

Abstract
As a member of the epidermal growth factor receptor (EGFR) family, ERBB3 plays an essential role in development and disease independent of inherently inactive kinase domain. Recently, ERBB3 has been found to bind to ATP and has catalytic activity in vitro. However, the biological function of ERBB3 kinase activity remains elusive in vivo. Here we have identified the physiological function of inactivated ERBB3 kinase activity by creating Erbb3‐K740M knockin mice in which ATP cannot bind to ERBB3. Unlike Erbb3 knockout mice, kinase‐inactive Erbb3K740M homozygous mice were born in Mendelian ratios and showed normal development. After dextran sulfate sodium‐induced colitis, the kinase‐inactive Erbb3 mutant mice showed normal recovery. However, the outgrowth of ileal organoids by neuregulin‐1 treatment was more attenuated in Erbb3 mutant mice than in WT mice. Moreover, in combination with the ApcMin mouse, the proportion of polyps less than 1 mm in diameter in mutant mice was higher than in control mice and an increase in the number of apoptotic cells was observed in polyps from mutant mice compared with polyps from control mice. Taken together, the ERBB3 kinase activity contributes to the outgrowth of ileal organoids and intestinal tumorigenesis, and the development of ERBB3 kinase inhibitors, including epidermal growth factor receptor family members, can be a potential way to target colorectal cancer.
Keywords: ApcMin mouse, ERBB3, intestinal organoid, intestinal polyp, tyrosine kinase activity
ERBB3 kinase activity contributes to the outgrowth of ileal organoids and intestinal tumorigenesis, and the development of ERBB3 kinase inhibitors, including epidermal growth factor receptor family members, can be a potential way to target colorectal cancer

Abbreviations
- IEC
intestinal epithelial cell
- qRT‐PCR
quantitative RT‐PCR
1. INTRODUCTION
The epidermal growth factor receptor (EGFR) family, including ERBB1 (EGFR), ERBB2, ERBB3, and ERBB4, is a tyrosine kinase receptor that plays an essential role in development and disease.1 The EGFR family is abnormally expressed and is associated with metastasis in colorectal cancers.2, 3, 4 Unlike EGFR and ERBB2 as targets for disease control and cancer therapy, ERBB3 has been underestimated because of its inherently inactive kinase domain.1 However, ERBB3 is often overexpressed in colorectal cancers and associated with EGFR and ERBB2 expression.5, 6, 7 Upregulation of ERBB3 can induce resistance of therapeutic inhibitors designed to counteract EGFR and ERBB2 signaling.3, 8
In early biochemical studies, the ERBB3 intracellular domain has not been associated with ATP and has no autophosphorylation capacity.9, 10, 11 Therefore, it was thought that it could be activated only by heterodimerization with other ERBB receptors. Once neuregulins (NRGs) bind to ERBB3, ERBB2 can phosphorylate the ERBB3 C‐terminal tyrosine residues by transphosphorylation, but the mechanism by which kinase‐inactive ERBB3 phosphorylates ERBB2 remains unknown.12, 13 Recently, the purified ERBB3 intracellular domain has been identified as binding to ATP with an affinity similar to that of other active kinases and as having specific autophosphorylation activity.14 In addition, ERBB3 can form homodimerization and heterodimerization in cell membranes.15 Moreover, kinase‐inactive human ERBB3(K723M) shows low levels of phosphorylation, indicating that the total ERBB3 phosphorylation includes ERBB3 autophosphorylation.14, 15 Although this activity of ERBB3 kinase is weaker than that of EGFR, it can phosphorylate other ERBBs and the ligand‐induced ERBB3 kinase activity is elevated by the ERBB2‐ERBB3 heterodimer.15
Regardless of the kinase activity, the genetic ablation in mice reveals the biological function of ERBB3 under normal physiological and pathological conditions. Homozygous Erbb3 knockout mice die because of an abnormality of the nervous system during pregnancy,16 and the additional defects in development and poor differentiation of several organs have been found in Erbb3 knockout embryos.17 Mammary gland‐specific Erbb3 knockout mice show that ERBB3 is essential for the formation of ducts during pregnancy and lactation by regulating the survival and differentiation of breast epithelial cells and is also necessary for promoting breast tumor formation.18, 19, 20, 21 The liver‐specific removal of Erbb3 retards tumor formation in a hepatocarcinogenesis mouse model and diminishes liver fibrosis.22, 23 Intestine‐specific Erbb3 ablation results in more damage of colonic epithelial cells and slow recovery of epithelial cells in a model of colitis induced by dextran sulfate sodium (DSS).24, 25 Moreover, in an ApcMin intestinal tumor mouse model, intestine‐specific Erbb3 knockout mice have less cellular proliferation within polyps in the intestine than do control mice.24
Previous studies have reported the in vivo function of impaired kinase activity in EGFR family members. Homozygous mice Egfrwa2 with V743G substitution are alive and reproducible but have defects in the eyes and skin26 and a feeding disorder.27 In addition, homozygous EgfrWa5 mice with D833G substitution in the DFG motif of the kinase domain die during pregnancy.28 The homozygous K750M kinase‐dead Erbb2 mice die at E10.5 because of defects in cardiac trabeculation and in the neural system.29 However, the physiological function of ERBB3 kinase activity remains an open question in vivo. In this study, we generated Erbb3K740M with K740M substitution knockin mice in which ATP could not bind to ERBB3, and sought to unveil the function of ERBB3 kinase activity in embryonic development, DSS‐induced colitis, ileal organoid formation, and tumorigenesis in the ApcMin mouse model.
2. MATERIALS AND METHODS
2.1. Generation of Erbb3 knockin mice with mutation of the ATP‐binding site
Detailed information on the generation of knockin mice using the CRISPR/Cas9 tool is described in Data S1. We undertook PCR with DNA lysates from live pups’ toes with Erbb3‐Ex19S1, 5′‐CTCCCTCCACTTCAAACTTT‐3′; Erbb3‐Ex19AS1, and 5′‐TACAGATGAAGTCCCTTCTT‐3′ primers. The PCR products were subcloned in a pGEM‐T Easy Vector System (Promega), and we verified the knockin allele of Erbb3 (Erbb3K740M, the kinase‐inactive mutation of Erbb3) by sequencing. We bred selected founder mice to identify germline transmission of the knockin allele, and further confirmed the F1 generation by PCR sequencing. Subsequent generations of mice carrying the Erbb3K740M allele were maintained on the B6 background, and genotyping was carried out by agarose gel electrophoresis of the PCR product after NlaIII digestion. The research ethics regulation and guidelines were approved by the Institutional Animal Care and Use Committee (IACUC) of Ewha Womans University.
