


Abstract
As the largest ethnic group in the world, the Han Chinese population is nonetheless underrepresented in global efforts to catalogue the genomic variability of natural populations. Here, we developed the PGG.Han, a population genome database to serve as the central repository for the genomic data of the Han Chinese Genome Initiative (Phase I). In its current version, the PGG.Han archives whole-genome sequences or high-density genome-wide single-nucleotide variants (SNVs) of 114 783 Han Chinese individuals (a.k.a. the Han100K), representing geographical sub-populations covering 33 of the 34 administrative divisions of China, as well as Singapore. The PGG.Han provides: (i) an interactive interface for visualization of the fine-scale genetic structure of the Han Chinese population; (ii) genome-wide allele frequencies of hierarchical sub-populations; (iii) ancestry inference for individual samples and controlling population stratification based on nested ancestry informative markers (AIMs) panels; (iv) population-structure-aware shared control data for genotype-phenotype association studies (e.g. GWASs) and (v) a Han-Chinese-specific reference panel for genotype imputation. Computational tools are implemented into the PGG.Han, and an online user-friendly interface is provided for data analysis and results visualization. The PGG.Han database is freely accessible via http://www.pgghan.org or https://www.hanchinesegenomes.org.
INTRODUCTION
With the continuous development and cost reduction of sequencing technology (1), many large-scale genome sequencing projects have been launched in Western countries to support personalized medicine (PM), such as the UK10K project (2), the Estonian Genome Project (EGP) (3) and the NHLBI Trans-Omics for Precision Medicine (TOPMed) program (4). Some genomic resources have also been created for a few East Asian populations, including Japanese, Korean and Vietnamese (5–8), but the Han Chinese population has been largely underrepresented. The Han Chinese population is the largest ethnic group in East Asia, and in the world, comprising ∼20% of the global human population, ∼92% of Mainland Chinese, ∼92% of Hongkongers, ∼97% of Taiwanese, ∼74% of Singaporeans, ∼23.4% of Malaysians and ∼21.4% of the San Francisco's population (9). Most genetic investigations, however, have focused on in European and African populations. For example, the majority of the genome-wide association studies (GWASs) have been predominantly conducted on populations with European ancestry (2,10–13). Reference genomes of natural populations are well-established and have demonstrated their power in disease-mapping studies for populations with European ancestry (14), but are still lacking for the Han Chinese population. Some previous studies have also included Han Chinese samples, although either the sample size (15–23) or the geographical coverage was limited (24,25).
There were recently efforts attempting to analyze shallow (<2×) or ultra-shallow (∼0.05–0.1×) sequencing data of 100 000 samples or more collected from resources for other purposes, such as the data that were originally generated for Non-Invasive Prenatal Testing (NIPT), in which a sufficient number of high-quality variants on an individual level could not be attained, thus limiting their usefulness for population stratification/structure analysis and further association studies (26). The overarching issue in utilizing NIPT whole-genome sequencing data in surveys of genetic variation at the population scale is their sparse and ultra-shallow (∼0.05×) sequencing coverage at each sample level. In particular, individual genotypes could not be determined on a genome-wide level, making the data roughly equivalent to the deep-sequencing of a few hundred of samples to high coverage (30×) and thereby limiting their usefulness for population stratification/structure analysis due to the failure to acquire sufficient numbers of high-quality variants on an individual level. For example, it is not feasible to investigate cryptic population structures if individual genotypes cannot be determined on a genome-wide level. In addition, the false positive rate (FPR) of the SNP calling for NIPT data can be very high—as much as 32%, as estimated by a previous study (26).
Here, we developed a population genomic database specific to the Han Chinese population, namely the PGG.Han, and constructed a reference panel of 114 783 Han Chinese individuals (the Han100K), with whole-genome deep-sequenced or high-density genome-wide single-nucleotide variants (SNVs) genotyped or imputed. The PGG.Han has very clear applications/functions in the practice of scientific research; in particular, it provides the first and only-available reference data for (i) a population-structure-aware shared control for genotype–phenotype association studies (e.g. GWASs) and (ii) a Han-Chinese-specific reference panel for genotype imputation. The PGG.Han also provides a variety of online analysis tools, including ancestry inference, genotype imputation, and a routine GWAS pipeline. User-friendly interfaces and online analysis reports greatly facilitate users in the application of genomic data to evolutionary and medical studies of the Han Chinese, as well as East Asian populations.
DATA INTEGRATION
The PGG.Han archives two types of genomic data: whole-genome sequencing data and high-density genotyping data (Figure 1). The whole genomes of ∼12 000 Han Chinese individuals have been sequenced using next-generation sequencing technology and are archived in the PGG.Han, including three deep-sequencing datasets (∼30–80×) (n = 319) (16–17,20,22), and two low-pass sequencing datasets (∼1.7×, n = 11 670; and ∼4×, n = 208) (18,21).
Figure 1.
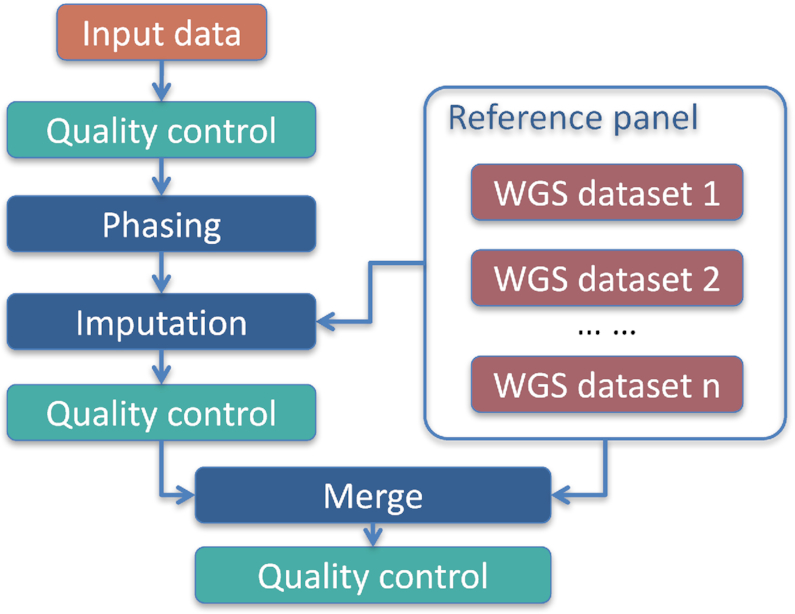
Sketch map of data integration in the PGG.Han. Using all the whole-genome sequencing data of the Han Chinese samples as a reference panel, the genotype data of 102 586 samples were carefully imputed. The imputation results and the whole-genome sequencing data were further integrated. Strict quality control was applied throughout the process. WGS, whole-genome sequencing.
The deep-sequencing datasets (16,20) represent 28 Han Chinese dialect groups across China. The genomes of 11 670 female Han Chinese were sequenced by the CONVERGE project as a control group for the investigation of major depressive disorders (18). Although in a much lower coverage (∼1.7×), this dataset provides a catalog of 25 057 223 variants for the Han Chinese population and is also included in the PGG.Han.
The high-density genome-wide SNP data of 102 586 samples were collected from previous GWAS projects, with only non-patient (control) data being retained. Using all the whole-genome sequencing data of the Han Chinese samples as reference data, the genotype data of the 102 586 samples were carefully imputed. We then integrated these data into a dataset and performed strict quality control (Figure 1), including individual filtering and variant filtering. The final dataset contains 8 056 973 genome-wide SNPs shared by all of the 100K Han Chinese samples, of which the geographical origins are known for 56 308 of these samples.
DATABASE CONTENT
Overview
The database consists of two major functional modules: (i) visualizing the population structure and querying allele frequencies of subgroups, and (ii) online analysis tools (Figure 2). The population structure follows the visual display mode of PGG.Population (27), including genetic affinity as measured by FST (28), genetic coordinates of populations based on principal component analysis (PCA) (29), and ancestry composition based on ADMIXTURE analysis (30). The presence of population structure can lead to false positive or false negative results in genotype–phenotype association studies (31), but with the help of the PGG.Han data, which contain the fine-scale genetic structure of the Han Chinese population, this effect can be minimized or avoided as much as possible. In order to illuminate the allele frequency information, both tables and interactive maps are provided. It is convenient to query the frequency distribution of each locus in natural populations or in genetic subgroups of the Han Chinese. Since the PGG.Han focuses on the Han Chinese population, we also provide a way to query the allele frequency of each variant in the worldwide population by linking to the PGG.SNV (https://www.pggsnv.org), a sister database of the PGG.Han that provides various information concerning single-nucleotide variants.
Figure 2.

Sitemap of the PGG.Han. The database consists of two major functional modules: (i) visualizing the population structure and querying allele frequencies of subgroups (red boxes); (ii) online analysis tools (blue boxes). Each analysis tool can be used independently and combined freely. The red arrow represents the recommended workflow. The imputation step is optional and should be contingent upon the details of the dataset of interest. AF, allele frequency; Ref. allele, reference allele.
As a significant feature, essential computational functions are embedded in the PGG.Han, including three analysis pipelines, i.e. ancestry inference, genotype imputation and GWAS, with the shared control. It is well known that a population-specific panel or a control dataset is essential for improving the accuracy of imputation or GWAS results (31,32). Accordingly, we designed and recommend the following workflow for analyzing the users’ data (Figure 2). The first step is ancestry inference, which yields the genetic background and subgroup affinity of each individual. For Han Chinese individuals, the ancestry inference is refined to the detailed scale of six genetic subgroups. In the second step, users can select an ancestry-matching reference panel for imputation based on the ancestral inference results. Finally, we provide a population-structure-aware shared control of the Han Chinese for genotype-phenotype association studies. An online GWAS analysis task can be easily be submitted to the analysis platform of the PGG.Han. In addition, each of the three modules can run independently with a great deal of flexibility.
Visualization population structure and allele frequencies
Based on the genetic relationships among provincial populations, six major subgroups have been identified (Supplementary Figure S1): Northwest Han (NWH), Northeast Han (NEH), Central China Han (CCH), Southwest Han (SWH), Southeast Han (SEH) and South Coast Han (SCH). Visualization of the population structure and allele frequency distribution maps are shown from two different perspectives, i.e. provincial populations and genetic subgroups. The interactive page design offers a user-friendly way to acquire information. Overall, the main difference in the Han population is between northern and southern subgroups, consistent with previous observations (9,33–34) (Supplementary Figure S1), with southern subgroups exhibiting greater divergence than northern subgroups.
The detailed population structures in the form of high-quality SNV data provide researchers with helpful panels for investigating variant allele frequencies within Han Chinese subgroups. For example, ALDH2 is a protein-coding gene involved in the process of alcohol metabolism (35). rs671 is a missense variant on this gene (NP_000681.2: p.Glu504Lys), which decreases enzyme activity and shortens the enzyme half-life (36). This change manifests as acute alcohol sensitivity (OMIM: 610251) and has a high frequency among East Asian ethnic groups. In current public datasets, the only information available is that the frequency of the rs671 missense variant in the Han Chinese from Beijing (CHB) is 0.160 and its frequency in Han Chinese from South China (CHS) is 0.271 (21). The PGG.Han, however, provides more detailed and enlightening information. For example, even populations from Guangdong and Guangxi, which are both southern Han Chinese, have considerably different rs671 missense variant frequencies (Guangdong: 0.291; Guangxi: 0.230). Moreover, not all northern Han Chinese subgroups have lower frequencies than southern Han Chinese subgroups. For example, the frequency of the rs671 missense variant in Gansu is 0.230 and the frequency in Jilin is 0.216, while the frequency in the Yunnan sub-population is 0.191. Therefore, the PGG.Han provides a richer picture of the population stratification and genomic diversity of the Han Chinese population, which is essential for future evolutionary and medical studies.
ONLINE ANALYSIS
Data upload and quality check
There are two ways to upload data to the PGG.Han. For small datasets (<100 MB), users can upload the data directly on the Web via HTTPS protocol, while FTP is suggested for uploading large datasets. The data uploaded to the server by users are fully protected and accessible to the user only. Users can delete their data at any time from the server. Uploaded data will first be subjected to a quality check. The quality check is a functional module used to determine whether the input data meet the requirements for subsequent data analyses. This module primarily consists of data conversion, summary statistics, and routine data quality control. All data that pass the quality control check can then be used for other analyses.
Ancestry inference
The function of ancestry inference is provided to analyze the genetic ancestry of each individual from different perspectives (Supplementary Figure S2). In order to determine the population affiliation of an individual, we use the hierarchical clustering analysis by representative reference populations based on nested AIMs panels (34). We also quantitatively estimate the proportion of each ancestral component of each individual in the context of global populations. Moreover, proportions of the archaic ancestry are estimated and provided as an independent result using ADMIXTOOLS (30). The haplogroups of mtDNA and/or the Y chromosome of an individual are determined using HaploGrep2 (37,38) as well as our own self-designed algorithm (20), and are displayed on a map with migration routes, which illuminate the possible genetic history of a particular individual. Furthermore, for the Han Chinese samples, we provide more detailed ancestral inference results. For instance, for a given Han Chinese sample, we determine which province is its most likely geographical source. Visual charts are provided for each module.
Genotype imputation
Genotype imputation, or simply imputation in the context of the PGG.Han database, is used to estimate the unobserved genotypes and to replace the missing genotypes in a given dataset. Our imputation service is designed to meet three different requests for imputation: (i) achieving the best imputation result for the Han population data with reference panels based on our NGS datasets of Han Chinese; (ii) performing classical imputation tasks with public reference panels of global populations; (iii) estimating and replacing the missing genotypes in the data provided by users. Compared with other imputation services (Supplementary Table S1) (14,39), we provide a few more imputation tools (39–43) to meet users’ different needs. Various reference panels (14,18–19,21) allow users to optimize the results. Notably, the largest reference panel of the Han Chinese population (the Han100K) is also provided, which we suggest employing for the genotype imputation analysis of samples of Han Chinese or East Asian origin. Output files can be downloaded or directly used for other analyses on our website (Supplementary Figure S3).
GWAS
The genome-wide association study (GWAS) is an approach used to find genetic variations associated with a particular trait or disease by scanning the genome-wide genetic markers (typically SNPs) of many samples. Here, we provide a platform for GWAS analysis, as well as the largest scale of control data for the Han Chinese population (the Han100K Project). Our pipeline includes data quality control procedures (Supplementary Figure S4) and implements two analysis modes, as provided by Plink1.9 (44), i.e. ‘linear’ or ‘logistic,’ which can be identified automatically depending on the data format of the provided phenotypes. Summary statistics of the association analysis for each site are documented in a plain text file, which can be downloaded by users. In addition, a Manhattan plot and an QQ-plot are also provided in each online report (Supplementary Figure S5).
FUTURE DIRECTIONS
In the current version, we have compiled the genome data of >100 000 Han Chinese samples covering most geographical divisions in China, as well as Singapore. We not only provide detailed information regarding fine-scale genetic structure and allele frequency, but also furnish three online analysis tools, including ancestry inference, genotype imputation, and population-structure-aware control data for GWAS. We expect the PGG.Han to serve as a generally applicable Han Chinese genome reference not only for researchers specifically interested in the Han Chinese population, but also for those in different fields who need to validate their analysis in East Asian populations.
Overall, as the central repository for the genomic data of the Han Chinese Genome Project (Phase I), the PGG.Han will be a useful database and an efficient platform for the practice of both evolutionary and medical studies of the Han Chinese as well as other East Asian populations.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to all of the participants in the Han100K Project. We also thank Dr Charleston Chiang for sharing with us the sample information from the CONVERGE data. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program [2016YFC0906403]; Strategic Priority Research Program [XDB13040100]; Key Research Program of Frontier Sciences [QYZDJ-SSW-SYS009] of the Chinese Academy of Sciences (CAS); National Natural Science Foundation of China (NSFC) [91731303, 31771388, 31961130380, 31711530221]; National Science Fund for Distinguished Young Scholars [31525014]; UK Royal Society-Newton Advanced Fellowship [NAF\R1\191094]; Program of Shanghai Academic Research Leaders [16XD1404700]; Shanghai Municipal Science and Technology Major Project [2017SHZDZX01]; Zhangjiang Special Project of the National Innovation Demonstration Zone [ZJ2018-ZD-013]; S.X. also gratefully acknowledges the support of the UK Royal Society-Newton Mobility Grants (IE160943); ‘Wanren Jihua’ Project. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of this manuscript.
Conflict of interest statement. None declared.
REFERENCES
- 1. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Consortium U.K., Walter K., Min J.L., Huang J., Crooks L., Memari Y., McCarthy S., Perry J.R., Xu C., Futema M. et al.. The UK10K project identifies rare variants in health and disease. Nature. 2015; 526:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Metspalu A., Kohler F., Laschinski G., Ganten D., Roots I.. The Estonian Genome Project in the context of European genome research. Dtsch. Med. Wochenschr. 2004; 129(Suppl. 1):S25–28. [DOI] [PubMed] [Google Scholar]
- 4. Brody J.A., Morrison A.C., Bis J.C., O’Connell J.R., Brown M.R., Huffman J.E., Ames D.C., Carroll A., Conomos M.P., Gabriel S. et al.. Analysis commons, a team approach to discovery in a big-data environment for genetic epidemiology. Nat. Genet. 2017; 49:1560–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tadaka S., Katsuoka F., Ueki M., Kojima K., Makino S., Saito S., Otsuki A., Gocho C., Sakurai-Yageta M., Danjoh I. et al.. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 2019; 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le V.S., Tran K.T., Bui H.T.P., Le H.T.T., Nguyen C.D., Do D.H., Ly H.T.T., Pham L.T.D., Dao L.T.M., Nguyen L.T.. A Vietnamese human genetic variation database. Hum. Mutat. 2019; doi:10.1002/humu.23835. [DOI] [PubMed] [Google Scholar]
- 7. Yasuda J., Katsuoka F., Danjoh I., Kawai Y., Kojima K., Nagasaki M., Saito S., Yamaguchi-Kabata Y., Tadaka S., Motoike I.N. et al.. Regional genetic differences among Japanese populations and performance of genotype imputation using whole-genome reference panel of the Tohoku Medical Megabank Project. BMC Genomics. 2018; 19:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J., Weber J.A., Jho S., Jang J., Jun J., Cho Y.S., Kim H.M., Kim H., Kim Y., Chung O. et al.. KoVariome: Korean National Standard Reference Variome database of whole genomes with comprehensive SNV, indel, CNV, and SV analyses. Sci. Rep. 2018; 8:5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu S., Yin X., Li S., Jin W., Lou H., Yang L., Gong X., Wang H., Shen Y., Pan X. et al.. Genomic dissection of population substructure of Han Chinese and its implication in association studies. Am. J. Hum. Genet. 2009; 85:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gudbjartsson D.F., Helgason H., Gudjonsson S.A., Zink F., Oddson A., Gylfason A., Besenbacher S., Magnusson G., Halldorsson B.V., Hjartarson E. et al.. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015; 47:435–444. [DOI] [PubMed] [Google Scholar]
- 11. Hehir-Kwa J.Y., Marschall T., Kloosterman W.P., Francioli L.C., Baaijens J.A., Dijkstra L.J., Abdellaoui A., Koval V., Thung D.T., Wardenaar R. et al.. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun. 2016; 7:12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. MacArthur J., Bowler E., Cerezo M., Gil L., Hall P., Hastings E., Junkins H., McMahon A., Milano A., Morales J. et al.. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017; 45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maretty L., Jensen J.M., Petersen B., Sibbesen J.A., Liu S., Villesen P., Skov L., Belling K., Theil H.C., Jmg I.. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature. 2017; 548:87. [DOI] [PubMed] [Google Scholar]
- 14. McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K. et al.. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016; 48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bergström A., McCarthy S.A., Hui R., Almarri M.A., Ayub Q., Danecek P., Chen Y., Felkel S., Hallast P., Kamm J. et al.. Insights into human genetic variation and population history from 929 diverse genomes. 2019; 27 June 2019, preprint: not peer reviewed 10.1101/674986. [DOI] [PMC free article] [PubMed]
- 16. Lu J., Lou H., Fu R., Lu D., Zhang F., Wu Z., Zhang X., Li C., Fang B., Pu F. et al.. Assessing genome-wide copy number variation in the Han Chinese population. J. Med. Genet. 2017; 54:685–692. [DOI] [PubMed] [Google Scholar]
- 17. Lan T., Lin H., Zhu W., Laurent T., Yang M., Liu X., Wang J., Wang J., Yang H., Xu X. et al.. Deep whole-genome sequencing of 90 Han Chinese genomes. Gigascience. 2017; 6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai N., Bigdeli T.B., Kretzschmar W.W., Li Y., Liang J., Hu J., Peterson R.E., Bacanu S., Webb B.T., Riley B. et al.. 11,670 whole-genome sequences representative of the Han Chinese population from the CONVERGE project. Sci. Data. 2017; 4:170011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A. et al.. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016; 538:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu D., Lou H., Yuan K., Wang X., Wang Y., Zhang C., Lu Y., Yang X., Deng L., Zhou Y. et al.. Ancestral origins and genetic history of Tibetan highlanders. Am. J. Hum. Genet. 2016; 99:580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sung W.K., Zheng H., Li S., Chen R., Liu X., Li Y., Lee N.P., Lee W.H., Ariyaratne P.N., Tennakoon C. et al.. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012; 44:765–769. [DOI] [PubMed] [Google Scholar]
- 23. Zhang C., Lu Y., Feng Q., Wang X., Lou H., Liu J., Ning Z., Yuan K., Wang Y., Zhou Y. et al.. Differentiated demographic histories and local adaptations between Sherpas and Tibetans. Genome Biol. 2017; 18:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu D., Dou J., Chai X., Bellis C., Wilm A., Shih C.C., Soon W.W.J., Bertin N., Khor C.C., DeGiorgio M. et al.. Large-scale whole-genome sequencing of three diverse Asian populations in Singapore. 2018; 10.1101/390070, 11 August 2018, preprint: not peer reviewed. [DOI] [PubMed]
- 25. Lin J.C., Fan C.T., Liao C.C., Chen Y.S.. Taiwan Biobank: making cross-database convergence possible in the Big Data era. Gigascience. 2018; 7:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu S., Huang S., Chen F., Zhao L., Yuan Y., Francis S.S., Fang L., Li Z., Lin L., Liu R. et al.. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral Infections, and Chinese population history. Cell. 2018; 175:347–359. [DOI] [PubMed] [Google Scholar]
- 27. Zhang C., Gao Y., Liu J., Xue Z., Lu Y., Deng L., Tian L., Feng Q., Xu S.. PGG.Population: a database for understanding the genomic diversity and genetic ancestry of human populations. Nucleic Acids Res. 2018; 46:D984–D993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weir B.S., Cockerham C.C.. Estimating F-statistics for the analysis of population structure. Evolution. 1984; 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- 29. Abraham G., Qiu Y., Inouye M.. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017; 33:2776–2778. [DOI] [PubMed] [Google Scholar]
- 30. Alexander D.H., Novembre J., Lange K.. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009; 19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchini J., Cardon L.R., Phillips M.S., Donnelly P.. The effects of human population structure on large genetic association studies. Nat. Genet. 2004; 36:512–517. [DOI] [PubMed] [Google Scholar]
- 32. Mitt M., Kals M., Parn K., Gabriel S.B., Lander E.S., Palotie A., Ripatti S., Morris A.P., Metspalu A., Esko T. et al.. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 2017; 25:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J., Zheng H., Bei J.X., Sun L., Jia W.H., Li T., Zhang F., Seielstad M., Zeng Y.X., Zhang X. et al.. Genetic structure of the Han Chinese population revealed by genome-wide SNP variation. Am. J. Hum. Genet. 2009; 85:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qin P., Li Z., Jin W., Lu D., Lou H., Shen J., Jin L., Shi Y., Xu S.. A panel of ancestry informative markers to estimate and correct potential effects of population stratification in Han Chinese. Eur. J. Hum. Genet. 2014; 22:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haft D.H., DiCuccio M., Badretdin A., Brover V., Chetvernin V., O’Neill K., Li W., Chitsaz F., Derbyshire M.K., Gonzales N.R. et al.. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018; 46:D851–D860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weissensteiner H., Pacher D., Kloss-Brandstatter A., Forer L., Specht G., Bandelt H.J., Kronenberg F., Salas A., Schonherr S.. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016; 44:W58–W63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Oven M. PhyloTree Build 17: growing the human mitochondrial DNA tree. Forensic Sci. Int.: Genet. Supp. Ser. 2015; 5:e392–e394. [Google Scholar]
- 39. Das S., Forer L., Schonherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M. et al.. Next-generation genotype imputation service and methods. Nat. Genet. 2016; 48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Delaneau O., Zagury J.-F., Robinson M., Marchini J., Dermitzakis E.. Integrative haplotype estimation with sub-linear complexity. 2018; 10.1101/493403, 13 December 2018, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 41. Browning B.L., Zhou Y., Browning S.R.. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018; 103:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Durbin R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics. 2014; 30:1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Howie B.N., Donnelly P., Marchini J.. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLos Genet. 2009; 5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J.. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015; 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.