


Abstract
The introduction of non-bridging phosphorothioate (PS) linkages in oligonucleotides has been instrumental for the development of RNA therapeutics and antisense oligonucleotides. This modification offers significantly increased metabolic stability as well as improved pharmacokinetic properties. However, due to the chiral nature of the phosphorothioate, every PS group doubles the amount of possible stereoisomers. Thus PS oligonucleotides are generally obtained as an inseparable mixture of a multitude of diastereoisomeric compounds. Herein, we describe the introduction of non-chiral 3′ thiophosphate linkages into antisense oligonucleotides and report their in vitro as well as in vivo activity. The obtained results are carefully investigated for the individual parameters contributing to antisense activity of 3′ and 5′ thiophosphate modified oligonucleotides (target binding, RNase H recruitment, nuclease stability). We conclude that nuclease stability is the major challenge for this approach. These results highlight the importance of selecting meaningful in vitro experiments particularly when examining hitherto unexplored chemical modifications.
INTRODUCTION
The use of synthetic oligonucleotides as a therapeutic modality has gained tremendous momentum since the first proof of concept experiments in the late 1970s (1). As a consequence, therapeutic nucleic acids are today actively investigated in the context of various mechanisms of action (2–4) and are meanwhile clinically validated (4,5). All of these oligonucleotides share a need for improved pharmacological properties compared to unmodified RNA and DNA. Thus, chemical modifications have been instrumental for the progress of the field (4,6,7). Among the manifold modifications that have been developed, one of the most abundant is the introduction of phosphorothioate (PS) linkages, where one of the non-bridging oxygen atoms within the phosphate groups is replaced by sulfur (8,9). The PS modification renders oligonucleotides more stable to nucleolytic degradation and confers a substantial pharmacokinetic benefit through increased protein binding (10). These crucial features were the basis for developing the first generation of oligonucleotide therapeutics. On the other hand, diastereoisomeric mixtures of phosphorothioates usually produce lower affinity compared to native diesters resulting in lower potency in a cell-free system. The discovery of high affinity nucleic acid analogs like LNA has been shown to compensate for that, and today the combination of LNA nucleosides and phosphorothioates in one oligonucleotide partner both high affinity and good pharmacokinetic properties in one (11–13).
Replacement of a phosphate with a phosphorothioate creates a chiral center at the phosphorus atom (Sp or Rp, Figure 1A). Hence, the introduction of every PS linkage into an oligonucleotide results in the creation of two sets of diastereoisomers and, in the absence of a stereocontrolled synthesis, an N-mer phosphorothioate oligonucleotide is obtained as an inseparable mixture of 2N−1 diastereoisomers, all of which have different and potentially opposing physico- and biochemical properties (13). Recently, new methods for the stereospecific synthesis of PS linkages have been developed giving access to single compounds rather than diastereoisomeric mixtures (14–16). This allows for the selection of stereoisomerically pure compounds with potentially superior drug properties (13–15). On the one hand, the use of diastereoisomeric mixtures of phosphorothioate oligonucleotides as drug candidates is appealing. Since a lower number of building blocks are needed and because their chemical synthesis is well developed, use of mixtures offers a distinct cost of goods advantage. On the other hand, despite some reports to the contrary (17) an increasing body of evidence suggests that within the mixture of a large number of possible diastereoisomers a few isomers have dramatically improved drug properties (13–15). Additionally, all observable properties of a mixture are the averaged properties across its members. While some diastereoisomers are likely to be mainly responsible for the activity, many others are much less active at best or even, e.g. toxic at worst. This very much complicates the chemical optimization of lead sequences.
Figure 1.
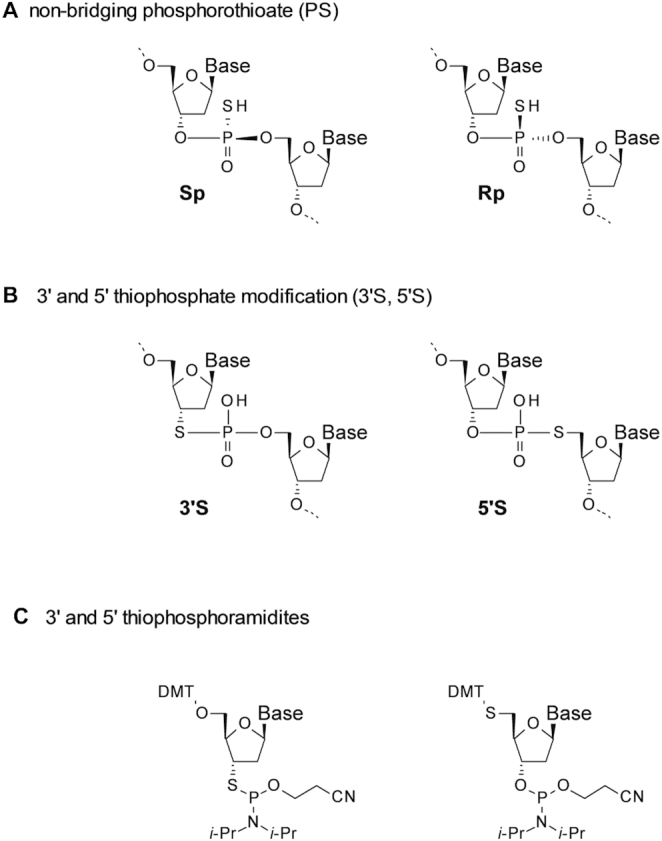
(A) The two diastereoisomers of a phosphorothioate (PS) linkage. (B) Non-chiral 3′S and 5′S thiophosphate linkages. (C) Phosphoramidite building blocks for the synthesis of 3′S (19–30) and 5′S thiophosphate (31–38) linkages.
However, identifying some of the few stereoisomers dramatically outperforming the diastereoisomeric mixture from generally 104–106 possibilities remains a complex challenge.
One potential way of reducing this diastereoisomeric complexity and keeping sulfur as a composition of the internucleoside linkage is the introduction of 3′ or 5′ thiophosphate modifications (Figure 1B). Here, the sulfur atom is moved from the non-bridging to the bridging position (i.e. the 3′ or 5′ carbon atom of the adjacent ribose ring) (18,19). Each replacement of a PS-linkage in an oligonucleotide by one of these symmetrical 3′ or 5′ thiophosphates removes one chiral center, therefore reducing the library complexity by 50%.
Incorporation of 3′S and 5′S linkages into oligonucleotides is possible by standard solid phase oligonucleotide synthesis using appropriate phosphoramidite monomers (Figure 1C (20–38)) and the hybridization behavior as well as the conformational properties of such oligonucleotides have been reported (18–36). 3′S and 5′S thiophosphates have also been applied in mechanistic studies with enzymes and ribozymes (39–50) and in site selective chemical strand cleavage (31,32,34,35). In one study investigating the 5′S modification in RNase H activating gapmers a hitherto unexplained discrepancy between in vitro and in vivo activity was observed (37,38). However, detailed studies on therapeutic oligonucleotides bearing such linkages are still missing.
Herein, we describe the synthesis of LNA gapmers containing 3′S and 5′S phosphorothioate linkages and report their activity as antisense oligonucleotides in cell based in vitro systems and in vivo. Based on a detailed investigation of the individual factors contributing to their mechanism of action (hybridization to complementary RNA, RNase H activation, nucleolytic stability) we offer a hypothesis rationalizing our in vitro and in vivo findings. In doing so we showcase the importance of carefully choosing the right in vitro systems.
