

Abstract
Objective
Hepatitis D virus (HDV) is a circular RNA virus coinfecting hepatocytes with hepatitis B virus. Chronic hepatitis D results in severe liver disease and an increased risk of liver cancer. Efficient therapeutic approaches against HDV are absent.
Design
Here, we combined an RNAi loss-of-function and small molecule screen to uncover host-dependency factors for HDV infection.
Results
Functional screening unravelled the hypoxia-inducible factor (HIF)-signalling and insulin-resistance pathways, RNA polymerase II, glycosaminoglycan biosynthesis and the pyrimidine metabolism as virus-hepatocyte dependency networks. Validation studies in primary human hepatocytes identified the carbamoyl-phosphatesynthetase 2, aspartate transcarbamylase and dihydroorotase (CAD) enzyme and estrogen receptor alpha (encoded by ESR1) as key host factors for HDV life cycle. Mechanistic studies revealed that the two host factors are required for viral replication. Inhibition studies using N-(phosphonoacetyl)-L-aspartic acid and fulvestrant, specific CAD and ESR1 inhibitors, respectively, uncovered their impact as antiviral targets.
Conclusion
The discovery of HDV host-dependency factors elucidates the pathogenesis of viral disease biology and opens therapeutic strategies for HDV cure.
Keywords: antiviral therapy, hepatitis D, screening, liver
Significance of this study.
What is already known on this subject?
Chronic hepatitis D is the most severe form of viral hepatitis.
Efficient therapeutic strategies are absent.
Hepatitis D virus is a small hepatitis B virus satellite virus.
Knowledge about HDV–hepatocyte interactions is limited.
HDV host-dependency factors are largely unknown.
What are the new findings?
A RNAi screen identified oestrogen receptor 1 (ESR1) and CAD as novel host factors for HDV infection.
The inhibition of CAD restricts HDV infection through uridine depletion.
ESR1 and CAD are functionally linked by transcriptional activation of gene expression.
ESR1 and CAD inhibitors fulvestrant and PALA, respectively, specifically inhibit HDV replication in a dose-dependent manner in human hepatocytes.
CAD and ESR1 are previously undiscovered targets for antiviral therapies.
How might it impact on clinical practice in the foreseeable future?
The discovery of HDV host-dependency factors opens the door for novel therapeutic strategies against chronic hepatitis D - a major unmet medical need.
Fulvestrant and PALA-like molecules are candidate compounds for HDV antivirals to enter preclinical development.
Introduction
Hepatitis delta virus (HDV) infects human hepatocytes and causes acute acerbation of liver disease in patients chronically infected with hepatitis B virus (HBV).1 Among HBV-infected patients, 5%–10% are coinfected with HDV.2 HDV coinfection or surinfection in HBV carriers induces more severe disease compared with infection with HBV alone. This includes a greater likelihood of experiencing liver failure and a more rapid progression to liver cirrhosis, with a markedly increased risk of developing hepatocellular carcinoma.3 Consequently, chronic hepatitis D is considered as one of the most severe forms of viral hepatitis. Treatment with HBV nucleos(t)ide analogues is not effective at reducing HDV replication or disease. Moreover, the virus is only poorly controlled by the current interferon (IFN)-based therapies.3 To date, no treatment allows efficient cure of hepatitis D with eradication of the virus and its associated disease.4–6 Thus, there is a major unmet medical need for curative therapies.