2.2. Cell staining
Both BrdU and TUNEL staining were undertaken as described previously.30 The number of BrdU‐ or TUNEL‐positive cells per crypt was counted from 20 well‐shaped crypts per mouse. The number of BrdU‐ or TUNEL‐positive cells in polyps was counted in a given area at 400× magnification and the average number of stained cells was obtained from 2 to 8 fields of size‐matched polyps from each mouse.
2.3. Dextran sulfate sodium treatment and histological scoring
Mice aged 2–3 months were treated with 2.5% (w/v) DSS (Cat No. 160 110; MP Biomedicals) in drinking water. After 5 days, 2.5% DSS was replaced by drinking water for recovery. The body weight, stool consistency, and rectal bleeding were recorded daily. To evaluate the function of ERBB3 kinase, mice stayed in recovery for either 5 or 25 days. At the end of the experiment, the small intestine and colon were isolated and fixed in neutral buffered formalin, followed by embedding in paraffin and sectioning. We used H&E‐stained sections for histological scoring using the previously described criteria (described in Data S1).31
2.4. Disease activity index measurement
We analyzed the body weight, stool consistency, and rectal bleeding daily in DSS‐treated mice. The disease activity index (DAI) was the combined scores of weight loss, stool consistency, and rectal bleeding adapted as previously described.32 Each condition was graded on a scale of 0 to 4: weight loss percentage (0, none; 1, 0%‐10%; 2, 10%‐15%; 3, 15%‐20%; 4, more than 20%); stool consistency (0, normal; 2, loose stool; 4, diarrhea); and rectal bleeding (0, normal; 2, hemoccult; 4, cross bleeding).
2.5. Quantitative RT‐PCR
Total RNA preparation and quantitative RT‐PCR (qRT‐PCR) were carried out as described previoulsy.30 The primer sequences used for qRT‐PCR are detailed in Data S1. Reactions were run on an ABI 7900HT Fast Real‐Time PCR system (Thermo Fisher Scientific).
2.6. In vivo phosphorylation analysis
We genotyped the neonatal pups (postnatal day 1). Next day, the male neonatal pups were injected s.c. with recombinant human NRG1‐β1 (1 µg/g body weight) (396‐HB‐050; R&D Systems) or PBS (10 µg/g body weight) as the vehicle. After 10 minutes, skin and liver tissues were harvested and frozen in liquid nitrogen. We homogenized the tissues (50‐100 mg) in 10 volumes of RIPA buffer supplements with proteinase and phosphatase inhibitors by means of a glass‐Teflon homogenizer on ice.28 We measured protein concentration by using a Bradford Assay (Bio‐Rad Lab).
2.7. Western blot analysis
Equal amounts of protein extracts were separated by SDS‐PAGE (7.5%), then were transferred to PVFD membranes. The detailed procedure for western blotting was described previously.24 The primary Abs are listed in Data S1. The protein‐band densities were quantified by image analysis software ImageJ (http://imagej.nih.gov).
2.8. Small intestinal crypt isolation and organoid culture
As described previously, we isolated small intestinal crypts from the ileum region.33, 34 Briefly, the distal ileum was washed with ice‐cold PBS. The lengthwise opened intestine was cut into pieces (less than 5 mm) and incubated in chelating buffer (2 mmol/L EDTA) with horizontal shaking for 30 minutes at 4°C. Ileum pieces were shaken by hand to dissociate the crypt from the basement membrane in dissociation buffer (D‐sorbitol 54.9 mmol/L, sucrose 43.4 mmol/L). The crypt suspension was filtered by means of a cell strainer and pelleted by centrifuge. Five hundred whole crypts were suspended in 50 µL of a mixture containing 50% Matrigel (BD Biosciences) and 50% IntestiCul Organoid Growth Medium (Corning) and slowly ejected into 24‐well plates. After polymerization, 750 µL IntestinCul media was added and replaced completely every 3 days. For organoid passaging, 7‐9 days after seeding, the primary organoids were disrupted by being passed through a 26.5‐gauge needle‐equipped syringe in cold PBS. The dissociated organoids solution was centrifuged and embedded in Matrigel as above.
2.9. Organoid measurement, treatment, and analysis
To evaluate the efficiency of organoid growth, we counted total organoid numbers/well under a light microscope at day 5 of culture in double‐blinded fashion. We measured the size of organoids using the ImageJ program. The structure of the organoid was evaluated by counting the number of buds: sphere, no budding; organoid 1, 1 bud; organoid 2, 2 buds; organoid 3+, 3 or more buds.35 To examine the outgrowth of organoids by ligand treatment, we treated organoids with recombinant human epidermal growth factor (EGF; 10 ng/mL) (236‐EG‐200; R&D Systems) and recombinant human NRG1‐β1 (10 ng/mL) for 3 days. For phosphorylation analysis, we homogenized organoid pellets in 50 µL RIPA buffer supplemented with proteinase and phosphatase inhibitors followed by western blotting. To examine the short‐term effect of ligands on phosphorylation, we treated organoids with ligands for 10 minutes before preparing the protein extract.
2.10. Macroadenoma counts and histopathological evaluation
We removed fecal material from the colon and small intestine of 2‐month‐old mice by flushing with PBS. Longitudinally opened small intestine and colon were splayed flat on 3MM paper. We counted polyp (>0.3 mm) numbers and diameters using a dissecting microscope and in‐scope micrometer in double‐blinded fashion. For histopathological evaluation, Swiss‐rolled ileum was fixed, embedded in paraffin, and then stained in 5‐μm sections with H&E. Histopathology of the polyps was classified as low grade, high grade, and adenocarcinoma following a previous report.36
2.11. Statistical analysis
We undertook statistical analysis by using GraphPad Prism 7.0 software. For significant analysis, we used Student’s t test or 2‐way ANOVA with Sidak’s multiple comparisons test. P < .05 was considered statistically significant.
3. RESULTS
3.1. Erbb3K740M/K740M knockin mice show normal development
Based on previous reports for EGFR and ERBB2 kinase‐inactive mutation,37, 38, 39 we selected a conserved lysine 740 amino acid of mouse ERBB3 as a site for mutagenesis. In vitro, mutant human ERBB3 with this lysine replaced by a methionine had strongly reduced autophosphorylation.14, 15 We introduced knockin of the K740M (Lys‐AAA to Met‐ATG) sequence in mouse Erbb3 using the CRISPR‐Cas9 system in 1‐cell embryos. Of note, the codon sequence Ile‐ATC (I739) was replaced by Ile‐ATT to generate the NlaIII restriction enzyme site in the knockin allele (Figure 1A). NlaIII digestion of the PCR product of genomic DNA showed a 151‐bp fragment for WT, 151‐bp, 75‐bp and 76‐bp for Erbb3 +/K740M (Erbb3 +/M) heterozygous, and 75‐bp and 76‐bp for Erbb3K740M/K740M (Erbb3 M/M) homozygous mice (Figure 1B). In addition, qRT‐PCR analysis to compare the relative Erbb3 mRNA levels in the liver and colon tissues revealed that the Erbb3 mRNA level was the same in both WT and Erbb3K740M/K740M mice (Figure 1C). Western blot analysis showed that the ERBB3 proteins in Erbb3K740M/K740M mice were not significantly different from those in the WT mice, suggesting that the half‐life of mutant ERBB3 protein is similar to that of WT ERBB3 (Figure 1D).
