

Abstract
Purpose of review
Our purpose is to review recent publications in the area of marked human HDL deficiency, HDL particles, coronary heart disease (CHD), amyloidosis, the immune response, and kidney disease.
Recent findings
Lack of detectable plasma apolipoprotein (apo) A-I can be due to DNA deletions, rearrangements, or nonsense or frameshift mutations within the APOA1 gene resulting in a lack of apoA-I secretion. Such patients have marked HDL deficiency, normal levels of triglycerides and LDL cholesterol, and can have xanthomas and premature CHD. ApoA-I variants with amino acid substitutions, especially in the region of amino acid residues 50–93 and 170–178, have been associated with amyloidosis. Patients with homozygous Tangier disease have defective cellular cholesterol efflux due to mutations in the adenosine triphosphate-binding cassette transporter A1, detectable plasma apoA-I levels and preβ-1 HDL in their plasma. They have decreased LDL cholesterol levels and can develop neuropathy and premature CHD. Patients with lecithin : cholesterol acyltransferase deficiency have both preβ-1 and α-4 HDL present in their plasma and develop corneal opacities, anemia, proteinuria, and kidney failure.
Summary
Patients with marked HDL deficiency can have great differences in their clinical phenotype depending on the underlying defect.
Keywords: apolipoprotein A-I deficiency, ATP-binding cassette A1 transporter dysfunction, coronary heart disease, high-density lipoprotein deficiency, lecithin : cholesterol acyltransferase deficiency, Tangier disease
Introduction
Decreased HDL cholesterol less than 40 mg/dl has been defined as an independent risk factor for coronary heart disease (CHD) by the National Cholesterol Education Program. About 15% of families with premature CHD have dyslipidemia (elevated triglycerides and decreased HDL cholesterol), about 15% have combined hyperlipidemia [elevated triglycerides and LDL cholesterol, and decreased HDL cholesterol], and about 5% have isolated low HDL. Patients with CHD and low HDL cholesterol often are overweight or obese with elevated insulin levels.
Our purpose here is to review recent publications regarding patients with undetectable apoA-I and compare and contrast them with findings in patients with homozygous and heterozygous Tangier disease and lecithin : cholesteryl acyltransferase (LCAT) deficiency. In addition, we will briefly review recent research on apoA-I variants and their relationship to amyloidosis, as well as the relationships between HDL and susceptibility to infection.
ApoA-I containing HDL can be readily quantified by two-dimensional gel electrophoresis followed by immunoblotting with antibodies specific for apoA-I. A gel and a diagram of the particles are shown in Fig. 1, with very small discoidal preβ-1 HDL, small discoidal α-4 migrating HDL, medium spherical α-3 migrating HDL, large spherical α-2 migrating HDL, and very large α-1 migrating HDL. CHD patients often have significant decreases in the large α-1 and α-2 HDL, and modest increases in the small preβ-1 HDL and α-4 HDL. In Fig. 2 we see these same gels in patients with homozygous apoA-I deficiency (undetectable apoA-I containing particles), Tangier disease (only preβ-1 HDL), and LCAT deficiency (preβ-1 HDL and α-4 HDL). In Fig. 3 we can clearly see that other apolipoproteins such as apoA-IV and apoE are found on their own HDL particles, separate from those containing apoA-I. Models of apoA-I containing HDL particles are shown in Fig. 4. The two small particles, preβ-1 and α-4 HDL, are discs containing apoA-I without apoA-II. The medium-sized α-3 and α-2 HDL are spherical particles containing both apoA-I and apoA-II, with α-2 HDL also containing serum amyloid A protein. The large α-1 HDL particle is also spherical and contains apoA-I without apoA-II.
Figure 1. In the left panel the plasma apoA-I pattern obtained after two-dimensional gel electrophoresis is shown. To the right a schematic diagram of all the A-I containing HDL particles is shown.
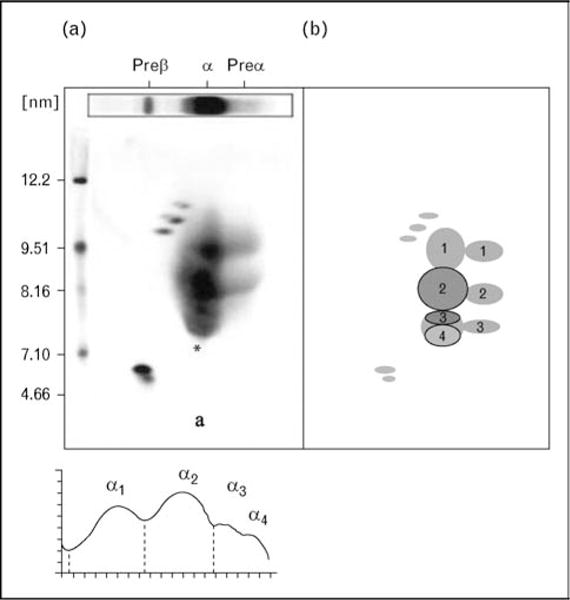
Particles are separated by size (diameter in nm) in the vertical dimension and by charge in the horizontal dimension into particles of preβ, α, and preα mobility.
Figure 2. A composite of the HDL gel patterns is shown.
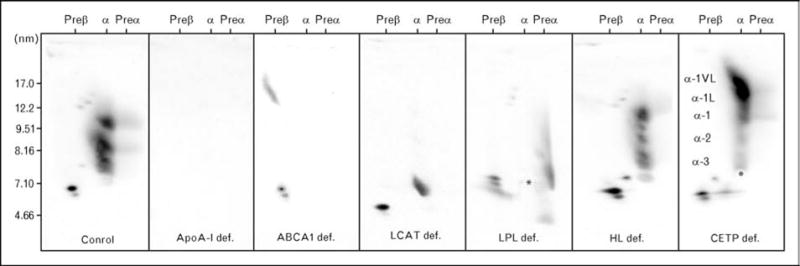
A composite is shown of the HDL gel patterns observed in a normal subject (far left), a homozygote with apoA-I deficiency (second gel from left), a Tangier homozygote (third gel from left), and a homozygote with LCAT deficiency (fourth from left). Also shown are gels from patients with lipoprotein lipase (LPL) deficiency (fifth from left), hepatic lipase deficiency (second from right), and cholesteryl ester transfer protein (CETP) deficiency (farthest to the right).
Figure 3.
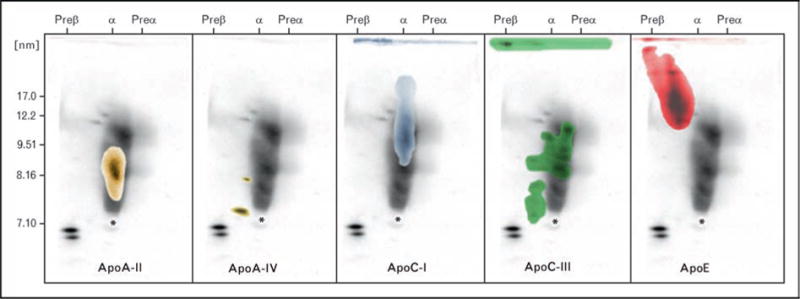
In this figure we have depicted results obtained when apoA-I HDL gels are also immunoblotted with antibodies containing apoA-II (far left), apoA-IV (second from left), apoC-I (third from left), apoC-III (second from right) and apoE (far right)
Figure 4. Models of apoA-I containing HDL particles are shown.
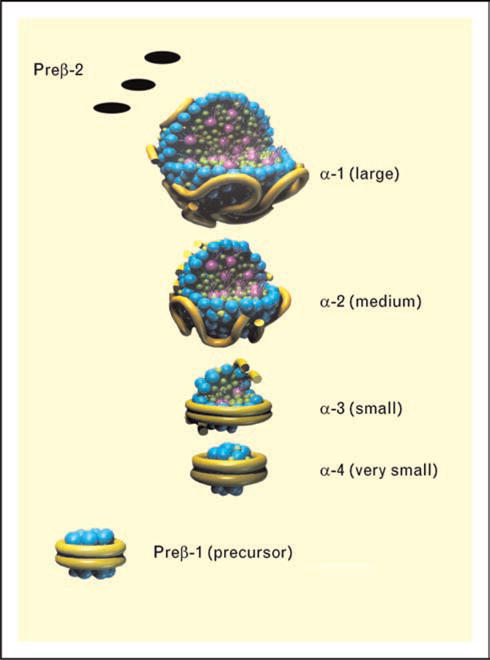
The two small particles, preβ-1 and α-4 HDL are discs containing apoA-I without apoA-II. Medium-sized α-3 and α-2 HDL are shown, which are spherical particles containing both apoA-I and apoA-II, with α-2 HDL also containing serum amyloid A protein. The large spherical α-1 HDL particle is also shown
Familial apolipoprotein A-I deficiency states
There are a number of forms of familial apolipoprotein A-I deficiency: those characterized by a lack of plasma apoA-I, apoC-III, and apoA-IV, those characterized by a lack of apoA-I and apoC-III, and those characterized by a lack of apoA-I only. These states are briefly reviewed below.
Apolipoprotein A-I/C-III/A-IV deficiency
Schaefer and colleagues in 1982 described a kindred in which the index case had marked HDL deficiency, undetectable plasma apoA-I levels, low-triglyceride levels, normal LDL cholesterol levels, and severe premature coronary artery disease. The female index case presented with severe coronary artery disease and died during coronary artery bypass surgery at age 43 years. The patient’s defect was a deletion of the entire APOA1/C3/A4 gene complex. The patient had evidence of mild malabsorption of fat and fat-soluble vitamins, probably due to the apoA-IV deficiency. The patient’s triglycerides were probably low due to a lack of apoC-III. Heterozygotes had plasma HDL cholesterol, apoA-I, apoA-IV, and apoC-III, levels that were about 50% of normal (reviewed in [1–3]).
Familial apolipoprotein A-I/C-III deficiency
Norum and colleagues in 1982 described two sisters with marked HDL deficiency, undetectable plasma apoA-I and apoC-III, and planar xanthomas. They had premature CHD and had coronary artery bypass surgery at ages 29 and 30 years. They had no evidence of fat malabsorption, their triglyceride levels were low, and their LDL cholesterol levels were normal. The defect was a DNA rearrangement affecting the adjacent APOA1 and APOC3 genes. They had enhanced clearance of very LDL apoB, probably due to a lack of apoC-III, with inhibition of lipolysis (reviewed in [1–3]).
Familial apolipoprotein A-I deficiency
In 1991 Matsunaga and colleagues described a 56-year-old Japanese woman with premature CHD, planar xanthomas, normal triglyceride and LDL cholesterol levels, marked HDL deficiency, and undetectable plasma apoA-I levels. The defect was an APOAI codon 84 nonsense mutation, resulting in a lack of normal apoA-I production. Ng et al. in 1994 reported a kindred in which the 34-year-old proband had undetectable plasma apoA-I levels, marked HDL deficiency, tendinous xanthomas, premature CHD, cerebellar ataxia, and elevated LDL cholesterol levels. The defect was a codon −2 APOA1 nonsense mutation. Four other homozygotes in this kindred were found with a similar phenotype, two of whom had CHD in their 30s. The authors concluded that the combined hyperlipidemia in this kindred was probably not related to the APOA1 gene mutation.
We recently reported another kindred with the same mutation as in the prior kindred (−2 codon) in two homozygous brothers who had CHD at 38 and 39 years of age, with striking tubo-eruptive and planar xanthomas (see Fig. 5), corneal arcus and corneal opacification, and HDL cholesterol levels less than 5 mg/dl [1]. LDL cholesterol and triglyceride levels in the two probands were normal. Severe coronary artery calcification (see Fig. 6), a complete obstruction of his right coronary artery and a 90% narrowing of his left anterior descending coronary artery (see Fig. 7) were documented in the proband, who underwent successful coronary artery bypass surgery. This patient developed coronary atherosclerosis similar to that which is observed in the general population, albeit the two homozygotes developed the disease in their 30s [2]. In contrast homozygous familial hypercholesterolemia often develop severe aortic or coronary ostial atherosclerosis [2] (see Fig. 8). In plasma the two homzygotes had no detectable apoA-I containing HDL, but relatively normal amounts of apoE and apoA-IV containing HDL were found to be present [1]. Multiple heterozygotes were found in this kindred with HDL cholesterol levels of less than 30 mg/dl, and they had less than 50% of normal α-1, α-2, and preβ-1 HDL levels (reviewed in [1–3]).
Figure 5.
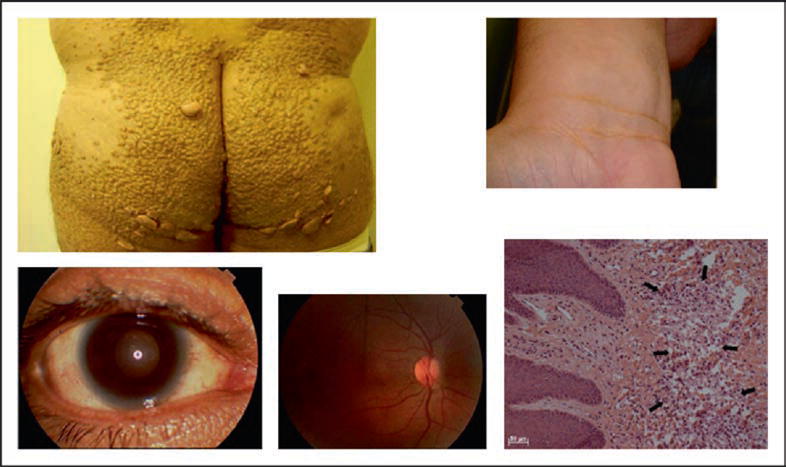
Findings in homozygous familial apolipoprotein deficiency are shown: tubo-eruptive xanthomas (upper left), with microscopy (lower right); planar xanthomas in the wrist area (upper right), moderate corneal opacification (lower left), and a normal eye fundus (lower middle)
Figure 6.
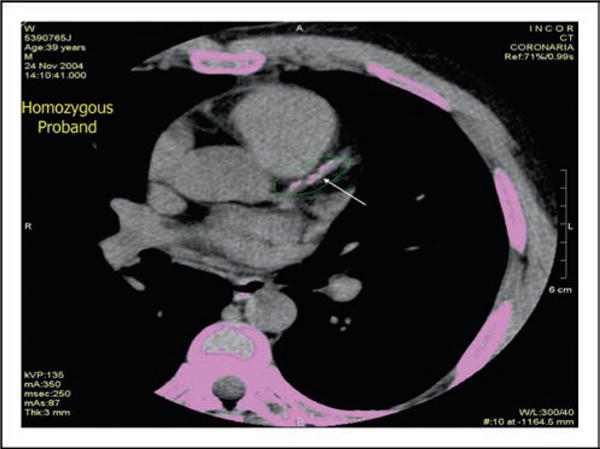
Computed tomographic image of the heart of a 39-year-old patient with homozygous familial apolipoprotein A-I deficiency indicating marked coronary calcification in the left anterior descending coronary artery is shown
Figure 7.
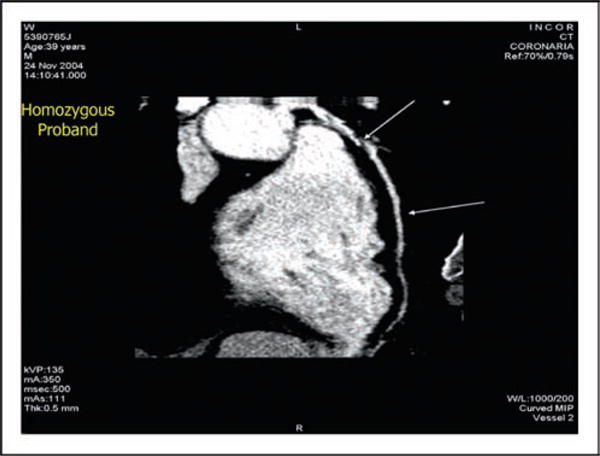
Coronary angiography of a 39-year-old patient with homozygous apoA-I deficiency is shown documenting complete obstruction of the left anterior descending coronary artery
Figure 8. A multidetector 64 slice computed tomographic angiographic image of a 39-year-old patient with homozygous familial apoA-I deficiency with premature coronary artery is shown.
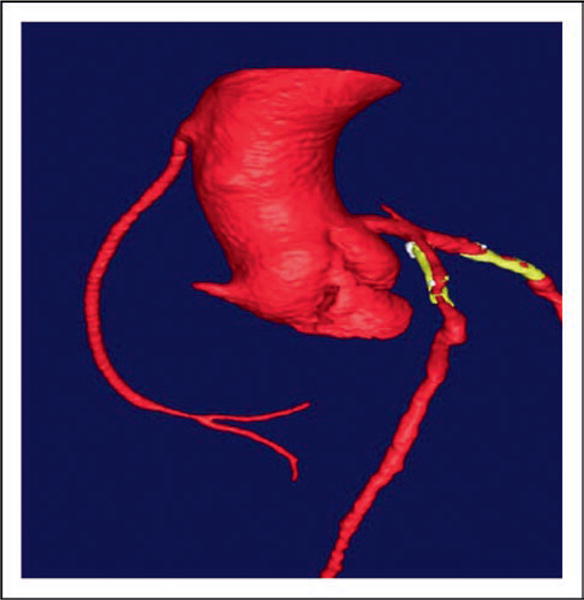
The aorta was normal, but significant narrowing of the left anterior descending coronary artery and the diagonal branches were noted, and the right coronary artery was totally obstructed, with a vein graft to the distal right coronary artery
Wada and colleagues [4] reported a 64-year-old male patient with severe three vessel coronary disease with marked plasma HDL cholesterol (4 mg/dl) and apoA-I deficiency (5 mg/dl), with no detectable apoA-I containing HDL particles being noted by two-dimensional gel electrophoresis in plasma. Heterozygotes had levels of both large α-1 HDL and very small preβ-1 HDL levels that were about 35% of normal. The defect was an APOA1 deletion of two successive adenine residues in codon 138, resulting in a frameshift mutation at amino acid residues 138–178.
Defective adenosine triphosphate-binding cassette transporter A1 (Tangier disease)
Tangier disease was described by Fredrickson and colleagues in 1961 in two young siblings from Tangier Island in the Chesapeake Bay. The patients were noted to have very low levels of plasma of HDL, moderate hypertriglyceridemia, and decreased levels of triglyceride-enriched LDL. They had hepatosplenomegaly and enlarged orange tonsil (see Fig. 9). Cholesterol-laden macrophages were found in cells in the tonsils, bone marrow, nerves, and smooth muscle (see Fig. 10). The heterozygous parents had approximately half normal levels of HDL cholesterol. Other kindreds were described, and in some cases neuropathy was noted. Schaefer and colleagues noted that homozygotes had markedly increased fractional catabolism of HDL proteins, while heterozygotes had enhanced clearance. These authors also noted that Tangier homozygotes had an increased risk of developing premature CHD in their 50s and 60s (see Fig. 11). They speculated that these patients may not develop CHD earlier because their LDL cholesterol levels were about 50% of normal.
Figure 9.
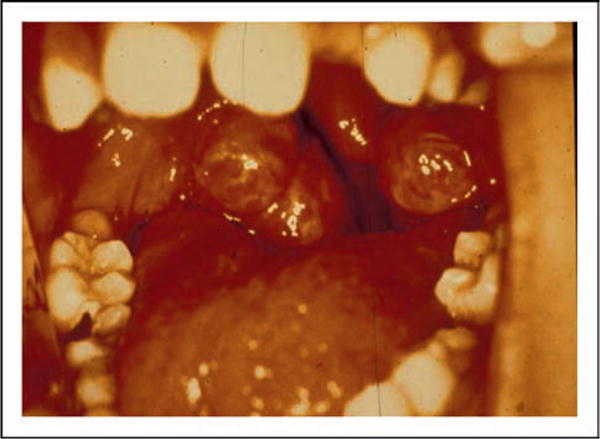
A photograph of the enlarged orange tonsils from a patient with homozygous Tangier disease is shown
Figure 10.
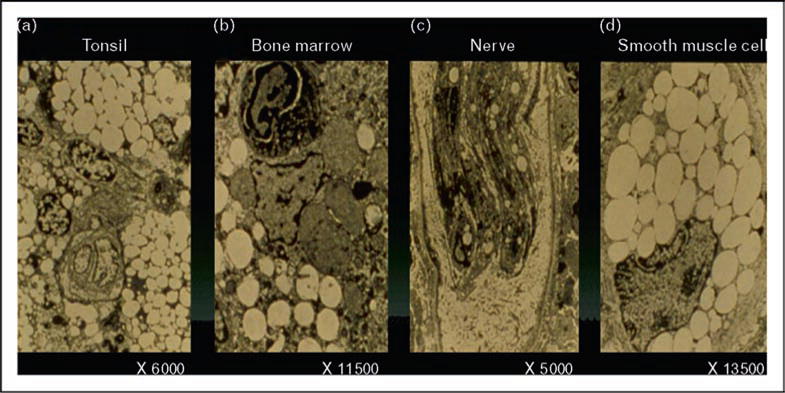
Electron micrographs of the cholesteryl ester deposition in the macrophages in the tonsils, bone marrow, nerve cells, and smooth muscle cells in a patient with homozygous Tangier disease are shown
Figure 11.
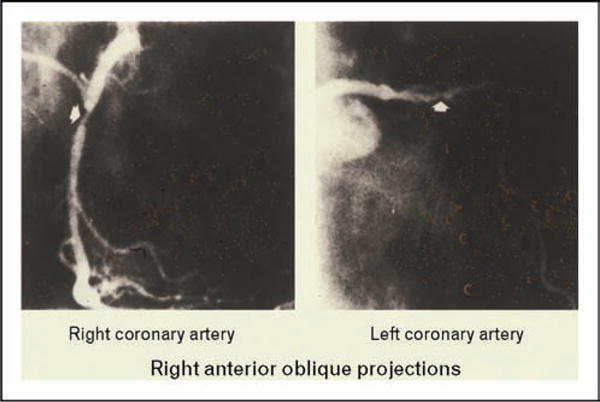
A coronary angiogram in a 56-year-old patient with homozygous Tangier disease is shown documenting the presence of significant coronary artery disease in the right and left anterior descending coronary arteries
In the 1990s Francis and Oram, as well as Schmitz and Assmann, independently noted that Tangier fibroblasts were defective in their ability to efflux cellular cholesterol and phospholipids onto HDL or apolipoprotein A-I. In 1998 the chromosomal locus (9q31) for Tangier disease was reported by Rust, Assmann, and colleagues. Subsequently it was reported by Schmitz and colleagues that the adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) was involved in the efflux of cellular cholesterol and phospholipid to HDL and apoA-I. In 1999 and 2000 six different research groups reported various mutations in ABCA1 as causes of homozygous Tangier disease, with the first three reports by Rust, Assmann and colleagues, Bodzioch, Schmitz and colleagues, and Brooks, Hayden and colleagues appearing simultaneously in 1999 in Nature Genetics.
We were one of the groups reporting these mutations, and we also documented that Tangier homozygotes had only preβ-1 HDL present in their plasma, while heterozygotes had a lack of large α-1 and α-2 HDL particles, normal preβ-1 HDL, and only 50% of normal cellular cholesterol efflux. We also noted that intestinal cholesterol absorption was normal in a Tangier homozygote, and that the LDL apoB values were about 50% reduced due to enhanced fractional catabolism of small triglyceride-rich, cholesteryl ester-poor LDL. We have speculated that the lack of uptake of cholesterol from cells by HDL in these patients, not only results in the rapid clearance of very small HDL particles by the kidney, but also relative enrichment of the core of LDL by beta-carotene, resulting in enhanced uptake by the reticulo-endothelial cells resulting in the orange coloration of these tissues. Recently Favari et al. [5] have reported that preβ-1 HDL serves as an efficient acceptor of cellular cholesterol via both the ABCA1 and the ABCG1 transporters.
Familial lecithin : cholesterol acyltransferase deficiency
LCAT deficiency was first described by Norum and Gjone in 1967 in a 33-year-old woman living in Norway, who presented with severe corneal opacities, hyperlipidemia, anemia, proteinuria, and normal kidney function. A kidney biopsy revealed foam cells in the glomerular tufts. Plasma cholesterol and triglyceride levels were moderately increased, with most of the cholesterol being unesterified. Two of her sisters were similarly affected, and all three were found to have a marked deficiency of LCAT activity, lacking the ability to transfer a fatty acid from phosphatidylcholine or lecithin to cholesterol to form cholesteryl ester and lysolecithin. Kindreds in other countries were subsequently described. These patients were found to have not only very low levels of HDL cholesterol, but also were noted to have elevations in free cholesterol-enriched VLDL, which had β instead of preβ mobility on electrophoresis. Moreover their LDL was found to be large and heterogeneous and enriched in free cholesterol, phospholipids, and triglyceride, with a very low cholesteryl ester content. These particles have also been found to be low in apoB and enriched in the C apolipoproteins.
The corneal opacification observed in familial LCAT deficiency presents early in life and consists of numerous, minute, grayish dots in the entire corneal stroma and is much more striking than that reported for Tangier disease or the apoA-I deficiency states. The opacification is especially marked near the limbal area, forming a circular band resembling arcus senilis. Surprisingly vision is usually not impaired. Recently the corneal opacification in a patient with familial LCAT deficiency was imaged with high-resolution Fourier domain optical coherence tomography and noncontact Rostock confocal laser scanning microscopy [6]. These studies revealed a thinned epithelium, focal disruptions of Bowman’s layer, and focal areas of hyperreflexivity with reduced and irregular ketatocytes throughout the stroma.
The anemia in these patients is moderate with hemoglobin levels of around 10 g/dl and is associated with enhanced fractional clearance of red cells. The renal disease presents as proteinuria early in life, and increases in the fourth or fifth decades of life as renal function deteriorates. Atherosclerosis has been reported in some patients with familial LCAT deficiency with aortic, carotid, and femoral atherosclerosis but CHD prior to age 60 years has not been reported.
McLean and colleagues cloned and sequenced the LCAT gene for LCAT in 1986, and since that time multiple mutations have been reported. Hypercatabolism of HDL proteins, especially apoA-II, has been reported in homozygotes. We have documented that homozygotes have apoA-I in plasma present only in preβ-1 and α-4 discoidal HDL particles. Two-dimensional gel electrophoresis of whole plasma followed by immunoblotting with specific antibody for apoA-I can therefore readily be used to distinguished homozygous apoA-I deficiency, ABCA1 deficiency, or LCAT deficiency (see Fig. 2).
Baass and colleagues in Montreal recently reported a novel LCAT defect (102delG) causing a frameshift at codon 61 and abolishing LCAT activity. The two pro-bands had low HDL cholesterol levels, marked corneal opacification, anemia, proteinuria, and renal insufficiency [7]. These investigators also documented that APOE genotype could modify the lipid phenotype in these patients. Park et al. [8] reported the finding of two mutations in a proband with familial LCAT deficiency: an insertion of the nucleotide G in exon 2 at codon Leu212 causing a frameshift and the introduction of 16 new amino acids before the stop codon. Calabresi et al. [9] have reported that LCAT activity is not required for normal cellular cholesterol efflux from macrophages. We have reported with Calabresi that heterozygotes for LCAT deficiency have less than 50% of normal large α-1 HDL, but two-fold increases in very small preβ-1 HDL.
Based on our examination of patients with these and other inborn errors of HDL metabolism, we have developed a schema of HDL particle metabolism as shown in Fig. 12. In the first step free apoA-I is secreted and picks up phospholipid (PL) to form very small discoidal preβ-1 HDL. This step does not occur in familial apoA-I deficiency. In the second step small discoidal preβ-1 HDL picks up free cholesterol and phospholipid from cells via the ATP-binding cassette transporter A1 (ABCA1) to form small discoidal α-4 HDL particles. This step does not occur in homozygous Tangier disease. In the third step very small discoidal α-4 HDL is converted to small spherical α-3 HDL via the action of lecithin : cholesterol acyltransferase (LCAT). The step does not occur in homozygous LCAT deficiency. During this process α-3 HDL becomes spherical with cholesteryl ester and triglyceride in its core, and apoA-I, phospholipids and free cholesterol on its surface. During this process α-3 HDL also picks up liver-derived lipidated apoA-II on its surface. In the fourth step small α-3 HDL is converted to medium-sized α-2 HDL by picking up surface (free cholesterol and phospholipid) and core (triglycerides) components from triglyceride-rich lipoproteins (TRL), as well as cellular-free cholesterol and phospholipid via ATP-binding cassette transporter G1 (ABCG1). Then there is further esterification of the free cholesterol via LCAT to form spherical medium-sized α-2 HDL. In the fifth step medium-sized α-2 HDL picks up more cholesterol phospholipid from cells via ABCG1 with further esterification to form very large α migrating HDL. In the sixth step these very large HDL particles, which are only seen in CETP deficiency, are rapidly converted into large α-1 HDL after either donating their cholesteryl ester to TRL in exchange for triglyceride via cholesteryl ester transfer protein (CETP). Large α-1 HDL can also donate cholesteryl ester to TRL in exchange for triglyceride. In the seventh step large α-1 can deliver cholesteryl ester to the liver via scavenger receptor-B1 (SR-B1). During the sixth and seventh steps, apoA-I can be lost from large HDL particles and recycle back to preβ-1 or α-4 HDL and go through the cycle again. In the eighth step large α-1 HDL can be converted into medium-sized α-2 HDL by the action of hepatic lipase, a phospholipase which breaks down phospholipids on the surface of HDL. In the ninth step endothelial lipase (EL) and secretory phospolipase A2 (sPLA2) can act on large α-1 HDL to remove phospholipid and form very small preβ-1HDL. The final and tenth step in HDL particle metabolism is the catabolism or clearance of free apoA-I, preβ-1 or α-4 HDL by cubulin in the kidney, or the recycling of the apoA-I back through the HDL maturation cycle.
Figure 12.
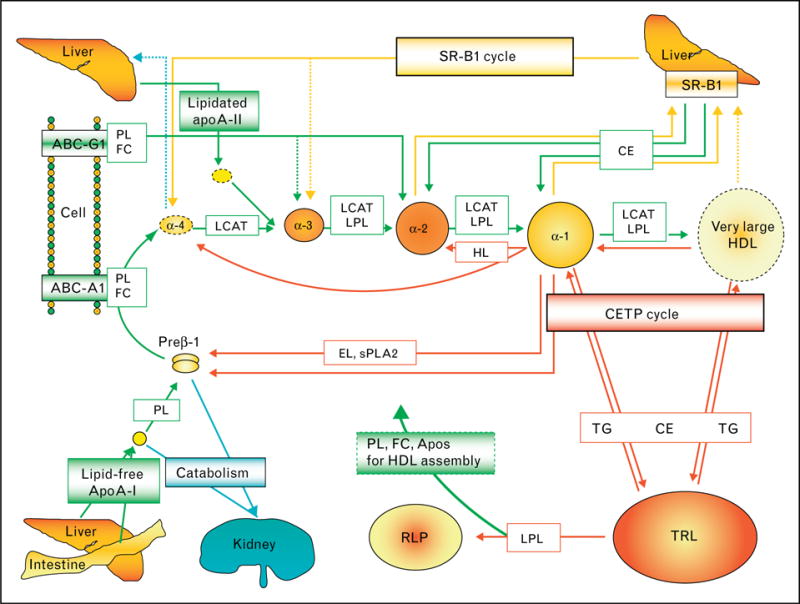
A diagram depicting our view of HDL particle metabolism is shown
Apolipoprotein A-I variants and amyloidosis
Apolipoprotein A-I-derived amyloidosis may present either as a nonhereditary form with wild-type protein deposits in atherosclerotic plaques or as a hereditary form due to germline mutations in the APOA1 gene [10••]. At present many apoA-I variants are known, and 13 have been associated with amyloidosis (see Table 1). Eriksson et al. [10••] have recently described six patients with ApoA-I amyloidosis due to APOA1 germline mutations that affect the larynx, small intestine, large intestine, heart, liver, kidney, uterus, ovary, or pelvic lymph nodes. In each patient, the amyloid showed a characteristic green birefringence when viewed under polarized light after Congo red staining and was immunoreactive with antibodies against apoAI. Sequence analyses revealed one known mutation (p.Leu75Pro) and three novel APOA1 mutations that included gene variations leading to two different frameshifts (p.Asn74fs and p.Ala154fs) and one amino acid exchange (p.Leu170Pro). Thirteen of the now 16 known amyloidogenic mutations are localized in two hot-spot regions that span residues 50–93 and 170–178 of the apoA-I amino acid sequence (see Table 1 for summary of APOA1 variants). These mutations result in the formation of apoA-I–amyloid protein complexes.
Table 1.
Missense/nonsense mutations in human apoA-I
| Codon | Change | AA change | Codon | Phenotype |
|---|---|---|---|---|
| 1 | CAG-TAG | Gln-Term | −2 | Low-HDL, CHD |
| 3 | TGG-TAG | Trp-Term | 8 | Low-HDL |
| 5 | CGA-CTA | Arg-Leu | 10 | Low-HDL |
| 7 | GGC-CGC | Gly-Arg | 26 | Amyloidosis |
| 9 | AGA-ACA | Arg-Thr | 27 | Low-HDL? |
| 11 | CAG-TAG | Gln-Term | 32 | Low-HDL |
| 13 | TGG-CGG | Trp-Arg | 50 | Amyloidosis |
| 15 | CTG-CGG | Leu-Arg | 60 | Amyloidosis |
| 17 | CTC-CCC | Leu-Pro | 64 | Amyloidosis |
| 19 | CTG-CCG | Leu-Pro | 75 | Amyloidosis |
| 21 | CAG-TAG | Gln-Term | 84 | Low-HDL |
| 23 | CTG-CCG | Leu-Pro | 90 | Amyloidosis |
| 25 | GAG-TAG | Glu-Term | 136 | Low-HDL |
| 27 | CTG-CGC | Leu-Arg | 141 | Low-HDL, CHD |
| 29 | CTG-CGG | Leu-Arg | 144 | Low-HDL |
| 31 | CGC-CAC | Arg-Cys | 151 | Low-HDL |
| 33 | GCC-CCC | Arg-Pro | 153 | Low-HDL, CHD |
| 35 | GTG-GAG | Val-Glu | 156 | Low HDL |
| 37 | CTG-CGC | Leu-Arg | 159 | Low-HDL |
| 39 | CTG-CCG | Leu-Pro | 159 | Low-HDL, CHD |
| 41 | CGC-CTC | Arg-Leu | 160 | Low-HDL |
| 43 | CCC-CGC | Pro-Arg | 165 | Low-HDL |
| 45 | CTG-CCG | Leu-Pro | 170 | Amyloidosis |
| 47 | CGC-TGC | Arg-Cys | 173 | Low-HDL |
| 49 | CGC-CCC | Arg-Pro | 173 | Amyloidosis |
| 51 | TTG-TCG | Leu -Ser | 174 | Amyloidosis |
| 53 | GCC-CCC | Ala-Pro | 175 | Amyloidosis |
| 55 | CTT-CAT | Leu-His | 178 | Amyloidosis |
| 57 | CTT-CCT | Leu-Pro | 178 | Low-HDL, CHD |
Apolipoprotein A-I, HDL particles, and the immune response
In patients with sepsis, low levels of plasma apoA-I and HDL cholesterol are predictors of mortality [11]. It has now clearly been shown that resistance to trypanosoma brucei is due to HDL apoA-I forming a complex with apoL-1 and haptoglobin-related protein [12••].
Conclusion
Severe deficiency of plasma HDL (HDL cholesterol <10 mg/dl) can be due to marked hypertriglyceridemia or liver failure. In the absence of these conditions it is due to genetic forms of apoA-I deficiency, ABCA1 dysfunction, or LCAT deficiency as previously discussed. All these disorders can be readily diagnosed by the use of two-dimensional gel electrophoresis to determine apoA-I-containing HDL particles. Homozygotes with apoA-I deficiency lack any apoA-I-containing HDL, while heterozygotes have at least 50% reductions in very small preβ-1 and large α-1 HDL. Homozygotes with Tangier disease have only preβ-1 HDL in their plasma, while heterozygotes have normal levels of preβ-1 HDL, but less than 50% of normal large α-1 HDL. Homozygotes with LCAT deficiency have only preβ-1 and α-4 HDL in their plasma, while heterozygotes have two-fold increases in preβ-1 HDL and less than 50% of normal large α-1 HDL. A diagram of our view of HDL particle metabolism is shown in Fig. 12 [13]. Premature CHD (<60 years of age) is clearly found in homozygous apoA-I deficiency, often found in Tangier disease and has not been reported in LCAT deficiency.
The role of apoA-I variants in causing complexes with amyloid and amyloidosis is fascinating, as is the role of apoA-I in the immune response and in complexing with two other proteins (apoL-1 and haptoglobin-related protein) resulting in immunity from African trypanosomiasis.
Acknowledgments
Supported by research grants to the Lipid Metabolism Laboratory at Tufts University from the National Institutes of Health, Bethesda, MD (HL-60935, HL 74753 and PO50HL083813) and the US Department of Agriculture, Washington DC (contract 53-3K06-5-10) (Drs Schaefer and Asztalos) and research grants to INCOR (Dr Santos).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 378).
- 1.Santos RD, Schaefer EJ, Asztalos BF, et al. Characterization of high density lipoprotein particles in familial apolipoprotein A-I deficiency. J Lipid Res. 2008;46:349–357. doi: 10.1194/jlr.M700362-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Santos RD, Miname MH, Martinez LR, et al. Noninvasive detection of aortic and coronary atherosclerosis in homozygous familial hypercholesterolemia by 64 slice multidetector row computed tomography angiography. Atherosclerosis. 2008;197:910–915. doi: 10.1016/j.atherosclerosis.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 3.Santos RD, Miname L, Asztalos BF, et al. Clinical presentation, laboratory values, and coronary heart disease risk in marked high density lipoprotein deficiency states. J Clin Lipidol. 2008;2:237–247. doi: 10.1016/j.jacl.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada M, Iso T, Asztalos BF, et al. Marked high density lipoprotein deficiency due to apolipoprotein A-I Tomioka (codon 138 deletion) Atherosclerosis. 2009;207:157–161. doi: 10.1016/j.atherosclerosis.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 5.Favari E, Calabresi L, Adorni MP, et al. Small discoidal prebeta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry. 2009;48:11067–11074. doi: 10.1021/bi901564g. [DOI] [PubMed] [Google Scholar]
- 6.Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin–cholesterol acyltransferase deficiency. Cornea. 2009;28:1061–1064. doi: 10.1097/ICO.0b013e31819839ae. [DOI] [PubMed] [Google Scholar]
- 7.Baass A, Wassef H, Tremblay M, et al. Characterization of a new LCAT mutation causing familial LCAT deficiency (FLD) and the role of APOE as a modifier gene in FLD phenotype. Atherosclerosis. 2009;207:452–457. doi: 10.1016/j.atherosclerosis.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Park CW, Lim MH, Youn DY, et al. Two novel frameshift mutations in lecithin–cholesterol acyltransferase (LCAT) gene associated with a familial LCAT deficiency phenotype. Atherosclerosis. 2009;206:346–348. doi: 10.1016/j.atherosclerosis.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Calabresi L, Favari E, Moleri E, et al. Functional LCAT is not required for macrophage cholesterol efflux to human serum. Atherosclerosis. 2009;204:141–146. doi: 10.1016/j.atherosclerosis.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 10••.Eriksson M, Schönland S, Yumlu S, et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009;11:257–262. doi: 10.2353/jmoldx.2009.080161. This is an excellent review of the role of apoA-I variants in the pathogeneisis of familial amyloidosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barlage S, Gnewuch C, Liebisch G, et al. Changes in HDL-associated apolipoproteins relate to mortality in human sepsis and correlate to monocyte and platelet activitation. Intensive Care Med. 2009;3:1877–1885. doi: 10.1007/s00134-009-1609-y. [DOI] [PubMed] [Google Scholar]
- 12••.Pays E, Vanhollebeke B. Human innate immunity against African trypanosomes. Curr Opin Immunol. 2009;21:493–498. doi: 10.1016/j.coi.2009.05.024. This is an excellent review of the precise role which apoA-I plays in interacting with apoL-1 and haptoglobin-related protein to convey resistance to African trypanosomiasis in humans. [DOI] [PubMed] [Google Scholar]
- 13.Asztalos BF, Brunzell J. The kinetics and remodeling of HDL particles: lessons from inborn errors of lipid metabolism. In: Schaefer EJ, editor. High density lipoproteins, dyslipidemia, and coronary heart disease. New York: Springer Publishing; 2010. pp. 33–44. [Google Scholar]