HDV is a small, circular RNA HBV satellite virus related to plant viroids using HBV envelope proteins to assemble its infectious particles.1 5 6 Consequently, it can only be propagated by HBV-infected hepatocytes producing HBV envelope proteins (Hepatitis B surface antigen or HBsAg). The HDV genome is a circular 1.7 kb single-stranded negative-sense RNA molecule (for a review on HDV virology and pathogenesis, see ref 5). The HDV life cycle starts with virus entry into hepatocyte after attachment to heparan sulfate proteoglycans (HSPG) at the cell surface.5 7 As HBV and HDV share the same envelope proteins, they appear to share the same entry pathway and receptor(s), including the sodium taurocholate cotransporting polypeptide (NTCP) and glypican 5 (reviewed in refs 5 6 8). The HDV genome is then imported in the nucleus, and three types of RNAs are transcribed by the host RNA polymerase II: replication is initiated by synthesis of an antigenomic RNA that serves as a template for the production of new HDV genomic RNA and HDV mRNA from genomic RNA.5 mRNA transcripts contain one unique open reading frame encoding the hepatitis delta antigen (HDAg) that exists in two forms (small version [S-HDAg] and large version [L-HDAg]) which play differential roles in the HDV life cycle.5 6 Indeed, S-HDAg is essential to HDV replication, while prenylation of L-HDAg leads to the inhibition of HDV replication and is crucial for binding to HBV envelope proteins and virion assembly.6 In the nucleus, L-HDAgs bind to the de novo HDV genomic RNA to form a new ribonucleoprotein, which is exported to the cytoplasm and interacts with HBsAgs at the endoplasmic reticulum to form new infectious virions that are then secreted to propagate further rounds of HDV infection.6 While the molecular virology of HDV has been elucidated, the role of host-dependency factors for HDV infection remains largely unknown.5
A unique feature of HDV is the apparent lack of non-structural protein encoded by the viral genome (including viral polymerase), making it highly dependent of the host machinery for its replication. Targeting host factors is an emerging concept in the treatment of infectious diseases including hepatitis viruses.9–12 Two host compounds targeting HDV factors have been developed up to clinical proof of concept: these include Myrcludex B, a small peptide targeting HBV/HDV entry factor NTCP, and the prenylation inhibitor lonafranib.4 13 14 Both compounds demonstrated a significant clinical antiviral effect in chronically infected patients,4 13 14 supporting the validity of the concept of host-targeting antivirals for HDV infection. However, data on long-term safety, potential resistance in subsets of patients and sustained virological response remain to be determined. Thus, complementary approaches targeting other host factors and/or steps of the HDV life cycle are needed for curative therapies.
Taking advantage of our recently developed robust HDV infection system for the study of HDV–host factor interactions7 combined with a dual screening approach, we aimed to uncover HDV host-dependency factors as antiviral targets.
Results
A high-throughput RNAi loss-of-function screen uncovers hepatocyte host-dependency factors required for HDV infection
To identify host factors required for HDV infection, we performed a high-throughput loss-of-function screen using the HDV-susceptible cell line Huh-106.7 Since one key goal was the discovery of druggable host targets, we used a library comprising siRNAs targeting 7567 genes, which have been selected as targets for therapeutics, including kinases, proteases, phosphatases, G protein-coupled receptor, ion channels, ubiquitin ligases, proteins involved in senescence, autophagy, DNA repair and targetable nuclear receptors. Huh-106 cells were transfected with siRNAs pools 48 hours before inoculation with HDV (figure 1A) with an optical immunostaining-based viral protein (HDAg) readout 7 days after infection. The silencing of expression of HDV host factor NTCP was used as functional positive control (figure 1B-C). The results of the primary screen are presented in online supplementary table S1.
Figure 1.

Identification of host-dependency factors for HDV infection using a high-throughput RNAi screen. (A). Approach with flow chart of the screen. NTCP-overexpressing Huh7 cells (termed Huh-106) were transfected with pools of four siRNAs per target of the Dharmacon ‘Druggable genome’ library 48 hours before HDV infection. Infection was assessed after 7 days by immunofluorescence. Each siRNA pool was tested in triplicate. As controls, Huh-106 cells were transfected with a non-targeting siRNA control (siCtrl), siRNA targeting SLC10A1 (siNTCP) and PLK1 (siPLK1, lethal for the cells) expression. Representative images of HDV infection in Huh-106 cells from the primary screen are shown in B. As readout for infection, cells were stained with an anti-HDAg antibody and cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). (C) General effect of the siNTCP on HDV infection at the screen level. Results are expressed as means ± SD % HDV-positive cells from siCtrl-treated cells (n=318) and siNTCP-treated cells (n=265). ***p value <0.001 (unpaired Student’s t-test). (D). Distribution of host-factor candidates according to their inhibitory effect on HDV infection. The top 5% (% HDV positive cells <8.3) were selected as candidates for further work-up. (E). Selection of candidate genes from the primary screen. From the 311 genes inducing a >45% decrease in HDV infection after silencing, candidate genes were selected depending on their robustness (p value and false discoery rate (FDR) <0.05), their expression in the liver (Illumina Body Map tool) and their toxicity. Toxicity was evaluated as the percentage of viability compared with the siCtrl, quantified by counting the DAPI-positive nuclei in the wells at the end of infection. From the 194 selected candidates, RNF130 was present twice (subset 8 and subset 10), and two pseudogenes (tAKR/AKR1C6P and LOC402164) were removed. One hundred and ninety-one candidates (top 2.5%) were further worked up. HDV, hepatitis D virus; NTCP, sodium taurocholate cotransporting polypeptide.