Figure 1.
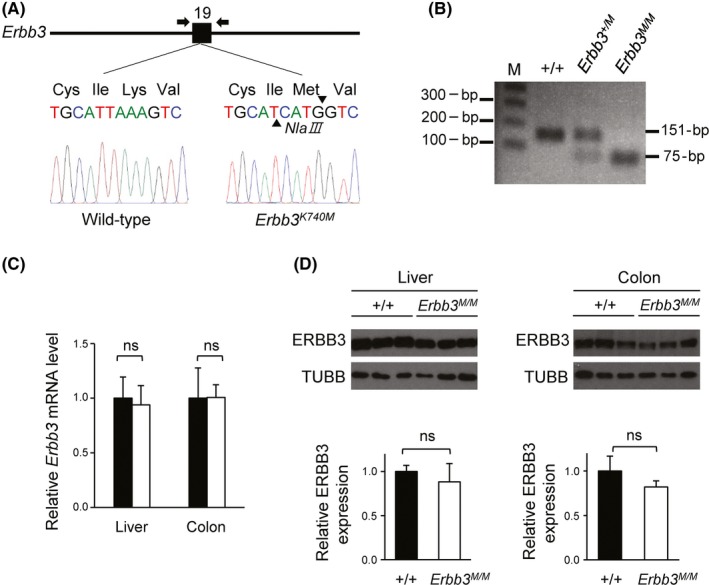
Genotyping of Erbb3K740M mutant mice and analysis of ERBB3 expression. A, Exon 19 of Erbb3 gene is labeled 19. Arrows indicate primers for PCR genotyping. Targeting Erbb3 with the K740M mutation (Erbb3K740M), in which K740M stands for Lys to Met substitution of 740th amino acids. B, Erbb3 PCR products from genomic DNA of WT Erbb3+/+, heterozygote Erbb3+/K740M (Erbb3+/M), and Erbb3K740M/K740M (Erbb3M/M) mice were cut with NlaIII restriction enzyme and analyzed with agarose gel electrophoresis (151‐bp product for WT allele; 75‐, 76‐bp for mutant allele; M, 1 kb plus DNA ladder). C, Quantitative RT‐PCR of Erbb3 mRNA expression level from the liver and colon of WT (n = 3) and Erbb3K740M/K740M mice (n = 3). D, Western blotting of ERBB3 protein from the liver and colon of WT (n = 6) and Erbb3K740M/K740M mice (n = 6). Left, liver; right, colon. ns, statistically insignificant
In contrast to the embryonic lethality of Erbb3 knockout mice,16, 17 Erbb3K740M/K740M mice were born in the Mendelian ratio and had no obvious abnormal phenotype. Overall histology was similar in the ileum and colon between WT and Erbb3K740M/K740M mice (Figure 2A). The Erbb3K740M/K740M mice showed normal length of the small intestine and colon compared to the WT mice (Figure S1). After BrdU injection, the BrdU‐positive cells per crypt were counted to evaluate the effect on intestinal cell proliferation. The number of BrdU‐positive cells per crypt in the ileum and colon were not significantly different between the 2 groups (Figure 2B). The TUNEL assay revealed that apoptosis was also not significantly different in the ileum or colon of WT and Erbb3K740M/K740M mice (Figure 2C). Overall, kinase‐inactive Erbb3 mutant mice showed normal intestinal homeostasis similar to that of intestinal epithelial cell (IEC)‐specific Erbb3 knockout mice.24
Figure 2.
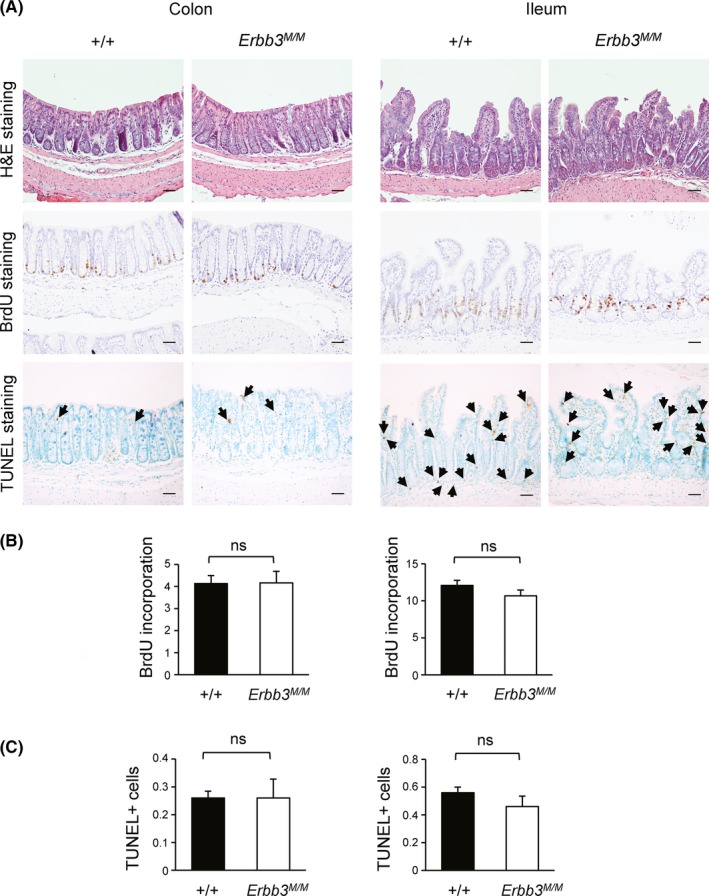
Cell proliferation analysis of Erbb3K740M/K740M mice in the intestine. A, Colon and ileum sections were examined by staining with H&E, immunostaining with anti‐BrdU Ab, or TUNEL staining (100× magnification). Arrows indicate TUNEL‐positive cells. Scale bar = 100 μm. B, Number of BrdU‐positive cells per crypt in WT (n = 5) and Erbb3K740M/K740M mice (n = 5) is shown as mean ± SEM. Left, colon; right, ileum. C, Number of TUNEL‐positive cells per crypt in WT (n = 5) and Erbb3K740M/K740M mice (n = 5) is shown as mean ± SEM. Left, colon; right, ileum. ns, statistically insignificant
3.2. In vivo tyrosine phosphorylation of the ERBB family is not affected in Erbb3K740M/K740M mice
In vitro, the isolated ERBB3 intracellular domain was able to autophosphorylate, and the endogenous full‐length ERBB3 significantly upregulated ERBB3 tyrosine kinase activity when stimulated with NRG1.14, 15 In order to validate the ERBB3 kinase activity in vivo, WT and Erbb3K740M/K740M littermates were treated with either PBS or NRG1, and tissue extracts from skin were analyzed with western blotting. The NRG1‐induced overall tyrosine phosphorylation levels (PY20) were similar between the WT and Erbb3K740M/K740M mice (Figure 3). Moreover, tyrosine phosphorylation of ERBB3 (pERBB3‐Tyr1197), EGFR (pEGFR‐Tyr1068), and ERBB2 (pERBB2‐Tyr877) were not different between the 2 groups (Figure 3). Total levels of EGFR, ERBB2, and ERBB3 were similar between the 2 groups. There was little attenuation of ERBB3 phosphorylation in Erbb3K740M/K740M mice compared to WT in the liver (Figure S2).
