

Abstract
Birt‐Hogg‐Dubé (BHD) syndrome is associated with the development of hereditary renal cell carcinoma (RCC) and is caused by a germline mutation in the folliculin gene. Most cases of BHD syndrome‐associated RCC (BHD‐RCC) are less aggressive than sporadic clear cell RCC and multifocal. Therefore, it is critical to distinguish BHD‐RCC from its sporadic counterparts to identify and monitor affected families and to preserve renal function for as long as possible. The World Health Organization/International Society of Urological Pathology consensus classification defined distinct entities for certain hereditary RCC; however, BHD‐RCC was not included in this classification. Although the clinical features and molecular mechanisms of BHD‐RCC have been investigated intensively over the last two decades, pathologists and urologists occasionally face difficulties in the diagnosis of BHD‐RCC that require genetic testing. Affected patients usually have miscellaneous benign disorders that often precede renal carcinogenesis. In the present review, we summarize the current understanding of the histopathological features of BHD‐RCC based on our epidemiological studies of Japanese families and a literature review. Pathological diagnostic clues and differential diagnosis of BHD‐RCC from other hereditary RCC are also briefly discussed.
Keywords: Birt‐Hogg‐Dubé syndrome, folliculin, hereditary cancer, histopathology, renal cell carcinoma
Birt‐Hogg‐Dubé (BHD) syndrome is associated with the development of hereditary renal cell carcinoma. We summarize the current understanding of the histopathological features of BHD syndrome‐associated renal cell carcinoma based on our epidemiological studies of Japanese families and a literature review.

1. INTRODUCTION
Hereditary cancer syndromes predispose patients and affected family members to multifocal and metachronous tumors. As genomic research has improved our understanding of the pathogenic events in each patient, hereditary cancers constitute an area of highly unmet medical needs. Comprehensive genomic approaches have increased the ability to identify hereditary cancers. As the carcinogenic mechanisms of hereditary cancers are now understood, therapeutic strategies to target the signaling cascades affected by the responsible genes have improved. As individuals with hereditary cancers potentially develop multifocal and metachronous disorders, it is important to preserve the functions of the affected organ(s) when the cancer is low grade.
The kidney is an essential organ for maintaining body fluid, electrolyte balance, blood pressure and hormone secretion. Although most cases of renal cell carcinoma (RCC) are sporadic, increasing numbers of previously undiagnosed hereditary cancers have been re‐evaluated.1 Birt‐Hogg‐Dubé (BHD) syndrome‐associated RCC (BHD‐RCC) is a newly emerging hereditary cancer whose genomic features and renal pathology have been evaluated intensively over the 20 years. We collected clinicopathological information from 220 independent Japanese families with BHD syndrome through consultation with the Japanese Society of Pathology (http://pathology.or.jp/) and BHD‐NET Japan (http://www.bhd-net.jp/). In this review, we summarize the histopathology of BHD‐RCC based on data from these Japanese families. The diagnostic conundrum and differential diagnosis of BHD‐RCC from other hereditary RCC are also discussed.
2. BIRT‐HOGG‐DUBÉ SYNDROME GENERAL INFORMATION
Birt‐Hogg‐Dubé syndrome, also called Hornstein‐Knickenberg syndrome, is an autosomal dominant disorder characterized by development of skin fibrofolliculomas, lung cysts and RCC.2, 3, 4 In 2002, the susceptibility gene, folliculin (FLCN), was identified on chromosome 17p11.2,5 and FLCN consists of 14 exons (OMIM: 607273, NCBI reference sequence: NM_144997.6). Most affected individuals with BHD syndrome harbor germline mutations in the coding region of FLCN between exons 4 and 14. Pathogenic variants have been uploaded to several academic databases including Leiden Open Variation Database (https://databases.lovd.nl/shared/genes/FLCN) and The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FLCN). We recently described the locations and patterns of FLCN mutations in Japanese families in another review regarding the lung pathology of this disease.6
3. CLINICAL FEATURES OF BHD‐RCC
Clinical information related to BHD syndrome has been collected and reviewed by several study groups, including the European BHD Consortium and National Cancer Institute (NCI) in the USA.7, 8 Most Caucasians with BHD syndrome (85%‐90%) have skin papules as the major complaint, whereas the incidence of pneumothorax is less frequent (24%).9 In contrast, skin manifestations are not the major complaint in the Japanese population.10 The fibrous papules in Japanese patients are generally subtle or non‐detectable; therefore, skin manifestations alone are unlikely to raise suspicion of BHD syndrome in Japanese individuals unless other disorders such as pneumothorax or RCC are present. Physicians should consider the race‐associated differences in the frequency of pneumothorax and conspicuity of skin papules. Renal carcinogenesis seems to be related to age, and the median age at RCC diagnosis in BHD patients is 46‐56 years.8, 10, 11 The morbidity rate of RCC in Caucasian populations is 27%‐34% according to an NCI study.12 Multifocal RCC usually alert physicians of the possibility of hereditary RCC. In a substantial number of cases, this possibility is not recognized at the time of the initial RCC resection, and not until the development of metachronous RCC. In our analysis of the Japanese population, 25.8% (31/120) of previously undiagnosed probands had RCC histologically characteristic of BHD syndrome,10 which prompted pathologists and urologists to consider genetic testing. According to our most recent data, 64.3% (27/42) of probands and their first‐degree relatives with RCC had multiple lesions at the time of genetic diagnosis. Others with a single RCC at the time of genetic diagnosis developed de novo tumors during surveillance.
4. HISTOPATHOLOGICAL FEATURES OF BHD‐RCC
In 2002, for the first time, Pavlovich et al13 described the detailed histopathological findings of BHD‐RCC based on 130 tumors obtained from 30 patients. This landmark study showed that most BHD‐RCC are histologically either hybrid oncocytic/chromophobe tumors (HOCT) or chromophobe RCC. In our epidemiological study of the Japanese population, the most frequent histological type was chromophobe RCC (43.6%), followed by HOCT (34.5%).10 In addition to HOCT and chromophobe RCC, affected patients are at risk of developing clear cell RCC, papillary RCC and angiomyolipoma, although the frequencies of each of these histological types are less than 10%.4, 10, 11
Gross pathology of BHD‐RCC varies depending on size and histology. HOCT and chromophobe RCC are indistinguishable macroscopically, and thus should be evaluated microscopically. Both tumor types show clearly demarcated margins, and fibrous capsules are either lacking or incomplete. Sporadic oncocytoma and chromophobe RCC are mahogany brown and beige in color, respectively (Figure 1A,B). HOCT and chromophobe BHD‐RCC are a more yellowish color under formalin‐fixed tissue conditions (Figure 1C). Central degeneration is occasionally observed, but necrosis is rare. The tumor may have a radial scar, which is otherwise common in sporadic oncocytomas. We experienced an unusual nodular scar that seemed to be an obsolete RCC.
Figure 1.
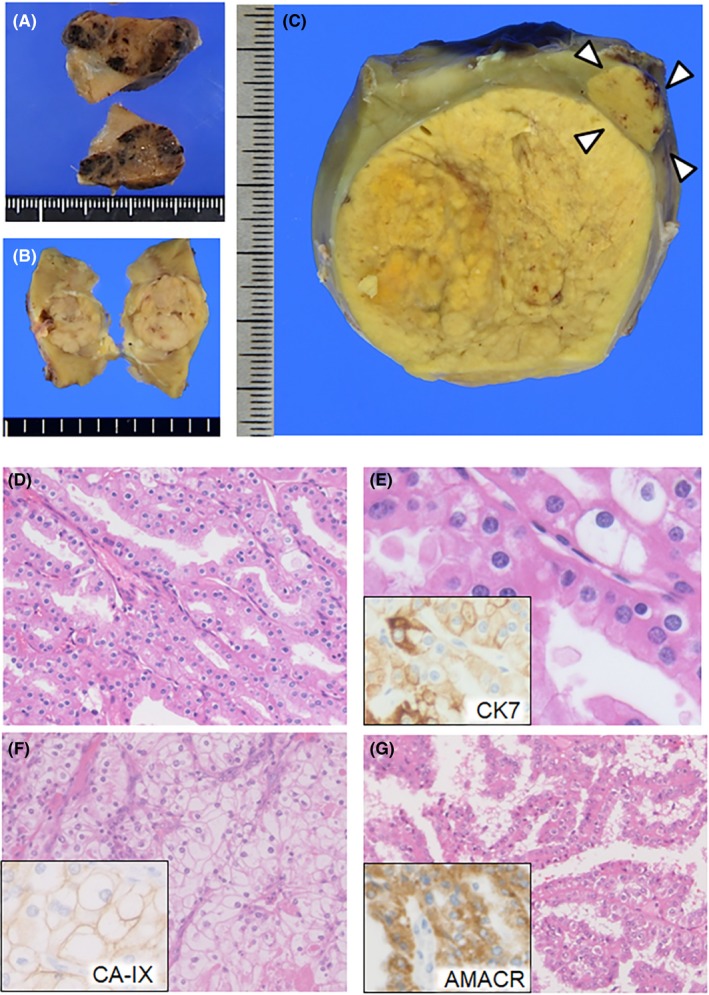
Macro‐ and microscopic features of Birt‐Hogg‐Dubé (BHD) syndrome‐associated renal cell carcinoma (RCC). A, Surface section of a sporadic oncocytoma. A mahogany‐brown nodule is observed. B, Gross section of a sporadic chromophobe RCC. A beige nodule is observed. C, Gross section of hybrid oncocytic/chromophobe tumors from a 34‐year‐old woman with BHD syndrome. In the vicinity of the main tumor, an independent tumor is detected (arrowheads). Both tumors are a yellowish color and clearly demarcated. D, Hybrid oncocytic/chromophobe tumor with a tubular and cord‐like structure developed in a 44‐year‐old man with BHD syndrome. E, Higher magnification of (D). The tumor cells have round nuclei with perinuclear halo and oncocytoma‐like granular cytoplasm with cell‐cell boundaries. Inset: immunostaining of cytokeratin 7 (CK7) in a serial section. F, Clear cell RCC developed in a 58‐year‐old man with BHD syndrome. Histological features are indistinguishable from those of its sporadic counterparts. Inset: immunostaining of carbonic anhydrase IX (CA‐IX) in a serial section. G, Papillary RCC developed in a 56‐year‐old man with BHD syndrome. Tumor cells show eosinophilic granular cytoplasm. Inset: immunostaining of α‐methylacyl‐CoA racemase (AMACR) in a serial section
According to the World Health Organization (WHO) classification, HOCT is currently defined as a subtype of chromophobe RCC, which is a malignant neoplasm. HOCT is described as having an “overlapping histology between oncocytoma and chromophobe RCC” (Figure 1D,E). The International Society of Urological Pathology (ISUP) lists three types of HOCT carcinogenesis: (i) sporadic; (ii) associated with renal oncocytosis/oncocytomatosis; and (iii) associated with BHD syndrome.14 As HOCT is a rare tumor, pathologists may be the first to recognize the possibility of BHD syndrome if the renal specimens of previously undiagnosed patients are submitted for histological diagnosis. In contrast to sporadic chromophobe RCC that occasionally converts into the sarcomatoid type, BHD‐RCC with a chromophobe component rarely shows sarcomatoid transformation.15 Although some radiological liver/lung metastases under stable conditions have been reported, patients with HOCT associated with BHD syndrome who receive curative surgery generally have favorable prognoses.11 The clear cell and papillary types appear to be more aggressive and often result in poor prognoses8, 16 (Figure 1F,G). Correct diagnosis of BHD‐RCC is important for early detection and preservation of renal function in patients who develop multiple/bilateral tumors. Therefore, if the resected specimen shows a HOCT appearance on histology, pathologists and urologists are encouraged to consult hereditary RCC specialists for consideration of genetic testing.
Hybrid oncocytic/chromophobe tumors can occur in patients without BHD syndrome, which is known as sporadic HOCT (SHOCT). A recent study of 25 patients with SHOCT showed that six patients developed multifocal lesions (24%), two of whom had poor prognoses.17 The differential diagnosis between BHD syndrome‐associated HOCT and SHOCT requires genetic testing; however, failure to detect mutations in the FLCN coding exons by Sanger sequencing does not necessarily exclude the possibility of BHD syndrome. For FLCN mutation‐negative cases, either multiplex ligation‐dependent probe amplification or next‐generation sequencing‐based analysis may be necessary. Therefore, HOCT and SHOCT are unlikely to be differentiated during routine medical care. Thorough examination including detection of lung cysts and skin lesions, genetic testing, and long‐term follow up to monitor onset of BHD syndrome‐associated manifestations are needed.
Hybrid oncocytic/chromophobe tumors and chromophobe BHD‐RCC show a similar immunostaining pattern: positivity for cytokeratin 7 (CK7), kidney‐specific cadherin, E‐cadherin, CD82 and S100‐A1 and negativity for carbonic anhydrase IX (CA‐IX).18 CK7 immunoreactivity is occasionally focal. BHD‐RCC of the clear‐cell type shows CA‐IX positivity and CK7 negativity, whereas BHD‐RCC of the papillary type shows α‐methylacyl‐CoA racemase positivity.16, 18 These staining patterns of BHD‐RCC are similar to those of its sporadic counterparts. These findings support that HOCT and chromophobe RCC are probably derived from distal tubules or collecting ducts, whereas clear cell RCC is derived from proximal tubules in patients with BHD syndrome, as discussed later.
5. CYTOGENETIC FEATURES OF BHD‐RCC
A pioneering cytogenetic analysis showed distinctive gene expression patterns in BHD‐RCC such as high expression of parvalbumin, a distal convoluted tubule marker.19 The expression patterns in BHD‐RCC are more similar to those of sporadic chromophobe RCC and oncocytoma than those of other RCC types which supports the immunostaining pattern. Gene expression profiling of BHD‐RCC showed that HOCT and chromophobe RCC lack the mutations in driver genes (TP53, CDKN1A, RB1, PTEN and MTOR) that characterize sporadic chromophobe RCC.20 No copy number variations were observed among chromosomes in any of the HOCT or chromophobe RCC cases, except for two chromophobe RCC cases with a 3q gain.20, 21 These findings highlight a distinct difference between these BHD‐RCC types and sporadic chromophobe RCC (classic type), which are characterized by copy number losses in chromosomes 1, 2, 6, 8, 10, 13, 17 and 21.20, 22 HOCT and chromophobe BHD‐RCC show a disomic pattern on FISH using probes targeting the centromeric regions of chromosomes 2, 6 and 17, whereas sporadic chromophobe RCC show a predominantly monosomic pattern.23 Sporadic oncocytomas occasionally show copy number losses in chromosomes 1 and Y, as well as ERCC2 and C2CD4C mutations.24, 25 A recent study showed that SHOCT also have copy number losses in chromosomes 1 and X/Y but lack recurrent mutations.17 Collective cytogenetic studies have improved our understanding of the unique features of BHD‐RCC but have also shown a limitation in the histology‐based differential diagnosis of BHD‐RCC from its sporadic counterparts. Our clear cell BHD‐RCC cases (n = 2) showed no copy number alterations at 3p with wild‐type von Hippel‐Lindau tumor suppressor gene (VHL),21 which is different from its sporadic counterparts, characterized by 3p copy number loss and VHL mutations.26 However, the study by Pavlovich et al13 showed that two of eight cases of clear‐cell BHD‐RCC had 3p loss of heterozygosity and missense mutations in VHL. Therefore, whether VHL mutations play a key role in clear‐cell BHD‐RCC cannot be concluded until a larger number of cases have been evaluated.
According to epidemiological studies, 19.2%‐45% of affected individuals develop renal cysts.10, 27 Most renal cysts are benign, but tumors may develop within the cyst walls (Figure 2A,B). We experienced four dialysis patients who developed multifocal tumors in polycystic kidneys; three of these patients underwent bilateral nephrectomy, which showed HOCT, chromophobe, clear cell and papillary adenomatous features within the multifocal neoplastic lesions (Figure 2C‐F). The wide array of histological variations associated with BHD‐RCC observed in these patients indicates an intimate association between FLCN insufficiency and polycystic changes. Mice with Flcn inactivation in kidney epithelial cells developed polycystic changes and renal dysfunction.28 Although the morbidity of chronic renal failure is low in FLCN mutation carriers,10 renal function may be threatened not only by multiple tumors but also by polycystic changes.
Figure 2.
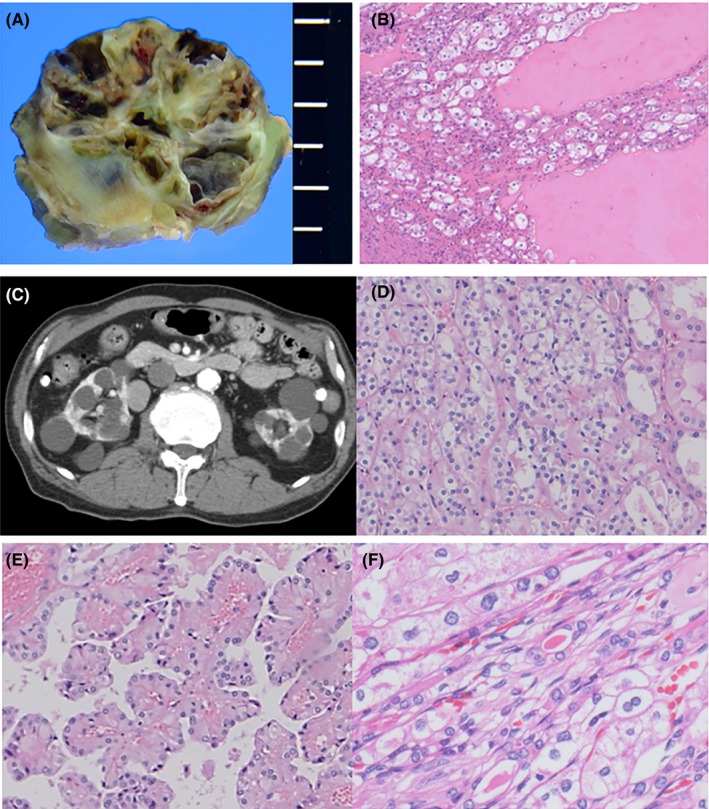
Macro‐ and microscopic features of cystic tumors. A, Gross section of a cystic tumor in a 61‐year‐old man. B, Microscopic features of (A). Tumor cells composed of oncocytic and clear cells are present along the cyst walls. The cysts are filled with eosinophilic exudate. C, Abdominal computed tomography scan of a 65‐year‐old man with chronic renal failure and dialysis. Polycystic lesions are present in both kidneys. D‐F, Microscopic features of the resected kidneys in (C). Tumors are composed of hybrid (D), papillary adenomatous (E) and chromophobe‐like morphology with raisinoid nuclei and/or binucleation (F)
6. DIFFERENTIATION OF BDH‐RCC FROM OTHER HEREDITARY RENAL NEOPLASMS
The differential diagnosis of BDH‐RCC is discussed in relation to tuberous sclerosis complex (TSC); hereditary leiomyomatosis and renal cell cancer (HLRCC); and succinate dehydrogenase (SDH)‐deficient RCC.