gutjnl-2018-317065supp001.pdf (3.8MB, pdf)
Using the hit selection algorithm described in online supplementary material and methods and figure 1E, we identified 191 host factor genes showing a robust correlation between gene silencing and level of HDV infection, moderate toxicity of gene-specific siRNA and their expression in the liver (figure 1D, E and online supplementary table S2). The threshold of hit selection (described in figure 1E) was based on the functional effect of SLC10A1 silencing (approximately 45% decrease in HDV infection, online supplementary table S1). The hit rate of 191 genes corresponds to 2.5% of total genes, which is similar to screens for other virus host factor-dependency screens.15 16
gutjnl-2018-317065supp003.pdf (104.2KB, pdf)
Functional pathway analysis using KEGG pathways identified a network of host factors belonging to HSPG biosynthesis, including EXT1 and EXT2 (figure 2A, B). These genes encode two major exostosins, key HSPG-related enzymes exhibiting glycosyltransferase activities and involved in the elongation of heparan sulfate chains.17 Moreover, several subunits of the RNA polymerase II were identified in our screen (figure 2B), including specific subunits such as POLR2G or POLR2I, encoding Rpb7 and Rpb9, which play key roles in the initiation of transcription and binding to DNA template for an accurate selection of the transcription start sites, respectively.18 19 The identification of HSPG enzymes and RNA polymerase II as known HDV-related pathways5 7 validates our approach for host factor discovery (figure 2). Importantly, the screen identified several previously unknown HDV-related networks, such as the hypoxia inducible factor 1 alpha (HIF-1α) signalling pathway, known to enhance the replication of several RNA and DNA viruses such as Epstein-Barr virus and HIV.20 21 Interestingly, the HIF-1α signalling pathway also plays a key role in hepatocarcinogenesis and liver tumour progression22 23 through its ability to target the expression of oncogenic genes such as the proliferation-specific transcription factor Forkhead box M1.24 HIF-1α overexpression in HCC has been correlated with worse clinical outcomes and is considered as a poor prognosis factor and molecular target for liver disease therapy.24 Interestingly, the highly significant scoring of insulin resistance-related pathways highlights the importance of hepatocyte metabolism as host-dependency factors on HDV infection (figure 2).
Figure 2.

Pathway analysis of HDV host factors identified within the RNAi screen. The identified host genes were subjected to functional enrichment pathway analysis through ToppGene Suite (https://toppgene.cchmc.org) using Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Pathways scoring at a FDR value of < 0.05 were considered significant. (A) Representation of the number of genes and FDR values of different KEGG pathways significantly enriched within the primary siRNA screen candidates. (B) Individual genes contributing to the enrichment of the significant KEGG pathways were further analyzed for protein-protein interactions using STRING database. Interaction networks were represented using Cytoscape 3.6.0.
Finally, we identified several host factors involved in the biosynthesis of pyrimidine, including CANT1, ENTPD5 and CAD.
The pyrimidine biosynthesis enzyme CAD is a key host factor for HDV infection and antiviral target in human hepatocytes
Given its high impact on HDV infection, functional relevance, liver expression and targetability, we focused on CAD, encoding the carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase (CAD), an enzyme playing a key role in the pyrimidine biosynthesis (online supplementary table S2; figure 2).25 Silencing of CAD led to a robust decrease in HDV infection (figure 3A). CAD is a trifunctional enzyme catalysing the first steps of pyrimidine biosynthesis.26 Interestingly, CAD expression is known to be regulated by activated estrogen receptor alpha (or estrogen receptor 1, ESR1), which binds to the transcription factor SP1,27 two other host-dependency factors uncovered by the RNAi screen (figure 3A). These data suggest a key role of the pyrimidine biosynthesis pathway and its associated nuclear proteins for the HDV life cycle.
Figure 3.

CAD is a key host factor required for HDV infection. (A) Functional validation of host factors belonging to the pyrimidine biosynthesis using perturbation studies. Results are presented as means ± SD % HDV infection compared with control siRNA (siCtrl, set at 100%) from three independent screens (n=9, primary screen and two validation screens performed in triplicate). ***p value <0.001 (unpaired Student’s t-test compared with siCtrl samples). (B–D) CAD is required for HDV infection. Huh-106 cells were reverse-transfected with four individual siRNAs targeting CAD mRNA or with the pool of four siRNAs. Silencing efficacy was assessed by western blot after 2 days. (B) One representative experiment is shown. Cells were then infected with HDV, and virus infection was assessed after 7 days by quantitative reverse transcription (qRT)-PCR. Results are expressed as means ± SEM % HDV infection compared with siCtrl (set at 100%) from three independent experiments (n=8). (C) Alternatively, HDV infection was assessed by immunofluorescence (IF) using a patient-derived anti-HDAg antibody. (D) One representative experiment using the pool of siCAD is shown. (E) HDV infection has no effect on CAD expression in PHH. PHH were treated with preS1 peptide (preS1) or a peptide control (Ctrl) for 1 hour prior to infection with HDV for 7 days. Results are expressed as means ± SD % HDV infection (assessed by HDV RNA levels) or CAD expression compared with control peptide-treated cells (Ctrl, set at 100%) from four independent experiments (n=8). (F–G) The CAD inhibitor PALA dose-dependently inhibits HDV infection in Huh-106 cells (F) and PHH (G). Cells were treated with PALA at the indicated concentrations 24 hours before infection with HDV. Cells were then cultured for 7 days in presence of PALA. HDV infection was assessed by qRT-PCR. Cell viability was assessed by Presto Blue. Results are expressed as means ± SD % HDV infection or cell viability compared with untreated cells (0, set at 100%) from three independent experiments (Huh-106 [F], n=9) or as means ± SEM % HDV infection or cell viability from three independent experiments (PHH [G], n=8). CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase; HDV, hepatitis D virus; PALA, N-(phosphonoa cetyl)-L-aspartic acid; PHH, primary human hepatocyte.
We next investigated the functional role of CAD in HDV infection. We first performed additional silencing studies using individual CAD-specific siRNA. As shown in figure 3B–D, a strong reduction of HDV RNA was observed after transfection of Huh-106 cells with the four individual siRNAs composing the siRNA pool, validating the key role of CAD expression in HDV infection. Notably, CAD expression was not significantly affected by HDV infection (figure 3E).
Aiming to characterise the potential of CAD as an antiviral target, we synthetised a specific inhibitor of CAD, sparfosic acid or N-(phosphonoacetyl)-L-aspartic acid (PALA).26 PALA is an aspartic acid derivative, which inhibits the aspartate carbamoyltransferase activity of the enzyme. We then investigated the antiviral activity of PALA in HDV-infected Huh-106 cells. As shown in figure 3F, PALA dose dependently inhibits HDV infection with an IC50=1.2 µM. Notably, a decrease in cell viability was observed after 7 days of culture.
To validate this result in the most physiological model for HDV infection, we investigated the antiviral activity of PALA using HDV infection of primary human hepatocytes (PHHs). Interestingly, while no toxicity was observed in PHH even at high doses (100 µM), the IC50 of PALA was slightly higher in PHH (figure 3G), which may reflect differences in uridine pools and/or CAD expression in rapidly dividing Huh7-based cells versus non-dividing PHH in cell culture.28
To investigate the mechanism of action of CAD–HDV interaction, we performed a rescue experiment using PALA and uridine-supplemented medium. Northern blot detection of HDV genomic RNA in infected cells revealed that the addition of uridine in PALA-treated cells restored HDV infection, validating the functional relevance of pyrimidine pathway for HDV replication (figure 4A). Notably, PALA at a concentration of 2.5 µM induced a strong decrease in HDV infection, which was efficiently rescued by uridine treatment (figure 4B, C). This dose was then selected for further functional studies. Importantly, no direct interaction was observed between CAD and the HDV antigens (figure 4D). As the inhibition of pyrimidine pathway has been shown to stimulate the innate antiviral response with subsequent inhibition of viral propagation,29 we next assessed the expression of IFNB1 following PALA treatment in Huh-106 cells. As shown in figure 4E, no IFNB1 induction was observed after treatment with either PALA or Brequinar, targeting dihydroorotate dehydrogenase (DHODH), suggesting an absence of innate immune stimulation by pyrimidine inhibitors in hepatocyte-derived cells.