Figure 3.
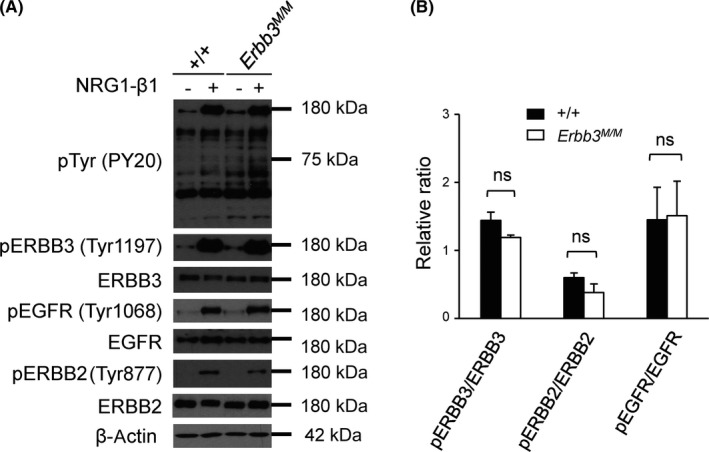
Neuregulin‐1 (NRG1)‐induced tyrosine phosphorylation of ERBB3 in vivo. A, Two‐day‐old WT (n = 3) and Erbb3K740M/K740M pups (n = 3) were treated with either PBS (−) or NRG1‐β1 (+) for 10 minutes before harvesting skin tissues. Tissue extracts were analyzed by western blotting using indicated Abs. Photographs are representative images of 3 independent experiments. B, Relative phosphorylated (p) band intensities were normalized with total protein levels (n = 3). ns, statistically insignificant
3.3. Erbb3K740M/K740M mice show normal recovery following DSS‐induced colitis
The IEC‐specific Erbb3 deletion mice show greater histological damage following DSS treatment and delayed recovery.24, 25 To clarify the contribution of ERBB3 kinase activity in those processes, Erbb3K740M/K740M mice were treated with 2.5% DSS for 5 days to induce colitis and then allowed to recover on normal drinking water for either a 5‐day period (short‐term recovery) or a 25‐day period (long‐term recovery). The body weight loss and DAI showed no significant difference between the WT and Erbb3K740M/K740M mice (Figure 4A,B). After a 5‐day recovery, the small intestinal length or colonic length of WT and Erbb3K740M/K740M mice were similar (Figure 4C). Histological analysis of DSS‐induced colitis by H&E staining of proximal, middle, and distal colonic sections showed no significant difference (Figure 4D), and all of the inflammation scores, epithelial regeneration scores, crypt damage scores, and total histological scores were similar between the WT and Erbb3K740M/K740M mice in short‐term recovery (Figure 4E). There were also no significant differences in body weight loss (except on days 10 and 12), DAI, or histological scores in long‐term recovery (Figure S3), indicating that ERBB3 kinase activity is not likely necessary for regeneration following intestinal damage.
Figure 4.
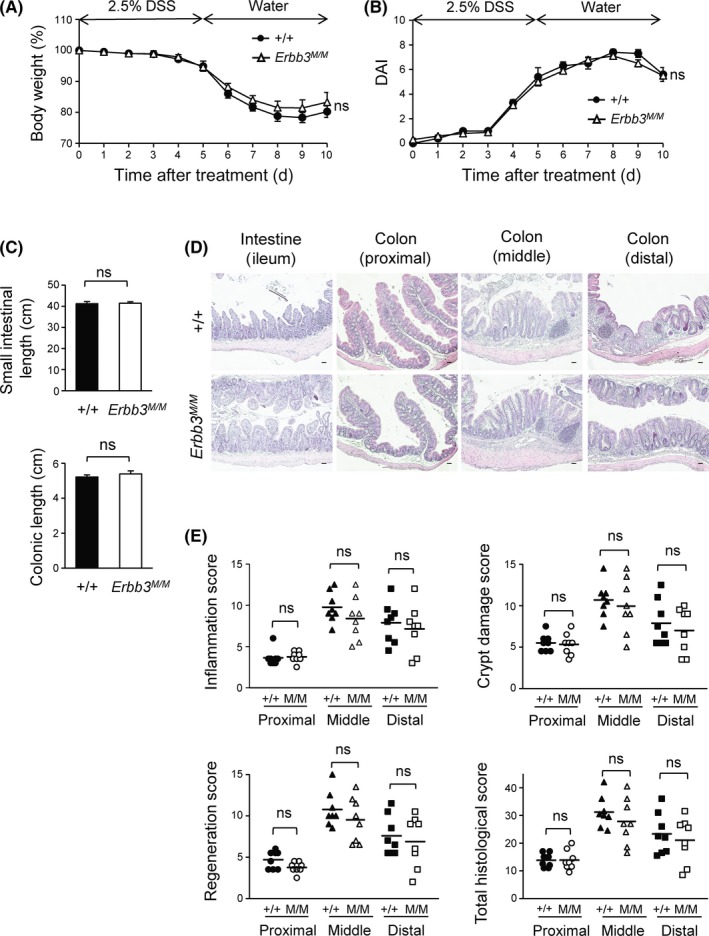
Effects of dextran sulfate sodium (DSS) on Erbb3K740M/K740M mice. Wild‐type (n = 8) and Erbb3K740M/K740M (n = 8) adult mice were treated with 2.5% DSS for 5 d and switched to normal drinking water; mice were killed for analysis at day 10. A, Body weight loss during DSS treatment is expressed as a percentage of the weight on the day of first exposure to DSS. B, Disease activity index (DAI) score was graded daily in WT and Erbb3K740M/K740M mice. C, Small intestinal (top) and colonic (bottom) length of WT and Erbb3K740M/K740M mice. Data are represented as mean ± SEM of each genotype. D, Colon and ileum sections were stained with H&E. E, Inflammation score, epithelial regeneration score, crypt damage score, and total histological score of the colon are shown. Each dot represents the score of 1 individual mouse, and horizontal lines are means for each genotype. ns, statistically insignificant
3.4. Neuregulin‐1‐β1‐induced outgrowth of ileum organoids is inhibited in Erbb3K740M/K740M mice
Although ERBB3 is dispensable for the proliferation and survival of IEC in vivo, the abundant expression of ERBB3 in the IEC might have some function in intestinal homeostasis.24 To investigate the function of ERBB3 kinase activity in the intestine, we isolated and cultured the ileum crypts in a 3‐D environment to form organoids, which are an ideal model to track morphology change under ligand treatment. In accordance with the in vivo proliferation result, the organoids from the ileum of Erbb3K740M/K740M mice showed organoid growth, size, and budding numbers similar to those of WT mice (Figure 5A,B). Exogenous treatment with NRG1‐β1 for 3 days, but not EGF, promoted outgrowth of organoids and disrupted the architecture of organoids from WT mice, consistent with a previous study.40 However, Erbb3K740M/K740M organoids treated with NRG1‐β1 failed to increase in size and complexity (Figure 5C,D; Figure S4). Also, the phospho‐ERBB3(Tyr1197) protein level was significantly reduced in organoids from Erbb3K740M/K740M mice compared to WT mice after NRG1‐β1 treatment (Figure 5E). Neuregulin‐1‐β1 has poor effects on phospho‐EGFR(Tyr1068) levels. In contrast, short‐term (10‐minute) treatment with NRG1‐β1 resulted in similar activation of phospho‐ERBB3(Tyr1197) protein levels in both WT and Erbb3K740M/K740M mice (Figure S5), indicating that ERBB3 kinase activity affects phosphorylation of ERBB3 only in long‐term NRG1‐β1 treatment.
Figure 5.
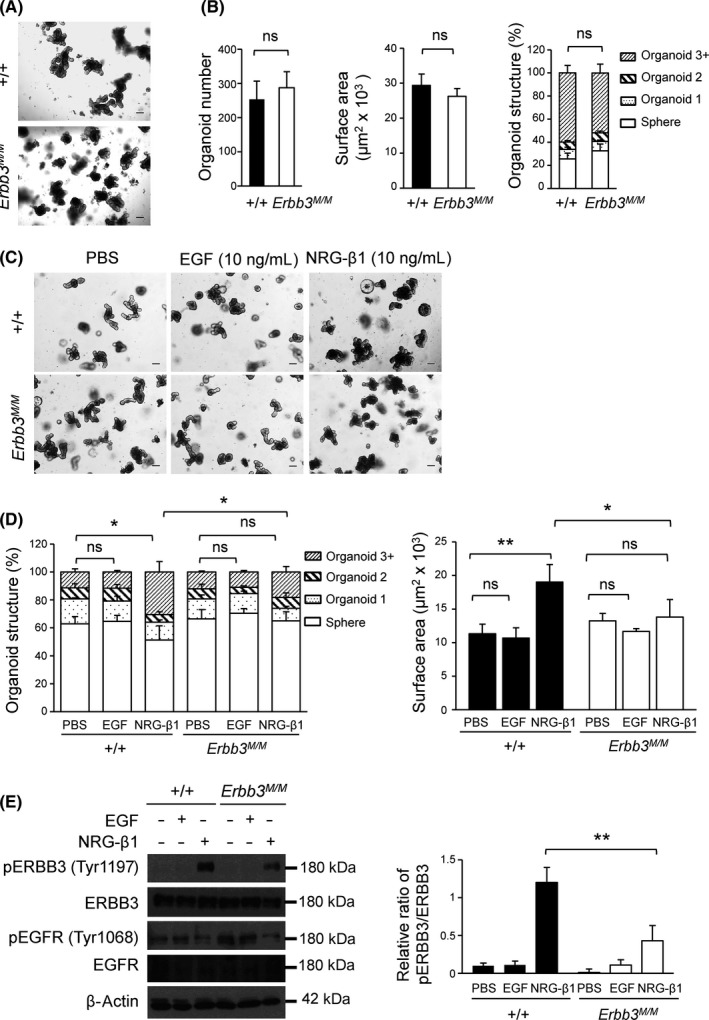
Effect of neuregulin‐1 (NRG1) treatment on ileum organoids’ outgrowth in Erbb3K740M/K740M mice. A, Representative images of organoids cultured for 5 d from WT and Erbb3K740M/K740M mice. B, Organoid number, surface area, and structure from WT (n = 5) and Erbb3K740M/K740M (n = 7) mice at day 5 are shown. C, Representative image of organoids treated with PBS, EGF (10 ng/mL), and NRG1‐β1 (10 ng/mL) for three days. D, Quantitation of organoid surface area and structure of WT (n = 3) and Erbb3K740M/K740M (n = 3) mice after 3 days of treatment with ligands. E, Protein extracts from WT and Erbb3K740M/K740M organoids treated with PBS, epidermal growth factor (EGF), and NRG1‐β1 for 3 days were prepared and analyzed by western blotting with indicated Abs. Photographs are representative images of 3 independent experiments (left), and the relative phosphorylated band intensities were normalized with total protein levels (right) (n = 3). Values are means ± SEM. *P < .05 or **P < .01. ns, statistically insignificant. Scale bar = 100 µm
3.5. Effect of ERBB3 kinase activity on ApcMin tumor development
Because IEC‐specific knockout of Erbb3 results in a dramatic reduction of the number and size of polyps in ApcMin/+ tumor mice,24 the WT and Erbb3K740M mice were crossed with ApcMin mice to find out whether ERBB3 kinase activity contributes to intestinal tumorigenesis. The polyp numbers in the small intestine, but not in the colon of Erbb3K740M/K740M, was significantly reduced more than in the control mice (Figure 6A,B). The tumor sizes in the small intestine of control and Erbb3K740M/K740M mice were not significantly different (Figure 6C). But the fraction of tumors less than 1 mm in diameter were 78.0 ± 2.1% in Erbb3K740M/K740M, significantly higher than the 71.0 ± 2.4% in control mice (Figure 6D). Conversely, the fraction of tumors between 1 mm and 2 mm in diameter was smaller in Erbb3K740M/K740M mice than in control mice (21.3 ± 2.1% and 27.7 ± 2.3%, respectively), and the fraction of tumors between 2 mm and 3 mm in diameter was also smaller in Erbb3K740M/K740M mice than in control mice (0.46 ± 0.1% and 1.1 ± 0.19%, respectively), indicating that ERBB3 kinase activity contributes to polyp growth from the early stages. In control mice, high‐grade adenomas showed more distorted glandular structures. In contrast, most high‐grade adenomas in Erbb3 mutant mice showed less distorted glandular structures with multiple crypts and scattered goblet cells (Figure 6E). The number of low‐grade adenomas was superior to high‐grade adenomas in both control and Erbb3 mutant mice (Figure 6F). The number of low‐grade adenomas was not significantly different between the control (10.2 ± 2.2) and Erbb3 mutant mice (9.5 ± 2.1), but the high‐grade adenomas showed a significant decrease in the Erbb3 mutant (1.8 ± 0.7) compared to the control mice (4.7 ± 1.2). Adenocarcinoma was not found in either group. The number of BrdU‐positive cells in intestinal polyps of Erbb3K740M/K740M mice showed no significant difference compared with those of control mice (Figure 6G). In contrast, the number of TUNEL‐positive cells in intestinal polyps of Erbb3K740M/K740M mice was higher than that of control mice (12.8 ± 3.7 and 9.6 ± 1.9, respectively) (Figure 6H), showing that loss of ERBB3 kinase activity leads to increased apoptosis in intestinal polyps.
