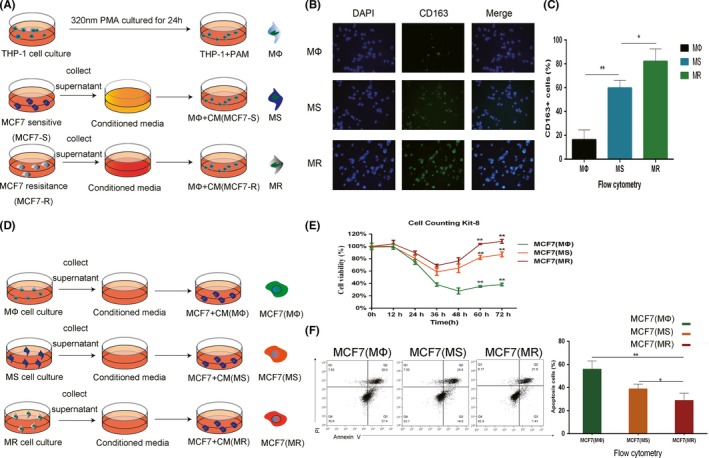
Crosstalk between tumor‐associated macrophages (TAM) and MCF7 cells. A, Procedure used to obtain macrophages in different microenvironments (MΦ, TAM, TAM from a tamoxifen‐sensitive tumor microenvironment (TME) [MS], and TAM from tamoxifen‐resistant TME [MR]). B, CD163 immunofluorescence (IF) staining in MΦ, MS, and MR macrophages. Single channels and merged images are shown. C, CD163 expression of MΦ, MS, and MR macrophages was analyzed by flow cytometry. D, Procedure used to elucidate the effects of conditioned medium (CM) from different macrophages on MCF7 cells. E, Relative viability of MCF7 (MΦ), MCF7 (MS), and MCF7 (MR) cells treated with 5 μmol/L tamoxifen. F, Apoptosis of MCF7 (MΦ), MCF7 (MS), and MCF7 (MR) cells was analyzed by flow cytometry after adding 5 μmol/L tamoxifen for 24 h. *P < .05, **P < .01
To examine the differences in the macrophages, cellular CD163 immunofluorescence and flow cytometry analysis were carried out on the MΦ, MS, and MR. CD163 is considered a polarization maker for TAM and M2. We found that CM from breast cancer cell lines increased CD163 expression in MΦ, indicating M2 polarization. CD163 expression was higher and M2 polarization was more significant in MR than in MS (Figure 1B,C). These observations indicate that macrophages can be polarized to assume different phenotypes in different microenvironments.
3.2. Conditioned medium from M2‐polarized macrophages induces MCF7 tamoxifen resistance
To investigate the function of macrophages of different phenotypes, supernatants of MΦ and tumor‐related macrophages, MS and MR, were added to tamoxifen‐sensitive MCF7 cells for 24 hours to obtain MCF7(MΦ), MCF7(MS), and MCF7(MR) cells (Figure 1D). The cells were then treated with 5 μmol/L tamoxifen. Apoptotic flow cytometry analysis was carried out and cell viability was assessed using the CCK‐8 assay (Figure 1E,F). Significant resistance to tamoxifen was observed in MCF7 cells cultured in CM of TAM (MS, MR), with MCF7(MR) being the most tamoxifen resistant. Thus, we conclude that the increased M2 polarization of TAM increased their ability to resist apoptosis and promote endocrine resistance. Endocrine‐resistant cancer cells promote M2 polarization of macrophages, which, in turn, promotes endocrine‐resistance in cancer cells.
3.3. CCL2 secreted by TAM is associated with endocrine resistance in MCF7 cells
We hypothesize that the different abilities of TAM to promote endocrine resistance is due to the different levels of secreted cytokines. After reviewing the relevant literature, we selected seven cytokines (CXCL‐1, CCL2, CCL5, interleukin [IL]‐6, IL‐8, IL‐17, and CXCL‐5) that may promote endocrine resistance in breast cancer cells.37, 38, 39, 40, 41, 42, 43 Transcription of these seven cytokines in the three macrophages (MΦ, MS, and MR) was assessed by RT‐PCR to identify the cytokines whose transcription levels were consistent with the trends from the CCK‐8 and apoptotic flow cytometry assays. Only CCL2 was consistent with the previous results and the difference was statistically significant (Figure 2A). IL‐6 also has the ability to promote endocrine resistance, and the transcription in MS and MR is different. However, our results showed that the transcription of IL‐6 in MΦ and MS was not statistically significant. Secretion of IL‐6 could not explain the difference between MΦ and MS in promoting drug resistance of endocrine tumors. Therefore, IL‐6 was not chosen as the main object of the present study. To exclude the effect of autocrine CCL2 from cancer cells, we measured CCL2 expression levels in MCF7‐S and MCF7‐R cells by ELISA, and did not detect significant levels. We then determined the CCL2 concentration in CM from the three macrophages. With M2 polarization of macrophages, the concentration of secreted CCL2 increased significantly. Concentration of CCL2 was positively correlated with the trend of endocrine resistance in MCF7 cells (Figure 2B). Thus, we conclude that CCL2 is associated with endocrine resistance promoted by TAM. Two hormone receptor‐positive cell lines, MCF7 and T47D, were selected to test the effect of CCL2. We confirmed that CCR2, the receptor of CCL2, was expressed in both cell lines (Figure S1). CCL2 (100 nm/mL) was added to the two cell lines and the cells were cultured for 24 hours. Subsequently, tamoxifen (5 μmol/L) was added to all four groups (MCF7, MCF7+CCL2, T47D, and T47D+CCL2) and the cells were cultured for 24 hours. Cell viability was assessed using the CCK‐8 assay (Figure 2C,D). The four groups were also analyzed by flow cytometry to detect apoptosis (Figure 2E,F). We found that CCL2 promotes endocrine resistance and reduces apoptotic proportion in the two hormone receptor‐positive cell lines.