MATERIALS AND METHODS
Oligonucleotide synthesis and purification
Oligonucleotides were synthesized using a MerMade 12 automated DNA synthesizer by Bioautomation. Syntheses were conducted on a 1 μmol scale using a controlled pore glass support (500Å) bearing an universal linker. Standard phosphoramidite synthesis procedures were used for unmodified LNA- and DNA-phosphoramidites. 2′,3′-Dideoxy-3′-mercapto phosphoramidites show lower coupling efficiencies than standard phosphoramidites. As a consequence, couplings were performed using repeated couplings (up to 10x) using reduced equivalents of building blocks (4.4 eq). In case of 2′,5′-dideoxy-5′-mercapto phosphoramidites DMT deprotection in the subsequent synthesis cycle in order to liberate the thiol is challenging and was performed using 5 applications of 10% (v/v) trifluoroacetic acid in the presence of 30% (v/v) of triethylsilane in CH2Cl2. Removal of the nucleobase protecting groups and cleavage from the solid support was achieved under standard conditions using 32% aqueous ammonia at 55°C for a minimum of 8 h. Crude DMT-on oligonucleotides were purified either using a solid phase extraction cartridge (Oasis HLB 6cc extraction cartridges from waters) or by preparative RP-HPLC purification (C18 column, NH4OAc/CH3CN buffer system) followed by DMT removal with 80% aqueous acetic acid and ethanol precipitation. Compounds were characterized by reversed phase ultra performance liquid chromatography coupled to high resolution electrospray mass spectrometry.
For synthetic procedures on monomer synthesis as well as detailed conditions for solid phase synthesis please refer to Supplementary Data.
T m measurements
To 10 mM phosphate buffer, 100 mM NaCl and 0.1 mM EDTA (pH 7.0) were added LNA gapmers (to a final concentration of 1.5 mM) and their complementary RNA sequences (to a final concentration of 1.5 mM). Samples were heated to 96°C for 3 min and then slowly cooled to room temperature over a period of 30 min. Thermal melting curves were recorded at 260 nm on a Lambda 40 UV/VIS Spectrometer equipped with a PTP6 Peltier Temperature Programmer using a temperature gradient that was increased by 1°C/min from 25°C to 96°C and then decreased to 25°C. The first derivatives of both curves were used to determine the melting temperature (Tm). Values are reported as averages between heating and cooling curves.
Gapmer-mediated cleavage of RNA by human recombinant RNase H1
LNA gapmers (15 pmol) were mixed with 5′FAM labelled RNA (45 pmol) in 13 μl of water. After the addition of 6 μl of annealing buffer (200 mM KCl, 2 mM EDTA, pH 7.5) the solution was heated to 90°C for 2 min. The sample was allowed to reach room temperature, followed by the addition of RNAse H1 enzyme (0, 15 U) in 3 μl of 750 mM KCl, 500 mM Tris–HCl, 30 mM MgCl2, 100 mM dithiothreitol, pH 8.3). The resulting mixture was kept at 37°C for 30 min at which time the reaction was stopped by the addition of 4 μl of EDTA solution (0.25 M). Anion exchange high performance liquid chromatography (AIE-HPLC) analysis was performed on a Dionex DNA pac 100 column (2 × 250 mm) using Buffer A (10 mM NaClO4, 1 mM EDTA, 20 mM Tris–HCl, pH 7.8) and Buffer B (1 M NaClO4, 1 mM EDTA, 20 mM Tris–HCl, pH 7.8) with the following gradient: 0–22% B in 22 min at 0.25 ml/min, 22–100% B in 3 min at 0.25 ml/min, 100% B for 5 min at 0.25 ml/min. Fluorescence of the FAM label was detected at 518 nm following excitation at 494 nm. 15 μl of the above reaction mixture was added to 200 μl of buffer A and an injection volume of 50 μl was subjected to chromatographic analysis. Peaks corresponding to the intact full length RNA as well as peaks corresponding to cleaved fragments were integrated and %full length target RNA was calculated based on the areas thus obtained.
Treatment with mouse liver homogenate
Liver samples were collected from NMRI mice and homogenized in a homogenisation buffer (25 mM Tris, 150 mM KCl, 1.5 mM KCl pH 7.5 (adjusted with 1 M NaOH)). The homogenate was spun at 12 000 g for 10 min. To the supernatant, LNA gapmers were added to a concentration of 22.9 μM. Samples were incubated for 48 h at 37°C in UPLC vials. The solution was then analysed by reversed phase ultra performance liquid chromatography coupled with mass spectrometry (UPLC-MS). Analyses were performed on a Waters Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 150 mm) using Buffer A (2.5% methanol in 0.2 M hexafluoro isopropanol, 16.3 M triethyl amine in water) and Buffer B (60% methanol in 0.2 M hexafluoro isopropanol, 16.3 M triethyl amine in water) at a flow rate of 0.5 ml/min with the following gradient program: 10% B for 0.5 min, 10–30% B in 4.5 min, 30% B in 1 min, 30–100% B in 1 min, 100–10% in 1 min. Temperature 65°C. Injection volume 2 μl. Components were detected based on absorption at 260 nm. Areas of full-length and degradation products were integrated, and longer chain fragments were interpreted based on electrospray mass spectrometry analysis. The areas of the degradation products were adjusted with respect to their base composition.
In vivo study
In vivo experiments were conducted according to the European standards and protocols were approved by the Danish National Committee for Ethics in Animal Experiments. Inbred C57BL/6JBomTac female mice weighing 20 ± 2 g (arithmetic mean ± standard deviation) were obtained from Taconic (Denmark). The animals were housed in groups of 5, water and a standard diet was supplied ad libitum. Mice were dosed intravenously on days 0 and anesthetized (70% CO2/30% O2) before termination by cervical dislocation on day 7. The treatment groups (n = 5) received either 0.9% saline or saline-formulated gapmer (1 mg/kg) administered by intravenous injection. At the end of the study, blood was sampled for serum analysis, and livers and kidneys were sampled in RNA later and analyzed by qPCR.
mRNA isolation and qPCR measurements from tissue samples
Total RNA from liver was isolated using the RNeasy kit (Qiagen) and quantification of messenger RNA (mRNA) was done using TaqMan assays (Applied Biosystems). The reverse transcription reaction was carried out with random decamers, 0.5 mg total RNA and the M-MLV reverse transcriptase enzyme (Ambion) according to protocol for first strand complementary DNA (cDNA) synthesis. Depending on expression levels, cDNA was subsequently diluted five times in nuclease-free water before addition to the RT-PCR reaction mixture. The Applied Biosystems 7500/7900/ViiA real-time PCR instruments were used for amplification. Within each study, mRNA levels were normalized to actin, beta (Actb) or glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and presented as fold changes relative to average levels in saline controls.
Cell culture, oligonucleotide treatment, mRNA isolation and qPCR and oligonucleotide content analysis: rat hepatocytes
Hepatocytes were isolated from 10 to 14 weeks old male Wistar rats by a two-step collagenase liver perfusion method as previously described (51). Freshly isolated primary rat hepatocytes were plated in collagen-l coated 96-well plates and treated in Williams Medium E containing 10% fetal bovine serum without antibiotics. Cells were treated with LNA solutions in the indicated concentrations in full cell culture medium. After an incubation time of 24 h, the cells were washed three times with PBS containing Ca2+ and Mg2+ and lysed with 165 ul PureLink Pro lysis buffer. Total RNA was isolated using the PureLink Pro 96 RNA Kit from Thermo Fisher according to the manufacturer's instructions and RT-qPCR was performed using the LightCycler Multiplex RNA Virus Master (Roche) with Primer Probe Sets for RnApoB (Invitrogen). The obtained data was normalized to total RNA amounts determined via Ribogreen quantification. Intracellular concentrations of the LNA gapmers were determined using a hybridization based ELISA assay as described earlier (52). All data points were performed in duplicates and data is given as the average thereof.
Cell culture, oligonucleotide treatment, mRNA isolation and qPCR analysis: human lung carcinoma cells
Human lung carcinoma cells (NCI-H460) were cultivated up to passage 15 in RPMI-1640 ATCC formulation (Gibco A10491) + 10% fetal bovine serum. For experimental use the cells were plated at a density of 20k cells per well into collagen-l coated 96-well plates and cultivated overnight before treatment. The treatment with LNA gapmers was performed in full cell culture medium for 24 h. Sample preparation and analytics were performed in analogy to hepatocyte samples described above. All data points were performed in technical triplicates and data is given as the average thereof. IC50 values were fitted with GraphPad Prism with a constrained HillSlope set to 1.
RESULTS
In vitro activity of LNA gapmers in human lung carcinoma cells
We started our investigations by preparing LNA gapmers bearing single 3′S linkages (Table 1). The oligonucleotides were generated by means of solid phase oligonucleotide synthesis using the phosphoramidite method. The required phosphoramidite building blocks were readily accessible from the appropriately protected nucleoside alcohols by synthetic routes similar to those reported in the literature (19–29). The modifications were incorporated into a LNA gapmer that was designed to target Malat1 (metastasis associated lung adenocarcinoma transcript 1) (53).
Table 1.
Prepared anti-Malat1-gapmers with their sequence and the corresponding in vitro IC50 value for unassisted uptake in H460 cells at 24 h (IC50 values are given as value ± std. error)
| Entry | Denotation | Sequence | IC50 [nM] |
|---|---|---|---|
| 1 | anti-Malat1-gapmer (control) | GAGttacttgccaAmCT | 93 ± 10 |
| 2 | anti-Malat1-gapmer-1 | GAGt(3′S)tacttgccaAmCT | 647 ± 205 |
| 3 | anti-Malat1-gapmer-2 | GAGtt(3′S)acttgccaAmCT | 785 ± 144 |
| 4 | anti-Malat1-gapmer-3 | GAGtta(3′S)cttgccaAmCT | 1129 ± 469 |
| 5 | anti-Malat1-gapmer-4 | GAGttac(3′S)ttgccaAmCT | 8833 ± 4660 |
| 6 | anti-Malat1-gapmer-5 | GAGttact(3′S)tgccaAmCT | 936 ± 174 |
| 7 | anti-Malat1-gapmer-6 | GAGttactt(3′S)gccaAmCT | 1345 ± 573 |
| 8 | anti-Malat1-gapmer-7 | GAGttacttg(3′S)ccaAmCT | 1932 ± 496 |
| 9 | anti-Malat1-gapmer-8 | GAGttacttgc(3′S)caAmCT | 934 ± 180 |
| 10 | anti-Malat1-gapmer-9 | GAGttacttgcc(3′S)aAmCT | 410 ± 58 |
| 11 | anti-Malat1-gapmer-10 | GAGttacttgcca(3′S)AmCT | 340 ± 79 |
Positions of 3′S linkages are indicated by (3′S) between bold nucleotides; A, G, mC, T represent LNA nucleotides; a, g, c, t represent DNA nucleotides; all other linkages were prepared as non-bridging phosphorothioates.
A set of otherwise fully phosphorothioated gapmers was prepared, where 3′S linkages were walked along the gap region (Table 1). These designs should not only allow for an assessment of the effect of this modification on the activity of the oligonucleotides, but also reveal any potential positional dependence.
The in vitro potency of the LNA gapmers was investigated in human lung carcinoma cells (H460). The assay conditions were gymnotic, i.e. the LNAs were added without any transfection reagents. Malat1 levels were recorded 24 h after incubation at five concentrations (Figure 2). Corresponding IC50 values are given in Table 1. Figure 2 shows that all the tested gapmers dose dependently lower Malat1 expression levels. A reduction of >50% compared to the untreated control is observed for all the tested molecules at the highest concentrations (P < 0.0001 in all cases). However, the potencies differ significantly compared to the control compound (anti-Malat1-gapmer) as well as among the modified oligonucleotides. In general, the modified gapmers are more potent, the closer the modification is located towards the flank region (wings). Particularly, moving the 3′S modification from the 5′ end towards the center of the gap results in a progressive drop in activity cumulating in anti-Malat1-gapmer-4, where significant target reduction is only observed at the highest concentration (10 μM).
Figure 2.

In vitro activity of anti-Malat1-gapmers bearing 3′S modifications in the gap region (H460 cells, no transfection, 24 h treatment, data were generated in technical triplicates).
In vivo potency of LNA gapmers
Encouraged by the data obtained from the Malat1 in vitro study, we decided to investigate the impact of 3′S modifications of gapmers to another target in an in vivo setting. Here, we tested the 3′S modifications in LNA gapmers designed to target apolipoprotein B (ApoB). ApoB is highly expressed in liver and has been widely used as a reference target for Antisense and RNAi mediated knockdown (52,54). Four LNA gapmers (anti-ApoB-gapmer-1 to anti-ApoB-gapmer-4, Table 2) were synthesized, each bearing a 3′S linkage at different positions within the gap regions.
Table 2.
Prepared 3′S-modified anti-ApoB-gapmers and their respective sequences
| Entry | Denotation | Sequence |
|---|---|---|
| 1 | anti-ApoB-gapmer (control) | GmCattggtatTmCA |
| 2 | anti-ApoB-gapmer-1 | GmCatt(3′S)ggtatTmCA |
| 3 | anti-ApoB-gapmer-2 | GmCattg(3′S)gtatTmCA |
| 4 | anti-ApoB-gapmer-3 | GmCattggt(3′S)atTmCA |
| 5 | anti-ApoB-gapmer-4 | GmCattggta(3′S)tTmCA |
Positions of 3′S linkages are indicated by (3′S) between bold nucleotides; A, G, mC, T represent LNA nucleotides; a, g, c, t represent DNA nucleotides; all other linkages were prepared as non-bridging phosphorothioates
The designs were chosen to investigate a potential positional dependence of 3′S modifications introduced. Oligonucleotides were administered intravenously to female C57BL/6JBomTac mice at a dose level of 1 mg/kg (5 mice/group; single administration). After 7 days animals were sacrificed, kidneys and livers sampled and analyzed by qPCR (Figure 3). While the parent oligonucleotide (anti-ApoB-gapmer) efficiently reduced ApoB mRNA levels both in liver and in kidney (P = 0.0012 and P < 0.0001 respectively), the 3′S modified analogues were much less active. In kidney the modified compounds showed some target reduction, albeit much less than the unmodified parent (P = 0.0012–0.018, Figure 3, right). In liver, however, the target organ of these anti-ApoB-gapmers, no target reduction was observed (Figure 3, left).
Figure 3.

In vivo activity of anti-ApoB-gapmers in mice liver (left) and kidney (right). Oligonucleotides were dosed 1 mg/kg iv and mice were sacrificed after 7 days (n = 5 per compound).
In order to further investigate the underlying cause of these observations, we investigated the individual properties required of a therapeutic oligonucleotide for in vivo activity (target binding, RNAse H activation and stability).
Target binding
To further investigate the properties of 3′S and 5′S modified LNA gapmers, we decided to prepare a full set of compounds where both, the 3′S modification as well as the 5′S modification, is moved through the anti-ApoB-gapmer sequence in a positional manner (Table 3).
Table 3.
Prepared anti-ApoB-gapmers with their sequence and their melting temperature versus their complementary RNA strand. Tm is given as the average between the heating and the cooling curve
| Entry | Denotation | Modification | Sequence | Tm (°C) |
|---|---|---|---|---|
| 1 | anti-ApoB-gapmer (control) | None | GmCattggtatTmCA | 59.4 |
| 2 | anti-ApoB-gapmer-5 | 3′S | GmCa(3′S)ttggtatTmCA | 60.0 |
| 3 | anti-ApoB-gapmer-6 | 3′S | GmCat(3′S)tggtatTmCA | 62.5 |
| 4 | anti-ApoB-gapmer-1 | 3′S | GmCatt(3′S)ggtatTmCA | 61.0 |
| 5 | anti-ApoB-gapmer-2 | 3′S | GmCattg(3′S)gtatTmCA | 62.5 |
| 6 | anti-ApoB-gapmer-7 | 3′S | GmCattgg(3′S)tatTmCA | 62.5 |
| 7 | anti-ApoB-gapmer-3 | 3′S | GmCattggt(3′S)atTmCA | 62.5 |
| 8 | anti-ApoB-gapmer-4 | 3′S | GmCattggta(3′S)tTmCA | 61.9 |
| 9 | anti-ApoB-gapmer-8 | 3′S | GmCattggtat(3′S)TmCA | 63.5 |
| 10 | anti-ApoB-gapmer-9 | 5′S | GmC(5′S)attggtatTmCA | 56.4 |
| 11 | anti-ApoB-gapmer-10 | 5′S | GmCa(5′S)ttggtatTmCA | 58.5 |
| 12 | anti-ApoB-gapmer-11 | 5′S | GmCat(5′S)tggtatTmCA | 57.4 |
| 13 | anti-ApoB-gapmer-12 | 5′S | GmCatt(5′S)ggtatTmCA | 56.4 |
| 14 | anti-ApoB-gapmer-13 | 5′S | GmCattg(5′S)gtatTmCA | 59.0 |
| 15 | anti-ApoB-gapmer-14 | 5′S | GmCattgg(5′S)tatTmCA | 58.5 |
| 16 | anti-ApoB-gapmer-15 | 5′S | GmCattggt(5′S)atTmCA | 59.5 |
| 17 | anti-ApoB-gapmer-16 | 5′S | GmCattggta(5′S)tTmCA | 59.0 |
Positions of 3′S and 5′S linkages are indicated by (3′S) and (5′S) between bold nucleotides; A, G, mC, T represent LNA nucleotides; a, g, c, t represent DNA nucleotides; all other linkages were prepared as non-bridging phosphorothioates.
As a first experiment, we wanted to confirm the ability of these compounds to bind to their complementary RNA sequences. Synthesized compounds including their design, sequences and melting temperatures are given in Table 3. In agreement with what has been reported in the literature (20–36), an increase in the DNA·RNA duplex stability of ∼0.5–2.5°C is observed for the singly 3′S modified compounds compared to the reference (Table 3, entries 2–9). Also in line with literature reports (37,38), single 5′S modifications turned out to have either no destabilizing effect for some designs or a destabilizing effect of up to −3°C for other designs against complementary RNA (Table 3, entries 10–17).
RNAse H activation
We next set out to investigate the ability of oligonucleotides bearing 3′S or 5′S thiophosphate linkages to recruit RNase H. Towards this end, modified compounds were mixed with a threefold excess of their 5′ dye labelled target RNA and exposed to human RNase H1 at 37°C. After an incubation time of 30 min samples were analyzed by ion exchange chromatography. We compared modified anti-ApoB-gapmers bearing both 3′S as well as 5′S modifications in the same position of the sequence in order to assess differences between the two modifications (Table 4). The investigated compounds and the percentage of intact target RNA left are given in Table 4. The control anti-ApoB-gapmer turns out to be highly efficient in recruiting RNase H and only 5.8% of the target RNA remained intact after 30 min of incubation (Table 4, entry 1). All of the 3′S or 5′S modified gapmers were able to successfully recruit RNase H but performed somewhat less efficiently than the control (9.8–17.3% of intact target RNA left, entries 2–7). Surprisingly, one particular oligonucleotide (anti-ApoB-gapmer-16, entry 8) showed only a very low RNase H recruitment with more than half of the target RNA remaining.
Table 4.
Anti-ApoB-gapmers investigated in the human RNase H1 cleavage assay and the percentage of intact target RNA after an incubation time of 30 min (single experiment)
| Entry | Denotation | Modification | Sequence | % Full length target RNA |
|---|---|---|---|---|
| 1 | anti-ApoB-gapmer (control) | none | GmCattggtatTmCA | 5.8 |
| 2 | anti-ApoB-gapmer-5 | 3′S | GmCa(3′S)ttggtatTmCA | 17.3 |
| 3 | anti-ApoB-gapmer-10 | 5′S | GmCa(5′S)ttggtatTmCA | 9.9 |
| 4 | anti-ApoB-gapmer-6 | 3′S | GmCat(3′S)tggtatTmCA | 13.6 |
| 5 | anti-ApoB-gapmer-11 | 5′S | GmCat(5′S)tggtatTmCA | 11.4 |
| 5 | anti-ApoB-gapmer-7 | 3′S | GmCattgg(3′S)tatTmCA | 10.0 |
| 6 | anti-ApoB-gapmer-14 | 5′S | GmCattgg(5′S)tatTmCA | 11.3 |
| 7 | anti-ApoB-gapmer-4 | 3′S | GmCattggta(3′S)tTmCA | 9.8 |
| 8 | anti-ApoB-gapmer-16 | 5′S | GmCattggta(5′S)tTmCA | 56.9 |
Positions of 3’S and 5’S linkages are indicated by (3’S) and (5′S) between bold nucleotides; A, G, mC, T represent LNA nucleotides; a, g, c, t represent DNA nucleotides; all other linkages were prepared as non-bridging phosphorothioates.
Nuclease stability
In order to investigate the metabolic stability of these compounds, we exposed anti-ApoB-gapmers 1–16 (Table 3) to diluted mouse liver homogenate and incubated for 48 h. The samples were analyzed by HPLC-MS. Interestingly, distinct differences between the gapmers were observed with respect to the relative amount of intact oligonucleotide left (Figure 4, left). Within the 3′S series, best stability is observed when the modification is placed at the ends of the gap region, while modifications within the center of the gap lead to significantly lower stabilities. In a similar manner 5′S modifications at the 5′ end of the gap resulted in the best relative stability compared to any of the other 5′S modifications. Furthermore, inspection of the HPLC–MS chromatograms for any well-defined degradation products revealed a distinct picture (the chromatogram of anti-ApoB-gapmer-6 is given in Figure 4, right, as an illustrative example). For most of the investigated anti-ApoB-gapmers 1–16 defined longer chain degradation products were observable. MS analysis revealed that the observed fragments almost exclusively corresponded to cleavage of the oligonucleotide at the 3′S or 5′S modified internucleoside linkages.
Figure 4.

Left: Relative amounts of intact gapmers left after 48 h treatment with diluted mouse liver homogenate. Right: Representative chromatogram of anti-ApoB-gapmer-6 after incubation. Major observable degradation products derive from cleavage at the 3′S modification (single experiment).
In vitro activity in primary rat hepatocytes
We next decided to run additional activity studies in a cellular system using primary rat hepatocytes. Cells were treated with anti-ApoB-gapmers 1–16 at a treatment concentration of 2 μM for 24 h. The corresponding mRNA levels compared to an untreated control are shown in Figure 5 (light grey bars). We also determined the intracellular gapmer concentration (Figure 5, dark grey bars). No conceivable target reduction was observed for most of the examined oligonucleotides bearing a 3′S modification (Figure 5, 3′S-series). This lack of activity is well reflected by the fact, that none of these compounds were significantly detected intracellularly (anti-ApoB-gapmers 1–4, 6 and 7). In the cases, where a significant amount of gapmer was found, target reduction was also detected (anti-ApoB-gapmers 5 and 8, P = 0.0002 and P = 0.0004). Although anti-ApoB-gapmers bearing a 5′S-modification overall were more active (Figure 5, 5′S-series), a similar conclusion can be drawn for that modification as well: poor overall activity is observed in hepatocytes with higher intracellular concentrations correlating well with better LNA activity (anti-ApoB-gapmers 9 and 10, P < 0.0001 and P = 0.0017).
Figure 5.

In vitro target reduction and intracellular concentration of anti-ApoB-gapmers with 3′S as well as 5′S modifications in the gap region – tested in primary rat hepatocytes (no transfection, 24 h treatment). Light grey bars: target mRNA levels in % of untreated control; dark grey bars: intracellular concentration as determined by lysis of carefully washed cells followed by ELISA based quantification. Data were generated in technical duplicates.
Discussion
The introduction of sulfur into the 3′ position of a DNA nucleotide results in a conformational change of the ribose sugar ring both in dinucleotides as well as in longer duplexed oligonucleotides (27,30). While unsubstituted deoxyribose prefer a ‘south pucker’ (C2’-endo/C3’-exo), upon introduction of sulfur, for stereoelectronic reasons this equilibrium is shifted more towards a ‘north pucker’ (C2’-exo/C3’-endo, Figure 6) (27,28). Since RNase H naturally recognizes and cleaves duplexes of target RNA with DNA (i.e. ribose in ‘south’ conformation) and does not cleave duplexes of RNA with complementary sequences bearing modifications such as LNA (where the ribose is ‘locked’ in a ‘north’ configuration), we were curious to investigate the general impact of 3′S modification on the activity of oligonucleotides. With these considerations, we were delighted to see that all of our modified anti-Malat-1 gapmers showed activity in our initial in vitro experiments using lung carcinoma cells (Figure 2). However, our initial results also clearly showed a strong positional dependence of activity with generally lower activity the more the modification was positioned towards the center of the gap region of the oligonucleotide. This behavior is in line with a hampered recruitment of RNase H by 3′S modified oligonucleotides. Consequently, a higher impact is expected the more the modification is placed towards a potentially preferred cleavage site of RNase H in the center of the gap. Nonetheless, the data clearly demonstrated that LNA gapmers containing the 3′S modification in their gap region generally retain activity to a high degree, when they are positioned towards the flanks of the gapmers.
Figure 6.
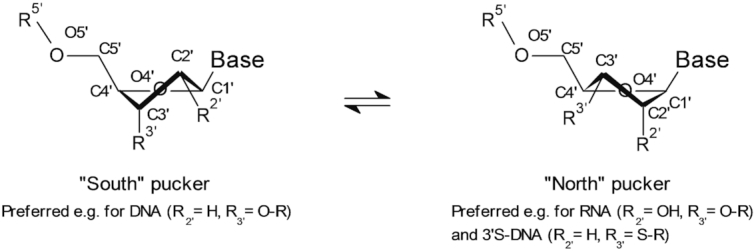
Schematic representation of ‘South’ (left) and ‘North’ (right) puckering of the ribose sugar ring. RNase H preferentially recognizes and cleaves RNA duplexed with oligonucleotides in a ‘South’ pucker.
Based on the promising in vitro potency of 3′S modified anti-Malat1-gapmers, we were optimistic that the introduction of 3′S modifications would also be tolerated in our ApoB in vivo study. Unfortunately, 3′S modified anti-ApoB-gapmers showed only little activity in kidney and no activity in the liver, the target organ of this anti-ApoB-gapmer sequence. On one hand, this observation for 3′S modified gapmers might be due to the aforementioned conformational impact of this modification on RNase H activity, since the shorter gap region in the anti-ApoB-gapmer compared to the anti-Malat1-gapmer-sequence suggests that RNAse H cleavage might be more strongly inhibited. On the other hand, the modification of the gapmers with 3′S linkages could also lead to dramatically different pharmacokinetic properties explaining differences between in vitro and in vivo properties. Indeed it has recently been reported that changes in as little as one single phosphate linkage in an antisense oligonucleotide can dramatically alter its properties such as toxicity or protein binding (55). For example, one would certainly expect an impact on the protein binding properties of an oligonucleotide when 3′S linkages are introduced. The enhanced unspecific protein binding by PS compared to phosphodiester is generally attributed to the negative charge delocalization at the thioate bond (9–11). As in 3′S (and 5′S) linkages the terminal sulfur is moved in a bridging position and the charge of the phosphate is mainly located on the terminal oxygen a lower proneness to protein binding is expected.
In addition, a surprising discrepancy between promising in vitro results and low in vivo activity has been reported for 5′S modified gapmers (38), where no such conformational inhibition of RNAse H is expected. The group of Obika reported a set of 5′S modified LNA gapmers targeting mouse PTEN mRNA bearing between one and two modifications. These sequences showed good in vitro activities in mice hepatic NMuLi cells comparable to the unmodified control. However, in a subsequent in vivo study in mice, little to no reduction in liver PTEN levels was observed (38).
To further investigate the underlying cause of the unsatisfactory in vivo behavior of 3′S and 5′S modified LNA gapmers, we synthesized a full set of compounds, where the 3′S as well as the 5′S modification is moved through the anti-ApoB-gapmer sequence in a positional manner. With these compounds in hand we carefully examined isolated features that are important for a productive antisense effect (target binding, RNase H activation and nuclease stability).
The ability of such compounds to successfully bind complementary RNA was confirmed by the determination of their corresponding melting temperatures (Tm). Melting temperatures in the same order of magnitude as the unmodified control were obtained for all of the modified compounds confirming their ability to bind to the RNA target. In agreement with literature (20–38), and in line with the structural considerations described above, generally a higher melting temperature for 3′S modified compounds was observed. In the case of 5′S modified gapmers no clear tendency was evident, which is in agreement with no general structural difference upon introduction of 5′S modifications. While these results could explain differences between individual molecules, the small differences in binding, would not explain the lack of in vivo activity of oligonucleotides bearing 3′S and 5′S modifications.
We next assessed the ability of these 3′S and 5′S modified LNA gapmers to recruit RNase H when mixed with the enzyme and RNA target. Eight compounds (Table 4) bearing 3′S and 5′S modifications in the same position of the sequence were compared in order to directly compare the effect of the two modifications on the activity of RNAse H. Interestingly, the introduction of single 3′S or 5′S modifications appears not to reveal a general preference by RNase H and the observed differences turn out to be dependent on the position of the phosphate in question within the sequence. While, for example, the 5′S modification appears to be more readily tolerated than the 3′S modification in the third phosphate group from the 5′ end of the sequence (Table 4, compare entries 2 and 3), the opposite appears to be true for the ninth phosphate group from the 5′ end (entries 7 and 8). In all the other positions examined, no clear bias towards any of the two modifications was observed. Interestingly, while the introduction of 5′S modifications did not alter the nature and the distribution of cleavage products (see Supplementary Data for details), when a 3′S linkage was introduced changes in the relative abundance of the major cleavage products were noticeable. This result suggests that in gapmers of this sort, the introduction of single 3′S modifications might be better tolerated by RNase H than initially expected, because of the availability of alternative cleavage sites. Obviously, this purely biochemical assay is only an approximation to the complex situation in vivo, where e.g. all involved concentrations are much lower. Nonetheless, the results show that the introduction of 3′S and 5′S modifications into gapmers does not exclude RNase H mediated target reduction and thus suggest that the observed lack of in vivo activity is highly likely not solely caused by a lower RNase H activity (e.g. entry 7 in Table 4 shows almost equal efficacy to the control but lacks in vivo activity completely in liver).
After demonstrating that 3′S and 5′S modified oligonucleotides are able to bind their target and do recruit RNase H, we next wanted to investigate if such compounds are stable enough in vivo to reach their site of action. Whereas the stability of 3′S and 5′S modified oligonucleotides against individual nucleases or mouse serum have been reported with somewhat inconclusive results (37,38,56), we decided to investigate the stability of our constructs versus a cellular extract. The use of liver homogenates has been reported to provide metabolic patterns comparable to the in vivo situation (57,58). The metabolism of unmodified anti-ApoB-gapmer has been demonstrated to predominantly result in the corresponding n-1 analogue resulting from 3′ exonucleolytic cleavage of the LNA A nucleotide at the 3′ end of the oligonucleotide (59). Further degradation by endo- or exonucleases is comparatively slow and typically results in only minor quantities of other metabolites. In contrast, when we exposed 3′S or 5′S modified gapmers to diluted mouse liver homogenate and analyzed the resulting mixtures by HPLC-MS a distinctly different behavior was observed. The major degradation product for most of the modified anti-ApoB-gapmers 1–16 were metabolites deriving from cleavage of the 3′S or 5′S modified linkage (Figure 4, right), while the expected cleavage of the LNA A nucleotide at the 3′ end is not observed at all under the chosen conditions. The fact that these species are present in substantially higher quantity than any other degradation products reveals a significantly lower metabolic stability of 3′S and 5′S linkages compared to regular PS thiophosphates. Thus, while treatment with liver homogenate might not be an exact model for the in vivo situation in liver, these results provide evidence, that besides a potential reduction in RNase H activity (see above) indeed instability against endonucleases in hepatic tissue may be the main contributor to the poor in vivo performance we observed for 3′S modified gapmers. This interpretation is also in line with the observed relative stabilities among the individual modified gapmers (Figure 4, left). The higher relative stability of 3′S and 5′S modified linkages when in close proximity to the LNA wings of the gapmer suggests that a locked ribose sugar is able to protect adjacent phosphate linkages from nucleolytic degradation. This offers an alternative explanation (besides RNase H activity) to why higher in vitro activities were observed for compounds where the modification was placed near to the LNA wings (Figure 2).
Based on this hypothesis we decided to run further activity studies of modified anti-ApoB-gapmers in primary rat hepatocytes with the hope to pick up the potential instability of 3′S and 5′S modified linkages in a cellular in vitro system and thus offer an in vitro rationale for their in vivo inactivity. Indeed, comparing target reduction (Figure 5, light grey bars), the determined intracellular gapmer concentrations in primary rat hepatocytes (Figure 5, dark grey bars) and their relative stability to mouse liver homogenate (Figure 4, left) reveals a striking trend. Compounds of higher nucleolytic stability (e.g. anti-ApoB-gapmer 5, 8 or 9) give rise to higher intracellular concentrations and, therefore, better target reduction in the cellular assay. This link of stability with potency for 3′S modified gapmers together with the observed lack of in vitro activity in this cell system further corroborates the hypothesis that insufficient nuclease stability is the main contributor to the poor in vivo performance of such compounds. Obviously, additional factors such as RNase H activity cannot be fully excluded. However, the present conclusions not only explain the observed lack of in vivo activity of 3′S modified gapmers but also offer an appealing explanation to why similar findings were made with 5′S modified compounds (38).
In summary, non-chiral 3′S-modifications, where the sulfur within the thiophosphate linkage is in a bridging rather than a non-bridging position, were investigated in LNA oligonucleotides for the first time. In these studies we have initially observed good in vitro activity. However, when these modifications were introduced into a different sequence with a different target, no in vivo efficacy or target reduction in a primary cell system was obtained. Investigations of the fundamental parameters for an oligonucleotide therapeutic (target binding (Tm), RNase H recruitment, nuclease stability) revealed a lack of nuclease stability as the most probable reason for this discrepancy. The findings presented herein may have important implications for future research on 3′S and 5′S modified oligonucleotides and beyond. When introducing 3′S and 5′S linkages into LNA gapmers, the position of the modification within the sequence (close to protecting LNA wings) is crucial. Furthermore, our work is an important case study how crucial it generally is to carefully chose the conditions of in vitro experiments in order to avoid seemingly inexplicable discrepancies to later in vivo experiments. The results presented here highlight the need for a careful selection of appropriate cell-lines or primary cells depending on the target tissue. For example, when targeting liver, use of primary hepatocytes simulating the degradation characteristics of liver tissue, certainly represents a more informative in vitro system. This aspect is particularly important when hitherto unexplored chemical modifications are investigated. In that respect, our results will assist a more informed compound design and experimental setup for future drug discovery efforts.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Erik Funder for his invaluable investigative efforts. We gratefully acknowledge Jörg Hörnschemeyer and Sigfried Stolz for compound analytics, Valesi Mukuka for formulation, Heidii Mazur for her help running the in vivo study and Charlotte Øverup, who performed the bioanalytic analysis of in vivo samples. We also thank Jon Bodnar for proofreading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Roche Postdoc Fellowship (RPF) program. Funding for open access charge: F. Hoffmann-La Roche AG.
Conflict of interest statement. J.D. is a member of a shareholder group with pooled voting rights of Roche.
REFERENCES
- 1. Zamecnik P.C., Stephenson M.L.. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bennett C.F., Baker B.F., Pham N., Swayze E., Geary R.S.. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017; 57:81–105. [DOI] [PubMed] [Google Scholar]
- 3. Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F.. RNA-targeted therapeutics. Cell Metab. 2018; 27:714–739. [DOI] [PubMed] [Google Scholar]
- 4. Shen X., Corey D.R.. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018; 46:1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stein C.A., Castanotto D.. FDA-Approved oligonucleotide therapies in 2017. Mol. Ther. 2017; 25:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khvorova A., Watts J.K.. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017; 35:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wan W.B., Seth P.P.. The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem. 2016; 59:9645–9667. [DOI] [PubMed] [Google Scholar]
- 8. Eckstein F. Phosphorothioate oligodeoxynucleotides: what is their origin and what is unique about them. Antisense Nucleic Acid Drug Dev. 2000; 10:117–121. [DOI] [PubMed] [Google Scholar]
- 9. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014; 24:374–387. [DOI] [PubMed] [Google Scholar]
- 10. Watanabe T.A., Geary R.S., Levin A.A.. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides. 2006; 16:169–180. [DOI] [PubMed] [Google Scholar]
- 11. Koch T. LNA antisense: a review. Curr. Phys. Chem. 2013; 3:55–68. [Google Scholar]
- 12. Veedu R.N., Wengel J.. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009; 6:321–323. [DOI] [PubMed] [Google Scholar]
- 13. Hagedorn P.H., Persson R., Funder E.D., Albaek N., Diemer S.L., Hansen D.J., Moller M.R., Papargyri N., Christiansen H., Hansen B.R. et al.. Locked nucleic acid: modality, diversity, and drug discovery. Drug Discov. Today. 2018; 23:101–114. [DOI] [PubMed] [Google Scholar]
- 14. Li M., Lightfoot H.L., Halloy F., Malinowska A.L., Berk C., Behera A., Schumperli D., Hall J.. Synthesis and cellular activity of stereochemically-pure 2′-O-(2-methoxyethyl)-phosphorothioate oligonucleotides. Chem. Commun. 2017; 53:541–544. [DOI] [PubMed] [Google Scholar]
- 15. Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H. et al.. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:845–851. [DOI] [PubMed] [Google Scholar]
- 16. Oka N., Wada T.. Stereocontrolled synthesis of oligonucleotide analogs containing chiral internucleotidic phosphorus atoms. Chem. Soc. Rev. 2011; 40:5829–5843. [DOI] [PubMed] [Google Scholar]
- 17. Wan W.B., Migawa M.T., Vasquez G., Murray H.M., Nichols J.G., Gaus H., Berdeja A., Lee S., Hart C.E., Lima W.F. et al.. Synthesis, biophysical properties and biological activity of second generation antisense oligonucleotides containing chiral phosphorothioate linkages. Nucleic Acids Res. 2014; 42:13456–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li N.S., Frederiksen J.K., Piccirilli J.A.. Synthesis, properties, and applications of oligonucleotides containing an RNA dinucleotide phosphorothiolate linkage. Acc. Chem. Res. 2011; 44:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaynor J.W., Cosstick R.. Synthesis, properties and application of nucleic acids containing phosphorothiolate linkages. Curr. Org. Chem. 2008; 12:291–308. [Google Scholar]
- 20. Evans K., Bhamra I., Wheelhouse R.T., Arnold J.R., Cosstick R., Fisher J.. Stabilization of a bimolecular triplex by 3′-S-phosphorothiolate modifications: an NMR and UV thermal melting investigation. Chem. Eur. J. 2015; 21:7278–7284. [DOI] [PubMed] [Google Scholar]
- 21. Piperakis M.M., Gaynor J.W., Fisher J., Cosstick R.. Thermal stabilisation of RNA.RNA duplexes and G-quadruplexes by phosphorothiolate linkages. Org. Biomol. Chem. 2013; 11:966–974. [DOI] [PubMed] [Google Scholar]
- 22. Gaynor J.W., Bentley J., Cosstick R.. Synthesis of the 3′-thio-nucleosides and subsequent automated synthesis of oligodeoxynucleotides containing a 3′-S-phosphorothiolate linkage. Nat. Protoc. 2007; 2:3122–3135. [DOI] [PubMed] [Google Scholar]
- 23. Bentley J., Brazier J.A., Fisher J., Cosstick R.. Duplex stability of DNA.DNA and DNA.RNA duplexes containing 3′-S-phosphorothiolate linkages. Org. Biomol. Chem. 2007; 5:3698–3702. [DOI] [PubMed] [Google Scholar]
- 24. Brazier J.A., Fisher J., Cosstick R.. Stabilization of the DNA I-motif structure by incorporation of 3′-S-phosphorothiolate linkages. Angew. Chem. Int. Ed. 2005; 45:114–117. [DOI] [PubMed] [Google Scholar]
- 25. Buckingham J., Sabbagh G., Brazier J., Fisher J., Cosstick R.. Control of DNA conformation using 3′-S-phosphorothiolate-modified linkages. Nucleosides Nucleotides Nucleic Acids. 2005; 24:491–495. [DOI] [PubMed] [Google Scholar]
- 26. Sabbagh G., Fettes K.J., Gosain R., O’Neil I.A., Cosstick R.. Synthesis of phosphorothioamidites derived from 3′-thio-3′-deoxythymidine and 3′-thio-2′,3′-dideoxycytidine and the automated synthesis of oligodeoxynucleotides containing a 3′-S-phosphorothiolate linkage. Nucleic Acids Res. 2004; 32:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beevers A.P., Fettes K.J., Sabbagh G., Murad F.K., Arnold J.R., Cosstick R., Fisher J.. NMR and UV studies of 3′-S-phosphorothiolate modified DNA in a DNA: RNA hybrid dodecamer duplex; implications for antisense drug design. Org. Biomol. Chem. 2004; 2:114–119. [DOI] [PubMed] [Google Scholar]
- 28. Beevers A.P., Fettes K.J., O’Neil I.A., Roberts S.M., Arnold J.R., Cosstick R., Fisher J.. Probing the effect of a 3′-S-phosphorothiolate link on the conformation of a DNA:RNA hybrid; implications for antisense drug design. Chem. Commun. 2002; 1458–1459. [DOI] [PubMed] [Google Scholar]
- 29. Fettes K.J., O’Neil I., Roberts S.M., Cosstick R.. Solid-phase synthesis of oligodeoxynucleotides containing 3′-S-phosphorothiolate linkages. Nucleosides Nucleotides Nucleic Acids. 2001; 20:1351–1354. [DOI] [PubMed] [Google Scholar]
- 30. Beevers A.P.G., Witch E.M., Jones B.C.N.M., Cosstick R., Arnold J.R.P., Fisher J.. Conformational analysis of 3′-S-PO3-linked ribo- and deoxyribodinucleoside monophosphates. Magn. Reson. Chem. 1999; 37:814–820. [Google Scholar]
- 31. Jahn-Hofmann K., Engels J.W.. Efficient solid phase synthesis of cleavable oligodeoxynucleotides based on a novel strategy for the synthesis of 5′-S-(4,4′-dimethoxytrityl)-2′-deoxy-5′-thionucleoside phosphoramidites. Helv. Chim. Acta. 2004; 87:2812–2828. [Google Scholar]
- 32. Jahn-Hofmann K., Holzhey N., Ellinger T., Engels J.W.. A new concept for DNA-arrays. Nucleosides Nucleotides Nucleic Acids. 2003; 22:1479–1482. [DOI] [PubMed] [Google Scholar]
- 33. Matulic-Adamic J., Haeberli P., DiRenzo A.B., Mokler V.R., Maloney L., Beigelman L., Usman N., Wincott F.E.. Synthesis and incorporation of 5′-Amino- and 5′-Mercapto-5′-Deoxy-2′-O-Methyl nucleosides into hammerhead ribozymes. Nucleosides Nucleotides. 1997; 16:1933–1950. [Google Scholar]
- 34. Kuimelis R.G., McLaughlin L.W.. Cleavage properties of an oligonucleotide containing a bridged internucleotide 5′-phosphorothioate RNA linkage. Nucleic Acids Res. 1995; 23:4753–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mag M., Luking S., Engels J.W.. Synthesis and selective cleavage of an oligodeoxynucleotide containing a bridged internucleotide 5′-phosphorothioate linkage. Nucleic Acids Res. 1991; 19:1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sproat B.S., Beijer B., Rider P., Neuner P.. The synthesis of protected 5′-mercapto-2′,5′-dideoxyribonucleoside-3′-O-phosphoramidites - uses of 5′-mercapto-oligodeoxyribonucleotides. Nucleic Acids Res. 1987; 15:4837–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Islam M.A., Fujisaka A., Mori S., Ito K.R., Yamaguchi T., Obika S.. Synthesis and biophysical properties of 5′-thio-2′,4′-BNA/LNA oligonucleotide. Bioorg. Med. Chem. 2018; 26:3634–3638. [DOI] [PubMed] [Google Scholar]
- 38. Islam M.A., Waki R., Fujisaka A., Ito K.R., Obika S.. In vitro and in vivo biophysical properties of oligonucleotides containing 5′-thio nucleosides. Drug Discov. Ther. 2016; 10:263–270. [DOI] [PubMed] [Google Scholar]
- 39. Shah S., Sanchez J., Stewart A., Piperakis M.M., Cosstick R., Nichols C., Park C.K., Ma X., Wysocki V., Bitinaite J. et al.. Probing the run-on oligomer of activated SgrAI bound to DNA. PLoS One. 2015; 10:e0124783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li N.S., Tuttle N., Staley J.P., Piccirilli J.A.. Synthesis and incorporation of the phosphoramidite derivative of 2′-O-photocaged 3′-S-thioguanosine into oligoribonucleotides: substrate for probing the mechanism of RNA catalysis. J. Org. Chem. 2014; 79:3647–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keenholtz R.A., Mouw K.W., Boocock M.R., Li N.S., Piccirilli J.A., Rice P.A.. Arginine as a general acid catalyst in serine recombinase-mediated DNA cleavage. J. Biol. Chem. 2013; 288:29206–29214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu J., Li N.S., Sengupta R.N., Piccirilli J.A.. Synthesis and biochemical application of 2′-O-methyl-3′-thioguanosine as a probe to explore group I intron catalysis. Bioorg. Med. Chem. 2008; 16:5754–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das S.R., Piccirilli J.A.. General acid catalysis by the hepatitis delta virus ribozyme. Nat. Chem. Biol. 2005; 1:45–52. [DOI] [PubMed] [Google Scholar]
- 44. Kuo L.Y., Piccirilli J.A.. Leaving group stabilization by metal ion coordination and hydrogen bond donation is an evolutionarily conserved feature of group I introns. BBA-Gene Struct. Expr. 2001; 1522:158–166. [DOI] [PubMed] [Google Scholar]
- 45. Yoshida A., Sun S., Piccirilli J.A.. A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat. Struct. Biol. 1999; 6:318–321. [DOI] [PubMed] [Google Scholar]
- 46. Brautigam C.A., Sun S., Piccirilli J.A., Steitz T.A.. Structures of normal single-stranded DNA and deoxyribo-3′-S-phosphorothiolates bound to the 3′-5′ exonucleolytic active site of DNA polymerase I from Escherichia coli. Biochemistry. 1999; 38:696–704. [DOI] [PubMed] [Google Scholar]
- 47. Weinstein L.B., Jones B.C., Cosstick R., Cech T.R.. A second catalytic metal ion in group I ribozyme. Nature. 1997; 388:805–808. [DOI] [PubMed] [Google Scholar]
- 48. Curley J.F., Joyce C.M., Piccirilli J.A.. Functional evidence that the 3′-5′ exonuclease domain of Escherichia coli DNA polymerase I employs a divalent metal ion in leaving group stabilization. J. Am. Chem. Soc. 1997; 119:12691–12692. [Google Scholar]
- 49. Kuimelis R.G., McLaughlin L.W.. Ribozyme-mediated cleavage of a substrate analogue containing an internucleotide-bridging 5′-phosphorothioate: evidence for the single-metal model. Biochemistry. 1996; 35:5308–5317. [DOI] [PubMed] [Google Scholar]
- 50. Burgin A.B. Jr, Huizenga B.N., Nash H.A.. A novel suicide substrate for DNA topoisomerases and site-specific recombinases. Nucleic Acids Res. 1995; 23:2973–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roth A., Boess F., Landes C., Steiner G., Freichel C., Plancher J.M., Raab S., de Vera Mudry C., Weiser T., Suter L.. Gene expression-based in vivo and in vitro prediction of liver toxicity allows compound selection at an early stage of drug development. J. Biochem. Mol. Toxicol. 2011; 25:183–194. [DOI] [PubMed] [Google Scholar]
- 52. Straarup E.M., Fisker N., Hedtjarn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Orum H., Hansen J.B., Koch T.. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010; 38:7100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A.. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007; 8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nordestgaard B.G., Nicholls S.J., Langsted A., Ray K.K., Tybjaerg-Hansen A.. Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 2018; 15:261–272. [DOI] [PubMed] [Google Scholar]
- 55. Migawa M.T., Shen W., Wan W.B., Vasquez G., Oestergaard M.E., Low A., De Hoyos C.L., Gupta R., Murray S., Tanowitz M. et al.. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019; 47:5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weinstein L.B., Earnshaw D.J., Cosstick R., Cech T.R.. Synthesis and characterization of an RNA dinucleotide containing a 3′-S-phosphorothiolate linkage. J. Am. Chem. Soc. 1996; 118:10341–10350. [Google Scholar]
- 57. Crooke R.M., Graham M.J., Martin M.J., Lemonidis K.M., Wyrzykiewiecz T., Cummins L.L.. Metabolism of antisense oligonucleotides in rat liver homogenates. J. Pharmacol. Exp. Ther. 2000; 292:140–149. [PubMed] [Google Scholar]
- 58. Baek M.-S., Yu R.Z., Gaus H., Grundy J.S., Geary R.S.. In vitro metabolic stabilities and metabolism of 2′-O-(methoxyethyl) partially modified phosphorothioate antisense oligonucleotides in preincubated rat or human whole liver homogenates. Oligonucleotides. 2010; 20:309–316. [DOI] [PubMed] [Google Scholar]
- 59. Husser C., Brink A., Zell M., Müller M.B., Koller E., Schadt S.. Identification of GalNAc-conjugated antisense oligonucleotide metabolites using an untargeted and generic approach based on high resolution mass spectrometry. Anal. Chem. 2017; 89:6821–6826. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.