

Abstract
AIM
To investigate the expression and possible role of the autophagy related protein p62 and LC3 in the retina based on a rat model of acute ocular hypertension.
METHODS
Fifty rats were randomized into five groups: control group A, B, C, and D. Groups A to D all received normal saline perfusion into the anterior chamber with pressure of 80 mm Hg for one hour, and retina tissue was obtained at 6, 12, 24 and 48h after perfusion respectively, to investigate the activation of autophagy following ischemia-reperfusion. The distribution and semi-quantification of autophagy related protein p62 and LC3 in the retina were detected using immunohistochemistry technique. The expression level of these two proteins was evaluated using Western blot.
RESULTS
The number of retinal ganglion cells (RGCs) decreased with increasing reperfusion time, and significant reduction in the retinal thickness was observed 48h after perfusion. In normal adult rats, LC3 protein was mainly expressed in the ganglion cell layer (GCL), and p62 protein was expressed in the nerve fiber layer (NFL), GCL, inner plexiform layer (IPL), inner nuclear layer (INL) and outer plexiform layer (OPL). In comparison to the control group, the expression level of LC3- II was higher in all the experimental groups (P<0.05), with the peak expression at 12h after reperfusion. Additionally, the expression level of p62 was higher in all the experimental groups than the control (P<0.05, except for group A), with the peak level occurred 24h after reperfusion.
CONCLUSION
Both p62 and LC3 show low level and uneven expression in the retina of normal adult rats. Acute ocular hypertension can lead to upregulation of LC3- II and p62 expression in the retina. Autophagy flux is damaged 12h after reperfusion, potentially resulting in further loss of RGCs.
Keywords: glaucoma, acute ocular hypertension, LC3, p62, autophagy
INTRODUCTION
Glaucoma is the leading cause of irreversible blindness worldwide, which had been estimated to affect 111 million people by 2040[1]. It is a progressive neurodegenerative disease characterized by degeneration or loss of retinal ganglion cells (RGCs) and its axons[2]. Previous studies have suggested that multiple factors contribute to the irreversible damage of visual function in glaucoma, among which elevated intraocular pressure (IOP) has been widely acknowledged as the most important and only modifiable risk factors for glaucoma[2]–[4]. However, continued worsening of visual function has been observed in glaucoma patients with normal IOP[5]. This may be partly explained by the cascading damage of RGCs caused by initial IOP rising. IOP elevation could directly affect retinal blood vessels and reduce retinal blood flow[6]. In addition, the reperfusion injury of the retina with reducing IOP may lead to further loss of RGCs. However to date, little is known about the specific mechanisms underlying the association between high IOP and cell death in the optic nerve. Recent studies suggested that autophagy was related to photoreceptor degradation and death in glaucoma pathogenesis.
Autophagy, also known as cell self-digestion, is a cell-protective process which could be stimulated in response to varying stressors including oxidative stress[7]. Under normal physiological condition, the intracellular autophagy keep a state of equilibrium. Once the autophagy balance is broken, autophagy will lose the ability of protecting cells and result in diseases. The role of autophagy in RGCs death is controversial in literature. It has been suggested that a certain level of autophagy can protect the RGCs, while excessively activated or inhibited autophagy may cause RGCs damage[8]–[9]. Few studies had reported that autophagy was activated in RGCs after acute IOP elevation[10]. The number of photoreceptor deaths decreased significantly with autophagy suppressed in a light damage mouse model[8]. In addition, changes in the level of autophagy can determine RGCs survival in traumatic optic nerve injury and glaucoma animal models[5]. Some studies have also confirmed that excessive activation of autophagy may lead to self-digestion and even cell death.
Challenge remains to explain the relationship between autophagy related pathways and glaucoma, and the main effect of autophagy in glaucoma pathogenesis. The goal of the present study was to investigate the expression and potential role of autophagy related protein p62 and LC3 in the retina in a rat model acute ocular hypertension. The change of autophagy level was evaluated at various time points after retina ischemia-reperfusion to better understand the possible pathological mechanism of optic nerve injury in glaucoma.
SUBJECTS AND METHODS
Ethical Approval
All animals were sacrified after the experiment by intraperitoneal injection of 1% pentobarbital (80 mg/kg). The sacrificed animals were sent to the local animal center for further disposal. Animal treatment and care were conducted according to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. This research was approved according to relevant laws and regulations of animal experiment and laboratory animal welfare committee of the Second Affiliated Hospital of Fujian Medical University.
Animals
Adult Male Sprague-Dawley (SD) rats (220-250 g) were purchased from the Fuzhou Wu's animal experiment Co., Ltd., (certificate number: SCXK/Shanghai 2012-0002). For procedures that might cause discomfort, rats were anesthetized by intraperitoneal injection of 1% pentobarbital (40 mg/kg). Topical analgesia was achieved using 0.4% oxybuprocaine hydrochloride eye drops (Santen, Osaka, Japan). Pupils were dilated with 1% tropicamide eye drops (Santen).
A total of 50 SD rats were used in this study and randomly divided into five groups: one control group and four experimental groups (groups A, B, C, and D). No intervention was administered in the control group. The right eyes of rats in the experimental groups were chosen as the experimental eyes in establishment of acute ocular hypertension model according to the procedure described by Odagiri et al[11]. In brief, the vertical distance between the liquid lever in infusion bottle and the animal eye was 110 cm H2O, with around 80 mm Hg the normal saline solution (0.9% 500 mL sodium chloride injection). A 27-gauge needle, connected to a reservoir containing 500 mL sterile saline, was inserted into the anterior chamber of the right eye. Using direct ophthalmoscope (Suzhou medical equipment factory, YZ-11), the pale of retina was observed. As slowly dropping the infusion bottle to the same plane as the rat's eye after 60-minute normal saline perfusion, the corneal edema gradually improving. IOP was raised to 110 mm Hg by elevating the reservoir 149.6 cm above the animal's eye. The color of the retina recovered as soon as the infusion needle was removed, and the corneal edema disappeared within 3min. Chloramphenicol eye drops and erythromycin eye ointment were applied to the experimental eyes after the experiment.
H&E Staining and Rerinal Ganglion Cells Counting
Rats in the experimental groups were sacrificed at 6h (group A), 12h (group B), 24h (group C) and 48h (group D) after removal of the infusion needle. The right eyeballs were enucleated. After 24-hour fixation in 4% paraformaldehyde, dehydration in graded alcohols, and washing with dimethylbenzene, the specimens were embedded into paraffin. Histological sections were then prepared on the polylysine pre-managed slides. The same procedures were applied to the control group. For each eye, three slices were randomly selected for routine hematoxylin-eosin staining (H&E), and the micromorphology of retina was evaluated using the microscope (Japan, SN-MD). Five high power field images for each slide at 400 time were taken using the medical image analysis system (Image J 1.46r, National Institutes of Health, USA) for RGCs counting (number/field), and the average of all three slices was calculated and recorded.
Immunohistochemistry
The prepared eyeball slides were cleared using xylene conventionally, endogenous enzymes inactivation was then administered using 3% hydrogen peroxide and antigen retrieval. Subsequently, the slides were incubated with primary antibodies against p62 and LC3 overnight at 4°C. PBS was used as negative control agent instead of primary antibodies. These sections were then incubated with secondary antibodies for 30min at 37°C, followed by coloring with the developer of 3,3′-diaminobenzidine (DAB) for 3-5min. Re-staining cell nucleus with hematoxylin after fully washed with running water. Five eyes of each group and 5 slices of each eye were randomly chosen for observing the expression of p62 and LC3 protein in the retina. Five high power field images for each slide were randomly selected for semi-quantifying the expression of proteins using Image-Pro Plus 6 color image analysis system (Image J 1.46r, National Institutes of Health, USA). The nuclei stained with brown yellow or brown were positive for p62 and LC3, and the value was presented by integral optical density (IOD).
Western Blot Analysis
Retina tissue was dissected from the sclera and then immediately homogenized in a glass-Teflon Potter homogenizer in an ice-cold lysis buffer containing 20 mmol/L Hepes, pH 7.5,10 mmol/L KCl,1.5 mmol/L MgCl2,1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L ethylene glycol tetraacetic acid (EGTA), 1 mmol/L DTT, 0.5% CHAPS, complete protease inhibitors. After 1-hour standing, the samples were centrifuged for 20min using the high-speed refrigerated centrifuge under 12 000 r/min. The supernatant was fully mixed with the protein sample buffer (5× sample buffer and 20× reducing agent), and boiled at 100°C for 5min. The protein was measured by standard bicinchoninic acid (BCA) method. After blocking with 5% skim milk for 2h, the membranes were incubated with rabbit polyclonal antibodies against LC3B and p62/ SQSTM1 polyclonal antibody overnight at 4°C and subsequently with appropriate secondary antibody (goat anti rabbit IgG/horseradish peroxidase) for 1h at room temperature. After reaction with ECL luminescent for 3min, the membranes were tableting into films for 3-minitue exposure. After the film was scanned, the images were analyzed by Quantity One software (American Bio-Rad company), which evaluates the relative amount of protein staining and quantifies the results in terms of optical density. The gray values of protein bands were calculated by normalization to β-actin which act as the internal reference.
Statistical Analysis
All data followed normal distribution and presented as mean±SD. The randomized controlled single factor intervention multilevel experimental design was adopted. Homogeneity of data variance in each group was confirmed by Levene's test. Differences among groups were compared using one-way ANOVA. Statistical analyses were performed using SPSS 17 (IBM Corporation, Armonk, NY) with a P value of <0.05 considered to be statistically significant.
RESULTS
Retina Structure of Normal Rats
The H&E staining images showed well distinguished layers of the retina in normal rats (Figure 1): the photoreceptor layer (PL) including the rods and cones, the external limiting membrane (ELM), the outer nuclear layer (ONL) consisting of 8-10 layers of compacted arranged cell nuclei, the outer plexiform layer (OPL), the inner nuclear layer (INL) composing of 3-5 layers of compacted arranged cell nuclei, the inner plexiform layer (IPL), the ganglion cell layer (GCL) which is a regular distributed monolayer, the nerve fiber layer (NFL) and the internal limiting membrane (ILM).
Figure 1. H&E staining of a normal rat's retina (×200) showing well distinguished retinal layers.
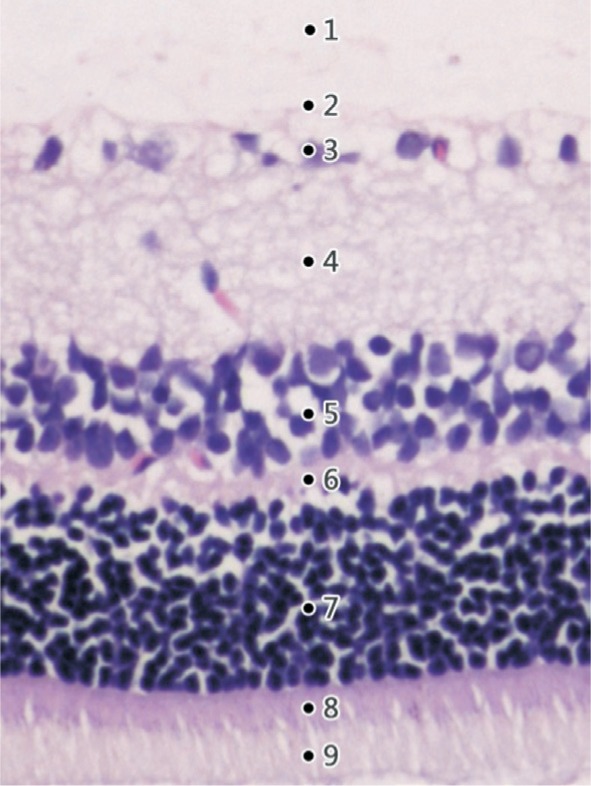
1: The inner limiting membrane (ILM); 2: The nerve fiber layer (NFL); 3: The ganglion cell layer (GCL); 4: The inner plexiform layer (IPL); 5: The inner nuclear layer (INL); 6: The outer plexiform layer (OPL); 7: The outer nuclear layer (ONL); 8: The external limiting membrane (ELM); 9: The photoreceptor layer (PL).
Retinal Structure of Experimental Rats
Figure 2 illustrates the retina H&E staining images in the control and experimental groups at different time points after reperfusion. The average numbers of RGCs in each group were presented in Table 1. Six hours after reperfusion (group A), slightly widening of the retinal tissue space was observed with no obvious change in the retinal thickness. Twelve hours after reperfusion (group B), retinal edema developed with irregular arrangement and loosen of cells in the NFL and IPL, and the number of RGCs decreased significantly as compared to the control group (percent reduction, 16.9%; P<0.05). At 24h after reperfusion (group C), thinning of retina was observed, especially for the IPL. There was no retina edema and number of RGCs further decreased (percent reduction, 29.2%; P<0.05). The thinning of retina and reduction in RGCs numbers (percent reduction, 35.4%; P<0.05) was more significant 48h after reperfusion (group D). The average number of RGCs was 19.5±1.03/500 µm in the control group, and showed an overall significant decreasing trend with prolonged reperfusion time (P<0.01).
Figure 2. Retinal structure in the control group and different experimental groups (H&E staining, 200×).

Table 1. Numbers of retinal ganglion cells in each group.
| Groups | Number of eyes | Average RGCs number/500 µm (mean±SD) | Percentage reduction |
| Control | 5 | 19.5±1.03 | |
| Group A | 4 | 17.6±0.89 | 9.74% |
| Group B | 5 | 16.2±1.47a | 16.9% |
| Group C | 5 | 13.8±0.73a | 29.2% |
| Group D | 4 | 12.6±0.75a | 35.4% |
| F | 110.56 | ||
| P | <0.01 |
aSignificant difference (P<0.05) between the experimental groups and the control group (one-way ANOVA, LSD test).
Distribution and Semi-quantitation of LC3 and p62
LC3 mostly expressed in the GCL in the retina of normal rats (control group). In comparison, immunohistochemical staining of LC3 was enhanced in all the experimental groups, and was strongest at 12h after reperfusion (group B; Figure 3). Semi-quantitative detection of LC3 showed significant differences between the control group and experimental groups (P<0.05; Figure 4).
Figure 3. Distribution of LC3 in rats' retina (immunohistochemical staining, 400×) with arrowheads pointing to brown RGCs.

A: An image of the retina from the control group; B: An image of the retina at 6h after reperfusion (group A); C: An image of the retina at 12h after reperfusion (group B); D: An image of the retina at 24h after reperfusion (group C); E: An image of the retina at 48h after reperfusion (group D).
Figure 4. Semi-quantitative expression level of LC3 in rats' retina (n=4).

aSignificant difference (P<0.05) between the experimental groups and the control group (one-way ANOVA, LSD test).
Expression of p62 was found in the NFL, GCL, IPL, INL and OPL in the control and all experimental groups. p62 staining was enhanced in all the experimental groups as compared to the control group (Figure 5). The higher p62 expression in experimental groups was statistically significant as demonstrated by the semi-quantitative analysis (P<0.05; Figure 6).
Figure 5. Immunohistochemical staining images showing p62 as scattered brown particles in NFL, GCL, IPL, INL, and OPL of retina (400×).

A: An image of the retina from the control group; B: An image of the retina at 6h after reperfusion (group A); C: An image of the retina at 12h after reperfusion (group B); D: An image of the retina at 24h after reperfusion (group C); E: An image of the retina at 48h after reperfusion (group D).
Figure 6. Semi-quantitative expression level of p62 in rats' retina (n=4).

aSignificant difference (P<0.05) between the experimental groups and the control group (one-way ANOVA, LSD test).
Expression of LC3-II and p62
As shown by the Western blot analysis, the expression level of LC3-II in the rat retina in group A was higher than that in the control group (0.095±0.01344 vs 0.02±0.00816, P<0.05; Figure 7). Expression of LC3-II reached the highest level at 12h after reperfusion (group B), followed by a decreasing trend with longer reperfusion time. The difference in LC3-II expression between the experimental groups and the control group was statistically significant (P<0.05). Meanwhile, the expression of p62 in the rat retina showed an increasing trend within 48h after reperfusion (Figure 8). The expression level of p62 was 0.745±0.04655 (mean±SD) in the control group, and was not significantly different with the level at 6h after reperfusion. However, at 12h after reperfusion, the expression level of p62 increased to 0.965±0.01291, which was significantly higher than the control group (P<0.05) and this higher expression lasted at least 48h after reperfusion.
Figure 7. Expression level of LC3-II in rats' retina of each group.

A: Western blot radioactive detected bands of LC3-I, LC3-II and β-actin; B: The amount of LC3-II in each group after normalized to β-actin (n=5). aSignificant difference (P<0.05) between the experimental groups and the control group (one-way ANOVA, LSD test).
Figure 8. Expression level of p62 in rats' retina of each group.

A: Western blot radioactive detected bands of p62 and β-actin; B: The amount of p62 in each group after normalized to β-actin (n=5). aSignificant difference (P<0.05) between the experimental groups and the control group (one-way ANOVA, LSD test).
DISCUSSION
In the present study, we demonstrated the activation of autophagy and its relationship with RGCs apoptosis in a rat model of acute ocular hypertension.
By comparing the retina structure and RGCs numbers between normal rats and rats in different experimental groups, we found that the degree of neuronal loss was related to the duration of ischemia-reperfusion. Retina thinning and reduction in RGCs were more obvious with longer reperfusion time. The underlying mechanisms for cell death after ischemia-reperfusion are largely unknown. Previous studies have reported that many kinds of programmed cell death (including cell apoptosis and autophagy) play an important role in the retina metabolism in glaucoma patients. Autophagy had also been suggested as a potential cause for RGCs death after retina ischemia-reperfusion (RIR).
Autophagy can be divided into chaperone-mediated autophagy (CMA), micro-autophagy and macro-autophagy according to the different combining forms of autophagosome and lysosome[12]. The present study mostly focused on the process of macro-autophagy. The core function of macro-autophagy is to degrade long-lived proteins and damaged organelles, thus ensuring the stability of intracellular environment. During autophagy, the complete organelles (such as mitochondria) and some cytoplasm are separated into the double layer coating vesicles to form autophagosomes, which will connect with the lysosomes and form autophagolysosomes. Finally, the hydrolase in autophagolysosomes degrades certain substances to reproduce new macromolecular products, which can be reused by cells[13]–[14]. A variety of proteins encoded by the autophagy associated gene (Atg) are involved in autophagy[10]. Microtubule- associated protein 1 light chain 3 (LC3) is currently known as the marker protein of autophagosome[15]. LC3 exists in two forms: LC3-I is usually found in the cytoplasm and LC3-II on the membrane. LC3- II is a hard-to-degrade protein formed by the binding of LC3-I with phosphatidyl ethanolamine (PE). Therefore, the expression level of LC3-II can be used to represent the volume of autophagy[13].
Earlier studies have shown that LC3 was expressed in RGCs and photoreceptors using immunohistochemical staining. Kim et al[16] reported that the expression of LC3-II in the RGCs significantly increased on the 1st, 3rd, 5th and 7th day after optic nerve damage and reached the highest level on the 3rd day based on an optic nerve disconnection rat model. This indicated that autophagy was activated in RGCs after optic nerve injury. Rodríguez-Muela et al[8] observed increased autophagy in RGCs 3-10d after optic nerve injury. In a chronic intraocular hypertension model, the expression of LC3-II and number of autophagosome increased at 1-4wk after IOP elevation. Additionally, the staining of LC3B in the GCL and IPL was also enhanced[9]. In a rat RIR model, Russo et al[17] reported no significant difference in the LC3-II expression between the normal retina and retina at 24h after reperfusion. However, another study reported significantly increased expression of LC3-II at 24h after reperfusion[10]. This discrepancy might be due to difference in IOP elevation degree. In the present study, LC3 immunoreactive particles was not found in the retina of normal rats, but found in the GCL in the rats with acute ocular hypertension with strongest staining at 12h after reperfusion, which was further confirmed by Western-blot analysis. Thus we suggest that autophagy is activated in the retina after retina ischemia-reperfusion damage.
The expression of LC3-II could not reflect the downstream process of autophagy, namely the autophagy flux[13]. Another multiple domain protein p62 is involved in this process. Therefore, the investigation of autophagy flux requires simultaneous detection of both p62 and LC3-II. The Phox and Bem1p (PB1) domain, located at the N end of p62, can attract and combine molecules with certain structures. Then it combines with LC3 by the LC3-interacting region (LIR) at the C terminal to accumulate the molecules to be degraded in the autophagosomes. The expression of p62 increases during the molecular accumulation process, and decreases as the enzymatic hydrolysis reaction starts[18]. Therefore, the decrease in p62 expression and the transformation of LC3 into LC3-II is considered to be an important indicator of autophagy flux. Previous studies have reported that compared with normal subjects, the level of p62 in the brain of Alzheimer's disease patients was higher[11]. Joshi et al[19] showed that the expression of p62 in the neurons of APP/PS1 transgenic mice increased significantly compared with the wild type mice, which proved that the clearance of p62 through autophagy was damaged in the AD animal model. Kitaoka et al[20] found that rapamycin decreased intracellular reactive oxygen species (ROS) production and increased cell viability and that 3-MA increased ROS production and reduced cell viability in RGC-5 cells. Another study showed that small amount of p62 could combine with mitochondria directly, and protect mitochondrial integrity. While, persistent high level of p62 can cause accumulation of reactive oxygen species and in pathological conditions. Kojima et al[21] reported that rapamycin has a protective effect on axons in tumor necrosis factor (TNF)-induced optic neurodegeneration.
In eukaryotic cells, the ubiquitin-protease line is a major way of protein metabolism, responsible for removing short-life proteins, various regulatory proteins, and defective proteins. Autophagy is responsible for degrading long-life proteins. In the normal process of growth and development, these two pathways can be interrelated by p62 to maintain cellular homeostasis. After combining with ubiquitin, p62 recruits ubiquitinated protein and affects the ubiquitin-proteasome process. Studies of knockout mice and drosophila models reported that p62 was the key protein of ubiquitinated protein aggregation[18]. Almost all neurodegeneration-related studies showed abnormal accumulation of ubiquitinated protein. It has been reported that inhibition of autophagy in neurons can lead to p62 accumulation and mitochondrial damage, which potentially lead to neuro-degeneration and nerve cell death[22]. p62 is also a stress response protein, which is upregulated with over aggregation of sodium arsenite, cadmium ion carrier, proteasome inhibition and abnormal proteins[23]. Conversely, the abnormal accumulation of p62 reduces the normal clearance of cells and leads to excessive accumulation of abnormal substances, posing detrimental effect to the retina.
The role of autophagy in the pathogenesis of many neurodegenerative diseases had been demonstrated in literature. Rapamycin, an autophagy inducer, can prevent A β oligomers from destroying synapses in rat hippocampal neurons. Salminen et al[24] proposed that the blocking of autophagy would cause mitochondrial dysfunction, which interfered with the clearance process of amyloid precursor protein and tau protein. These findings suggest that regulation of autophagy may delayed the progression of neurodegenerative diseases. It is reported RGC-5 cells can induce autophagy activation to protect themselves under starvation states in vitro[25]. Autophagy inducers, such as Rapamycin, may reduce the concentration of reactive oxygen species and increase cell viability in RGC-5 cells. Meanwhile, autophagy inhibitors play an opposite role in RGC-5 cells. Therefore, it is suggested that autophagy regulation can help improve the ability of RGCs to resist harmful external environment.
Few studies had investigated the expression and role of LC3 and p62 in glaucoma animal models. Based on the study of autophagy in neurodegenerative diseases, for the first time, the present study used the expression of LC3 and p62 to represent and investigate autophagic activation and autophagy flux in the rat model of the acute ocular hypertension RIR. In a normal autophagy process, the increased expression of LC3-II should be accompanied by p62 reduction. In the experimental groups of this study, the LC3-II in the retina increased significantly with prolonged reperfusion. However at the meantime, the expression of p62 also increased significantly. Immunohistochemical staining showed that the distribution of p62 and LC3 protein in the normal retina was uneven and mainly in the GCL, indicating low-level autophagy. In compared with the control group, the expression of p62 in the retina of the experimental groups was significantly higher and peaked at 24h after reperfusion, suggesting that the autophagic flux was interfered after ischemia and reperfusion[20]. There was a decreasing trend in the number of RGCs with longer reperfusion time, which suggested that abnormal accumulation of p62 and impaired autophagic flux leaded to further loss of RGCs. Given that p62 overexpression can activate caspase -8 to induce apoptosis, and p62 decline could reduce the death of human brain glioma cells, the optic nerve can be protected in the following two ways in the process of acute ocular hypertension ischemia and reperfusion. First, stimulates an appropriate expression of p62 to the tolerance of RGCs.The degree of p62 can reflect the autophagy flux, so stimulates an appropriate expression of p62 may enhance the tolerance of RGCs. Second, regulates p62-related siRNA transcription to increase the expression of autophagic proteins such as LC3- II, Beclin-1 and other related autophagic proteins, and promotes the ability of clearing abnormal proteins and organelles.
In conclusion, acute ocular hypertension could lead to upregulation of LC3- II and p62 expression in the retina. Autophagy flux was detected 12h after reperfusion, potentially resulting in further loss of RGCs. More autophagy related proteins need to be investigated in the future to explore the relationship between various regulatory pathways of autophagy and glaucomatous optic neuropathy to explore new therapeutic methods.
Acknowledgments
Authors' contributions: Wu YY conceived of the study, participanted in the study design and drafted the manuscript. Zheng BR participanted in the study design, carried out the experiments and drafted the manuscript. Chen WZ, Huang YH and Zhang Y helped to perform the experiments and to modify the manuscript; Guo MS conceived of the study and participanted in its design. All authors read and approved the final manuscript.
Foundation: Supported by the Natural Science Foundation of Fujian Province (No. 2016J01525).
Conflicts of Interest: Wu YY, None; Zheng BR, None; Chen WZ, None; Guo MS, None; Huang YH, None; Zhang Y, None.
REFERENCES
- 1.Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121(11):2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 2.MacGregor S, Ong JS, An JY, et al. Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat Genet. 2018;50(8):1067–1071. doi: 10.1038/s41588-018-0176-y. [DOI] [PubMed] [Google Scholar]
- 3.Choi J, Kook MS. Systemic and ocular hemodynamic risk factors in glaucoma. Biomed Res Int. 2015;2015:141905. doi: 10.1155/2015/141905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan MPY, Broadway DC, Khawaja AP, Yip JLY, Garway-Heath DF, Burr JM, Luben R, Hayat S, Dalzell N, Khaw KT, Foster PJ. Glaucoma and intraocular pressure in EPIC-Norfolk Eye Study: cross sectional study. BMJ. 2017;358:j3889. doi: 10.1136/bmj.j3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adeghate J, Rahmatnejad K, Waisbourd M, Katz LJ. Intraocular pressure-independent management of normal tension glaucoma. Surv Ophthalmol. 2019;64(1):101–110. doi: 10.1016/j.survophthal.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Xu JJ, Li YD, Song SZ, Cepurna W, Morrison J, Wang RK. Evaluating changes of blood flow in retina, choroid, and outer choroid in rats in response to elevated intraocular pressure by 1300 nm swept-source OCT. Microvasc Res. 2019;121:37–45. doi: 10.1016/j.mvr.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munemasa Y, Kitaoka Y. Autophagy in axonal degeneration in glaucomatous optic neuropathy. Prog Retin Eye Res. 2015;47:1–18. doi: 10.1016/j.preteyeres.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez-Muela N, Germain F, Mariño G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19(1):162–169. doi: 10.1038/cdd.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park HY, Kim JH, Park CK. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis. 2012;3:e290. doi: 10.1038/cddis.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One. 2011;6(7):e22514. doi: 10.1371/journal.pone.0022514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in α-synucleinopathy. Acta Neuropathol. 2012;124(2):173–186. doi: 10.1007/s00401-012-0975-7. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaaf MBE, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30(12):3961–3978. doi: 10.1096/fj.201600698R. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Munemasa Y, Kwong JM, Ahn JH, Mareninov S, Gordon LK, Caprioli J, Piri N. Activation of autophagy in retinal ganglion cells. J Neurosci Res. 2008;86(13):2943–2951. doi: 10.1002/jnr.21738. [DOI] [PubMed] [Google Scholar]
- 17.Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, Rotiroti D, Morrone LA, Bagetta G, Corasaniti MT. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144. doi: 10.1038/cddis.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caccamo A, Ferreira E, Branca C, Oddo S. P62 improves AD-like pathology by increasing autophagy. Mol Psychiatry. 2017;22(6):865–873. doi: 10.1038/mp.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer's disease-like pathology in the APP/ PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36(2):664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, Takagi H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis. 2013;4:e860. doi: 10.1038/cddis.2013.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kojima K, Kitaoka Y, Munemasa Y, Hirano A, Sase, Takagi H. Axonal protection by modulation of p62 expression in TNF-induced optic nerve degeneration. Neurosci Lett. 2014;581:37–41. doi: 10.1016/j.neulet.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Navone F, Genevini P, Borgese N. Autophagy and neurodegeneration: insights from a cultured cell model of ALS. Cells. 2015;4(3):354–386. doi: 10.3390/cells4030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moon JY, Cho SK. Nobiletin induces protective autophagy accompanied by ER-stress mediated apoptosis in human gastric cancer SNU-16 cells. Molecules. 2016;21(7):E914. doi: 10.3390/molecules21070914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, Hiltunen M. Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106-107:33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Chalasani ML, Kumari A, Radha V, Swarup G. E50K-OPTN-induced retinal cell death involves the Rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PLoS One. 2014;9(4):e95758. doi: 10.1371/journal.pone.0095758. [DOI] [PMC free article] [PubMed] [Google Scholar]