Figure 2.
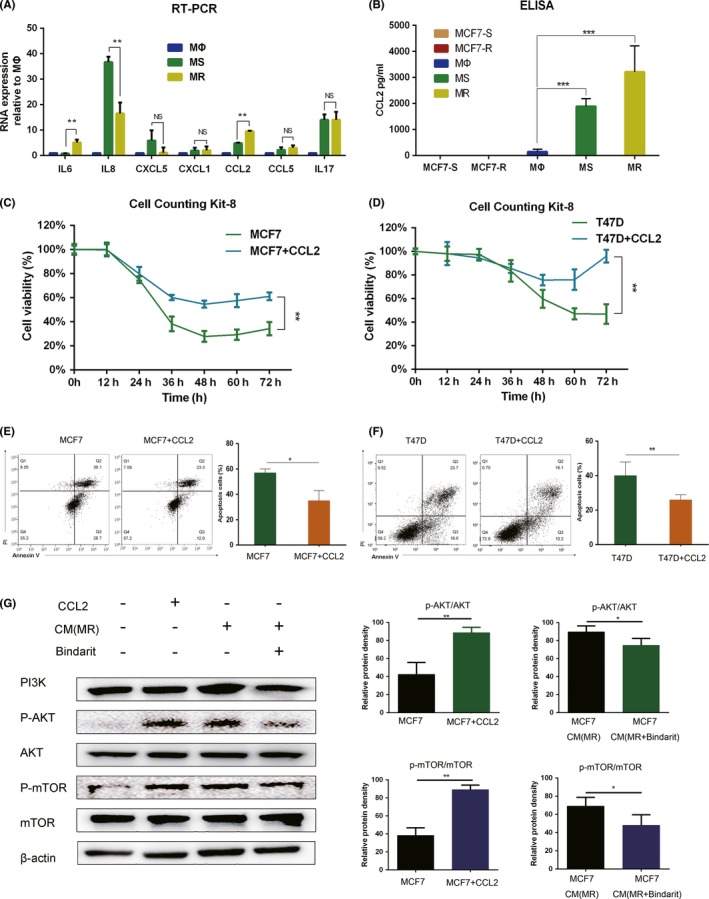
CC‐chemokine ligand 2 (CCL2) secreted by tumor‐associated macrophages (TAM) activates the PI3K/Akt/mTOR pathway to induce endocrine resistance in breast cancer cells. A, RT‐PCR shows the transcription of seven cytokines in three macrophages, MΦ, TAM from a tamoxifen‐sensitive tumor microenvironment (TME) (MS), and TAM from tamoxifen‐resistant TME (MR). Only the change trend of CCL2 is consistent with the previous experiment results, and the difference was statistically significant. B, CCL2 levels in the conditioned medium (CM) of tamoxifen‐sensitive MCF7 breast cancer cells (MCF7‐S), tamoxifen‐resistant MCF7 breast cancer cells (MCF7‐R), MΦ, MS, and MR cells. CCL2 was mainly produced by macrophages rather than breast cancer cells. C, Relative viability of MCF7 and MCF7 + CCL2 (100 nm/mL) cells treated with 5 μmol/L tamoxifen. D, Relative viability of T47D and T47D + CCL2 (100 nm/mL) cells treated with 5 μmol/L tamoxifen. E, Apoptosis of MCF7 and MCF7 + CCL2 (100 nm/mL) cells was analyzed by flow cytometry at 24 h after adding 5 μmol/L tamoxifen. F, Apoptosis of T47D and T47D + CCL2 (100 nm/mL) cells was analyzed by flow cytometry at 24 h after adding 5 μmol/L tamoxifen. G, Western blot analysis of the PI3K/Akt/mTOR pathway. CCL2 and CM of MR could activate the PI3K/AKT/mTOR pathway in MCF7 cells. When MR was treated with Bindarit for 24 h, activation of the PI3K/AKT/mTOR pathway by CM decreased. *P < .05, **P < .01, ***P < .001
3.4. CCL2 inhibits apoptosis and increases endocrine resistance by activating the PI3K/Akt/mTOR pathway
We have shown that CCL2 is associated with endocrine resistance in MCF7 and T47D cell lines. As the PI3K/Akt/mTOR signaling pathway is a classic pathway regulating cell proliferation, apoptosis, and endocrine resistance, we investigated the effect of CCL2 on the activation of this pathway. When MCF7 cells were treated with 100 nmol/L CCL2, phosphorylation of Akt and mTOR was significantly increased. Adding CM of MR to MCF7 cells also activated the PI3K/Akt/mTOR signaling pathway. When MR was treated with 300 nmol/L Bindarit (a CCL2 synthesis inhibitor), the ability of the CM (MR + Bindarit) to increase the levels of phosphorylated Akt and mTOR was weakened (Figure 2G). These results suggest that TAM promote endocrine resistance in breast cancer cells partly by secreting CCL2, which then activates the PI3K/Akt/mTOR pathway.
3.5. Tumor necrosis factor alpha and amino acid metabolism in CM activates the NF‐kB and mTORC1‐FOXK1 pathways of TAM promoting the secretion of CCL2
CCL2 secretion has been thought to be dependent on the transcription factor NF‐κB, which is activated by TNFα in TME.44 We tested whether breast cancer cells and macrophages produce TNFα by ELISA of CM(MCF7‐S), CM(MCF7‐R), CM(MΦ), CM(MS) and CM(MR). We found that MCF7‐S and MCF7‐R could secrete low concentrations of TNFα. The supernatant of breast cancer cells can promote autocrine secretion of TNFα in macrophages. However, there was no significant difference in TNFα level between CM(MS) and CM(MR), nor between CM(MCF7‐S) and CM(MCF7‐R) (Figure 3A). Further analysis of the activation of the NF‐κB pathway in MΦ, MS, and MR showed that activation of the NF‐κB pathway in TAM was significant, but that activation of the NF‐κB pathway in MS and MR was not statistically significant (Figure 3B). The results do not explain the difference in CCL2 secretion between MS and MR. The mTORC1‐FOXK1 pathway senses amino acid changes in CM and regulates CCL2 expression independent of NF‐κB signaling by dephosphorylating the transcription factor FOXK1.45 We tested the activation of the mTORC1‐FOXK1 pathway in MΦ, MS, and MR and found that activation of the mTORC1‐FOXK1 pathway was consistent with the secretion trend of CCL2 (Figure 3C). These results suggest that TNFα and amino acid metabolism in CM activate the NF‐κB and mTORC1‐FOXK1 pathways of TAM promoting the secretion of CCL2.
Figure 3.

Tamoxifen‐sensitive MCF7 breast cancer cells (MCF7‐S) and tamoxifen‐resistant MCF7 breast cancer cells (MCF7‐R) secrete tumor necrosis factor (TNF)α and induce activation of the nuclear factor kappa B (NF‐κB) and mTORC1‐FOXK1 pathways in tumor‐associated macrophages (TAM). A, TNFα levels in conditioned medium (CM) from MCF7‐S, MCF7‐R, MΦ, TAM from a tamoxifen‐sensitive tumor microenvironment (TME) (MS), and TAM from tamoxifen‐resistant TME (MR) cells. Level of TNFα in the CM of TAM was higher, but the difference between MS and MR was not significant (NS). B, Western blot analysis of the NF‐κB pathway. CM of MCF7‐S and MCF7‐R could activate the NF‐κB pathway of TAM. C, Western blot analysis of the mTORC1‐FOXK1 pathway. CM of MCF7‐S and MCF7‐R could activate the mTORC1‐FOXK1 pathway of TAM. Dephosphorylation of MR was more obvious than that of MS. Therefore, CM of MCF7‐R can promote activation of the mTORC1‐FOXK1 pathway more than MCF7‐S. *P < .05, **P < .01
3.6. Tumor‐associated macrophages promote THP‐1 and macrophage aggregation to the periphery of breast cancer cells by secreting CCL2
As a well‐known chemokine abundantly present in macrophages, CCL2 is responsible for the recruitment of monocytes.37, 38, 39, 40, 41, 42, 46 The concentration of CCL2 is positively correlated with its chemotaxis‐promoting potential. We have already determined that the levels of CCL2 secreted by MΦ, MS, and MR are different. The next question is whether the chemotaxis of THP‐1 cells induced by these three macrophages coincides with the secretion of CCL2. Results of the chemotaxis assay confirmed that the chemotaxis‐inducing effect of TAM toward THP‐1 cells was stronger than that of macrophages, the chemotaxis‐inducing effect of MR was stronger than that of MS, and Bindarit the inhibitor of CCL2 reverses the enhanced chemotaxis ability of MR (Figure 4A). To further validate this phenomenon in vitro, we analyzed tissue samples from 100 patients with ER‐positive breast cancer. Immunohistochemistry of serial pathological sections showed that a high expression of CCL2 in the paraneoplastic stroma was correlated with infiltration of CD163+ macrophages (r = 0.548, P < .001) (Figure 4B and Table S2). We propose that tumor cells stimulate M2 polarization of macrophages, which leads to increased secretion of CCL2, and a high concentration of CCL2 in turn promotes THP‐1 aggregation to form more M2‐like macrophages.
Figure 4.
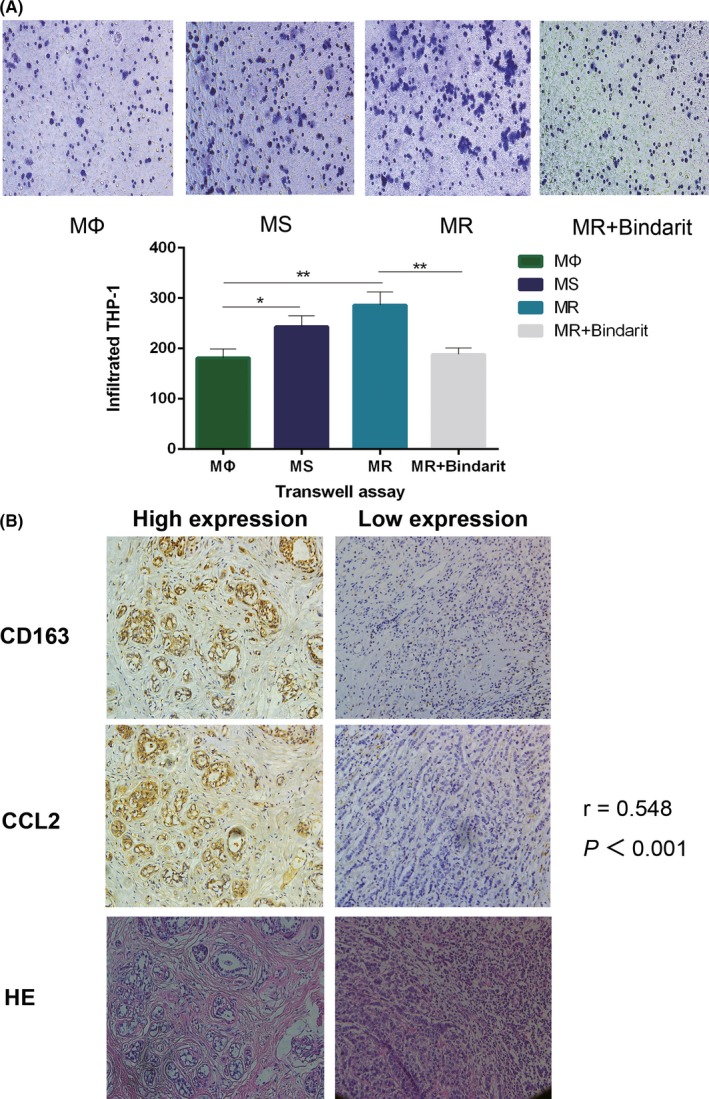
High concentration of CC‐chemokine ligand 2 (CCL2) recruits monocytes and macrophages to facilitate an endocrine‐resistant microenvironment in breast cancer. A, Representative photos of THP‐1 cells recruited by the conditioned medium (CM) of MΦ, TAM from a tamoxifen‐sensitive tumor microenvironment (TME) (MS), TAM from tamoxifen‐resistant TME (MR), and MR + Bindarit after the chemotaxis assay. Quantitative results show that the chemotaxis ability of TAM to recruit THP‐1 cells was enhanced, and TAM in the endocrine‐resistant environment had stronger chemotaxis ability. Bindarit reverses the enhanced chemotaxis ability of CM (MR). B, Representative images of immunohistochemical staining for CCL2 and CD163 and HE staining in serial sections from human breast cancer samples. The related correlation analyses showed r = .548. *P < .05, **P < .01
3.7. CCL2 expression in the stroma is correlated with poor PFS of patients with ER‐positive breast cancer
To determine the clinical relevance of CCL2 and endocrine resistance in the in vitro findings, we selected 100 patients with invasive ER‐positive breast cancer who received regular endocrine therapy at the Tumor Hospital of Harbin Medical University from 2004 to 2014 (Table S3). Pathological sections were stained by CCL2 immunohistochemistry, and the paraneoplastic stromal areas were observed by microscopy. Of the 29 patients with high expression of CCL2, 15 (51.7%) developed relapse and progression, with a median PFS of 25 months. Of the 71 patients with low expression of CCL2, 23 (32.3%) developed relapse and progression, with a median PFS of 65 months. The PFS curves were based on CCL2 expression in the paraneoplastic stroma (Figure 5A,B).
Figure 5.
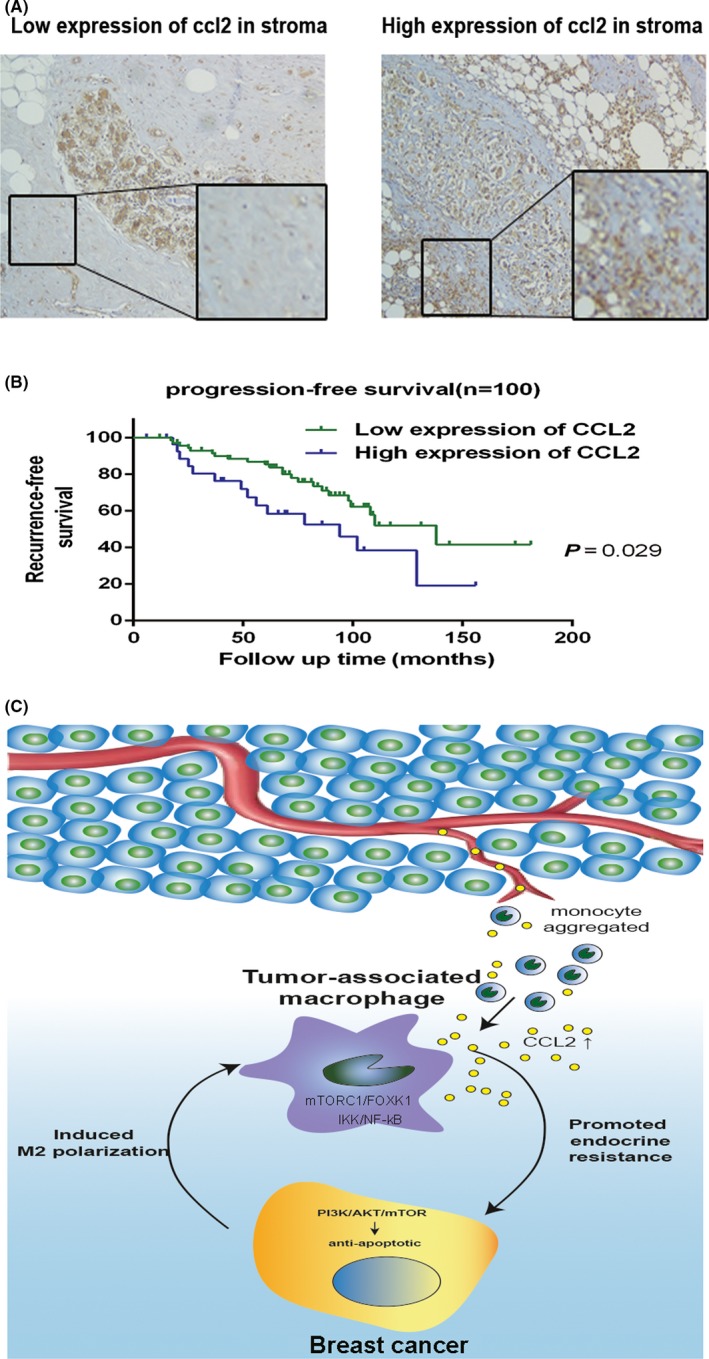
CC‐chemokine ligand 2 (CCL2) expression in the stroma correlated with poor recurrence‐free survival of patients with hormone receptor‐positive breast cancer. A, Different patterns of CCL2 immunohistochemical staining in the stroma of breast cancer cells. B, Kaplan‐Meier recurrence‐free survival curves of patients with hormone receptor‐positive breast cancer with low (n = 71) and high (n = 29) CCL2 expression in the stroma (P = .029). C, Proposed model for TAM‐secreted CCL2 activating the PI3K/Akt/mTOR pathway to promote a malignant cycle of endocrine resistance and recruitment of monocytes
Results showed that PFS of the CCL2 high‐expression group was significantly shorter than that of the CCL2 low‐expression group (P < .05). The prognostic value of each clinicopathological feature was assessed using Cox hazard regression analysis. CCL2 expression was found to be an independent risk factor for disease‐free survival (DFS) (HR = 2.323, P = .015) (Table 1). Taken together, these results indicate a crucial role of CCL2 in promoting endocrine resistance in breast cancer.
Table 1.
Univariate and multivariate analyses of clinicopathological features associated with PFS of patients with hormone receptor‐positive breast cancer
| Clinicopathological feature | Progression‐free survival | |||
|---|---|---|---|---|
| Univariate | Multivariate | |||
| HR (95%CI) | P | HR (95%CI) | P | |
| T‐staging | 0.737 | .357 | ||
| T1, T2, T3 | (0.385‐1.412) | |||
| N‐staging | 2.097 | <.001 | 2.290 | <.001 |
| N0, N1, N2, N3 | (1.484‐2.963) | (1.598‐3.282) | ||
| Histological grade | 0.755 | .528 | ||
| G1, G2, G3 | (0.315‐1.808) | |||
| PR status | 1.3 | .479 | ||
| Negative vs Positive | (0.629‐2.686) | |||
| Her‐2 status | 1.77 | .281 | 3.232 | .036 |
| Negative vs Positive (+++ or FISH+) | (0.627‐4.998) | (1.080‐9.668) | ||
| Chemotherapy | 1.151 | .77 | ||
| Yes vs No | (0.448‐2.956) | |||
| CCL2 expression | 2.003 | .038 | 2.323 | .015 |
| Low vs High | (1.041‐3.853) | (1.178‐4.581) | ||
4. DISCUSSION
Endocrine resistance in breast cancer is a major clinical challenge in treating patients with resistant disease. At present, most studies on the mechanism of endocrine resistance are focused on breast cancer cells, and only a few studies have focused on the role of other cells in the TME. TAM are an important component of the TME that play a role in angiogenesis, metastasis, and tumor malignancy.43, 47 Our previous studies have confirmed that TAM are correlated with tamoxifen resistance in patients with breast cancer.22 In the present study, we investigated the mechanisms underlying TAM‐mediated endocrine resistance in breast cancer cells.
We showed that endocrine‐resistant breast cancer cells can induce M2 polarization of TAM. M2‐polarized TAM, in turn, promote endocrine resistance in breast cancer cells. We found that breast cancer cells secrete trace amounts of TNFα and promote autocrine secretion of TNFα in macrophages, thereby activating the NF‐κB pathway and promoting CCL2 release. Endocrine‐resistant breast cancer cells activate the mTORC1‐FOXK1 pathway of macrophages by altering amino acid metabolism in the environment, further promoting M2 polarization and CCL2 secretion by macrophages.
We also found that TAM promote endocrine resistance by secreting CCL2, which activates the PI3K/Akt/mTOR signaling pathway. Therefore, CCL2 plays an important role in this malignant feedback loop. In addition, CCL2 promotes the aggregation of monocytes and macrophages in the TME.48 The transition from monocytes to TAM further promotes the formation of the endocrine‐resistant microenvironment. Analysis of tissue samples from patients with hormone receptor‐positive breast cancer confirmed a correlation between high expression of CCL2 in the paraneoplastic stroma and infiltration of CD163+ macrophages. Moreover, patients with high CCL2 expression in the stroma had a shorter PFS after endocrine therapy. This is consistent with the findings of our previous study that patients with high CD163+ macrophage infiltration tend to develop endocrine resistance.22 The finding that CCL2 promotes the formation of the TME and the malignant cycle of cells in the TME may partly explain why patients with endocrine resistance in the past are more likely to be resistant to new endocrine therapy.
Tumor‐associated macrophages can adjust their phenotype and function in response to local cues provided by the TME.49 Crosstalk between TAM and cancer cells is complex and involves exosomes, cytokines, and metabolites.50, 51, 52 In the present study, we focused on the cytokine CCL2 secreted by macrophages and examined its role in the endocrine resistance of breast cancer cells. We found that CCL2 activates the PI3K/Akt/mTOR pathway, which is a classical endocrine resistance pathway. However, the direct mechanism by which CCL2 activates this pathway was not determined. Another limitation of the present study was that due to the long culture time of drug‐resistant cell lines, the effects of endocrine‐resistant and ‐sensitive T47D cell lines on macrophages could not be repeated.
Cell components in the TME are complex. In addition to TAM, fibroblasts, dendritic cells, epithelial cells, neutrophils, and lymphocytes can also be tamed by tumor cells and promote a tumor malignant environment.53, 54, 55, 56, 57, 58 It has been reported that dendritic cells, fibroblasts, and epithelial cells tamed by cancer cells secrete CCL2.55, 56, 57, 58 These cells can also contribute to the positive feedback loop. However, there are only a few studies on the role of these cells in the endocrine resistance of breast cancer, and there is no report on the effect of endocrine‐resistant breast cancer cells on the differentiation of these cells. Future research should focus on the effect of other cell components in the TME on the endocrine resistance of breast cancer.
In conclusion, our results indicate that endocrine‐resistant tumor cells are more likely to promote M2 polarization of macrophages, whereas M2‐polarized TAM further promote endocrine resistance in tumor cells, thus forming a positive feedback loop between TAM and breast cancer cells (Figure 5C). CCL2 plays an important role in this positive feedback loop. Increased CCL2 secretion by M2‐polarized TAM activates the PI3K/Akt/mTOR pathway in tumor cells to promote endocrine resistance and formation of the endocrine‐resistant microenvironment by aggregating monocytes into the TME. These findings suggest that CCL2 and TAM may be potential novel therapeutic targets for patients with endocrine‐resistant breast cancer.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China (grant nos. 81730074 and 81672599) and the National Science Foundation of Heilongjiang Province of China for Returnees (grant no. LC2017037).
Li D, Ji H, Niu X, et al. Tumor‐associated macrophages secrete CC‐chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020;111:47–58. 10.1111/cas.14230
Dongbo Li and Hongfei Ji contributed equally to this study and should be considered co‐first authors.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. [DOI] [PubMed] [Google Scholar]
- 2. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631‐643. [DOI] [PubMed] [Google Scholar]
- 3. Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53(1):25‐71. [PubMed] [Google Scholar]
- 4. Ali S, Coombes RC. Endocrine‐responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2(2):101‐112. [DOI] [PubMed] [Google Scholar]
- 5. Zhou J, Teng R, Wang Q, et al. Endocrine resistance in breast cancer: current status and a perspective on the roles of miRNAs (review). Oncol Lett. 2013;6(2):295‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Briest S, Stearns V. Tamoxifen metabolism and its effect on endocrine treatment of breast cancer. Clin Adv Hematol Oncol. 2009;7(3):185‐192. [PubMed] [Google Scholar]
- 7. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309‐322. [DOI] [PubMed] [Google Scholar]
- 8. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582‐598. [DOI] [PubMed] [Google Scholar]
- 9. Nilendu P, Kumar A, Kumar A, Pal JK, Sharma NK. Breast cancer stem cells as last soldiers eluding therapeutic burn: a hard nut to crack. Int J Cancer. 2018;142(1):7‐17. [DOI] [PubMed] [Google Scholar]
- 10. Zhang M, Lee AV, Rosen JM. The cellular origin and evolution of breast cancer. Cold Spring Harb Perspect Med. 2016;7(3):a027128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Augustin HG, Koh GY. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science. 2017;357(6353):eaal2379. [DOI] [PubMed] [Google Scholar]
- 13. Clark JW, Chabner BA. Limits to Precision Cancer Medicine. N Engl J Med. 2017;376(1):96. [DOI] [PubMed] [Google Scholar]
- 14. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ginhoux F, Schultze JL, Murray PJ, Ochando I, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17:34‐40. [DOI] [PubMed] [Google Scholar]
- 16. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Obeid E, Nanda R, Fu Y‐X, Olopade OI. The role of tumor‐associated macrophages in breast cancer progression (review). Int J Oncol. 2013;43(1):5‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch Pharm Res. 2016;39:1588‐1596. [DOI] [PubMed] [Google Scholar]
- 19. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605‐612. [DOI] [PubMed] [Google Scholar]
- 20. Lau SK, Chu PG, Weiss LM. CD163: A specific marker of macrophages in paraffin‐embedded tissue samples. Am J Clin Pathol. 2004;122(5):794‐801. [DOI] [PubMed] [Google Scholar]
- 21. Escamilla J, Schokrpur S, Liu C, et al. CSF1 receptor targeting in prostate cancer reverses macrophage‐mediated resistance to androgen blockade therapy. Cancer Res. 2015;75(6):950‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xuan QJ, Wang JX, Nanding A, et al. Tumor‐associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathol Oncol Res. 2014;20(3):619‐624. [DOI] [PubMed] [Google Scholar]
- 23. Lee S, Lee E, Ko E, et al. Tumor‐associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1‐α and MMP‐9. Cancer Lett. 2018;437:25‐34. [DOI] [PubMed] [Google Scholar]
- 24. Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T‐lymphocyte chemoattractant. Proc Natl Acad Sci USA. 1994;91(9):3652‐3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C‐C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol. 1996;60(3):365‐371. [DOI] [PubMed] [Google Scholar]
- 26. Fader AN, Rasool N, Vaziri SA, et al. CCL2 expression in primary ovarian carcinoma is correlated with chemotherapy response and survival outcomes. Anticancer Res. 2010;30(12):4791‐4798. [PubMed] [Google Scholar]
- 27. McClellan JL, Davis JM, Steiner JL, et al. Linking tumor‐associated macrophages, inflammation, and intestinal tumorigenesis: role of MCP‐1. Am J Physiol Gastrointest Liver Physiol. 2012;303(10):G1087‐G1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morita Y, Zhang R, Leslie M, et al. Pathologic evaluation of tumor‐associated macrophage density and vessel inflammation in invasive breast carcinomas. Oncol Lett. 2017;14(2):2111‐2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Biswas SK, Gangi L, Paul S, et al. A distinct and unique transcriptional program expressed by tumor‐associated macrophages (defective NF‐kappaB and enhanced IRF‐3/STAT1 activation). Blood. 2006;107(5):2112‐2122. [DOI] [PubMed] [Google Scholar]
- 30. Ueno T, Toi M, Saji H, et al. Significance of macrophage chemoattractant protein‐1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res. 2000;6(8):3282‐3289. [PubMed] [Google Scholar]
- 31. Dowsett M. Overexpression of HER‐2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr Relat Cancer. 2001;8(3):191‐195. [DOI] [PubMed] [Google Scholar]
- 32. Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25(6):869‐898. [DOI] [PubMed] [Google Scholar]
- 33. Girault I, Lerebours F, Amarir S, et al. Expression analysis of estrogen receptor α coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin Cancer Res. 2003;9(4):1259‐1266. [PubMed] [Google Scholar]
- 34. Fan P, Yue W, Wang JP, et al. Mechanisms of resistance to structurally diverse antiestrogens differ under premenopausal and postmenopausal conditions: evidence from in vitro breast cancer cell models. Endocrinology. 2009;150(5):2036‐2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Masri S, Phung S, Wang X, et al. Genome‐wide analysis of aromatase inhibitor‐resistant, tamoxifen‐resistant, and long‐term estrogen‐deprived cells reveals a role for estrogen receptor. Cancer Res. 2008;68(12):4910‐4918. [DOI] [PubMed] [Google Scholar]
- 36. Thota K, Prasad K, Basaveswara Rao MR. Detection of cytochrome P450 polymorphisms in breast cancer patients may impact on tamoxifen therapy. Asian Pac J Cancer Prev. 2018;19(2):343‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soria G, Ben‐Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267(2):271‐285. [DOI] [PubMed] [Google Scholar]
- 38. Bai S, Mu Z, Huang Y, Ji P. Guanylate binding protein 1 inhibits osteogenic differentiation of human mesenchymal stromal cells derived from bone marrow. Sci Rep. 2018;8(1):1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li SW, Wang CY, Jou YJ, et al. SARS coronavirus papain‐like protease induces Egr‐1‐dependent up‐regulation of TGF‐β1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep. 2016;6(1):25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maher IE, Griffith JE, Lau Q, Reeves T, Higgins DP. Expression profiles of the immune genes CD4, CD8β, IFNγ, IL‐4, IL‐6 and IL‐10 in mitogen‐stimulated koala lymphocytes (Phascolarctos cinereus) by qRT‐PCR. PeerJ. 2014;2(3):e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seo EY, Cho MJ, Lee JH, et al. The differentially expressed genes by radiotherapy in the patients with uterine cervix cancer (Korean). J Korean Soc Ther Radiol Oncol. 2001;19(4):389‐396. [Google Scholar]
- 42. Chaturvedi U, Kalim S, Kumar R, et al. Cloning and expression of chicken granulocyte‐macrophage colony stimulating factor (GMCSF) gene. Indian J Exp Biol. 2010;48(12):1175‐1180. [PubMed] [Google Scholar]
- 43. Yan X, Jiao SC, Zhang GQ, Guan Y, Wang JL. Tumor‐associated immune factors are associated with recurrence and metastasis in non‐small cell lung cancer. Cancer Gene Ther. 2017;24(2):57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bussard KM, Mutkus L, Stumpf K, et al. Tumor‐associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1‐FOXK1 axis promotes recruitment of tumor‐associated macrophages. Cell Rep. 2017;21:2471‐2486. [DOI] [PubMed] [Google Scholar]
- 46. Ma K, Yang L, Shen R, et al. Th17 cells regulate the production of CXCL1 in breast cancer. Int Immunopharmacol. 2018;56:320‐329. [DOI] [PubMed] [Google Scholar]
- 47. Rafat M, Aguilera TA, Vilalta M, et al. Macrophages promote circulating tumor cell‐mediated local recurrence following radiation therapy in immunosuppressed patients. Cancer Res. 2018;78(15):4241‐4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amasyali B, Kose S, Kursaklioglu H, Barcin C, Kilic A. Monocyte chemoattractant protein‐1 in acute coronary syndromes: complex vicious interaction. Int J Cardiol. 2009;136(3):356‐357. [DOI] [PubMed] [Google Scholar]
- 49. Gosselin D, Link VM, Romanoski C, et al. Environment drives selection and function of enhancers controlling tissue‐specific macrophage identities. Cell. 2009;159(6):1327‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med. 2011;9(1):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cassetta L, Fragkogianni A, Sims AH, et al. Human tumor‐associated macrophage and monocyte transcriptional landscapes reveal cancer‐specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng P, Chen L, Yuan X, et al. Exosomal transfer of tumor‐associated macrophage‐derived miR‐21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kai M, Lihua Y, Ruwu S, et al. Th17 cells regulate the production of CXCL1 in breast cancer. Int Immunopharmacol. 2018;56:320‐329. [DOI] [PubMed] [Google Scholar]
- 54. Coffelt SB, Kersten K, Doornebal CW, et al. IL‐17‐producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang X, Shi H, Yuan X, et al. Tumor‐derived exosomes induce N2 polarization of neutrophils to promote gastric cancer cell migration. Mol Cancer. 2018;17(1):146‐152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsu YL, Hung JY, Tsai YM, et al. 6‐Shogaol, an active constituent of dietary ginger, impairs cancer development and lung metastasis by inhibiting the secretion of CC‐chemokine ligand 2 (CCL2) in tumor‐associated dendritic cells. J Agr Food Chem. 2015;63(6):1730‐1738 [DOI] [PubMed] [Google Scholar]
- 57. Loberg RD, Day LL, Harwood J, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8(7):578‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cackowski FC, Roodman GD. Perspective on the osteoclast: an angiogenic cell? Ann N Y Acad Sci. 2007;1117:12‐25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials