

Abstract
Drug repositioning is an emerging approach to developing novel cancer treatments. Vorinostat is a histone deacetylase inhibitor approved for cancer treatment, but it could attenuate its anticancer activity by activating the mTOR pathway. The HMG‐CoA reductase inhibitor fluvastatin reportedly activates the mTOR inhibitor AMP‐activated protein kinase (AMPK), and we thought that it would potentiate vorinostat's anticancer activity in renal cancer cells. The combination of vorinostat and fluvastatin induced robust apoptosis and inhibited renal cancer growth effectively both in vitro and in vivo. Vorinostat activated the mTOR pathway, as evidenced by the phosphorylation of ribosomal protein S6, and fluvastatin inhibited this phosphorylation by activating AMPK. Fluvastatin also enhanced vorinostat‐induced histone acetylation. Furthermore, the combination induced endoplasmic reticulum (ER) stress that was accompanied by aggresome formation. We also found that there was a positive feedback cycle among AMPK activation, histone acetylation, and ER stress induction. This is the first study to report the beneficial combined effect of vorinostat and fluvastatin in cancer cells.
Keywords: AMP‐activated protein kinase, endoplasmic reticulum stress, fluvastatin, histone acetylation, vorinostat
Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells via AMP‐activated protein kinase activation in concert with histone acetylation and endoplasmic reticulum stress induction.
1. INTRODUCTION
Histone deacetylase (HDAC) inhibitors have been shown in various cancer cells to promote histone acetylation and thereby induce apoptosis.1 Vorinostat became, in 2006, the first pan‐HDAC inhibitor approved for the treatment of cutaneous T‐cell lymphoma.2, 3 We have previously shown that the HDAC inhibitor panobinostat predisposes bladder cancer cells to apoptosis by inhibiting HDAC, but it also results in mTOR activity with consequent ribosomal protein S6 (S6) phosphorylation and these are antiapoptosis forces within a cell.4 Several recent studies have shown that combinations of an HDAC inhibitor and an mTOR inhibitor act cooperatively against renal cancer cells,5, 6 and S6 phosphorylation is thought to be especially relevant in renal cancer proliferation because it is associated with unfavorable prognosis in patients with renal cancer.7, 8 We therefore thought that vorinostat might phosphorylate S6 in renal cancer cells and thereby attenuate its ability to induce apoptosis.
Fluvastatin is one of the HMG‐CoA reductase inhibitors widely used for treating dyslipidemia and coronary artery disease.9, 10, 11 It also has anticancer activity12, 13, 14 and recently been shown to activate AMP‐activated protein kinase (AMPK),15 which acts against cancer by suppressing the mTOR pathway and inducing histone acetylation.16, 17, 18, 19, 20, 21
In the present study, we postulated that the combination of vorinostat and fluvastatin would suppress the vorinostat‐activated mTOR pathway and also enhance vorinostat‐induced histone acetylation, thereby killing renal cancer cells cooperatively.
2. MATERIALS AND METHODS
2.1. Cell cultures
Human renal cancer cells (ACHN, A498) and murine renal cancer cells (Renca) purchased from the ATCC were cultured in minimum essential medium (MEM) containing 10% FBS and 1.0% penicillin/streptomycin (Invitrogen) at 37°C under 5% CO2 in a humidified incubator.
2.2. Reagents
Vorinostat and fluvastatin purchased from Cayman Chemical, panobinostat purchased from LC Laboratories, belinostat purchased from Selleck Chemicals, and tunicamycin purchased from Enzo Life Sciences were dissolved in DMSO. Compound C dihydrochloride purchased from R&D Systems and cycloheximide purchased from Enzo Life Sciences were dissolved in distilled water. These reagents were stored at −80°C or −20°C until use.
2.3. Cell viability assay
Cell viability was evaluated by MTS assay (CellTiter 96 Aqueous kit; Promega) according to the manufacturer's protocol. Cells (5 × 103) were seeded into each well of a 96‐well culture plate 1 day before being treated under indicated conditions for 48 hours. After treatment, 20 μL MTS solution was added to the medium and the plates were incubated for 30‐60 minutes. The plates were then read at a wavelength of 490 nm in a microplate autoreader.
2.4. Clonogenic assay
Cells (0.5‐1 × 103) were seeded into each well of a 6‐well culture plate 1 day before being treated for 48 hours with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. The cells were then given fresh medium and cultured for 1‐2 weeks. The colonies were stained with Giemsa's solution after being fixed with 100% methanol and were quantified using the ImageJ plugin ColonyArea developed by Guzmán et al.22
2.5. In vitro cell invasion assay
The invasiveness of renal cancer cells was assessed by examining the invasion of 24 wells through Matrigel‐coated Transwell inserts with 8 μm pores (BD Biosciences) according to the manufacturer's protocol. Cells (2.0 × 105) in 500 mL serum‐free MEM were added to each insert, and 500 mL of MEM containing 10% fetal bovine serum with or without 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin was added to the bottom of each well. Forty‐eight hours later, the cells that had remained inside the inserts were removed and cells that had migrated through the inserts' membranes were fixed in methanol and stained with 1% toluidine blue (Kanto Chemicals) in 1% borax (Sigma). The cells were counted in 3 randomly chosen visual fields at ×200 magnification.
2.6. In vivo study
The experimental protocol for this in vivo experiment was approved by the institutional Animal Care and Use Committee of National Defense Medical College. Renca cells (1 × 107) were implanted s.c. into nude mice purchased from CLEA Japan and treatment was initiated 5 days later (day 1), when all the mice showed measurable tumors. The mice were divided into control and treatment groups (n = 5 per group). The treated mice received i.p. injections of either vorinostat (25 mg/kg) or fluvastatin (10 mg/kg) or both; the control mice received vehicle only. The injections were given once a day for 14 days (5 days on, 2 days off). Tumor volume and body weights were measured every 2 or 3 days. Tumor volumes were estimated using the following formula: volume = 0.5 × length × width2. After 14 days of treatment, the animals were killed in compliance with the ethical policy for animal experiments worldwide23 and the s.c. tumors were harvested.
2.7. Flow cytometry
Cells (1.0 × 105) were seeded into each well of a 12‐well culture plate 1 day before being cultured for 48 hours in medium with or without 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. The cells were then washed with PBS and harvested by trypsinization. For the annexin‐V assay, the cells were stained with annexin V and 7‐AAD following the protocol of the assay kit's manufacturer (Beckman Coulter). For cell cycle analysis, the cells were resuspended in citrate buffer, stained with propidium iodide, and then analyzed using a flow cytometer and CellQuest Pro Software (BD Biosciences). This cytometric analysis was carried out 3 times.
2.8. Western blot analysis
Cells were treated under the indicated conditions for 48 hours and whole cell lysates were obtained using RIPA buffer. Tumor specimens harvested from mice were homogenized using RIPA buffer, and whole cell lysates were obtained. Equal amounts of protein were separated by 12.5% SDS‐PAGE and transferred to nitrocellulose membranes. After the membranes were blocked by 5% skimmed milk, they were incubated overnight with the primary Abs: anti‐AMPK from Proteintech; anti‐phospho‐AMPK, anti‐phospho‐S6, anti‐S6, anti‐phospho‐eukaryotic translation initiation factor 4E‐binding protein 1 (p‐4EBP1), anti‐4EBP1, and anti‐endoplasmic reticulum resident protein (ERp) 44 from Cell Signaling Technology; anti‐glucose‐regulated protein (GRP) 78, anti‐cyclin D1, anti‐cyclin‐dependent kinase (CDK) 4, anti‐HDAC1, anti‐HDAC3, and anti‐HDAC6 from Santa Cruz Biotechnology; anti‐acetylated histone from Abcam; and anti‐actin from Millipore. Then the protein was detected by reaction with HRP‐tagged goat anti‐mouse or goat anti‐rabbit Ab (Bio‐Rad) and staining with chemiluminescence solution by using the ECL Plus system (GE Healthcare).
2.9. Immunohistochemical detection of active caspase 3
Tumor tissue samples derived from the mice models were fixed in 10% formalin, embedded in paraffin, and sliced into sections 4 μm thick. The sections were deparaffinized in xylene and rehydrated through graded alcohols and distilled water. After antigen retrieval, the sections were incubated in 10% normal goat serum in PBS for 1 hour at room temperature. They were then incubated overnight at 4°C with anti‐active caspase 3 Ab (Abcam), and this incubation was followed by incubation with HRP‐tagged anti‐rabbit Ab (Dako) for 1 hour at room temperature. The sections were developed in diaminobenzidine (Dako) and counterstained with hematoxylin.
2.10. Detection of aggresome formation
Cells (1.0 × 105) were seeded into each well of 2‐well chamber slides 1 day before treatment and incubated for 48 hours in media with or without 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. Cells were then fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton X before being incubated for 30 minutes with Hoechst 33342 and PROTEOSTAT dye (Enzo Life Sciences). Aggresomes and the nucleus were then detected using a fluorescence microscope (Carl Zeiss).
2.11. Statistical analysis
CalcuSyn software (Biosoft) was used for calculating the combination indexes according to the method developed by Chou and Talalay.24 The statistical significance of observed differences between samples was evaluated using the Mann‐Whitney U test (JMP Pro 14 software; SAS Institute), and differences for which P < .05 were considered statistically significant.
3. RESULTS
3.1. Anticancer activity of vorinostat in renal cancer cells
Vorinostat impaired renal cancer viability in a dose‐dependent manner (Figure 1A). Mechanistically, it induced not only histone acetylation but also endoplasmic reticulum (ER) stress evidenced by the increased expression of the ER stress markers GRP78 and ERp44 (Figure 1B). Of note, vorinostat activated the mTOR pathway by increasing the phosphorylation of S6, one of the pathway's most important downstream proteins associated with renal cancer proliferation.7, 8 Interestingly, vorinostat decreased the phosphorylation of 4EBP1, another downstream protein of the mTOR pathway, thus inhibiting another mTOR signaling cascade (Figure 1B). To confirm that HDAC inhibition activates the mTOR pathway, we then treated the cancer cells with other HDAC inhibitors. Both of the HDAC inhibitors panobinostat and belinostat inhibited the growth of renal cancer cells in a dose‐dependent manner (Figure 1C). As expected, each induced histone acetylation and ER stress but activated the mTOR pathway by increasing the phosphorylation of S6 (Figure 1D). Both also decreased the phosphorylation of 4EBP1, confirming that HDAC inhibition activated one downstream protein in the mTOR pathway while inhibiting another.
Figure 1.
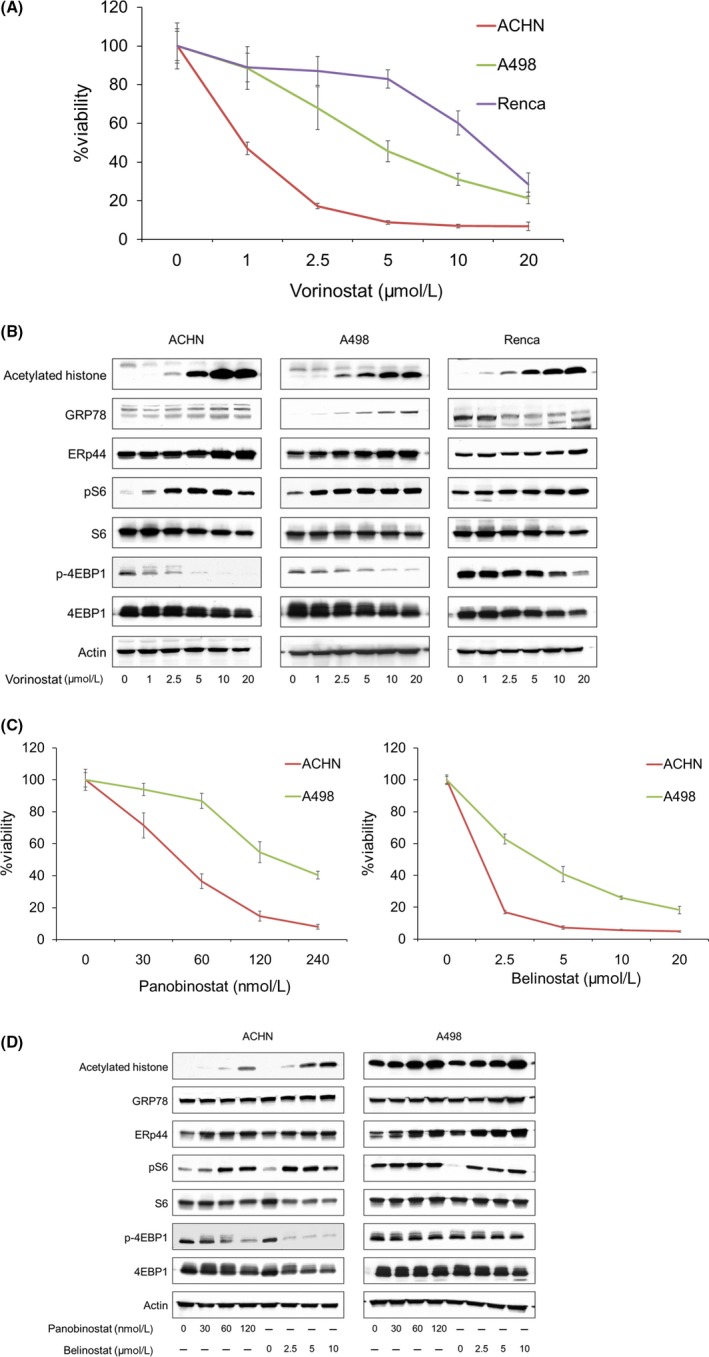
Anticancer activity of vorinostat in renal cancer cells. A, Cells were treated for 48 h with 1‐20 μmol/L vorinostat, and cell viability was measured using MTS assay. Mean ± SD, n = 12. B, Western blotting for acetylated histone, the endoplasmic reticulum (ER) stress markers glucose‐regulated protein (GRP) 78 and ER resident protein 44 (ERp44), S6 ribosomal protein (S6), and eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1). Cells were treated for 48 h with 1‐20 μmol/L vorinostat. Actin was used for the loading control. Representative blots are shown. C, Cells were treated for 48 h with 30‐240 nmol/L panobinostat or 2.5‐20 μmol/L belinostat, and cell viability was measured using MTS assay. Mean ± SD, n = 6. D, Western blotting for acetylated histone, GRP78, ERp44, S6, and 4EBP1. Cells were treated for 48 h with 30‐120 nmol/L panobinostat or 2.5‐10 μmol/L belinostat. Actin was used for the loading control. Representative blots are shown
3.2. Fluvastatin not only activated AMPK but induced histone acetylation
We next evaluated fluvastatin's anticancer activity and its mechanism of action in renal cancer cells. Fluvastatin reduced the viability of renal cancer cells in a dose‐dependent manner (Figure 2A). Mechanistically, it increased the phosphorylation of AMPK and decreased the phosphorylation of both S6 and 4EBP1 (Figure 2B), suggesting that it reduced renal cancer viability by inhibiting parts of the mTOR pathway through AMPK activation. We also found that fluvastatin induced histone acetylation in renal cancer cells (Figure 2B).
Figure 2.
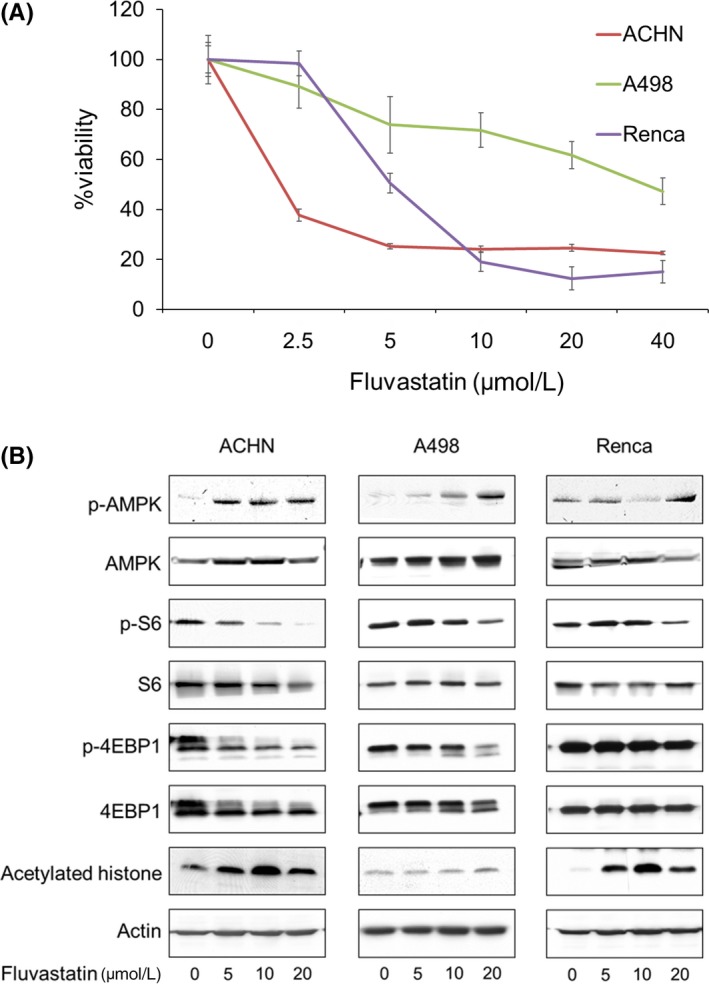
Fluvastatin not only activated AMP‐activated protein kinase (AMPK) but induced histone acetylation. A, Cells were treated for 48 h with 2.5‐40 μmol/L fluvastatin, and cell viability was measured using MTS assay. Mean ± SD, n = 12. B, Western blotting for AMPK, S6 ribosomal protein (S6), eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1), and acetylated histone. Cells were treated for 48 h with 5‐20 μmol/L fluvastatin. Actin was used for the loading control. Representative blots are shown
3.3. Vorinostat and fluvastatin in combination inhibited renal cancer growth in vitro and in vivo
The combination of vorinostat and fluvastatin inhibited renal cancer growth effectively (Figure 3A). The combined effect was synergistic in most of the treatment conditions (Figure 3B and Table 1). The clonogenic survival of renal cancer cells was also significantly inhibited by the combination (Figure 3C). Furthermore, the combination significantly impaired the cancer cells' invasiveness (Figure 3D).
Figure 3.
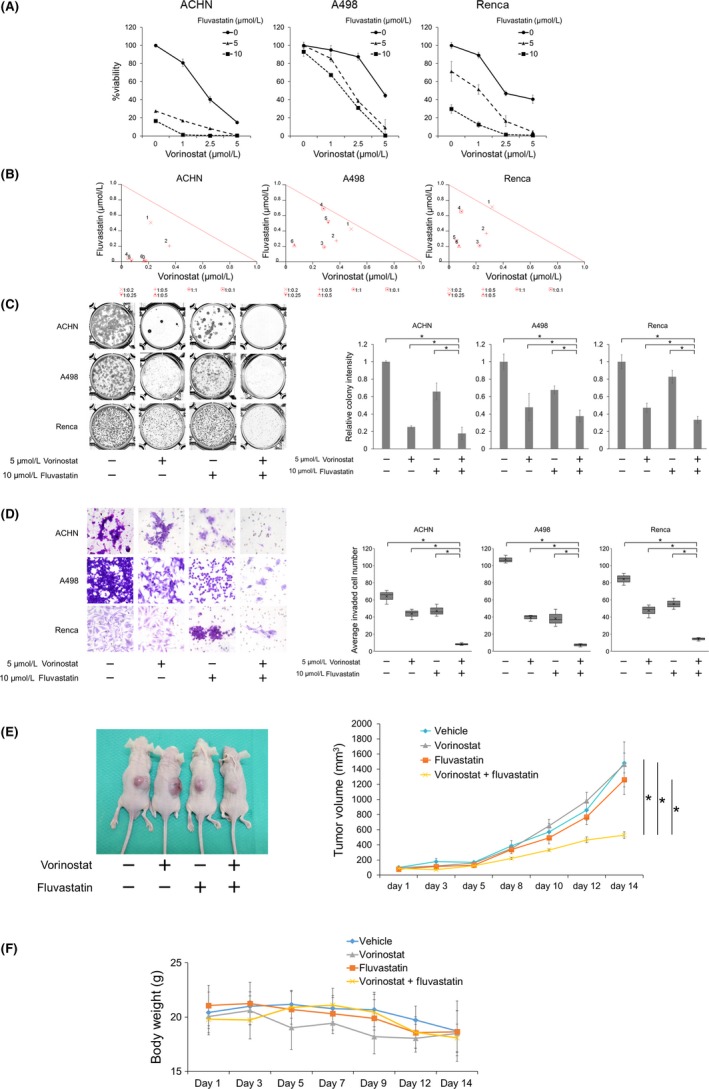
Combination of vorinostat and fluvastatin inhibited renal cancer growth in vitro and in vivo. A, Cells were treated for 48 h with 1‐5 μmol/L vorinostat and/or 5‐10 μmol/L fluvastatin, and cell viability was measured using MTS assay. Bars represent mean ± SD, n = 6. B, Isobologram analysis for the combination of vorinostat and fluvastatin. C, Clonogenic assay, in which 500‐1000 cells were treated for 48 h with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. The cells were then given fresh media and incubated for 1‐2 wk. Bar graphs show the relative colony intensity. Mean ± SD, n = 3. *P = .0495. D, Cells were treated for 48 h with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin, and cell invasion was evaluated using Matrigel invasion assay. Cells were counted in 3 randomly chosen visual fields at ×200 magnification. Box‐and‐whiskers plots show median (line within box), upper and lower quartiles (bounds of box), and minimum and maximum values (bars), n = 3. *P = .0495. E, A murine allograft model was established using Renca cells. The treated mice received i.p. injections of either vorinostat (25 mg/kg) or fluvastatin (10 mg/kg) or both; control mice received vehicle only. Injections were given once a day for 14 d (5 d on, 2 d off). Mean ± SE, n = 5. *P = .007 at day 14. F, Changes in the body weight. Mean ± SD, n = 5. Note that there is no significant difference in the body weight among groups at day 14
Table 1.
Combination indexes (CIs) for the combination of 1‐5 μmol/L vorinostat and 5‐10 μmol/L fluvastatin in renal cancer cells
| Fluvastatin (μmol/L) | Vorinostat (μmol/L) | ||
|---|---|---|---|
| 1 | 2.5 | 5 | |
| ACHN | |||
| 5 | 0.722 | 0.561 | 0.190 |
| 10 | 0.101 | 0.089 | 0.183 |
| A498 | |||
| 5 | 0.910 | 0.649 | 0.475 |
| 10 | 0.973 | 0.828 | 0.281 |
| Renca | |||
| 5 | 1.030 | 0.646 | 0.436 |
| 10 | 0.744 | 0.324 | 0.284 |
CI < 1 indicates synergy.
In murine s.c. allograft tumor models using Renca cells, a 14‐day treatment with the combination of fluvastatin and vorinostat suppressed tumor growth significantly (Figure 3E). Furthermore, the combination caused no remarkable weight loss (Figure 3F).
3.4. Combination of vorinostat and fluvastatin induced apoptosis cooperatively
The combination significantly caused the cancer cells to accumulate in the sub‐G1 fraction (Figure 4A), suggesting that it caused DNA fragmentation and induced apoptosis. Accordingly, the combination decreased the expression of cyclin D1 and CDK4 (Figure 4B). The induction of apoptosis was further confirmed by the annexin‐V assay: the combination significantly increased the percentage of the cell population that was annexin V‐positive (Figure 4C).
Figure 4.
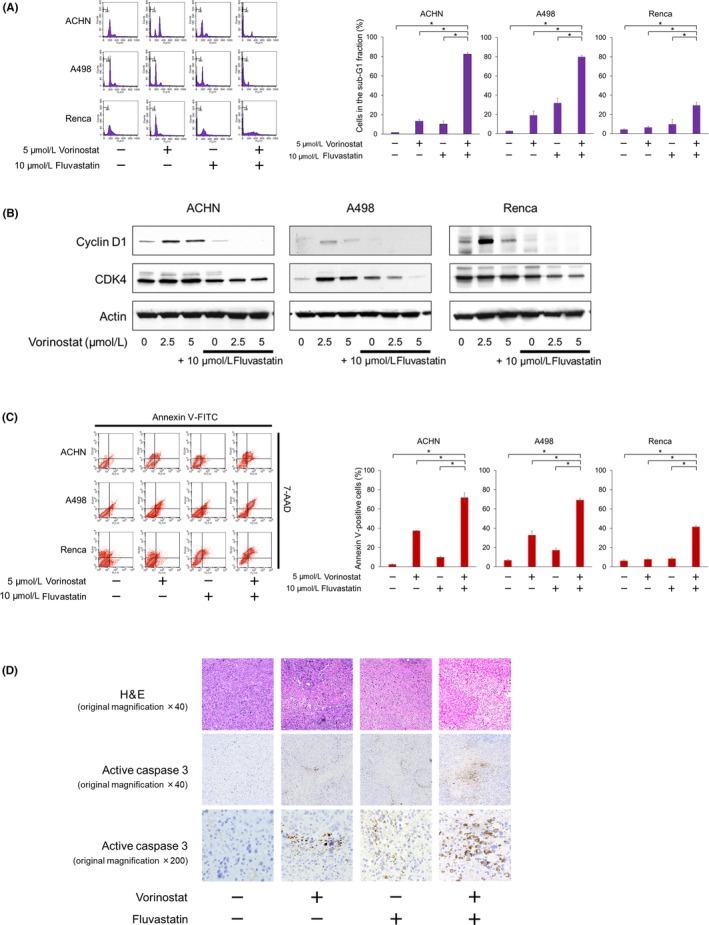
Combination of vorinostat and fluvastatin induced renal cancer cell apoptosis cooperatively. A, Cells were treated for 48 h with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. Changes in the cell cycle were evaluated using flow cytometry; 10 000 cells were counted. Bar graphs show the percentages of the cells in the sub‐G1 fraction. Data are expressed as mean ± SD from 3 independent experiments. *P = .0495. B, Western blotting for cyclin D1 and cyclin‐dependent kinase (CDK) 4. Cells were treated with 2.5‐5 μmol/L vorinostat and/or 10 μmol/L fluvastatin for 48 h. Actin was used for the loading control. Representative blots are shown. C, Cells were treated for 48 h with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. Apoptotic cells were detected by annexin‐V assay using flow cytometry. 10 000 cells were counted. Bar graphs show the percentages of apoptotic cells. Data are expressed as mean ± SD from 3 independent experiments. *P = .0495. D, Immunohistochemical analysis. After 14 d of treatment, the animals were killed and the s.c. tumors were harvested. Formalin‐fixed tumors were immunostained with anti‐active caspase 3 Ab
Immunohistochemical analyses of the tumor specimens revealed that the combination increased the expression of active caspase 3 more than each agent itself did (Figure 4D), suggesting that the combination would induce apoptosis cooperatively also in vivo.
3.5. Combination of vorinostat and fluvastatin inhibited the mTOR pathway
As expected, fluvastatin increased the phosphorylation of AMPK and decreased the vorinostat‐increased S6 phosphorylation (Figure 5), showing that fluvastatin indeed suppressed the vorinostat‐activated mTOR pathway. Further dephosphorylation of 4EBP1 is also evidence of the suppression of the mTOR pathway by fluvastatin. Thus, the combination was shown to inhibit the mTOR pathway.
Figure 5.
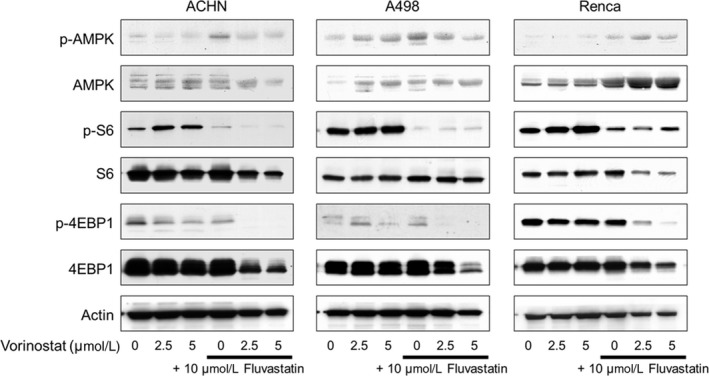
Combination of vorinostat and fluvastatin inhibited the mTOR pathway in renal cancer cells. Western blotting for AMP‐activated protein kinase (AMPK), S6 ribosomal protein (S6), and eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1). Cells were treated with 2.5‐5 μmol/L vorinostat and/or 10 μmol/L fluvastatin for 48 h. Actin was used for the loading control. Representative blots are shown
3.6. Pivotal role of AMPK in the combination's action
The AMPK inhibitor compound C attenuated not only the cytotoxicity of the combination (Figure 6A), but also its ability to induce apoptosis (Figure 6B). The increased phosphorylation of S6 proved that compound C inhibited the function of AMPK, and compound C reactivated the combination‐suppressed mTOR pathway evidenced by the increased phosphorylation of S6 and 4EBP1 (Figure 6C). These findings show that AMPK activation plays a pivotal role in the combination's action.
Figure 6.
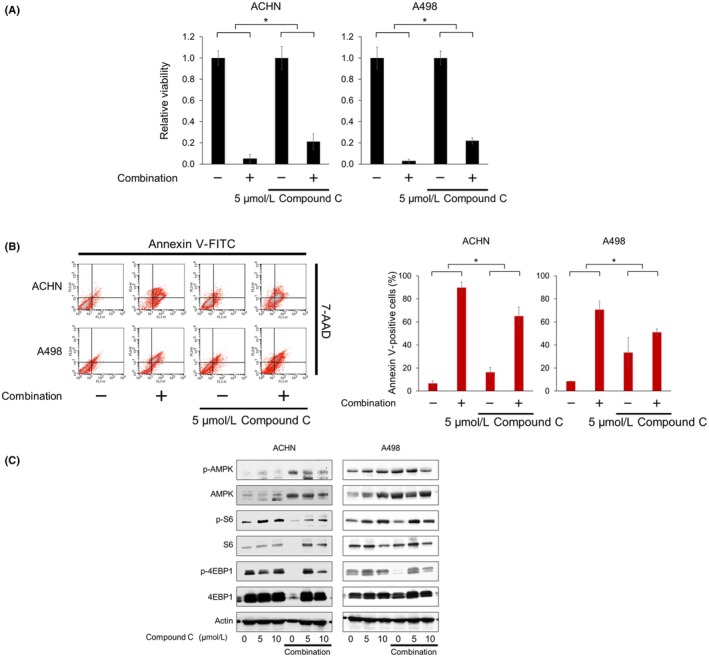
AMP‐activated protein kinase (AMPK) plays a pivotal role in vorinostat‐fluvastatin combination's action. A, Cells were treated with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μmol/L compound C for 48 h and renal cancer cell viability was measured using MTS assay. The viability of the control cells and that of the cells treated with compound C alone were both set at 1. Data are expressed as mean ± SD, n = 12. *P = .001. B, Cells were treated with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μmol/L compound C for 48 h. Apoptotic cells were detected by annexin‐V assay using flow cytometry; 10 000 cells were counted. Bar graphs show the increase in annexin V‐positive cells. Data are expressed as mean ± SD from 3 independent experiments. *P = .0495. C, Western blotting for AMPK, S6 ribosomal protein (S6), and eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1). Cells were treated for 48 h with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5‐10 μmol/L compound C. Actin was used for the loading control. Representative blots are shown
3.7. Vorinostat‐fluvastatin combination induced histone acetylation and ER stress cooperatively
Vorinostat is an HDAC inhibitor and induces histone acetylation. Fluvastatin was also shown to induce histone acetylation, as described above. Furthermore, AMPK activation itself is also known to induce histone acetylation.20, 21 We therefore thought that the combination of fluvastatin and vorinostat would induce histone acetylation cooperatively. Western blot analysis showed that vorinostat induced histone acetylation in a dose‐dependent manner and fluvastatin enhanced this acetylation (Figure 7A). Interestingly, the expression of HDACs was also decreased by the combination (Figure 7A). Because vorinostat itself induced both histone acetylation and ER stress (Figure 1B), we thought that the combination too would induce ER stress. As expected, the expression of the ER stress markers GRP78 and ERp44 was drastically increased by the combination, whereas fluvastatin or vorinostat alone increased their expression only moderately (Figure 7A).
Figure 7.
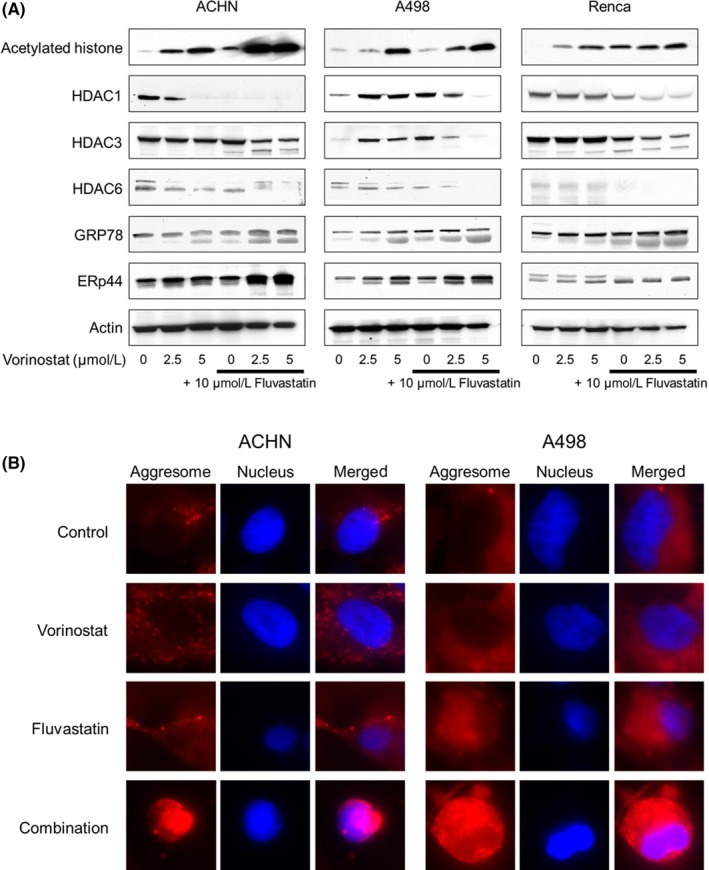
Combination of vorinostat and fluvastatin induced histone acetylation and endoplasmic reticulum (ER) stress cooperatively in renal cancer cells. A, Western blotting for acetylated histone, histone deacetylase (HDAC) 1, HDAC3, HDAC6, glucose‐regulated protein (GRP) 78, and ER resident protein 44 (ERp44). Cells were treated with 2.5‐5 μmol/L vorinostat and/or 10 μmol/L fluvastatin for 48 h. Actin was used for the loading control. Representative blots are shown. B, Aggresome detection after 48 h of treatment with 5 μmol/L vorinostat and/or 10 μmol/L fluvastatin. Blue, nucleus; red, aggresome. Original magnification, 1000×
Aggresomes are formed when ER stress triggers the aggregation of unfolded proteins,25, 26 so we next examined whether the combination facilitated aggresome formation. Notably, extensive aggresome formation was observed only when cells were treated with the combination (Figure 7B).
3.8. Vorinostat‐fluvastatin combination caused AMPK activation, histone acetylation, and ER stress in vivo
We then undertook western blot analysis using the specimens obtained in the in vivo experiment. The combination of vorinostat and fluvastatin increased the phosphorylation of AMPK and the expression of acetylated histone and GRP78 (Figure 8), indicating that the combination has the same mechanism of action in vivo that it does in vitro.
Figure 8.
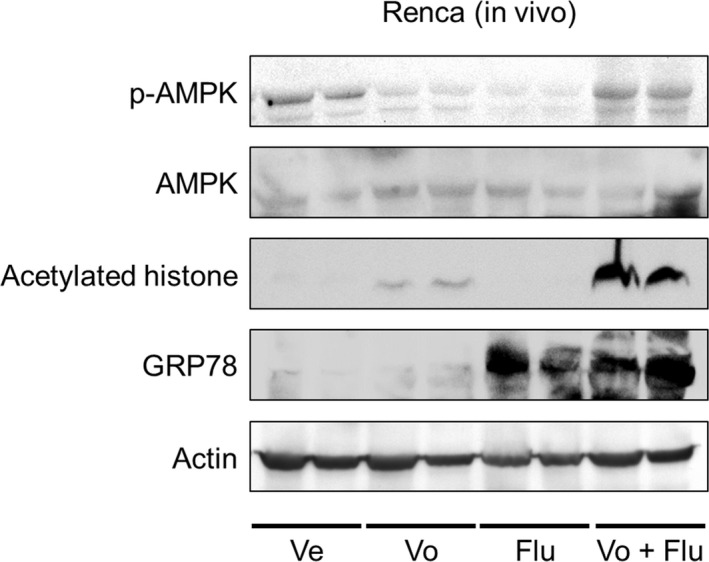
Combination of vorinostat and fluvastatin caused AMP‐activated protein kinase (AMPK) activation, histone acetylation, and endoplasmic reticulum stress in vivo. Western blotting for AMPK, acetylated histone, and glucose‐regulated protein (GRP) 78. After 14 d of treatment, the animals were killed and the s.c. tumors were harvested, lysed, and subjected to western blotting. Actin was used for the loading control. Representative blots are shown. Flu, fluvastatin‐treated mice; Ve, vehicle‐treated mice; Vo, vorinostat‐treated mice; Vo + Flu, combination‐treated mice
3.9. Cross‐talk among AMPK activation, histone acetylation, and ER stress induction
Because the combination of vorinostat and fluvastatin cooperatively induced histone acetylation and ER stress, we further investigated the contribution of ER stress induction to the combination's action. The protein synthesis inhibitor cycloheximide suppresses ER stress induction,27 so we examined whether it impaired the combination's action. Cycloheximide significantly attenuated the combination's ability to inhibit cell proliferation (Figure 9A) and induce apoptosis (Figure 9B). Western blot analysis indicated that cycloheximide inhibited the combination‐increased expression of ER stress markers, showing that it suppressed the ER stress induced by the combination (Figure 9C). Cycloheximide also inhibited the combination‐increased histone acetylation (Figure 9C). Interestingly, cycloheximide also suppressed the combination‐induced AMPK activation and thereby activated the combination‐suppressed mTOR pathway as evidenced by the increased phosphorylation of S6 and 4EBP1 (Figure 9D). In contrast, the AMPK inhibitor compound C attenuated the combination‐induced histone acetylation and ER stress (Figure 9E). Furthermore, the massive aggresome formation caused by the combination was inhibited by both compound C and cycloheximide (Figure 9F), which showed that both agents suppressed the combination‐induced ER stress.
Figure 9.
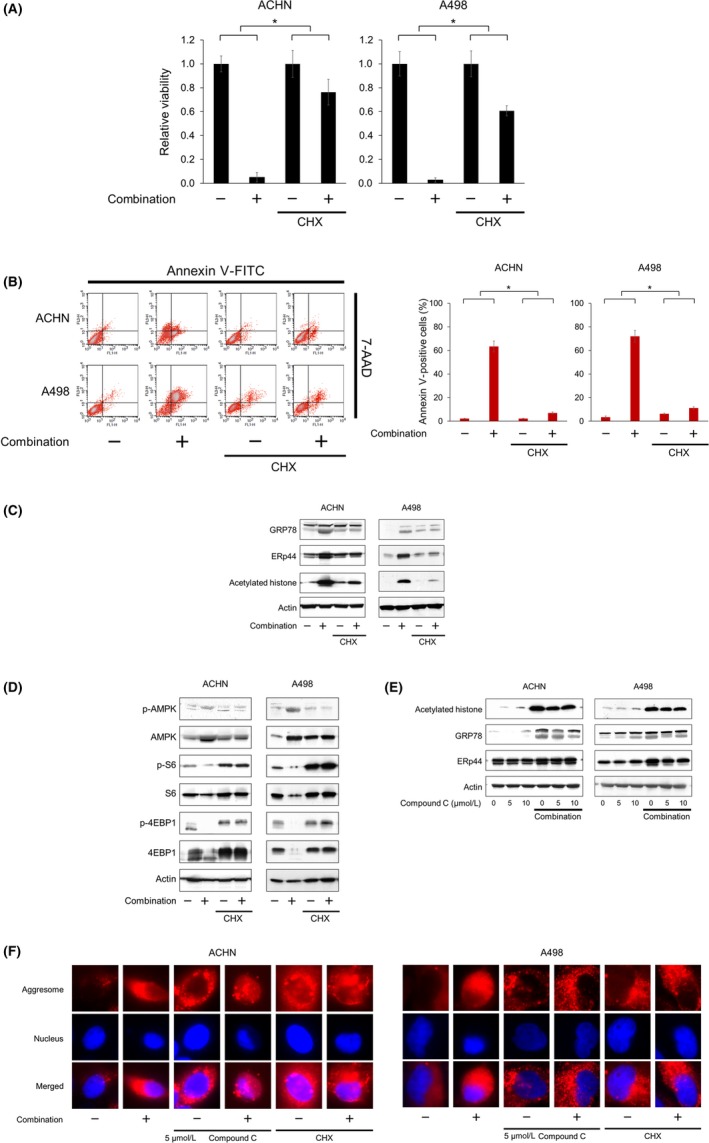
Cross‐talk among AMP‐activated protein kinase (AMPK) activation, histone acetylation, and endoplasmic reticulum (ER) stress induction. A, Renal cancer cells were treated with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μg/mL cycloheximide (CHX) for 48 h and cell viability was measured using MTS assay. The viability of the control cells and that of the cells treated with CHX alone were both set at 1. Data are expressed as mean ± SD, n = 12. *P = .001. B, Cells were treated with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μg/mL CHX for 48 h. Apoptotic cells were detected by annexin‐V assay using flow cytometry; 10 000 cells were counted. Bar graphs show the increase in annexin V‐positive cells. Data are expressed as mean ± SD from 3 independent experiments. *P = .0495. C, Western blotting for glucose‐regulated protein (GRP) 78, ER resident protein 44 (ERp44), and acetylated histone. Cells were treated for 48 h with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μg/mL CHX. Actin was used for the loading control. Representative blots are shown. D, Western blotting for AMPK, S6 ribosomal protein (S6), and eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1). Cells were treated for 48 h with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μg/mL CHX. Actin was used for the loading control. Representative blots are shown. E, Western blotting for acetylated histone, GRP78, and ERp44. Cells were treated for 48 h with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5‐10 μmol/L compound C. Actin was used for the loading control. Representative blots are shown. F, Aggresome detection after 48 h of treatment with 5 μmol/L vorinostat and 10 μmol/L fluvastatin with or without 5 μmol/L compound C or 5 μg/mL CHX. Blue, nucleus; red, aggresome. Original magnification, 1000×
We inferred from these results that there was cross‐talk among ER stress induction, histone acetylation, and AMPK activation.
4. DISCUSSION
Histone deacetylase inhibitors are considered to be innovative anticancer drugs,28, 29, 30 but their efficacy as single agents is limited, especially in solid tumors.31, 32, 33, 34, 35, 36 We have found that some HIV protease inhibitors and proteasome inhibitors enhance the activity of HDAC inhibitors.37, 38, 39, 40 We recently found that mTOR activation could be one of the important mechanisms of bladder cancer cells' resistance to the HDAC inhibitor panobinostat.4 In the present study, we postulated that vorinostat might induce mTOR activation, attenuating its anticancer activity in renal cancer cells, and that mTOR inhibition would overcome this attenuation.
Vorinostat indeed activated the mTOR pathway by increasing the phosphorylation of S6. The experiments with panobinostat and belinostat, hydroxamic acid‐based HDAC inhibitors like vorinostat,41, 42 provided further evidence that HDAC inhibition itself causes this phosphorylation. S6 phosphorylation reportedly regulates protein synthesis, glucose homeostasis, and cell size,43 but its molecular mechanism of action has not been clarified.44 Activation of mTOR is also an important mechanism of drug resistance,45, 46, 47, 48 so inhibiting the mTOR pathway is a reasonable approach to enhancing vorinostat's anticancer activity. Accordingly, we first treated renal cancer cells with vorinostat and the mTOR inhibitor temsirolimus and found that temsirolimus indeed enhanced the cytotoxicity of vorinostat (Figure S1A). However, the combined effect was only slightly synergistic in the limited treatment conditions (Figure S1B and Table S1). We therefore thought that only directly inhibiting mTOR would be insufficient to enhance the anticancer activity of vorinostat.
In the present study, we used the HMG‐CoA reductase inhibitor fluvastatin. HMG‐CoA reductase inhibitors reportedly have antiproliferative effects in various cancer cells,12, 13, 14, 49, 50, 51, 52 but they do not have demonstrable clinical anticancer activity (Table 2).53, 54, 55, 56, 57, 58, 59 One of fluvastatin's important mechanisms of action is AMPK activation.15 Statins are reported to activate AMPK by causing its phosphorylation through liver kinase B1 activation.60, 61 AMPK plays a key role in the regulation of energy balance.16 Because AMPK also controls cellular metabolism essential for cancer progression,62, 63 AMPK activation is crucial in the regulation of cancer cell growth and proliferation.16 We searched The Cancer Genome Atlas database by using the UCSC Cancer Browser UCSC Xena (https://xena.ucsc.edu/welcome-to-ucsc-xena/) and found that the expression of AMPK genes PRKAA1 and PRKAA2 was higher in normal tissue than cancer tissue (Figure S2A) and that renal cancer patients with higher expression of these genes had significantly longer overall survival (Figure S2B). These results also support the idea that activating AMPK is a promising way to treat renal cancer. To further develop this AMPK‐targeting strategy, the combined effect of vorinostat and other clinically available AMPK activators should be investigated. Our preliminary results showed that the antipsychotic olanzapine64 enhanced vorinostat's cytotoxicity only slightly (Figure S3 and Table S2), whereas the antidiabetic metformin65 synergized with vorinostat by a mechanism similar to that of fluvastatin (Figures S4‐6 and Table S3).
Table 2.
Clinical trials using statins in patients with various types of cancer
| Statin | Cancer type | Disease stage | Number of patients, statin/control | Study design | Median OS, statin/control (mo) | P value | Reference |
|---|---|---|---|---|---|---|---|
| Pravastatin | Small‐cell lung cancer | Limited or extensive disease | 422/424 | Phase III | 10.7/10.6 | .76 | Seckl et al53 |
| Simvastatin | Non‐ADC NSCLC | Advanced | 36/32 | Phase II | 10.0/7.0 | .93 | Lee et al54 |
| Simvastatin | Any | Brain metastases | 25/25 | Phase III | 3.4/3.0 | .88 | El‐Hamamsy et al55 |
| Simvastatin | Colorectal cancer | Metastatic | 134/135 | Phase III | 15.3/19.2 | .83 | Lim et al56 |
| Simvastatin | Gastric cancer | Metastatic | 120/124 | Phase III | 11.6/11.5 | .82 | Kim et al57 |
| Simvastatin | Pancreatic cancer | Locally advanced or metastatic | 58/56 | Phase II | 6.6/8.9 | .74 | Hong et al58 |
| Simvastatin | NSCLC | Locally advanced or metastatic | 52/54 | Phase II | 13.6/12.0 | .49 | Han et al59 |
ADC, adenocarcinomatous; NSCLC, non‐small‐cell lung cancer; OS, overall survival.
Activation of AMPK not only suppresses the mTOR pathway16, 17, 18, 19 but also induces histone acetylation.20, 21 We found that the AMPK activation played a pivotal role in the combination's action by showing that the AMPK inhibitor compound C impaired the combination's anticancer effects. Interestingly, compound C also inhibited the combination‐induced histone acetylation, confirming that AMPK activation played a role in regulating histone acetylation.
The combination of vorinostat and fluvastatin also induced ER stress. ER stress is caused by the accumulation of unfolded proteins, and profound ER stress inhibits the growth of malignant cells and causes their apoptosis.66, 67 The ER stressor tunicamycin reduced renal cancer cell viability in a dose‐dependent manner (Figure S7A). Furthermore, we have previously reported that ER stress‐inducing drug combinations killed urological cancers effectively.68, 69, 70, 71 The ER stress induction was also found to be crucial in the combination's action because the ER stress inhibitor cycloheximide significantly reduced combination‐caused apoptosis and the combination's cytotoxicity.
Our study showed that AMPK activation enhanced vorinostat‐induced histone acetylation and ER stress and that the AMPK inhibitor compound C attenuated the combination‐induced histone acetylation and ER stress. Similarly, the ER stressor tunicamycin caused AMPK activation and histone acetylation (Figure S7B), whereas the ER stress inhibitor cycloheximide attenuated the combination‐induced AMPK activation and histone acetylation. Both compound C and cycloheximide inhibited massive aggresome formation by the combination, which confirmed that both agents suppressed the combination‐induced ER stress. Furthermore, the HDAC inhibitors vorinostat, panobinostat, and belinostat all caused histone acetylation and ER stress (Figures 1B and D). These findings are compatible with those of previous studies, which showed that AMPK activation induces histone acetylation,4, 20, 21 ER stress induction is associated with calcium/calmodulin‐dependent kinase (CaMKK)‐beta, which is an activator of AMPK,72, 73, 74 ER stress induction causes histone acetylation in urological cancer cells,68, 69, 70 and decreased HDAC function causes ER stress by acetylating molecular chaperones and suppressing their function, thereby leading to an increased amount of unfolded proteins.75, 76, 77 This cross‐talk causes a positive feedback cycle, suppressing cancer growth (Figure 10).
Figure 10.
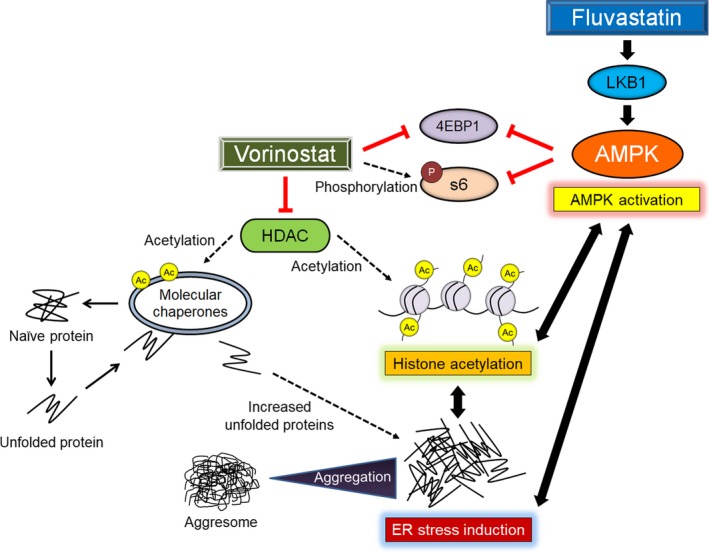
Mechanisms of action of the vorinostat‐fluvastatin combination in renal cancer cells. 4EBP1, eukaryotic translation initiation factor 4E‐binding protein 1; Ac, acetylation; AMPK, AMP‐activated protein kinase; ER, endoplasmic reticulum; HDAC, histone deacetylase; LKB1, liver kinase B1; S6, S6 ribosomal protein
Finally, safety is an important issue when clinical application of the combination is considered. The combination caused no remarkable weight loss in vivo. Furthermore, the combined effect was weaker in normal cells, even though its mechanisms of action were similar to those in cancer cells (Figures S8‐10 and Table S4). Thus, the vorinostat‐fluvastatin combination would be a safe combination therapy. However, careful clinical trials are needed before its clinical application.
DISCLOSURE
None.
Supporting information
ACKNOWLEDGMENTS
None.
Okubo K, Isono M, Miyai K, Asano T, Sato A. Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells. Cancer Sci. 2020;111:112–126. 10.1111/cas.14225
REFERENCES
- 1. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769‐784. [DOI] [PubMed] [Google Scholar]
- 2. Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T‐cell lymphoma. Expert Opin Investig Drugs. 2007;16(7):1111‐1120. [DOI] [PubMed] [Google Scholar]
- 3. Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T‐cell lymphoma. Oncologist. 2007;12(10):1247‐1252. [DOI] [PubMed] [Google Scholar]
- 4. Okubo K, Isono M, Asano T, et al. Metformin augments panobinostat's anti‐bladder cancer activity by activating AMP‐activated protein kinase. Transl Oncol. 2019;12(4):669‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahalingam D, Medina EC, Esquivel JA 2nd, et al. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16(1):141‐153. [DOI] [PubMed] [Google Scholar]
- 6. Park H, Garrido‐Laguna I, Naing A, et al. Phase I dose‐escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget. 2016;7(41):67521‐67531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knoll M, Macher‐Goeppinger S, Kopitz J, et al. The ribosomal protein S6 in renal cell carcinoma: functional relevance and potential as biomarker. Oncotarget. 2016;7(1):418‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li S, Kong Y, Si L, et al. Phosphorylation of mTOR and S6RP predicts the efficacy of everolimus in patients with metastatic renal cell carcinoma. BMC Cancer. 2014;28:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garnett WR. A review of current clinical findings with fluvastatin. Am J Cardiol. 1996;788(6A):20‐25. [DOI] [PubMed] [Google Scholar]
- 10. Scripture CD, Pieper JA. Clinical pharmacokinetics of fluvastatin. Clin Pharmacokinet. 2001;40(4):263‐281. [DOI] [PubMed] [Google Scholar]
- 11. Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. J Am Med Assoc. 2002;287(24):3215‐3222. [DOI] [PubMed] [Google Scholar]
- 12. Paragh G, Kertai P, Kovacs P, et al. HMG CoA reductase inhibitor fluvastatin arrests the development of implanted hepatocarcinoma in rats. Anticancer Res. 2003;23(5A):3949‐3954. [PubMed] [Google Scholar]
- 13. Chan KK, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res. 2003;9(1):10‐19. [PubMed] [Google Scholar]
- 14. Demierre MF, Higgins PD, Gruber SB, et al. Statins and cancer prevention. Nat Rev Cancer. 2005;5(12):930‐942. [DOI] [PubMed] [Google Scholar]
- 15. Kim JH, Lee JM, Kim JH, et al. Fluvastatin activates sirtuin 6 to regulate sterol regulatory element‐binding proteins and AMP‐activated protein kinase in HepG2 cells. Biochem Biophys Res Commun. 2018;503(3):1415‐1421. [DOI] [PubMed] [Google Scholar]
- 16. Faubert B, Vincent E, Poffenberger M, et al. The AMP‐activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165‐170. [DOI] [PubMed] [Google Scholar]
- 17. Jeon S, Hay N. The double‐edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015;38(3):346‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang W, Guan K. AMP‐activated protein kinase and cancer. Acta Physiol (Oxf). 2009;196(1):55‐63. [DOI] [PubMed] [Google Scholar]
- 19. Zadra G, Batista J, Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol Cancer Res. 2015;13(7):1059‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang M, Galdieri L, Vancura A. The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol Cell Biol. 2013;33(23):4701‐4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salminen A, Kauppinen A, Kaarniranta K. AMPK/Snf1 signaling regulates histone acetylation: impact on gene expression and epigenetic functions. Cell Signal. 2016;28(8):887‐895. [DOI] [PubMed] [Google Scholar]
- 22. Guzmán C, Bagga M, Kaur A, et al. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE. 2014;9(3):e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Workman P, Aboagye EO, Balkwill F, et al. Committee of the national cancer research institute. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102(11):1555‐1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou TC. Drug combination studies and their synergy quantification using the Chou‐Talalay method. Cancer Res. 2010;70(2):440‐446. [DOI] [PubMed] [Google Scholar]
- 25. Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143(7):1883‐1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nawrocki ST, Carew JS, Pino MS, et al. Aggresome disruption: a novel strategy to enhance bortezomib‐induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66(7):3773‐3781. [DOI] [PubMed] [Google Scholar]
- 27. Ram BM, Ramakrishna G. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim Biophys Acta. 2014;1843(11):2497‐2512. [DOI] [PubMed] [Google Scholar]
- 28. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349‐352. [DOI] [PubMed] [Google Scholar]
- 29. Huang H, Reed CP, Zhang JS, et al. Carboxypeptidase A3 (CPA3): A novel gene highly induced by histone deacetylase inhibitors during differentiation of prostate epithelial cancer cells. Cancer Res. 1999;59(12):2981‐2988. [PubMed] [Google Scholar]
- 30. Gallinari P, Di Marco S, Jones P, et al. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195‐211. [DOI] [PubMed] [Google Scholar]
- 31. Blumenschein GR Jr, Kies MS, Papadimitrakopoulou VA, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26(1):81‐87. [DOI] [PubMed] [Google Scholar]
- 32. Modesitt SC, Sill M, Hoffman JS, et al. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol. 2008;109(2):182‐186. [DOI] [PubMed] [Google Scholar]
- 33. Woyach JA, Kloos RT, Ringel MD, et al. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine‐refractory thyroid carcinoma. J Clin Endocrinol Metab. 2009;94(1):164‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tan J, Cang S, Ma Y, et al. Novel histone deacetylase inhibitors in clinical trials as anti‐cancer agents. J Hematol Oncol. 2010;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hainsworth JD, Infante JR, Spigel DR, et al. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest. 2011;29(7):451‐455. [DOI] [PubMed] [Google Scholar]
- 37. Sato A, Asano T, Ito K, et al. Suberoylanilide hydroxamic acid (SAHA) combined with bortezomib inhibits renal cancer growth by enhancing histone acetylation and protein ubiquitination synergistically. BJU Int. 2012;109(8):1258‐1268. [DOI] [PubMed] [Google Scholar]
- 38. Sato A, Asano T, Ito K, et al. Vorinostat and bortezomib synergistically cause ubiquitinated protein accumulation in prostate cancer cells. J Urol. 2012;188(6):2410‐2418. [DOI] [PubMed] [Google Scholar]
- 39. Isono M, Sato A, Okubo K, et al. Ritonavir interacts with belinostat to cause endoplasmic reticulum stress and histone acetylation in renal cancer cells. Oncol Res. 2016;24(5):327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okubo K, Isono M, Asano T, et al. Panobinostat and nelfinavir inhibit renal cancer growth by inducing endoplasmic reticulum stress. Anticancer Res. 2018;38(10):5615‐5626. [DOI] [PubMed] [Google Scholar]
- 41. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato A. Vorinostat approved in Japan for treatment of cutaneous T‐cell lymphomas: status and prospects. Onco Targets Ther. 2012;5:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ruvinsky I, Sharon N, Lerer T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19(18):2199‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meyuhas O. Ribosomal protein S6 phosphorylation: four decades of research. Int Rev Cell Mol Biol. 2015;320:41‐73. [DOI] [PubMed] [Google Scholar]
- 45. Grasso S, Tristante E, Saceda M, et al. Resistance to selumetinib (AZD6244) in colorectal cancer cell lines is mediated by p70S6K and RPS6 activation. Neoplasia. 2014;16(10):845‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burchert A, Wang Y, Cai D, et al. Compensatory PI3‐kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774‐1782. [DOI] [PubMed] [Google Scholar]
- 47. Rozengurt E, Soares HP, Sinnet‐Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. 2014;13(11):2477‐2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mishall KM, Beadnell TC, Kuenzi BM, et al. Sustained activation of the AKT/mTOR and MAP kinase pathways mediate resistance to the Src inhibitor, dasatinib, in thyroid cancer. Oncotarget. 2017;8(61):103014‐103031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sivaprasad U, Abbas T, Dutta A. Differential efficacy of 3‐hydroxy‐3‐methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol Cancer Ther. 2006;5(9):2310‐2316. [DOI] [PubMed] [Google Scholar]
- 50. Kusama T, Mukai M, Iwasaki T, et al. Inhibition of epidermal growth factor‐induced RhoA translocation and invasion of human pancreatic cancer cells by 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitors. Cancer Res. 2001;61(12):4885‐4891. [PubMed] [Google Scholar]
- 51. Li HY, Appelbaum FR, Willman CL, et al. Cholesterol‐modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood. 2003;101(9):3628‐3634. [DOI] [PubMed] [Google Scholar]
- 52. Sassano A, Katsoulidis E, Antico G, et al. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res. 2007;67(9):4524‐4532. [DOI] [PubMed] [Google Scholar]
- 53. Seckl MJ, Ottensmeier CH, Cullen M, et al. Multicenter, phase III, randomized, double‐blind, placebo‐controlled trial of pravastatin added to first‐line standard chemotherapy in small‐cell lung cancer (LUNGSTAR). J Clin Oncol. 2017;35(14):1506‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee Y, Lee KH, Lee GK, et al. Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced nonadenocarcinomatous non‐small cell lung cancer. Cancer Res Treat. 2017;49(4):1001‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. El‐Hamamsy M, Elwakil H, Saad AS, et al. A randomized controlled open‐label pilot study of simvastatin addition to whole‐brain radiation therapy in patients with brain metastases. Oncol Res. 2016;24(6):521‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lim SH, Kim TW, Hong YS, et al. A randomised, double‐blind, placebo‐controlled multi‐centre phase III trial of XELIRI/FOLFIRI plus simvastatin for patients with metastatic colorectal cancer. Br J Cancer. 2015;113(10):1421‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim ST, Kang JH, Lee J, et al. Simvastatin plus capecitabine‐cisplatin versus placebo plus capecitabine‐cisplatin in patients with previously untreated advanced gastric cancer: a double‐blind randomised phase 3 study. Eur J Cancer. 2014;50(16):2822‐2830. [DOI] [PubMed] [Google Scholar]
- 58. Hong JY, Nam EM, Lee J, et al. Randomized double‐blinded, placebo‐controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemother Pharmacol. 2014;73(1):125‐130. [DOI] [PubMed] [Google Scholar]
- 59. Han JY, Lee SH, Yoo NJ, et al. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non‐small cell lung cancer. Clin Cancer Res. 2011;17(6):1553‐1560. [DOI] [PubMed] [Google Scholar]
- 60. Ma L, Niknejad N, Gorn‐Hondermann I, et al. Lovastatin induces multiple stress pathways including LKB1/AMPK activation that regulate its cytotoxic effects in squamous cell carcinoma cells. PLoS ONE. 2012;7(9):e46055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang SH, Lin HY, Chang VH, et al. Lovastatin overcomes gefitinib resistance through TNF‐α signaling in human cholangiocarcinomas with different LKB1 statuses in vitro and in vivo. Oncotarget. 2015;6(27):23857‐23873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267‐277. [DOI] [PubMed] [Google Scholar]
- 63. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2(10):881‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He M, Zhang Q, Deng C, et al. Hypothalamic histamine H1 receptor‐AMPK signaling time‐dependently mediates olanzapine‐induced hyperphagia and weight gain in female rats. Psychoneuroendocrinology. 2014;42:153‐164. [DOI] [PubMed] [Google Scholar]
- 65. Galdieri L, Gatla H, Vancurova I, et al. Activation of AMP‐activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J Biol Chem. 2016;291(48):25154‐25166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu Y, Ye Y. Proteostasis regulation at the endoplasmic reticulum: a new perturbation site for targeted cancer therapy. Cell Res. 2011;21(6):867‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mimnaugh EG, Xu W, Vos M, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum‐derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004;3(5):551‐566. [PubMed] [Google Scholar]
- 68. Sato A, Asano T, Okubo K, et al. Ritonavir and ixazomib kill bladder cancer cells by causing ubiquitinated protein accumulation. Cancer Sci. 2017;108(6):1194‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sato A, Asano T, Okubo K, et al. Nelfinavir and ritonavir kill bladder cancer cells synergistically by inducing endoplasmic reticulum stress. Oncol Res. 2018;26(2):323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Isono M, Sato A, Asano T, et al. Delanzomib interacts with ritonavir synergistically to cause endoplasmic reticulum stress in renal cancer cells. Anticancer Res. 2018;38(6):3493‐3500. [DOI] [PubMed] [Google Scholar]
- 71. Okubo K, Sato A, Asano T, et al. Nelfinavir induces endoplasmic reticulum stress and sensitizes renal cancer cells to TRAIL. Anticancer Res. 2018;38(8):4505‐4514. [DOI] [PubMed] [Google Scholar]
- 72. Deshmukh RR, Dou QP. Proteasome inhibitors induce AMPK activation via CaMKKβ in human breast cancer cells. Breast Cancer Res Treat. 2015;153(1):79‐88. [DOI] [PubMed] [Google Scholar]
- 73. Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin‐dependent protein kinase kinase‐beta is an alternative upstream kinase for AMP‐activated protein kinase. Cell Metab. 2005;2(1):9‐19. [DOI] [PubMed] [Google Scholar]
- 74. Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin‐dependent protein kinase kinase‐beta acts upstream of AMP‐activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21‐33. [DOI] [PubMed] [Google Scholar]
- 75. Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280(29):26729‐26734. [DOI] [PubMed] [Google Scholar]
- 76. Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor‐alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13(16):4882‐4890. [DOI] [PubMed] [Google Scholar]
- 77. Baumeister P, Dong D, Fu Y, et al. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor‐induced apoptosis. Mol Cancer Ther. 2009;8(5):1086‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials