


Abstract
The extracellular matrix (ECM) is a complex and dynamic meshwork of cross-linked proteins that supports cell polarization and functions and tissue organization and homeostasis. Over the past few decades, mass-spectrometry-based proteomics has emerged as the method of choice to characterize the composition of the ECM of normal and diseased tissues. Here, we present a new release of MatrisomeDB, a searchable collection of curated proteomic data from 17 studies on the ECM of 15 different normal tissue types, six cancer types (different grades of breast cancers, colorectal cancer, melanoma, and insulinoma) and other diseases including vascular defects and lung and liver fibroses. MatrisomeDB (http://www.pepchem.org/matrisomedb) was built by retrieving raw mass spectrometry data files and reprocessing them using the same search parameters and criteria to allow for a more direct comparison between the different studies. The present release of MatrisomeDB includes 847 human and 791 mouse ECM proteoforms and over 350 000 human and 600 000 mouse ECM-derived peptide-to-spectrum matches. For each query, a hierarchically-clustered tissue distribution map, a peptide coverage map, and a list of post-translational modifications identified, are generated. MatrisomeDB is the most complete collection of ECM proteomic data to date and allows the building of a comprehensive ECM atlas.
INTRODUCTION
The extracellular matrix (ECM), a complex and dynamic meshwork of cross-linked proteins, is a fundamental component of multicellular organisms (1). The ECM provides architectural, mechanical, and biochemical signals, interpreted by cell-surface receptors and controlling cellular processes fundamental to development and homeostasis such as adhesion, migration, survival and differentiation (2,3). Alterations in the composition and organization of the ECM cause or accompany the development of diseases such as fibrosis, cardio-vascular and musculoskeletal diseases, and cancers (4–6).
Because of its complexity and insolubility, the ECM has been difficult to analyze biochemically and, until recently, we did not have a detailed understanding of its composition and changes therein. We and others have contributed to pioneer the development of proteomic and computational approaches to characterize the ECM composition—or matrisome—of normal and diseased tissues (7–13). Using an in-silico screen, we previously defined the matrisome as the ensemble of genes encoding core ECM proteins (including glycoproteins, collagens, and proteoglycans) and ECM-associated proteins (including ECM-affiliated proteins, ECM regulators, and secreted factors known or suspected to bind core ECM proteins) (7,10). The in-silico definition of the matrisome has offered a powerful way to annotate ECM genes and proteins in large datasets, including proteomic datasets. Indeed, over the past few years, mass spectrometry has emerged as the method of choice to characterize experimentally the ECM composition of tissues (14–16) and has been shown to offer novel translational avenues from biomarker to novel therapeutic target discovery (17–20). We and others have shown that any given tissue comprises well over 150 ECM and ECM-associated proteins and that there are characteristic differences in the ECM composition of different tissues (14,15). In 2016, we released a first version of MatrisomeDB (14), a database integrating experimental data on the proteomic characterization of the ECM of eight normal tissues and three tumor types from five studies (7,21–24). Since our original publications, the scientific community has adopted and built upon our methods and tools and the number of publications reporting matrisome analyses has significantly increased. This prompted us to develop an updated version of MatrisomeDB presented here, that features added functionalities and integrates data from 17 published studies.
DATABASE INFRASTRUCTURE
Data sets included
We curated the recent ECM proteomics literature (15) and identified 17 studies reporting the characterization of the ECM protein composition of normal and diseased tissues for which the underlying mass spectrometry data were publicly available (7,9,21–35) (Table 1 and Supplementary Table S1). Altogether, these studies include datasets on the ECM of 15 tissue types including normal murine or human tissues, six cancer types (different grades or stages of primary breast, colorectal and lung cancers, melanomas, or insulinomas, and metastases), other diseases, including vascular defects such as carotid plaques and varicose veins, and lung and liver fibroses (Table 1 and Supplementary Table S1, columns G-I). We have also integrated data on the ECM made by cells in culture (29,34), which is of increased interest to the bioengineering community for the purposes of designing more relevant in-vitro cell culture systems and of tissue regeneration (36,37). The deposition of proteomic data to public repositories, such as MassIVE, PRIDE (38) or any members of the ProteomeXchange consortium (39), is now strongly encouraged if not required. We were thus able to retrieve and curate over 2000 raw files deposited to public repositories or personally shared with us (Supplementary Table S1, columns A–C). Of note, all 17 studies were selected because they specifically focused on the analysis of the ECM compartment of the tissues studied and thus, all include a step aimed at enriching ECM proteins through decellularization (15). However, these studies vary in their experimental design, some having been performed using label-free and other label-based quantitative proteomics (reviewed in 15), some having been performed with various levels of protein and peptide fractionations, and all having been acquired on different instruments. Despite this heterogeneity, the compilation of the data in MatrisomeDB represents, to date, the largest aggregation of ECM proteomic datasets and, we believe, will be valuable to the scientific community.
Table 1.
List of datasets included in MatrisomeDB (adapted from (15))
| Tissue type | Dataset identifier | Reference |
|---|---|---|
| Human triple-negative breast cancer samples and matched adjacent mammary gland samples; Human normal omental samples from patients with non-metastatic ovarian cancer and high-grade-serous-ovarian-cancer-derived omental metastasis samples | PXD005554 | Naba et al. (9) |
| Human normal colon and normal liver; primary colorectal tumors and liver metastases | MSV000078555 | Naba et al. (22) |
| Human kidney glomeruli | PXD000456 | Lennon et al. (23) |
| Human eye: retinal blood vessel, inner limiting membrane, and lens capsule basement membranes | PXD001025 | Uechi et al. (24) |
| Human normal liver and fibrotic and cirrhotic livers from hepatitis C (HCV) patients | Personal communication | Baiocchini et al. (25) |
| Human normal and varicose saphenous veins | PXD002555 | Barallobre-Barreiro et al. (26) |
| Human symptomatic and asymptomatic atherosclerotic plaque samples | PXD005130 | Langley et al. (29) |
| Human patent and craniosynostotic cranial sutures | PXD003215 | Lyon et al. (30) |
| Human prostate and ECM produced by normal and tumor-associated prostate fibroblasts | PXD006562; PXD006563 | Ojalill et al. (34) |
| Primary lung adenocarcinoma and lymph node metastases from KrasG12D; p53−/− mice | PXD003517 | Gocheva et al. (27) |
| Murine liver fibrosis induced by carbon tetrachloride (CCl4) or diethoxycarbonyl dihydrocollidine (DCC) | Personal communication | Klaas et al. (28) |
| Murine liver fibrosis induced by carbon tetrachloride (CCl4); livery injury induced by ethanol or LPS | PXD006521 | Massey et al. (31) |
| Murine normal mammary gland, lung, and lymph node tissues; primary mammary tumors (4T1 cells) and derived lung and lymph node metastases | PXD006579 | Mayorca-Guiliani et al. (32) |
| Murine pancreatic islets and insulinomas from RIP1-Tag2 mice | MSV000080124 | Naba et al. (33) |
| Murine normal lung and bleomycin-induced lung fibrosis | PXD001765 | Schiller et al. (35) |
| Xenografts: Poorly (A375 cells) and highly (MA2 cells) metastatic human melanomas grown in mice | MSV000078494 | Naba et al. (7) |
| Xenografts: Poorly (MDA-MB-231 cells) and highly (MDA-MB-231_LM2 cells) metastatic human mammary tumors grown in mice | MSV000078535 | Naba et al. (21) |
Mass spectrometry data search and processing
All raw data files were converted and searched using uniform parameters and against the same and most recent reference proteome database (UniProt 04/17/2019) (40). The reference databases used contain 95 943 human proteoforms and 62 407 mouse proteoforms. 572 common contaminating proteins were also appended to the reference database. The raw data were searched by the ProluCID search engine (41) using an open-database search strategy (42). Specifically, here, we allowed for each study a set of corresponding fixed post-translational modifications (PTMs), e.g. carbamidomethylation, TMT labeling, iTRAQ labeling, and performed an open database search (42) that includes acetylation, methylation, deamidation. In addition, we included a set of PTMs of structural and functional importance in ECM proteins (43,44), including serine, threonine and tyrosine phosphorylation (45,46), proline hydroxylation (47,48), and citrullination (49,50) (Figure 2B). High-mass-tolerant open database search with +/- 500 Da precursor tolerance allowed us to not only identify the abovementioned PTMs but also unknown or unsuspected PTMs which are reported as mass shift such as (+28.99). The search results were then filtered with 1% protein false discovery rate (FDR) using DTASelect (51). Importantly, by using the same reference databases, uniform search parameters, and FDR-filter level, search results can now be compared among different studies, tissues, and disease states. The search results were further annotated with corresponding original data source and repository location (Supplementary Table S1, columns A-E), tissue classification, disease state, and sample type (Supplementary Table S1, columns F-I), matrisome classification (7,10), and confidence score (see below). Last, the data were deposited into a MySQL database and can be searched directly from our web interface. Of note, our pipeline allows us to update the protein reference databases as new versions are released as well as include new datasets as they become available (see Future directions).
Figure 2.

Peptide coverage map. (A) Peptide coverage map of Nidogen 1 in the glomerular basement membrane dataset. The color code indicates the peptide-spectrum match frequency from high (dark blue) to low (light blue). (B) Peptide coverage map of Nidogen 1 across all the datasets (11 tissues and 31 sample types) in which Nidogen 1 was detected. The color code indicates the peptide-spectrum match frequency from high (dark blue) to low (light blue).
DATABASE FEATURES AND FUNCTIONALITIES
MatrisomeDB query
To better assist the community with the use of the database, we have developed an intuitive interface available at http://www.pepchem.org/matrisomedb, which allows users to search, visualize and analyze the data. Search can be performed by inputting a gene name or a string of gene names separated by commas directly in the search box (see online Tutorial). Users can also select from different option boxes, specific matrisome categories, a specific organism, and/or one or several tissues, and retrieve all matrisome proteins meeting the selected search parameters. Search results are displayed in a table with the following columns: (i) gene symbol, the clickable link takes users to the GeneCards page (52) of that entry; (ii) UniProt identifier, the clickable link takes users to the UniProt page (40) of that entry; (iii) protein description, the clickable link displays peptide coverage maps (see below); (iv) a description of the samples and tissues in which the entry was detected as well as a reference to the primary research publications reporting the identification. The result table can be exported as .csv file (Figure 1, orange arrow). To facilitate the interrogation of MatrisomeDB, we provide a step-by-step tutorial accessible from the home page of MatrisomeDB. This tutorial will guide users to exploit MatrisomeDB to advance their research.
Figure 1.

Examples of database query result showing hierarchically-clustered protein distribution heatmaps. (A) Confidence-score-based heatmap generated upon querying ‘COL4’ and selecting ‘HUMAN’ in the Species option box. Results show the tissue distribution of the 6 collagen-IV protein chains encoded by the COL4A1, COL4A2, COL4A3, COL4A4, COL4A5 and COL4A6 genes. The color code indicates the confidence score from high (dark blue) to low (light yellow). Clicking on the heatmap itself or on the link located above the heatmap (red arrow) will open a detailed heatmap and a link to download the data in .csv format. The ‘Export result as CSV’ button (orange arrow) allows users to download the complete results. (B) Total-Ion-Current-based heatmap generated upon querying ‘COL4’ and selecting ‘HUMAN’ in the Species option box, and accessible from the detailed heatmap page. The color code indicates the confidence score from high (dark blue) to low (light yellow).
Hierarchically-clustered protein distribution heatmap
Query of MatrisomeDB generates a clustered heatmap of the tissue distribution of all the proteins retrieved from each database query (Figure 1) based on a confidence score calculated as described below. The proteins and tissues are clustered using unbiased hierarchical clustering based on the Euclidean distance. Each tissue annotation is color-coded based on the organ system it is a part of (Supplementary Table S1, columns G and H). The colors of the heatmap are based on a confidence score of all peptide-to-spectrum matches (PSMs), defined as:
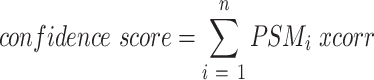 |
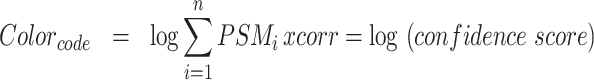 |
From this clustered heatmap, users can visualize the distribution of a given protein or group of proteins across tissues and across studies (see examples Figure 1). Clicking on the heatmap will take users to a detailed view of the heatmap from where they can download the values used to build the heatmap as a .csv file. On this detailed view page, users can also access a second heatmap built using the total ion current (TIC), calculated as the sum of raw MS1 signal intensities of all identified peptides corresponding to a given protein. In order to build the TIC-based heatmap, natural logarithms of TIC were summed and clustered by tissue and protein name. Note that whereas the confidence score can be used to compare proteins detected across different studies, we do not recommend the use of the TIC values to do so, since they are dependent on the instrument on which data were acquired and the quality of the samples, and this varies from study to study.
Peptide coverage map
A peptide coverage map can be viewed by clicking on the ‘protein description’ returned by the search engine. It is provided for each identified protein isoforms and shows the sequence and percent coverage for each sample (Figure 2A) as well as across all tissues (Figure 2B). The colors of the highlighted part of the sequence corresponds to the total number of identified PSMs, which can be used in the future to design selected reaction monitoring (SRM) experiments (53–55). The map also highlights the following post-translational modifications detected experimentally: lysine and proline hydroxylations; phosphorylation of serine, threonine and citrullination (Figure 2), which play significant roles in the proper folding and function of ECM proteins (see Introduction).
RESULTS
MatrisomeDB includes 847 human and 791 mouse matrisome proteoforms and 368 877 human and 638 221 mouse matrisome-protein-derived peptide-to-spectrum matches, detected across 15 different normal murine or human tissues, six cancer types including different grades or stages of primary breast, colorectal and lung cancers, melanomas, and insulinomas and metastases, normal and diseased vascular tissues such as carotid plaques and varicose veins, and human and murine samples of lung and liver fibroses.
Experimental coverage of the in-silico predicted matrisome
Using characteristic features of ECM proteins, we previously predicted computationally the matrisome, e.g. the ensemble of 1000+ genes encoding core structural ECM proteins and ECM-associated proteins (7,10). In the initial version of MatrisomeDB, we reported the experimental identification of ∼73% of the core matrisome components (including 100% of the collagens) and 45% of the matrisome-associated components (14). Analysis of the data aggregated in the new release of MatrisomeDB revealed a significantly higher percentage coverage of the in-silico predicted matrisome. Namely, 86% of the core matrisome components and 58% of the matrisome-associated components have now been detected experimentally by ECM-focused proteomics strategies (Figure 3A). This increase benefited all matrisome categories, since the data included in the database permitted the identification of 161 of the 195 predicted ECM glycoproteins, all 44 collagens, 30 of the 35 predicted proteoglycans, 101 of the 171 predicted ECM-affiliated proteins, 173 or the 238 predicted ECM regulators, and 160 of the 344 predicted secreted factors (Figure 3A). This increase can be attributed in part to the larger number of tissues included in the database, since, certain ECM components are known to present a tissue-specific expression pattern. It can also be attributed to the fact that protein samples of different solubility were included in this new release of the database (Supplementary Table S1), which could explain the 23% increase in the detection of secreted factors which are more soluble in nature and smaller in size, so generate fewer peptides.
Figure 3.

Building an ECM Atlas. (A) Experimental coverage of the in-silico predicted matrisome. Bar chart represents, for each matrisome category (x axis), the percentage of the in-silico predicted genes encoding proteins detected in the proteomic studies included in MatrisomeDB (y axis). The actual number of proteins detected is indicated inside the bars. The percentages indicated above the bar chart indicate the increase in coverage with the updated database. (B) Hierarchically-clustered tissue distribution heatmap of all 30 proteoglycans detected and reported in MatrisomeDB. The color code indicates the confidence score from high (dark blue) to low (light yellow).
Building an ECM atlas
The interrogation of MatrisomeDB by selecting an entire matrisome category, for example the proteoglycans, results in the generation of a hierarchically-clustered tissue distribution heatmap of all 30 proteoglycans detected and thus can constitute the Proteoglycan atlas (Figure 3B). Similarly, the ECM atlas of any of the tissues or any of the organisms included in this database can be generated.
FUTURE DIRECTIONS
Future data extension
When we designed MatrisomeDB, we specifically made it expandable to easily allow the inclusion of new datasets. The entire processing pipeline, including reference databases and all required software, are encapsulated in a Docker container. This ensures that, in the future, as we add new datasets, we can process them with the same pipeline and get comparable results. This also ensures maximum data reproducibility. With each future upgrade of MatrisomeDB, we will construct new Docker containers and re-process all the data to ensure data integrity. Researchers interested in submitting their datasets for consideration for inclusion in an upcoming release of MatrisomeDB can do so via the ‘Submit your data’ tab at the top of MatrisomeDB’s home page.
Extension to include studies from other model organisms
Over the past couple of years, we and others have defined the matrisome of model organisms broadly used to study ECM-related mechanisms and diseases, namely the zebrafish (56), C. elegans (57), Drosophila (58) and planarians (59). Mass-spectrometry-based proteomics is also starting to emerge as a powerful method to study the ECM of these organisms and we can foresee including in MatrisomeDB such datasets in the future.
Extracting quantitative data from label-free and label-based proteomics studies
Label-free proteomics has traditionally used total intensity and spectral counts to quantify the relative abundance of proteins. In the future, we propose to include in the database the summed precursor-ion chromatographic peak area of all peptides contributing to a given protein. However, this poses the question of data normalization since the datasets were generated on different instruments, which will need to be resolved before implementation. Label-based quantitative proteomics is also now being employed to study in more details the abundance of ECM proteins tissues (reviewed in 12). Although, we included here two datasets generated using TMT- or iTRAQ-based proteomics (27,33), we did not fully exploit the sample-specific quantitative information of the multiplexed data beyond deriving a confidence score for the PSMs of these two studies. In the future, we will aim to deconvolute multiplexed label-based ECM proteomic studies. One way we will be able to do so is to exploit both MS1 and MS2 data and split the MS1 precursor ion abundance (combined from samples mixed) in proportion to the MS2 reporter ion abundance (individual for each sample).
Citing MatrisomeDB
For a general citation of MatrisomeDB, researchers should cite this article. In addition, the following citation format is suggested when referring to specific data obtained from MatrisomeDB: http://www.pepchem.org/matrisomedb.
Supplementary Material
ACKNOWLEDGEMENTS
AN would like to thank Richard Hynes (MIT) and Steve Carr (Broad Institute) for their support and mentorship over the years.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Physiology and Biophysics at the University of Illinois at Chicago (to A.N.); College of Pharmacy (to Y.G.); IT is the recipient of a Research Grant from the Honors College at the University of Illinois at Chicago; A.N. acknowledges the Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support towards the open access publishing fee for this article. Funding for open access charge: Department of Physiology and Biophysics at the University of Illinois at Chicago to Dr Naba and Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hynes R.O., Yamada K.M.. Extracellular matrix biology. Cold Spring Harbor Perspectives in Biology. 2012; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 2. Dzamba B.J., DeSimone D.W.. Extracellular matrix (ECM) and the sculpting of embryonic tissues. Curr. Top. Dev. Biol. 2018; 130:245–274. [DOI] [PubMed] [Google Scholar]
- 3. Rozario T., DeSimone D.W.. The extracellular matrix in development and morphogenesis: a dynamic view. Dev. Biol. 2010; 341:126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonnans C., Chou J., Werb Z.. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014; 15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karamanos N.K., Theocharis A.D., Neill T., Iozzo R.V.. Matrix modeling and remodeling: a biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019; 75–76:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iozzo R.V., Gubbiotti M.A.. Extracellular matrix: the driving force of mammalian diseases. Matrix Biol. 2018; 71–72:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A., Hynes R.O.. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteomics. 2012; 11:M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naba A., Clauser K.R., Hynes R.O.. Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. J. Vis. Exp. 2015; 101:e53057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naba A., Pearce O.M.T., Del Rosario A., Ma D., Ding H., Rajeeve V., Cutillas P.R., Balkwill F.R., Hynes R.O.. Characterization of the extracellular matrix of normal and diseased tissues using proteomics. J. Proteome Res. 2017; 16:3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naba A., Hoersch S., Hynes R.O.. Towards definition of an ECM parts list: an advance on GO categories. Matrix Biol. 2012; 31:371–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Didangelos A., Yin X., Mandal K., Baumert M., Jahangiri M., Mayr M.. Proteomics characterization of extracellular space components in the human aorta. Mol. Cell Proteomics. 2010; 9:2048–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hansen K.C., Kiemele L., Maller O., O’Brien J., Shankar A., Fornetti J., Schedin P.. An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage. Mol. Cell Proteomics. 2009; 8:1648–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilson R., Diseberg A.F., Gordon L., Zivkovic S., Tatarczuch L., Mackie E.J., Gorman J.J., Bateman J.F.. Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label-free quantitative proteomics. Mol. Cell Proteomics. 2010; 9:1296–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naba A., Clauser K.R., Ding H., Whittaker C.A., Carr S.A., Hynes R.O.. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016; 49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taha I.N., Naba A.. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019; 63:417–432. [DOI] [PubMed] [Google Scholar]
- 16. Randles M.J., Humphries M.J., Lennon R.. Proteomic definitions of basement membrane composition in health and disease. Matrix Biol. 2017; 57–58:12–28. [DOI] [PubMed] [Google Scholar]
- 17. Barallobre-Barreiro J., Lynch M., Yin X., Mayr M.. Systems biology-opportunities and challenges: the application of proteomics to study the cardiovascular extracellular matrix. Cardiovasc. Res. 2016; 112:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lindsey M.L., Jung M., Hall M.E., DeLeon-Pennell K.Y.. Proteomic analysis of the cardiac extracellular matrix: clinical research applications. Expert Rev. Proteomics. 2018; 15:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Socovich A.M., Naba A.. The cancer matrisome: from comprehensive characterization to biomarker discovery. Semin. Cell Dev. Biol. 2019; 89:157–166. [DOI] [PubMed] [Google Scholar]
- 20. Hsueh M.-F., Önnerfjord P., Kraus V.B.. Biomarkers and proteomic analysis of osteoarthritis. Matrix Biol. 2014; 39:56–66. [DOI] [PubMed] [Google Scholar]
- 21. Naba A., Clauser K.R., Lamar J.M., Carr S.A., Hynes R.O.. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife. 2014; 3:e01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naba A., Clauser K.R., Whittaker C.A., Carr S.A., Tanabe K.K., Hynes R.O.. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. 2014; 14:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lennon R., Byron A., Humphries J.D., Randles M.J., Carisey A., Murphy S., Knight D., Brenchley P.E., Zent R., Humphries M.J.. Global analysis reveals the complexity of the human glomerular extracellular matrix. JASN. 2014; 25:939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uechi G., Sun Z., Schreiber E.M., Halfter W., Balasubramani M.. Proteomic view of basement membranes from human retinal blood vessels, inner limiting membranes, and lens capsules. J. Proteome Res. 2014; 13:3693–3705. [DOI] [PubMed] [Google Scholar]
- 25. Baiocchini A., Montaldo C., Conigliaro A., Grimaldi A., Correani V., Mura F., Ciccosanti F., Rotiroti N., Brenna A., Montalbano M. et al.. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS One. 2016; 11:e0151736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barallobre-Barreiro J., Oklu R., Lynch M., Fava M., Baig F., Yin X., Barwari T., Potier D.N., Albadawi H., Jahangiri M. et al.. Extracellular matrix remodelling in response to venous hypertension: proteomics of human varicose veins. Cardiovasc. Res. 2016; 110:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gocheva V., Naba A., Bhutkar A., Guardia T., Miller K.M., Li C.M.-C., Dayton T.L., Sanchez-Rivera F.J., Kim-Kiselak C., Jailkhani N. et al.. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E5625–E5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klaas M., Kangur T., Viil J., Mäemets-Allas K., Minajeva A., Vadi K., Antsov M., Lapidus N., Järvekülg M., Jaks V.. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci. Rep. 2016; 6:27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langley S.R., Willeit K., Didangelos A., Matic L.P., Skroblin P., Barallobre-Barreiro J., Lengquist M., Rungger G., Kapustin A., Kedenko L. et al.. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Invest. 2017; 127:1546–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyon S.M., Mayampurath A., Rogers M.R., Wolfgeher D.J., Fisher S.M., Volchenboum S.L., He T.-C., Reid R.R.. A method for whole protein isolation from human cranial bone. Anal. Biochem. 2016; 515:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Massey V.L., Dolin C.E., Poole L.G., Hudson S.V., Siow D.L., Brock G.N., Merchant M.L., Wilkey D.W., Arteel G.E.. The hepatic ‘matrisome’ responds dynamically to injury: Characterization of transitional changes to the extracellular matrix in mice. Hepatology. 2017; 65:969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mayorca-Guiliani A.E., Madsen C.D., Cox T.R., Horton E.R., Venning F.A., Erler J.T.. ISDoT: in situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 2017; 23:890–898. [DOI] [PubMed] [Google Scholar]
- 33. Naba A., Clauser K.R., Mani D.R., Carr S.A., Hynes R.O.. Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci. Rep. 2017; 7:40495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ojalill M., Rappu P., Siljamäki E., Taimen P., Boström P., Heino J.. The composition of prostate core matrisome in vivo and in vitro unveiled by mass spectrometric analysis. Prostate. 2018; 78:583–594. [DOI] [PubMed] [Google Scholar]
- 35. Schiller H.B., Fernandez I.E., Burgstaller G., Schaab C., Scheltema R.A., Schwarzmayr T., Strom T.M., Eickelberg O., Mann M.. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol. Syst. Biol. 2015; 11:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kyburz K.A., Anseth K.S.. Synthetic mimics of the extracellular matrix: how simple is complex enough. Ann. Biomed. Eng. 2015; 43:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hussey G.S., Dziki J.L., Badylak S.F.. Extracellular matrix-based materials for regenerative medicine. Nat. Rev. Mater. 2018; 3:159. [Google Scholar]
- 38. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S. et al.. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017; 45:D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. The UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu T., Park S.K., Venable J.D., Wohlschlegel J.A., Diedrich J.K., Cociorva D., Lu B., Liao L., Hewel J., Han X. et al.. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics. 2015; 129:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chick J.M., Kolippakkam D., Nusinow D.P., Zhai B., Rad R., Huttlin E.L., Gygi S.P.. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015; 33:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karsdal M.A., Nielsen M.J., Sand J.M., Henriksen K., Genovese F., Bay-Jensen A.-C., Smith V., Adamkewicz J.I., Christiansen C., Leeming D.J.. Extracellular matrix remodeling: the common denominator in connective tissue diseases possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev. Technol. 2013; 11:70–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zeltz C., Gullberg D.. Post-translational modifications of integrin ligands as pathogenic mechanisms in disease. Matrix Biol. 2014; 40:5–9. [DOI] [PubMed] [Google Scholar]
- 45. Yalak G., Olsen B.R.. Proteomic database mining opens up avenues utilizing extracellular protein phosphorylation for novel therapeutic applications. J. Transl. Med. 2015; 13:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yalak G., Shiu J.-Y., Schoen I., Mitsi M., Vogel V.. Phosphorylated fibronectin enhances cell attachment and upregulates mechanical cell functions. PLoS One. 2019; 14:e0218893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rappu P., Salo A.M., Myllyharju J., Heino J.. Role of prolyl hydroxylation in the molecular interactions of collagens. Essays Biochem. 2019; 63:325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gjaltema R.A.F., Bank R.A.. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2017; 52:74–95. [DOI] [PubMed] [Google Scholar]
- 49. Stefanelli V.L., Choudhury S., Hu P., Liu Y., Schwenzer A., Yeh C.-R., Chambers D.M., Pesson K., Li W., Segura T. et al.. Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion. Matrix Biol. 2019; 82:86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sipilä K.H., Ranga V., Rappu P., Torittu A., Pirilä L., Käpylä J., Johnson M.S., Larjava H., Heino J.. Extracellular citrullination inhibits the function of matrix associated TGF-β. Matrix Biol. 2016; 55:77–89. [DOI] [PubMed] [Google Scholar]
- 51. Tabb D.L., McDonald W.H., Yates J.R.. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002; 1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stelzer G., Rosen N., Plaschkes I., Zimmerman S., Twik M., Fishilevich S., Stein T.I., Nudel R., Lieder I., Mazor Y. et al.. The GeneCards Suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016; 54:1.30.1–1.30.33. [DOI] [PubMed] [Google Scholar]
- 53. Lange V., Picotti P., Domon B., Aebersold R.. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2008; 4:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carr S.A., Abbatiello S.E., Ackermann B.L., Borchers C., Domon B., Deutsch E.W., Grant R.P., Hoofnagle A.N., Hüttenhain R., Koomen J.M. et al.. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell Proteomics. 2014; 13:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shi T., Song E., Nie S., Rodland K.D., Liu T., Qian W.-J., Smith R.D.. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016; 16:2160–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nauroy P., Hughes S., Naba A., Ruggiero F.. The in-silico zebrafish matrisome: A new tool to study extracellular matrix gene and protein functions. Matrix Biol. 2018; 65:5–13. [DOI] [PubMed] [Google Scholar]
- 57. Teuscher A.C., Jongsma E., Davis M.N., Statzer C., Gebauer J.M., Naba A., Ewald C.Y.. The in-silico characterization of the Caenorhabditis elegans matrisome and proposal of a novel collagen classification. Matrix Biol. Plus. 2019; 1:100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Davis M.N., Horne-Badovinac S., Naba A.. In-silico definition of the Drosophila melanogaster matrisome. 2019; 10.1101/722868, 02 August 2019, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 59. Cote L.E., Simental E., Reddien P.W.. Muscle functions as a connective tissue and source of extracellular matrix in planarians. Nat. Commun. 2019; 10:1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.