Patients with TSC harbor germline mutations in either TSC1 or TSC2. TSC1, TSC2 and FLCN all mediate mTOR pathway signaling, although their functions and interaction partners are different.29 TSC and BHD syndrome commonly affect the skin, lung and kidney. Approximately two‐thirds of TSC cases are the result of de novo TSC1/TSC2 pathogenic mutations, whereas BHD syndrome cases rarely develop de novo FLCN mutations.30, 31 Cutaneous angiofibromas are typical features of TSC but can also occur in BHD syndrome. Skin manifestations generally arise during childhood in TSC patients versus adulthood in BHD syndrome patients.7, 32 Lung cysts and pneumothoraces are observed in both conditions (Figure 3A,B). Patients with TSC carry a risk of developing typical cystic lung disease, namely lymphangioleiomyomatosis (LAM). TSC‐associated LAM cells in the lung show positive immunostaining of α‐smooth muscle actin (α‐SMA) and focally positive staining of Human Melanoma Black (HMB) 45 and the estrogen receptor. The inner surface of the lung cysts in BHD syndrome is lined by benign pneumocytes that show positive immunostaining of TTF‐1, napsin A, pro‐surfactant protein C and CAM5.2.6 Radiological comparisons of the lung cysts between the two disorders have been discussed in several reports.33, 34, 35 In the kidney, angiomyolipoma is the most frequent neoplasm in TSC, developing in up to 80% of affected individuals,36 and is composed of three cell types: vascular; adipose; and smooth muscle cells. HMB45 is an immunohistochemical marker of angiomyolipomas.37 Angiomyolipomas are benign in most TSC cases; however, they tend to increase in number and size and replace the renal parenchyma, leading to life‐threatening hemorrhage.38
Figure 3.
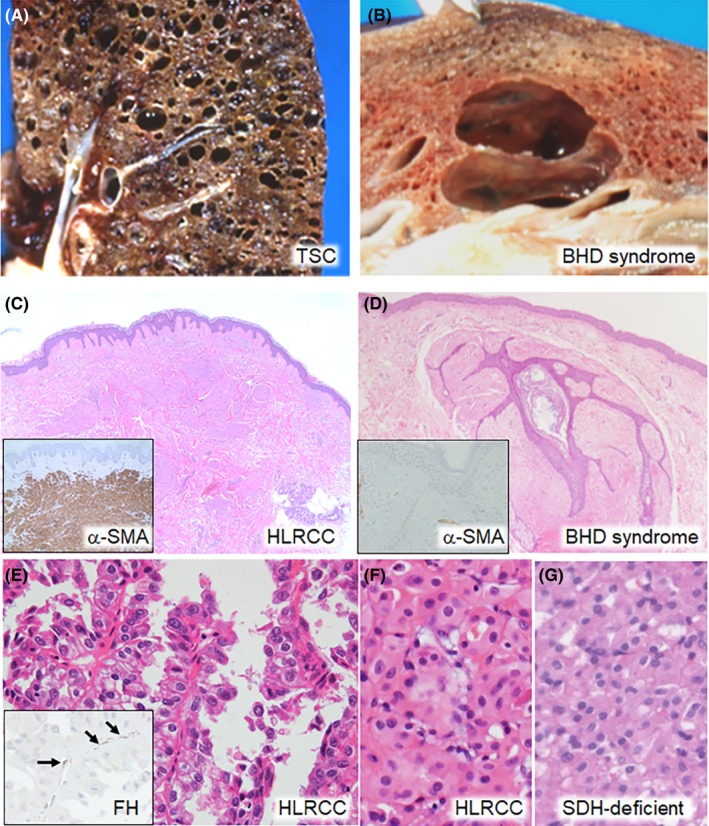
Differential diagnosis between Birt‐Hogg‐Dubé (BHD) syndrome and other hereditary diseases. A, Gross section of the lung of a 28‐year‐old woman with tuberous sclerosis complex. Numerous round cysts are observed in a diffuse arrangement. B, Gross section of the lung of an 81‐year‐old man with BHD syndrome. Two oval cysts extend along the interstitial tissue in a back to back way, compressing the alveolar structures between them. C, Histology of the skin tumor of a 75‐year‐old woman with hereditary leiomyomatosis and renal cell cancer (HLRCC). Proliferation of piloleiomyoma cells is observed in the dermic layer. Inset: immunostaining of α‐smooth muscle actin (α‐SMA). Tumor cells show diffuse staining. D, Histology of the skin tumor of a 54‐year‐old man with BHD syndrome. Proliferation of basaloid epithelial cells around a hair follicle and surrounding fibrous tissue is observed, which is consistent with fibrofolliculoma. Inset: immunostaining of α‐SMA. Fibrous tissue is negative. E, Histology of HLRCC‐associated RCC in a 49‐year‐old man. Type 2 papillary RCC with a high nuclear grade is observed. Inset: immunostaining of fumarate hydratase (FH). Capillary endothelial cells show FH staining (arrows), but the tumor cells are negative. F, G, Histology of HLRCC‐associated RCC in a 57‐year‐old man (F) and SDH‐deficient RCC in a 40‐year‐old man (G). Both patients were diagnosed by genetic testing. Oncocytic tumor cells are arranged in a solid sheet‐like pattern. They were indistinguishable from oncocytomas by H&E staining alone
Hereditary leiomyomatosis and renal cell cancer, also known as Reed's syndrome, is another autosomal dominant disorder caused by a germline mutation in fumarate hydratase (FH).39, 40 Affected individuals are at risk of developing cutaneous leiomyomatosis and RCC. HLRCC‐associated skin tumors are histologically benign piloleiomyomas (Figure 3C) showing diffuse immunostaining of α‐SMA and negative staining of FH. However, the stromal component of fibrofolliculomas in BHD syndrome shows positive immunostaining of CD34 and vimentin but rarely α‐SMA41, 42 (Figure 3D). The RCC component of HLRCC, in most cases, is highly aggressive and life‐threatening.29, 43 In addition, young adults and children occasionally develop RCC.44 The histology of HLRCC‐associated RCC was initially thought to be type 2 papillary architecture (Figure 3E), but later studies showed that these tumors occasionally show a mixture of solid, sieve‐like, tubular, cystic and papillary components.45 The morphological similarities between HLRCC‐associated RCC and other RCC types such as collecting duct, tubulocystic and renal medullary carcinomas have been re‐evaluated.46 Rarely, a low‐grade oncocytic morphology may be observed in HLRCC‐associated RCC that is reminiscent of SDH‐deficient RCC47 (Figure 3F,G). Individuals with germline mutations in the SDHB gene are at risk of developing SDH‐deficient RCC.48 Although the prognosis of SDH‐deficient RCC is not fully understood, this RCC type is regarded as low‐grade.47 FH and SDH subunits (SDHA‐D) are enzymes involved in the tricarboxylic acid cycle, and play important roles in the production of ATP.29 Therefore, the hypothesis that HLRCC‐associated RCC with an SDH‐deficient RCC‐like morphology has a more favorable course than that of HLRCC‐associated RCC with a high‐grade morphology requires further investigation. Immunohistochemically, HLRCC‐associated RCC is generally negative for FH and diffusely positive for 2‐succino cysteine.49 It should be noted that FH‐deficient RCC are not always associated with HLRCC.50
7. CONCLUSION
We suggest that BHD‐RCC, which consists of more than one histological type and shows unique cytogenetic features, may be defined more appropriately as an independent category rather than as a subtype of chromophobe RCC in the WHO classification. Reclassification of some other rare RCC, such as hereditary and sporadic FH‐deficient RCC, should be considered in the future. However, genetic counseling and testing for hereditary RCC are not covered by government subsidies in Japan. In addition, genetic discrimination may be a concern among suspected individuals. Medical circumstances occasionally discourage patients from receiving genetic testing, which prevents them from receiving the correct diagnosis from medical geneticists and pathologists and evidence‐based treatment from urologists. Legal protection of genetic information, such as the Genetic Information Nondiscrimination Act in the USA, should also be provisioned in Japan. Improvement of medical care for hereditary RCC patients and potential mutation carriers will be critical for their diagnosis and appropriate treatment.
DISCLOSURE
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
The authors thank all collaborators who provided the clinicopathological information for this study. This work was supported by JSPS KAKENHI Grant‐in‐Aid for Scientific Research: grant numbers 17K08745 (MF), 19K09694 (HH), 19K09717 (MY) and 17K11162 (YN).
Furuya M, Hasumi H, Yao M, Nagashima Y. Birt‐Hogg‐Dubé syndrome‐associated renal cell carcinoma: Histopathological features and diagnostic conundrum. Cancer Sci. 2020;111:15–22. 10.1111/cas.14255
REFERENCES
- 1. Hasumi H, Yao M. Hereditary kidney cancer syndromes: genetic disorders driven by alterations in metabolism and epigenome regulation. Cancer Sci. 2018;109:581‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674‐1677. [PubMed] [Google Scholar]
- 3. Hornstein OP, Knickenberg M. Perifollicular fibromatosis cutis with polyps of the colon–a cutaneo‐intestinal syndrome sui generis. Arch Dermatol Res. 1975;253:161‐175. [DOI] [PubMed] [Google Scholar]
- 4. Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt‐Hogg‐Dube syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393‐400. [PubMed] [Google Scholar]
- 5. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt‐Hogg‐Dube syndrome. Cancer Cell. 2002;2:157‐164. [DOI] [PubMed] [Google Scholar]
- 6. Furuya M, Nakatani Y. Pathology of Birt‐Hogg‐Dube syndrome: a special reference of pulmonary manifestations in a Japanese population with a comprehensive analysis and review. Pathol Int. 2019;69:1‐12. [DOI] [PubMed] [Google Scholar]
- 7. Menko FH, van Steensel MA, Giraud S, et al. Birt‐Hogg‐Dube syndrome: diagnosis and management. Lancet Oncol. 2009;10:1199‐1206. [DOI] [PubMed] [Google Scholar]
- 8. Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt‐Hogg‐Dube syndrome. Nat Rev Urol. 2015;12:558‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toro JR, Wei M‐H, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt‐Hogg‐Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Furuya M, Yao M, Tanaka R, et al. Genetic, epidemiologic and clinicopathologic studies of Japanese Asian patients with Birt‐Hogg‐Dube syndrome. Clin Genet. 2016;90:403‐412. [DOI] [PubMed] [Google Scholar]
- 11. Benusiglio PR, Giraud S, Deveaux S, et al. Renal cell tumour characteristics in patients with the Birt‐Hogg‐Dube cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis. 2014;9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pavlovich CP, Grubb RL 3rd, Hurley K, et al. Evaluation and management of renal tumors in the Birt‐Hogg‐Dube syndrome. J Urol. 2005;173:1482‐1486. [DOI] [PubMed] [Google Scholar]
- 13. Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt‐Hogg‐Dube syndrome. Am J Surg Pathol. 2002;26:1542‐1552. [DOI] [PubMed] [Google Scholar]
- 14. Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469‐1489. [DOI] [PubMed] [Google Scholar]
- 15. Houweling AC, Gijezen LM, Jonker MA, et al. Renal cancer and pneumothorax risk in Birt‐Hogg‐Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105:1912‐1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Furuya M, Hong SB, Tanaka R, et al. Distinctive expression patterns of glycoprotein non‐metastatic B and folliculin in renal tumors in patients with Birt‐Hogg‐Dube syndrome. Cancer Sci. 2015;106:315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruiz‐Cordero R, Rao P, Li L, et al. Hybrid oncocytic/chromophobe renal tumors are molecularly distinct from oncocytoma and chromophobe renal cell carcinoma. Mod Pathol. 2019;32:1698‐1707. [DOI] [PubMed] [Google Scholar]
- 18. Iribe Y, Kuroda N, Nagashima Y, et al. Immunohistochemical characterization of renal tumors in patients with Birt‐Hogg‐Dube syndrome. Pathol Int. 2015;65:126‐132. [DOI] [PubMed] [Google Scholar]
- 19. Klomp JA, Petillo D, Niemi NM, et al. Birt‐Hogg‐Dube renal tumors are genetically distinct from other renal neoplasias and are associated with up‐regulation of mitochondrial gene expression. BMC Med Genomics. 2010;3:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasumi H, Furuya M, Tatsuno K, et al. BHD‐associated kidney cancer exhibits unique molecular characteristics and a wide variety of variants in chromatin remodeling genes. Hum Mol Genet. 2018;27:2712‐2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iribe Y, Yao M, Tanaka R, et al. Genome‐wide uniparental disomy and copy number variations in renal cell carcinomas associated with Birt‐Hogg‐Dube syndrome. Am J Pathol. 2016;186:337‐346. [DOI] [PubMed] [Google Scholar]
- 22. Davis C, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kato I, Iribe Y, Nagashima Y, et al. Fluorescent and chromogenic in situ hybridization of CEN17q as a potent useful diagnostic marker for Birt‐Hogg‐Dube syndrome‐associated chromophobe renal cell carcinomas. Hum Pathol. 2016;52:74‐82. [DOI] [PubMed] [Google Scholar]
- 24. Durinck S, Stawiski EW, Pavía‐Jiménez A, et al. Spectrum of diverse genomic alterations define non‐clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yusenko MV, Kuiper RP, Boethe T, Ljungberg B, van Kessel AG, Kovacs G. High‐resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kluger N, Giraud S, Coupier I, et al. Birt‐Hogg‐Dube syndrome: clinical and genetic studies of 10 French families. Br J Dermatol. 2010;162:527‐537. [DOI] [PubMed] [Google Scholar]
- 28. Baba M, Furihata M, Hong S‐B, et al. Kidney‐targeted Birt‐Hogg‐Dube gene inactivation in a mouse model: Erk1/2 and Akt‐mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731‐741. [DOI] [PubMed] [Google Scholar]
- 31. Menko FH, Johannesma PC, van Moorselaar RJA, et al. A de novo FLCN mutation in a patient with spontaneous pneumothorax and renal cancer; a clinical and molecular evaluation. Fam Cancer. 2013;12:373‐379. [DOI] [PubMed] [Google Scholar]
- 32. Cardis MA, DeKlotz CMC. Cutaneous manifestations of tuberous sclerosis complex and the paediatrician's role. Arch Dis Child. 2017;102:858‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tobino K, Hirai T, Johkoh T, et al. Differentiation between Birt‐Hogg‐Dube syndrome and lymphangioleiomyomatosis: quantitative analysis of pulmonary cysts on computed tomography of the chest in 66 females. Eur J Radiol. 2012;81:1340‐1346. [DOI] [PubMed] [Google Scholar]
- 34. Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt‐Hogg‐Dube syndrome. Fam Cancer. 2013;12:387‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raoof S, Bondalapati P, Vydyula R, et al. Cystic lung diseases: algorithmic approach. Chest. 2016;150:945‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Warncke JC, Brodie KE, Grantham EC, et al. Pediatric renal angiomyolipomas in tuberous sclerosis complex. J Urol. 2017;197:500‐506. [DOI] [PubMed] [Google Scholar]
- 37. Stone CH, Lee MW, Amin MB, et al. Renal angiomyolipoma: further immunophenotypic characterization of an expanding morphologic spectrum. Arch Pathol Lab Med. 2001;125:751‐758. [DOI] [PubMed] [Google Scholar]
- 38. Amin S, Lux A, Calder N, Laugharne M, Osborne J, O'Callaghan F. Causes of mortality in individuals with tuberous sclerosis complex. Dev Med Child Neurol. 2017;59:612‐617. [DOI] [PubMed] [Google Scholar]
- 39. Reed WB, Walker R, Horowitz R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol. 1973;53:409‐416. [PubMed] [Google Scholar]
- 40. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA. 2001;98:3387‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kamada T, Yoshikawa Y, Shirase T, Furuya M, Yutaka Y. Perifollicular fibromas associated with Birt‐Hogg‐Dube syndrome. J Dermatol. 2015;42:1194‐1195. [DOI] [PubMed] [Google Scholar]
- 42. Misago N, Kimura T, Narisawa Y. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943‐951. [DOI] [PubMed] [Google Scholar]
- 43. Muller M, Ferlicot S, Guillaud‐Bataille M, et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. 2017;92:606‐615. [DOI] [PubMed] [Google Scholar]
- 44. Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y‐B, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome‐associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ohe C, Smith SC, Sirohi D, et al. Reappraisal of morphologic differences between renal medullary carcinoma, collecting duct carcinoma, and fumarate hydratase‐deficient renal cell carcinoma. Am J Surg Pathol. 2018;42:279‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith SC, Sirohi D, Ohe C, et al. A distinctive, low‐grade oncocytic fumarate hydratase‐deficient renal cell carcinoma, morphologically reminiscent of succinate dehydrogenase‐deficient renal cell carcinoma. Histopathology. 2017;71:42‐52. [DOI] [PubMed] [Google Scholar]
- 48. Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype‐phenotype‐proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41‐51. [DOI] [PubMed] [Google Scholar]
- 49. Trpkov K, Hes O, Agaimy A, et al. Fumarate hydratase‐deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. 2016;40:865‐875. [DOI] [PubMed] [Google Scholar]
- 50. Smith SC, Trpkov K, Chen Y‐B, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a frequent morphologic pattern of fumarate hydratase‐deficient renal cell carcinoma. Am J Surg Pathol. 2016;40:1457‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]