Figure 6.
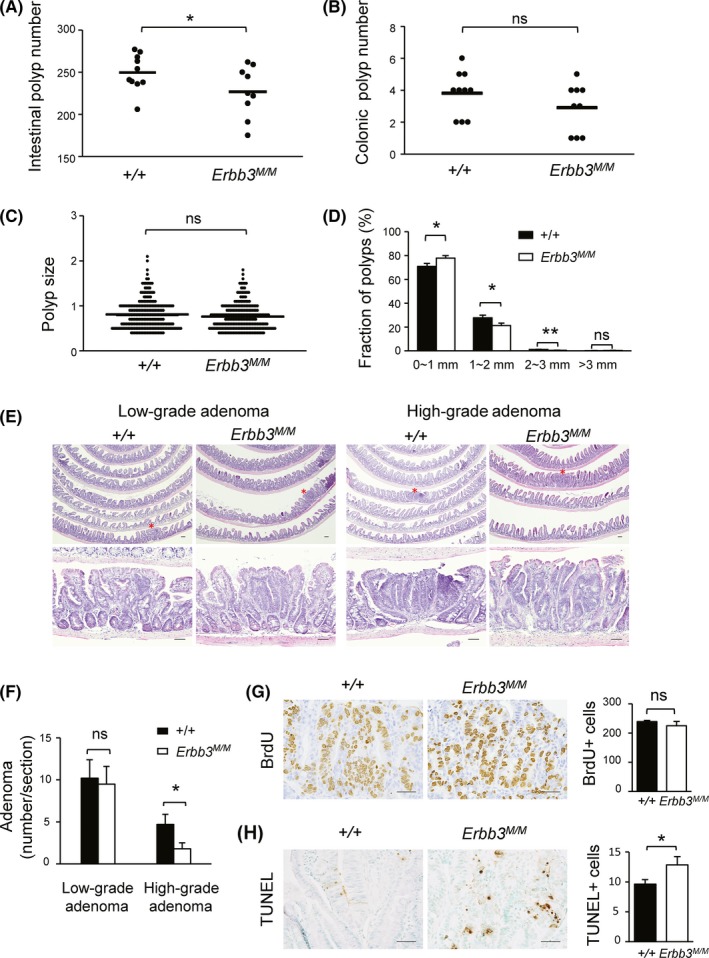
Effect of ERBB3 kinase activity on ApcMin intestinal tumorigenesis. A,B, Small intestinal polyp number (A) and colonic polyp number (B) in 2‐month‐old ApcMin mice. Each dot represents the total number of polyps from control (n = 10) and Erbb3K740M/K740M mice (n = 9). Horizontal lines are means for each genotype. C, Intestinal polyp size analysis. Each dot represents the size of each polyp, and horizontal lines are means for each genotype. D, Range of polyp size of control and Erbb3K740M/K740M mice. Closed bars indicate means ± SEM for control mice, and open bars indicate means ± SEM for Erbb3K740M/K740M mice. E, Representative H&E‐stained images of adenomas from control and Erbb3K740M/K740M mice. Adenomas marked with asterisks in the top panels are magnified in the bottom panels (200×). Scale bar = 50 µm. F, Numbers of low‐ and high‐grade adenomas in the ileum of control (n = 6) and Erbb3K740M/K740M mice (n = 6). G, Representative image of BrdU‐stained (left) and (H) TUNEL‐stained (left) intestinal polyp obtained from control and Erbb3K740M/K740M mice, respectively. Numbers of BrdU‐positive cells were counted in intestinal polyps from control (n = 4) and Erbb3K740M/K740M (n = 5) mice (G, right). Numbers of TUNEL‐positive cells were counted in intestinal polyps from control (n = 6) and Erbb3K740M/K740M mice (n = 7) (H, right). Values are means ± SEM. *P < .05 or **P < .01. Student’s t test, 1‐tailed. ns, statistically insignificant. Scale bar = 50 μm
4. DISCUSSION
In this study, we investigated the role of ERBB3 kinase activity in embryonic development, intestinal homeostasis, and tumorigenesis using Erbb3 kinase‐inactive mutant mice. Various mutant phenotypes were observed in the hypoactive EGFR kinase mutant mice, Egfrwa2/wa2, EgfrWa5/+, and kinase‐dead Erbb2K750M knockin mice.26, 28, 29 A dramatic impairment of mammary epithelial outgrowth in mice homozygous for the Erbb3 allele lacking the 7 known PI3K‐binding sites indicates that ERBB3‐associated PI3K activity is critical for mammary development.41 However, we observed that kinase‐inactive Erbb3 knockin homozygous mice were born and survived normally without any obvious defects, indicating that the kinase activity of ERBB3 is not crucial for embryonic development or survival. It is not surprising that the total phosphorylation and tyrosine phosphorylation of the ERBB family did not attenuate in kinase‐inactive Erbb3 homozygous mice. Contrary to the essential role of the ERBB family in development and disease regulation, ERBB3 kinase activity itself does not seem to participate in normal homeostasis.
ERBB3 is required not only for its protective role of the intestinal epithelium during DSS‐induced colitis but also for epithelium regeneration in both short‐term and long‐term recovery periods.24, 25 Both ERBB2 and ERBB3 were shown to be reparative elements to protect against colitis injury by means of promoting cell survival.25 In this study, unlike in IEC‐specific Erbb3 knockout mice, kinase‐inactive Erbb3 mice showed normal recovery from the DSS‐induced colitis, perhaps because kinase‐inactive ERBB3 still forms a heterodimer with ERBB2 or other EGFR family members that in turn trigger signals pathways for cell proliferation and survival regardless of ERBB3 kinase activity. Indeed, the dual inhibition of ERBB2/ERBB3 is known to be required to fully inhibit AKT activity.42
In the intestinal organoid model, it is known that NRG1‐β1 is superior to EGF in promotion of the hyperproliferation and dysregulated shape of ileal organoids.40 Epidermal growth factor is one of the essential factors for organoid culture,33 and the EGF concentration in the culture medium seems to be saturated as organoid outgrowth is not affected by the additional EGF treatment. Interestingly, the severe attenuation of organoid outgrowth in kinase‐inactive Erbb3 mutant mice suggests that ERBB3 kinase activities contribute to outgrowth of epithelial cells under NRG1‐β1 exposure, which is usually overexpressed in colon cancers.43 The phospho‐ERBB3 level of kinase‐inactive ERBB3 mutant was not reduced under short‐term stimulation by NRG1‐β1 either in vivo or ex vivo, but it failed to prolong this activated state in long‐term stimulation ex vivo. The kinase activity of ERBB3 seems to take a distinct role in maintaining its phosphorylation state instead of transphosphorylation by dimerization with other receptors in the presence of abundant NRG1‐β1. Epidermal growth factor receptor will form a heterodimer with ERBB3 only under an unsaturated level of EGF when EGF‐free EGFR molecules are available to interact with ERBB3.44 Our data showed that NRG1‐β1 induces a low EGFR phosphorylation, which indicates that EGFR phosphorylation is not advantageous in EGFR/ERBB3 heterodimers, because ERBB3 kinase activity is not strong enough to transphosphorylate EGFR.45, 46
When ERBB3 is heterodimerized with other ERBBs by NRG1 binding, ERBB3 itself is transphosphorylated and led to downstream activation of PI3K/AKT/mTOR and RAS/RAF/MEK/ERK signaling pathways.3, 8, 47 The oncogenic regulation of NRG1 and kinase activity of ERBBs in the intestinal epithelium are becoming strongly evident, and ERBB3 is a principal element in the regulation of the ERBB signaling pathway and a potential cancer‐drug target.48, 49 Oncogenic ERBB3 mutations in human cancer showed different patterns in ligand dependency, in which several mutants were oncogenic in the absence of NRG1, more stimulated by NRG1 treatment, or less sensitive to NRG1.50 Expression of NRG1 has been found to be higher in tumors than in normal colonic epithelial cells and contributes to malignant transformation.43, 47 These findings shed light on how ERBB3 kinase activity might have some functions in the presence of NRG1 ligands. The IEC‐specific Erbb3 knockout mice showed fewer and smaller polyps when crossed with ApcMin mice, emphasizing the critical roles in tumor formation and progression by ERBB3 signaling.24 Our finding fewer polyps and more small polyps in Erbb3 kinase‐inactive mice suggests that intrinsic ERBB3 kinase activity also contributes to early growth of polyps in ApcMin mice. Furthermore, an increase of apoptosis in intestinal polyps of Erbb3 kinase‐inactive mice similar to the IEC‐specific Erbb3 knockout mice indicates that ERBB3 kinase activity seems to support early tumor growth and survival given NRG1 overexpression in adenomas of ApcMin mice.
Taken together, we uncovered that the ERBB3 kinase activity contributes to intestinal adenoma formation in ApcMin mice and helps to induce ERBB3 phosphorylation, which is relevant to the outgrowth of ileal organoids in the presence of overexpressed NRG1. Several therapeutic mAbs targeting ERBB3 are under clinical evaluation,51 and the possibility of ERBB3 autophosphorylation in EGFR/ERBB2‐targeted therapeutic resistant tumors suggests that the development of kinase inhibitors for the pan‐EGFR family could also be an important therapeutic strategy in colorectal cancer.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (2018R1D1A1B07048622; 2014M3A9D5A01075128). AT‐QN was a beneficiary of the Ewha Global Partnership Program fellowship from Ewha Womans University.
Nguyen AT‐Q, Lee S‐Y, Chin HJ, Le QV‐C, Lee D. Kinase activity of ERBB3 contributes to intestinal organoids growth and intestinal tumorigenesis. Cancer Sci. 2020;111:137–147. 10.1111/cas.14235
REFERENCES
- 1. Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3‐8. [DOI] [PubMed] [Google Scholar]
- 2. Huang CW, Tsai HL, Chen YT, et al. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer. 2013;13:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism‐based cancer therapeutics. Cancer Cell. 2014;25:282‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schweiger T, Hegedus B, Nikolowsky C, et al. EGFR, BRAF and KRAS status in patients undergoing pulmonary metastasectomy from primary colorectal carcinoma: a prospective follow‐up study. Ann Surg Oncol. 2014;21:946‐954. [DOI] [PubMed] [Google Scholar]
- 5. Rajkumar T, Gooden CS, Lemoine NR, Gullick WJ, Goden CS. Expression of the c‐erbB‐3 protein in gastrointestinal tract tumours determined by monoclonal antibody RTJ1. J Pathol. 1993;170:271‐278. [DOI] [PubMed] [Google Scholar]
- 6. Maurer CA, Friess H, Kretschmann B, et al. Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol. 1998;29:771‐777. [DOI] [PubMed] [Google Scholar]
- 7. Kountourakis P, Pavlakis K, Psyrri A, et al. Clinicopathologic significance of EGFR and Her‐2/neu in colorectal adenocarcinomas. Cancer J. 2006;12:229‐236. [DOI] [PubMed] [Google Scholar]
- 8. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12:553‐563. [DOI] [PubMed] [Google Scholar]
- 9. Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL 3rd. Insect cell‐expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994;91:8132‐8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prigent SA, Gullick WJ. Identification of c‐erbB‐3 binding sites for phosphatidylinositol 3'‐kinase and SHC using an EGF receptor/c‐erbB‐3 chimera. EMBO J. 1994;13:2831‐2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sierke SL, Cheng K, Kim HH, Koland JG. Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB3 (HER3) receptor protein. Biochem J. 1997;322(Pt 3):757‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Riese DJ 2nd, van Raaij TM, Plowman GD, Andrews GC, Stern DF. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol. 1995;15:5770‐5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berger MB, Mendrola JM, Lemmon MA. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Lett. 2004;569:332‐336. [DOI] [PubMed] [Google Scholar]
- 14. Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692‐7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinkamp MP, Low‐Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo‐ and heterointeractions. Mol Cell Biol. 2014;34:965‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riethmacher D, Sonnenberg‐Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725‐730. [DOI] [PubMed] [Google Scholar]
- 17. Erickson SL, O'Shea KS, Ghaboosi N, et al. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2‐and heregulin‐deficient mice. Development. 1997;124:4999‐5011. [DOI] [PubMed] [Google Scholar]
- 18. Jackson‐Fisher AJ, Bellinger G, Breindel JL, et al. ErbB3 is required for ductal morphogenesis in the mouse mammary gland. Breast Cancer Res. 2008;10:R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cook RS, Garrett JT, Sanchez V, et al. ErbB3 ablation impairs PI3K/Akt‐dependent mammary tumorigenesis. Cancer Res. 2011;71:3941‐3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balko JM, Miller TW, Morrison MM, et al. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Proc Natl Acad Sci U S A. 2012;109:221‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams MM, Vaught DB, Joly MM, et al. ErbB3 drives mammary epithelial survival and differentiation during pregnancy and lactation. Breast Cancer Res. 2017;19:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scheving LA, Zhang X, Stevenson MC, et al. Loss of hepatocyte ERBB3 but not EGFR impairs hepatocarcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2015;309:G942‐G954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheving LA, Zhang X, Threadgill DW, Russell WE. Hepatocyte ERBB3 and EGFR are required for maximal CCl4‐induced liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2016;311:G807‐G816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee D, Yu M, Lee E, et al. Tumor‐specific apoptosis caused by deletion of the ERBB3 pseudo‐kinase in mouse intestinal epithelium. J Clin Invest. 2009;119:2702‐2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Dube PE, Washington MK, Yan F, Polk DB. ErbB2 and ErbB3 regulate recovery from dextran sulfate sodium‐induced colitis by promoting mouse colon epithelial cell survival. Lab Invest. 2012;92:437‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luetteke NC, Phillips HK, Qiu TH, et al. The mouse waved‐2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994;8:399‐413. [DOI] [PubMed] [Google Scholar]
- 27. Fowler KJ, Walker F, Alexander W, et al. A mutation in the epidermal growth factor receptor in waved‐2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc Natl Acad Sci U S A. 1995;92:1465‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee D, Cross SH, Strunk KE, et al. Wa5 is a novel ENU‐induced antimorphic allele of the epidermal growth factor receptor. Mamm Genome. 2004;15:525‐536. [DOI] [PubMed] [Google Scholar]
- 29. Chan R, Hardy WR, Laing MA, Hardy SE, Muller WJ. The catalytic activity of the ErbB‐2 receptor tyrosine kinase is essential for embryonic development. Mol Cell Biol. 2002;22:1073‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee SY, Kim H, Kim K, Lee H, Lee S, Lee D. Arhgap17, a RhoGTPase activating protein, regulates mucosal and epithelial barrier function in the mouse colon. Sci Rep. 2016;6:26923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238‐249. [PubMed] [Google Scholar]
- 33. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262‐265. [DOI] [PubMed] [Google Scholar]
- 34. Mahe MM, Aihara E, Schumacher MA, et al. Establishment of gastrointestinal epithelial organoids. Curr Protoc Mouse Biol. 2013;3:217‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang B, Rong X, Palladino END, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. 2018;22(206–20):e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsuneki M, Nakamura Y, Kinjo T, Nakanishi R, Arakawa H. Mieap suppresses murine intestinal tumor via its mitochondrial quality control. Sci Rep. 2015;5:12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Honegger AM, Dull TJ, Felder S, et al. Point mutation at the ATP binding site of EGF receptor abolishes protein‐tyrosine kinase activity and alters cellular routing. Cell. 1987;51:199‐209. [DOI] [PubMed] [Google Scholar]
- 38. Chen WS, Lazar CS, Poenie M, Tsien RY, Gill GN, Rosenfeld MG. Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor. Nature. 1987;328:820‐823. [DOI] [PubMed] [Google Scholar]
- 39. Qian X, LeVea CM, Freeman JK, Dougall WC, Greene MI. Heterodimerization of epidermal growth factor receptor and wild‐type or kinase‐deficient Neu: a mechanism of interreceptor kinase activation and transphosphorylation. Proc Natl Acad Sci U S A. 1994;91:1500‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moller Y, Morkel M, Schmid J, et al. Oncogenic Ras triggers hyperproliferation and impairs polarized colonic morphogenesis by autocrine ErbB3 signaling. Oncotarget. 2016;7:53526‐53539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lahlou H, Muller T, Sanguin‐Gendreau V, Birchmeier C, Muller WJ. Uncoupling of PI3K from ErbB3 impairs mammary gland development but does not impact on ErbB2‐induced mammary tumorigenesis. Cancer Res. 2012;72:3080‐3090. [DOI] [PubMed] [Google Scholar]
- 42. Ghasemi R, Rapposelli IG, Capone E, et al. Dual targeting of ErbB‐2/ErbB‐3 results in enhanced antitumor activity in preclinical models of pancreatic cancer. Oncogenesis. 2014;3:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zuo X, Xu M, Yu J, et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR‐delta/beta overexpression. J Natl Cancer Inst. 2014;106:dju052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Lengerich B, Agnew C, Puchner EM, Huang B, Jura N. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A. 2017;114:E2836‐E2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A. 2009;106:21608‐21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kovacs E, Das R, Wang Q, et al. Analysis of the role of the c‐terminal tail in the regulation of the epidermal growth factor receptor. Mol Cell Biol. 2015;35:3083‐3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127‐137. [DOI] [PubMed] [Google Scholar]
- 48. Zhang N, Chang Y, Rios A, An Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim Biophys Sin (Shanghai). 2016;48:39‐48. [DOI] [PubMed] [Google Scholar]
- 49. Hyman DM, Piha‐Paul SA, Won H, et al. HER kinase inhibition in patients with HER2‐ and HER3‐mutant cancers. Nature. 2018;554:189‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jaiswal BS, Kljavin NM, Stawiski EW, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603‐617. [DOI] [PubMed] [Google Scholar]
- 51. Mishra R, Patel H, Alanazi S, Yuan L, Garrett JT. HER3 signaling and targeted therapy in cancer. Oncol Rev. 2018;12:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials