

Abstract
Numerous studies indicate that non-coding RNAs (ncRNAs) have critical functions across biological processes, and single-nucleotide polymorphisms (SNPs) could contribute to diseases or traits through influencing ncRNA expression. However, the associations between SNPs and ncRNA expression are largely unknown. Therefore, genome-wide expression quantitative trait loci (eQTL) analysis to assess the effects of SNPs on ncRNA expression, especially in multiple cancer types, will help to understand how risk alleles contribute toward tumorigenesis and cancer development. Using genotype data and expression profiles of ncRNAs of >8700 samples from The Cancer Genome Atlas (TCGA), we developed a computational pipeline to systematically identify ncRNA-related eQTLs (ncRNA-eQTLs) across 33 cancer types. We identified a total of 6 133 278 and 721 122 eQTL-ncRNA pairs in cis-eQTL and trans-eQTL analyses, respectively. Further survival analyses identified 8312 eQTLs associated with patient survival times. Furthermore, we linked ncRNA-eQTLs to genome-wide association study (GWAS) data and found 262 332 ncRNA-eQTLs overlapping with known disease- and trait-associated loci. Finally, a user-friendly database, ncRNA-eQTL (http://ibi.hzau.edu.cn/ncRNA-eQTL), was developed for free searching, browsing and downloading of all ncRNA-eQTLs. We anticipate that such an integrative and comprehensive resource will improve our understanding of the mechanistic basis of human complex phenotypic variation, especially for ncRNA- and cancer-related studies.
INTRODUCTION
In recent decades, non-coding RNA (ncRNA) has gradually become a research hotspot because of its important roles in a wide range of biological processes. ncRNA can interact with various macromolecules, including DNA, RNA and proteins, and regulate gene expression at transcriptional, post-transcriptional and epigenetic levels (1,2). The number of identified ncRNAs has also sharply increased with the widespread usage of high-throughput technology in different cells and tissues. The latest GENCODE version 29 annotated more than 40 000 ncRNAs in the human genome. Many ncRNAs are important oncogenes or tumour suppressor genes, such as HULC (3) and HOXB-AS3 (4). However, compared with protein-coding genes, the function of most ncRNAs remains to be deciphered.
Single-nucleotide polymorphisms (SNPs), the most common type of human genetic variation, play important roles in human complex traits and diseases (5–7). Genome-wide association studies (GWAS) have found extensive SNPs associated with various traits and diseases. However, most GWAS-detected risk SNPs are located in the genomic non-coding regions (8), which indicates that ncRNAs may be possible causal targets of some GWAS loci (9). For example, rs6983267 on human chromosome 8q24.21 is a potential genetic biomarker of colorectal cancer predisposition and is located far from protein-coding genes. Recent studies demonstrated that rs6983267 may exert its role through influencing the expression of lncRNA CCAT2 (10,11). Therefore, investigation of the effects of SNPs on ncRNA expression will help to understand how risk alleles contribute towards tumorigenesis and cancer development.
Expression quantitative trait locus (eQTL) analysis links variations in gene expression to genotypes and has been considered a powerful tool to understand the effects and molecular mechanism of functional SNPs (12–15). However, most eQTL analyses are focused on the associations between genotypes and protein-coding genes; only a few ncRNA-related eQTL (ncRNA-eQTL) analyses have been performed at the genome-wide level (16). In recent years, although some ncRNA-related SNP databases such as LincSNP 2.0 (17), MSDD (18) and lncRNASNP2 (19) have been developed to explore the relationship of ncRNAs and SNPs, no database has been developed to specifically and comprehensively quantify the association between SNP and ncRNA expression. The Cancer Genome Atlas (TCGA) consortium (20) has generated DNA germline genotype datasets, transcriptome profiling and patient survival data for over 10 000 primary tumours in 33 cancer types. Using these valuable datasets, we previously performed eQTL analyses for all cancer types and developed the pancanQTL online database (21), which provides eQTLs of 20 531 genes. However, most of the genes in pancanQTL are protein-coding genes. Recently, the TCGA database has updated its gene expression profiles and provided expression profiles of more than 40 000 lncRNAs. In addition, the TCGA has also provided microRNA (miRNA) expression profiles. These data enable us to systematically analyse associations between SNPs and the gene expression of ncRNAs (including lncRNAs and miRNAs). Thus, in this study, using TCGA genotype data and the latest expression profiles, we developed a computational pipeline to systematically identify ncRNA-eQTLs across 33 cancer types. In addition, we linked ncRNA-eQTLs to known GWAS loci and patient survival times and identified thousands of GWAS-related eQTLs and survival-related eQTLs. Finally, we constructed a user-friendly database, ncRNA-eQTL (http://ibi.hzau.edu.cn/ncRNA-eQTL), for users to browse and download data.
DATA COLLECTION AND PROCESSING
Collection and processing of genotype data
Genotype data of 33 cancer types (full names of cancer types are shown in Supplementary Table S1) of each individual were obtained from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/), which were called by the Affymetrix SNP 6.0 array platform and included 898 620 SNPs (Figure 1A). To increase the power for eQTL discovery, we imputed autosomal variants for all samples using IMPUTE2 (22) in each cancer type, with 1000 Genomes Phase 3 as the reference panel (23). A two-step imputation procedure was used to improve the computation efficiency. TCGA genotype data were first pre-phased to certain haplotypes and then imputed from the reference panel into the estimated study haplotypes. After imputation, the following criteria (24) were employed to remove SNPs: (i) imputation confidence score, INFO < 0.4, (ii) minor allele frequency (MAF) < 5%, (iii) SNP missing rate > 5% for best-guessed genotypes at posterior probability ≥ 0.9 and (iv) Hardy–Weinberg equilibrium P-value < 1 × 10−6 estimated by Hardy–Weinberg R package (25) (Figure 1B).
Figure 1.

Flowchart of the ncRNA-eQTL database. (A) Data collection of sequencing, genotype and GWAS data. (B) Processing of sequencing and gene expression data. (C) Database content of four types of eQTLs (cis-eQTLs, trans-eQTLs, survival-eQTLs, GWAS-eQTLs).
Expression data processing and covariates
Gene expression profiles generated from RNA sequencing (RNA-seq) and small RNA sequencing (smRNA-seq) for each sample were obtained from the TCGA data portal (https://gdc-portal.nci.nih.gov/). The gene annotation, which was used for TCGA RNA-seq and miRNA-seq annotation, was downloaded from the GENCODE (version 22) website (https://www.gencodegenes.org/) and miRBase (http://www.mirbase.org/). According to the annotation, we removed all the protein-coding genes from the profiles. In each cancer type, lncRNAs with an average expression of ≥ 0.01 FPKM and miRNAs with an average expression of ≥0.01 TPM were retained. To minimize the effect of outliers on the regression scores, the expression values for each gene across all samples were transformed into a standard normal based on rank (24) (Figure 1B).
Global expression data may be influenced by several factors, such as batch effects (26) and genetic and non-genetic biases (27). Thus, covariates are often included to correct known and unknown confounders and increase the sensitivity of eQTL analyses (24). To adjust the global effect of population structure, we first used smartpca in the EIGENSOFT program (28) to perform principal component analysis (PCA) for each cancer type. The top five principal components in the genotype data were selected as covariates. To eliminate the possible batch effects and other confusions hidden in the expression data, we used PEER software (29) to select the first 15 PEER factors from the expression data as covariates. Other factors, such as tumour stage (30), gender (24) and age (12), were also counted as additional covariates (Figure 1A).
Identification of cis- and Trans-eQTL using Matrix eQTL
Cis-eQTL and trans-eQTL analyses were performed in our study. The cis-eQTLs were defined if the SNP was within 1 Mb from the gene transcriptional start site (TSS) and regulating the corresponding gene expression (24), and trans-eQTLs were defined if the eQTL was beyond that region or on another chromosome. To perform cis-eQTL analyses and trans-eQTL analyses, we first downloaded SNP annotations (GRCh38) (https://www.ncbi.nlm.nih.gov/projects/SNP) from the dbSNP database. We analysed the associations between each ncRNA and autosomal SNP through linear regression by employing a computationally efficient eQTL analysis called Matrix eQTL (31), controlling for population bias, sex, age, tumour stage and unobserved factors in the expression data for each cancer type (29,31). We defined eQTLs as SNPs with false discovery rates (FDRs) calculated by MatrixEQTL < 0.05 (Figure 1C).
Identification of GWAS-associated eQTLs
Overlaps between ncRNA-eQTLs and SNPs in GWAS regions were identified to explore the possible target genes of existing GWAS loci. To achieve that, we first downloaded all the known risk tag SNPs identified by GWAS from the National Human Genome Research Institute (NHGRI) GWAS Catalog (32) (http://www.ebi.ac.uk/gwas, accessed September 2018), a collection of data from GWAS for various human diseases and traits. Then, we obtained the SNPs in linkage disequilibrium (LD) with these tag SNPs from SNAP (33) (https://personal.broadinstitute.org/plin/snap/ldsearch.php). The parameters were set as follows: SNP dataset: 1000 Genomes; r2 (the square of the Pearson correlation coefficient of linkage disequilibrium) threshold: 0.5; population panel: CEU (Utah residents with northern and western European ancestry); distance limit: 500 kb. Finally, these GWAS tag SNPs and LD SNPs were mapped to ncRNA-eQTL results.
Identification of survival-associated eQTLs
To prioritize promising ncRNA-eQTLs, we identified ncRNA-eQTLs that may be associated with patient survival times. The clinical data, including patient overall survival times, were downloaded from the TCGA data portal. For each ncRNA-eQTL, we divided the samples into three different groups: AA (homozygous genotype), Aa (heterozygous genotype) and aa (homozygous genotype), and removed the group with samples fewer than three for reliable survival analysis and Kaplan–Meier (KM) P-value calculation. Then, the differences in survival times between the groups were detected by log-rank test, and KM curves were introduced to visualize the survival time difference of each group. In addition, survival-associated eQTLs were confirmed for ncRNA-eQTLs that fulfilled the condition of FDR < 0.05.
Database construction
We organized all results in MySQL (version 5.6) relation tables and constructed a web interface using HTML, CSS and PHP (version 5.4) running on an Apache web server (version 2.4.6). A highly flexible and editable plug-in for the jQuery JavaScript library called DataTables (https://datatables.net/) was integrated to display the data content in a dynamic way.
DATABASE CONTENT AND THE WEB INTERFACE
Database content and statistics of lncRNA-related eQTLs
Using genotype data and RNA-seq data, we first performed lncRNA-related cis-eQTL and trans-eQTL association analyses in each of the 33 cancer types independently. The sample sizes of cancer types ranged from 36 in cholangiocarcinoma (CHOL) to 1067 in breast invasive carcinoma (BRCA), with a median of 177 (Table 1). After genotype imputation, the incorporated SNPs ranged from 2 745 615 in BRCA to 5 078 753 in acute myeloid leukaemia (LAML), with a median of 4 525 414. There were 40 670 ncRNAs in the expression profiles of each cancer type. We filtered the ncRNAs with expression >0.01 FPKM, resulting in a median of 12 554 ncRNAs included in the analyses. All SNPs within the 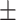 1 Mb region of the TSS of each gene were tested in cis-eQTL analyses, while others were used for trans-eQTL analyses.
1 Mb region of the TSS of each gene were tested in cis-eQTL analyses, while others were used for trans-eQTL analyses.
Table 1.
The summary of samples and lncRNA related eQTLs in this study
| Cis | Trans | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cancer type | No. of samples | No. of ncRNAs | No. of genotypes | Pairs | ncRNAs | eQTLs | Pairs | ncRNAs | eQTLs |
| ACC | 77 | 10 673 | 3 567 953 | 6906 | 229 | 6547 | 1030 | 49 | 934 |
| BLCA | 403 | 12 090 | 4 191 159 | 205 824 | 4077 | 156 228 | 29 840 | 935 | 23 714 |
| BRCA | 1067 | 13 170 | 2 745 615 | 498 969 | 8124 | 308 016 | 62 764 | 2 328 | 46 137 |
| CESC | 242 | 12 410 | 4 276 554 | 101 968 | 2626 | 82 242 | 20 779 | 735 | 17 555 |
| CHOL | 36 | 12 217 | 4 012 151 | 0 | 0 | 0 | 0 | 0 | 0 |
| COAD | 282 | 11 063 | 4 505 758 | 169 256 | 3558 | 129 931 | 25 618 | 859 | 20 423 |
| DLBC | 47 | 11 447 | 4 819 767 | 122 | 8 | 121 | 82 | 4 | 82 |
| ESCA | 148 | 19 921 | 4 431 385 | 44 450 | 1238 | 37 484 | 6177 | 218 | 4723 |
| GBM | 139 | 15 247 | 4 525 414 | 74 593 | 1743 | 61 211 | 6109 | 202 | 5518 |
| HNSC | 492 | 11 768 | 4 249 925 | 294 234 | 4698 | 217 470 | 38 283 | 1 075 | 31 312 |
| KICH | 65 | 11 736 | 3 755 519 | 9 289 | 265 | 8 075 | 382 | 26 | 320 |
| KIRC | 520 | 14 537 | 4 578 071 | 558 380 | 7216 | 380 619 | 57 962 | 1 492 | 47 020 |
| KIRP | 287 | 12 554 | 4 881 400 | 228 243 | 4350 | 175 762 | 28 242 | 905 | 22 640 |
| LAML | 96 | 19 856 | 5 078 753 | 44 537 | 1069 | 34 233 | 5570 | 148 | 4802 |
| LGG | 498 | 14 213 | 4 626 469 | 723 868 | 7844 | 465 249 | 71 517 | 1 595 | 54 899 |
| LIHC | 367 | 9691 | 4 157 271 | 171 255 | 3333 | 128 444 | 22 425 | 764 | 17 725 |
| LUAD | 506 | 13 624 | 4 384 017 | 347 537 | 5714 | 249 589 | 39 040 | 1 130 | 31 147 |
| LUSC | 495 | 14 319 | 3 745 439 | 321 036 | 5556 | 226 779 | 44 235 | 1 236 | 38 111 |
| MESO | 81 | 12 393 | 4 759 523 | 15 045 | 372 | 14 140 | 2152 | 97 | 1779 |
| OV | 251 | 16 660 | 2 966 217 | 102 136 | 3423 | 79 467 | 11 534 | 515 | 9317 |
| PAAD | 177 | 13 298 | 4 991 769 | 139 332 | 2623 | 112 129 | 16 561 | 494 | 13 926 |
| PCPG | 174 | 11 971 | 4 709 166 | 119 083 | 2571 | 93 599 | 16 630 | 552 | 14 066 |
| PRAD | 478 | 12 945 | 4 822 300 | 604 359 | 7181 | 412 073 | 69 412 | 1 686 | 56 307 |
| READ | 91 | 11 298 | 4 540 674 | 18 312 | 539 | 16 750 | 4645 | 153 | 4138 |
| SARC | 257 | 11 454 | 4 087 361 | 105 751 | 2607 | 85 278 | 17 121 | 600 | 14 261 |
| SKCM | 103 | 11 315 | 4 854 570 | 14 150 | 427 | 13 014 | 4856 | 159 | 4501 |
| STAD | 371 | 19 117 | 4 300 207 | 175 519 | 3637 | 128 258 | 21 276 | 635 | 15 847 |
| TGCT | 148 | 14 304 | 4 811 363 | 95 579 | 1989 | 79 971 | 11 477 | 325 | 9839 |
| THCA | 495 | 12 874 | 4 876 701 | 702 674 | 7426 | 464 482 | 57 645 | 1 353 | 44 077 |
| THYM | 119 | 13 223 | 4 930 920 | 92 773 | 1907 | 74 540 | 9355 | 330 | 7028 |
| UCEC | 173 | 11 548 | 4 957 767 | 44 594 | 1327 | 38 605 | 9933 | 411 | 8385 |
| UCS | 55 | 13 439 | 3 871 537 | 51 | 5 | 51 | 0 | 0 | 0 |
| UVM | 77 | 9182 | 4 692 767 | 15 620 | 405 | 14 530 | 3300 | 128 | 2993 |
| Total | 8817 | 26 839 | 7 169 904 | 6 045 445 | 98 087 | 4 294 887 | 715 952 | 21 139 | 573 526 |
We identified a total of 6 045 445 eQTL-lncRNA pairs in 33 cancer types of cis-eQTL analyses at a per-tissue FDR of < 0.05, which corresponded to a median P-value = 5.28 × 10−6. No cis-eQTL or trans-eQTL were identified in CHOL because of the low sample size of 36. Among other cancer types, the number of cis-eQTLs ranged from 51 in uterine carcinosarcoma (UCS) to 465 249 in lower grade glioma (LGG). GTEx multi-tissue eQTL studies have reported a directly proportional relationship of egenes (genes associated with eQTLs) with sample size, and no plateauing has been reported at a maximum 300 sample size (24,34). In our study, we also observed that the number of cis-eQTLs was significantly positively correlated with the sample size, even after adjusted numbers of genotype and ncRNAs (P-value = 6.12 × 10−8, Rs = 0.79, Supplementary Figure S1A), and a similar trend was exhibited for egenes (P-value = 8.66 × 10−10, Rs = 0.85, Supplementary Figure S1B).
The distribution of cis-eQTLs relative to the transcription start site shows that the majority of eQTLs are approximately symmetrically centred around the TSS (Supplementary Figure S1C), with 90% within ±300 kb of the TSS. Overall, 80.2% of lncRNA-eQTLs only regulate the expression of one lncRNA, and 19.8% of lncRNA-eQTLs can regulate the expression of more than two genes (Supplementary Figure S1D).
Across all cancer types, we identified a total of 715 952 eQTL–lncRNA pairs in trans-eQTL analyses at per-tissue FDR of <0.05, which corresponded to a median P-value = 3.45 × 10−13. The number of trans-eQTLs/egenes was also significantly positively correlated with the sample size (P-value = 5.52 × 10−9, R = 0.83 for eQTLs, P-value = 2.38 × 10−11, R = 0.88 for egenes, Supplementary Figure S1E and F).
Data summary of miRNA-related eQTLs
Using genotype data and miRNA-seq data, we further performed analyses of miRNA-related eQTLs (miRNA-eQTLs) across 33 cancer types. In the cis-eQTL analysis, we identified a total of 87 833 eQTL–miRNA pairs at a per-tissue FDR < 0.05, and the number of pairs ranged from six in lymphoid neoplasm diffuse large B-cell lymphoma (DLBC) to 11 779 in THCA (Supplementary Table S2). There was only one cis-regulated miRNA in UCS and DLBC, while 301 egenes were identified in THCA. For trans-eQTL analysis, a total of 5170 eQTL–miRNA pairs were identified, and the number of trans-eQTLs ranged from two in adrenocortical carcinoma (ACC) to 658 in lung squamous cell carcinoma (LUSC). The number of miRNA-related cis-eQTLs and trans-eQTLs was also significantly correlated with the number of samples (P-value = 1.41 × 10−7, R = 0.79 for cis and P-value = 7.75 × 10−5, R = 0.69 for trans, respectively).
Case study of eQTLs associated with patient survival times and GWAS loci
To prioritize promising ncRNA-eQTLs, we linked our eQTLs with TCGA patients’ clinical data and identified eQTLs that may be associated with overall survival times. We identified a total of 8235 lncRNA-eQTLs and 116 miRNA-eQTLs associated with patient overall survival times across 33 cancer types at FDR < 0.05. For example, rs10965295 and rs3131997 were significantly associated with patient overall survival times in LGG (Figure 2A), and these two SNPs could regulate CDKN2B-AS1 expression and hsa-miR-5699-5p in LGG, respectively (Figure 2B). These survival-eQTLs and related ncRNAs may play important roles in cancer development and could serve as predictive and prognostic biomarkers.
Figure 2.

Case study of eQTLs in survival-eQTLs and GWAS-eQTLs. (A) nc-eQTL rs10965295 and rs3131997 affect patient overall survival times in LGG. (B) egene CDKN2B-AS1 of rs10965295 and egene hsa-miR-5699–5p of rs3131997 were significantly differentially expressed among genotypes in LGG. (C) nc-eQTL rs17191861 located in BRCA GWAS locus. (D) egene LINC01235 of rs17191861 was significantly differentially expressed in paired tumour and normal samples.
To explore ncRNA-eQTLs and possible causal genes in known GWAS loci, we identified overlaps between ncRNA-eQTLs and SNPs in GWAS regions. A total of 45 826 tag SNPs were downloaded from the GWAS Catalog; 920 379 SNPs in LD with these tag SNPs were obtained, and all these SNPs were defined as GWAS SNPs. By mapping ncRNA-eQTLs to GWAS SNPs, we identified 253 080 lncRNA-eQTLs and 9252 miRNA-eQTLs overlapping with known disease/traits associated loci. To provide an example of a GWAS-eQTL application, we further mapped BRCA ncRNA-eQTL results to GWAS SNPs of corresponding breast cancer. Among breast GWAS SNPs, we found a total of 1989 eQTLs, which could regulate the expression of 161 ncRNAs. We further analysed the expression of these ncRNAs in tumours and their matched normal samples and found that several ncRNAs were significantly differentially expressed between paired tumour and normal samples at FDR < 0.05. These ncRNAs could be possible causal targets of BRCA GWAS loci. For example, Michailidou et al. found that rs77457752 on chromosome 9p23 is significantly associated with breast cancer risk (P-value = 8 × 10−7), but they did not report its possible target genes (35). rs77457752 is located in the intron of the lncRNA LINC00583. We retrieved the expression level of LINC00583 and found that it is not expressed in 91% of breast cancer samples. In the upstream and downstream regions of rs77457752, there are only a few genes (Figure 2C), and we did not find any correlated protein-coding genes of rs77457752 and its LD SNPs. In our results, we found that rs17191861, which is in LD with rs77457752 (LD r2 = 0.73), is significantly correlated with LINC01235. Differential expression analysis shows that the expression of LINC01235 in tumours is significantly lower than that of adjacent normal samples (P-value < 2 × 10−16, fold change = -2.44, Figure 2D), suggesting that LINC01235 may be a potential causal gene in this risk locus.
Web interface
To facilitate broad access to these ncRNA-eQTLs and associated data, we developed a user-friendly data portal, ncRNA-eQTL (http://ibi.hzau.edu.cn/ncRNA-eQTL/index.php) (Figure 3A), which includes two sub-databases, lncRNA-eQTL home and miRNA-eQTL home. The two sub-databases can be switched from the button on the top right of our data portal. Each sub-database provides four major datasets: cis-eQTLs, trans-eQTLs, survival-eQTLs and GWAS-eQTLs (Figure 3B). On the homepage, we designed a quick search option in which users can input their interested SNPs or genes. Then, four dynamic tables displaying cis-eQTLs, trans-eQTLs, survival-eQTLs and GWAS-eQTLs with related information will be presented (Figure 3C). The cis-eQTL and trans-eQTL table display SNP ID, SNP genomic position, SNP alleles, ncRNA, ncRNA position, β-value (effect size of SNP on gene expression), r-value (correlation coefficient between SNP and expression) and P-value of eQTLs. For each record, a vector diagram of a boxplot is provided to display the association between SNP genotypes and gene expression. The survival-eQTL table displays SNP ID, SNP genomic position, SNP alleles, log-rank test P-value and median survival times of different genotypes. For each record, a vector diagram of the Kaplan–Meier plot is embedded to display the association between SNP genotypes and overall survival times. The GWAS-eQTLs table will return the SNP information, gene information and related GWAS traits.
Figure 3.

Web interface of the ncRNA-eQTL database. (A) ncRNA-eQTL database header with navigation bar. (B) Four different searching modules we offer. (C) The results of a quick search. (D) Heat map of cis-eQTLs across 33 cancer types. The colour of each box indicates the values of the correlation coefficient (r). (E) Heat map of trans-eQTLs across 33 cancer types.
We also designed a ‘Pancan-eQTL’ page, where users can submit a batch of SNPs and/or gene symbols. Then, two interactive heat maps of cis-eQTL (Figure 3D) and trans-eQTL (Figure 3E) will display all the values of the correlation coefficient (r). Users can download all four datasets for each cancer type from the ‘Download’ section. The ‘Help’ page provides information for data collection, processing and result summary. ncRNA-eQTL welcomes any feedback by email to the address provided in the ‘Contact’ page. We have tested the database on various web browsers, including Chrome (recommended), Firefox, Opera, Windows Edge and Safari of macOS.
DISCUSSION
In summary, ncRNA-eQTL is a comprehensive ncRNA (lncRNA and miRNA)-related eQTL resource that uses large cancer samples to evaluate the effects of genetic variants on ncRNA expression. It comprises cis/trans-eQTLs, survival-eQTLs and GWAS-eQTLs and provides a user-friendly interface for users to query, browse and download data of interest. To the best of our knowledge, this is the first database specifically identifying ncRNA-eQTLs in multiple cancer types. In addition, we observed that the number of eQTLs increased with the sample size. The sample sizes of most of the other eQTL studies were below 300 (30,36), but in our analyses, 12 cancer types had more than 300 samples, indicating that the ncRNA-eQTL database would be the most comprehensive resource for ncRNA-eQTLs.
Two important features of our resource are linking ncRNA-eQTLs to patient survival times and known GWAS loci, which will help users narrow down their candidate eQTLs and egenes. Many researchers have been looking for genetic biomarkers related to disease susceptibility, development and prognosis and have committed to analysing the biological mechanisms behind genetic determinations. In our study, we found that thousands of genetic variants could influence cancer prognosis and provided their related non-coding egenes. By integrating ncRNA-eQTL data with known GWAS data, we identified thousands of ncRNA-eQTLs in known GWAS regions. ncRNAs and related genetic determinations are still poorly functionally characterized. These survival and GWAS-related ncRNA-eQTLs and egenes could be important candidates for further experimental validation. Our database will facilitate the fine mapping of post-GWAS and identification of therapeutic biomarkers for cancer. We believe that this valuable resource will be of significant interest to the research community, especially in the field of ncRNA and cancer-related studies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members in Zhang lab for helping in paper writing and website construction. Server support from the Hubei Key Laboratory of Agricultural Bioinformatics is also appreciated.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Huazhong Agricultural University Scientific & Technological Self-innovation Foundation [11041810351 to J.G., W.Z]. Funding for open access charge: Huazhong Agricultural University Scientific & Technological Self-innovation Foundation [11041810351].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dang Y., Wang Y., Ouyang X., Wang L., Huang Q.. High expression of IncRNA-PCNA-AS1 in human gastric cancer and its clinical significances. Clin. Lab. 2015; 61:1679–1685. [DOI] [PubMed] [Google Scholar]
- 2. Do H., Kim W.. Roles of oncogenic long non-coding RNAs in cancer development. Genomics Informatics. 2018; 16:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen S., Wu D.D., Sang X.B., Wang L.L., Zong Z.H., Sun K.X., Liu B.L., Zhao Y.. The lncRNA HULC functions as an oncogene by targeting ATG7 and ITGB1 in epithelial ovarian carcinoma. Cell Death Dis. 2017; 8:e3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang J.Z., Chen M., Chen Gao, X.C., Zhu S., Huang H., Hu M., Zhu H., Yan G.R.. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol. Cell. 2017; 68:171–184. [DOI] [PubMed] [Google Scholar]
- 5. Schork N.J., Fallin D., Lanchbury J.S.. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin. Genet. 2000; 58:250–264. [DOI] [PubMed] [Google Scholar]
- 6. Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J.. 10 years of GWAS Discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017; 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu C., Miao X., Huang L., Che X., Jiang G., Yu D., Yang X., Cao G., Hu Z., Zhou Y. et al.. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat. Genet. 2011; 44:62–66. [DOI] [PubMed] [Google Scholar]
- 8. Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A.. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hua J.T., Ahmed M., Guo H.Y., Zhang Y.Z., Chen S.J., Soares F., Lu J., Zhou S., Wang M., Li H. et al.. Risk SNP-Mediated Promoter-Enhancer switching drives prostate cancer through IncRNA PCAT19. Cell. 2018; 174:564–575. [DOI] [PubMed] [Google Scholar]
- 10. Kasagi Y., Oki E., Ando K., Ito S., Iguchi T., Sugiyama M., Nakashima Y., Ohgaki K., Saeki H., Mimori K. et al.. The expression of CCAT2, a novel long noncoding RNA transcript, and rs6983267 Single-Nucleotide polymorphism genotypes in colorectal cancers. Oncology. 2017; 92:48–54. [DOI] [PubMed] [Google Scholar]
- 11. Shah M.Y., Ferracin M., Pileczki V., Chen B., Redis R., Fabris L., Zhang X., Ivan C., Shimizu M., Rodriguez-Aguayo C. et al.. Cancer-associated rs6983267 SNP and its accompanying long noncoding RNA CCAT2 induce myeloid malignancies via unique SNP-specific RNA mutations. Genome Res. 2018; 28:432–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grundberg E., Small K.S., Hedman A.K., Nica A.C., Buil A., Keildson S., Bell J.T., Yang T.P., Meduri E., Barrett A. et al.. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 2012; 44:1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nica A.C., Montgomery S.B., Dimas A.S., Stranger B.E., Beazley C., Barroso I., Dermitzakis E.T.. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010; 6:e1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E. et al.. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013; 45:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M. et al.. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016; 48:481–487. [DOI] [PubMed] [Google Scholar]
- 16. Branco P.R., de Araujo G.S., Barrera J., Suarez-Kurtz G., de Souza S.J.. Uncovering association networks through an eQTL analysis involving human miRNAs and lincRNAs. Sci.Rep. 2018; 8:15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ning S., Yue M., Wang P., Liu Y., Zhi H., Zhang Y., Zhang J., Gao Y., Guo M., Zhou D. et al.. LincSNP 2.0: an updated database for linking disease-associated SNPs to human long non-coding RNAs and their TFBSs. Nucleic Acids Res. 2017; 45:D74–D78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yue M., Zhou D., Zhi H., Wang P., Zhang Y., Gao Y., Guo M., Li X., Wang Y., Zhang Y. et al.. MSDD: a manually curated database of experimentally supported associations among miRNAs, SNPs and human diseases. Nucleic Acids Res. 2018; 46:D181–D185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miao Y.R., Liu W., Zhang Q., Guo A.Y.. lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018; 46:D276–D280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gong J., Mei S., Liu C., Xiang Y., Ye Y., Zhang Z., Feng J., Liu R., Diao L., Guo A.Y. et al.. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018; 46:D971–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Howie B.N., Donnelly P., Marchini J.. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009; 5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Genomes Project C., Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. The GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graffelman J. Exploring diallelic genetic markers: the HardyWeinberg package. J. Stat. Softw. 2015; 64:1–23. [Google Scholar]
- 26. Kang H.M., Ye C., Eskin E.. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics. 2008; 180:1909–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams R.B., Cotsapas C.J., Cowley M.J., Chan E., Nott D.J., Little P.F.. Normalization procedures and detection of linkage signal in genetical-genomics experiments. Nat. Genet. 2006; 38:855–856. [DOI] [PubMed] [Google Scholar]
- 28. Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D.. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006; 38:904–909. [DOI] [PubMed] [Google Scholar]
- 29. Stegle O., Parts L., Piipari M., Winn J., Durbin R.. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012; 7:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ongen H., Andersen C.L., Bramsen J.B., Oster B., Rasmussen M.H., Ferreira P.G., Sandoval J., Vidal E., Whiffin N., Planchon A. et al.. Putative cis-regulatory drivers in colorectal cancer. Nature. 2014; 512:87–90. [DOI] [PubMed] [Google Scholar]
- 31. Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. MacArthur J., Bowler E., Cerezo M., Gil L., Hall P., Hastings E., Junkins H., McMahon A., Milano A., Morales J. et al.. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017; 45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O’Donnell C.J., de Bakker P.I.W.. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008; 24:2938–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. The GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michailidou K., Lindstrom S., Dennis J., Beesley J., Hui S., Kar S., Lemacon A., Soucy P., Glubb D., Rostamianfar A. et al.. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017; 551:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Popadin K., Gutierrez-Arcelus M., Dermitzakis E.T., Antonarakis S.E.. Genetic and epigenetic regulation of human lincRNA gene expression. Am. J. Hum. Genet. 2013; 93:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.