Figure 4.

Characterisation of CAD–HDV functional interaction. (A–C) Uridine complementation restores HDV infection in PALA-treated cells. Huh-106 cells were treated with PALA (1 µM or 10 µM) in presence or absence of 30 µM uridine 24 hours prior to infection with HDV, and compound treatment was maintained for 7 days. HDV infection was assessed by northern blot (A). HDV+ corresponds to approximately 5.107 HDV RNA genome equivalents extracted from HDV particles produced in Huh7 cells. rRNA corresponds to ribosomal RNA. One representative experiment is shown. Alternatively, Huh-106 cells were treated with PALA 2.5 µM in presence or absence of 30 µM uridine. HDV infection was assessed after 7 days by IF (B) or qRT-PCR (C). Results are expressed as means ± SEM % HDV infection compared with HDV-infected untreated cells (Ctrl, set at 100%) from three independent experiments (n=6). (D) Absent direct interaction between HDV antigens and CAD. Huh-106 were transfected with pSVL(D3) plasmid encoding the HDV genome. Three days after transfection, cells were lysed, and HDAg-specific co-immunoprecipitation (co-IP) was performed using an anti-HDAg antibody (Delta) or with a control antibody (Ctrl). HDV antigens and CAD expression in the original cell lysate (input), in Flow-through control samples (FT) and in IP eluates (IP) were assessed by western blot. One experiment is shown. (E) No IFN induction after inhibition of pyrimidine biosynthesis in Huh-106 cells. Huh-106 cells were treated with Brequinar or PALA at the indicated concentrations. Alternatively, Huh-106 cells were reverse-transfected with Poly(I:C) (100 ng). Cells were then lysed every day for 3 days, and IFNB1 expression was assessed by qRT-PCR. Results are expressed as means ± SD relative IFNB1 expression (log10) compared with untreated or non-transfected cells (0, all set at 1) from three independent experiments (n=6). CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase; HDV, hepatitis D virus; PALA, N-(phosphonoacetyl)-L-aspartic acid.
To further validate the involvement of the biological function of CAD in HDV infection, cell culture medium was then complemented using either glutamine (Glu; the initial substrate of CAD) or dihydroorotate (DHO; the final product of CAD) (figure 5A, as described in ref 25). As shown in figure 5B and online supplementary figure S1, while Glu treatment had no effect on intracellular HDV RNA, DHO treatment in PALA-treated cells restored HDV RNA to non-treated levels, suggesting that PALA antiviral activity is linked to CAD physiological function.
Figure 5.

PALA specifically inhibits HDV replication by targeting CAD biological function. (A) Schematic representation of reactions catalysed by CAD enzyme. (B) DHO treatment restores HDV infection in PALA-treated cells. Huh-106 cells were treated with PALA 2.5 µM and infected with HDV in presence or absence of L-glutamine (Glu) or DHO at the indicated concentrations. Infection was assessed after 7 days by northern blot detection of HDV genomic RNA. rRNA corresponds to ribosomal RNA. One experiment is shown. (C) PALA inhibits both HDV genomic and antigenomic production. Huh-106 cells were treated with PALA 2.5 µM 24 hours prior to infection with HDV. HDV genomic and antigenomic RNAs were detected by northern blot using specific probes 7 days after infection. HDV+ corresponds to approximately 5.107 HDV RNA genome equivalents extracted from HDV particles produced in Huh7 cells. One experiment is shown. (D) Kinetics of HDV infection by PALA. Huh-106 cells were pretreated with PALA at 2.5 µM and infected with HDV (conditions P#1 and P#2). Cells were then cultured in presence of PALA for 48 hours (P#1) or for 7 days (P#2) in presence or absence of uridine (30 µM). Alternatively, Huh-106 cells were infected with HDV with no PALA pretreatment (P#3). Sixteen hours after viral inoculation, cells were cultured in presence of PALA 2.5 µM with or without uridine (30 µM). HDV infection was assessed after 7 days by qRT-PCR. Results are expressed as means ± SEM % HDV infection compared with HDV-infected untreated cells (Ctrl, set at 100%) from three independent experiments (n=6). (E) PALA does not affect HDV binding. Huh-106 cells were cultured for 24 hours at 16°C in presence of HDV particles, which were pretreated or not with heparin (30 µg/mL). HDV binding was measured by qRT-PCR quantification of total HDV RNA bound to cells after 24 hours. Results are expressed as means ± SD % HDV binding relative to control untreated cells (Ctrl, set at 100%) from three independent experiments (n=9). (F–H) CAD inhibition or silencing does not affect HBV infection and replication. (F) HBV-producing HepAD38 cells were treated with PALA (10 µM) for 3 days. HBeAg and HBsAg secretion in culture supernatant was then quantified by chemiluminescent immunoassay (CLIA). Cell viability was assessed by Presto Blue. Results are expressed as means ± SEM % HBeAg production, HBsAg production or cell viability compared with untreated cells (0 µM, all set at 100%) from three independent experiments (n=6). (G) HepAD38 cells were treated with either tenofovir (TFV) or PALA at the indicated concentrations for 3 days. HBV replication was assessed by quantification of HBV DNA copies in the supernatant of treated cells by qPCR. Results are expressed as means ± SD % HBV DNA in the supernatant or cell viability compared with untreated cells (Ctrl, all set at 100%) from three independent experiments (n=9). (H) HepG2-NTCP cells were reverse-transfected with siCAD or siCtrl for 2 days prior to infection with HBV. HBV infection was assessed after 10 days by quantification of HBeAg and HBsAg production using chemiluminescent immunoassay (CLIA). Results are expressed as means ± SD % CAD expression, HBeAg production or HBsAg production compared with siCtrl-transfected cells (siCtrl, all set at 100%) from three independent experiments (n=6). CA, Carbamoyl aspartic acid; CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase; DHO: dihydroorotate; Glu, glutamine; HBV, hepatitis B virus; HDV, hepatitis D virus; PALA, N-(phosphonoacetyl)-L-aspartic acid; Pi, inorganic phosphate; UMP, uridine monophosphate.
gutjnl-2018-317065supp002.pdf (104.2KB, pdf)
To map the replication steps mediated by CAD and targeted by PALA, we performed northern blot analyses of HDV RNA allowing to quantify the HDV genome and antigenome in the presence or absence of PALA. As shown in figure 5C, PALA treatment effectively decreased the levels of both HDV genomic and antigenomic RNAs. Moreover, this inhibition was already observed during the early stages of viral replication (online supplementary figure S1). These data demonstrate that PALA inhibits all the steps of HDV replication including the synthesis of antigenomic RNA (figure 5C).
Next, we performed kinetic studies adding PALA preinfection, peri-infection and HDV postinfection (figure 5D). PALA potently inhibited HDV infection given at any time before, during and following initiation of replication. Interestingly, the inhibitory effect was most pronounced when PALA was added prior or during initiation of replication. Finally, to exclude effects on other steps of the early HDV life cycle, we studied the effect of PALA treatment on HDV binding to the Huh-106 cell surface. The absent effect on HDV binding further confirms HDV replication as the life cycle step targeted by PALA (figure 5E).
To test the specificity of the pyrimidine pathway for HDV infection, we treated HBV-producing HepAD38 cells with PALA. As shown in figure 5F, PALA did not modulate HBV antigen production as shown by an absent effect on HBeAg and HBsAg secretion. While tenofovir (TFV) treatment dose-dependently inhibited HBV replication, PALA had no effect on HBV DNA in the supernatant of HepAD38 cells (figure 5G). Finally, the silencing of CAD expression did not affect HBV infection in HepG2-NTCP cells (figure 5H). These results suggest that the pyrimidine/CAD pathway is relevant for HDV but not HBV replication. Collectively, our results identify CAD as a key HDV host factor involved in HDV replication and target for antiviral therapy.
A small molecule screen uncovers ESR1 inhibitor fulvestrant as host targeting agent
Next, we performed a small molecule screen using the Prestwick library, containing 1280 drugs approved by the Food and Drug Administration (FDA) (figure 6A). Among the molecules exhibiting an antiviral activity against HDV, we identified fluvastatin and cyclosporin A, two well-described NTCP inhibitors (online supplementary table S3), confirming the validity of the screen. Interestingly, ribavirin, a broad antiviral nucleoside analogue30 previously used for the treatment of chronic hepatitis C31 exhibits a marked antiviral activity against HDV (online supplementary table S3). In the same vein, nelfinavir, a protease inhibitor presenting antiretroviral activity32 was also identified in the drug screen (online supplementary table S3).
Figure 6.
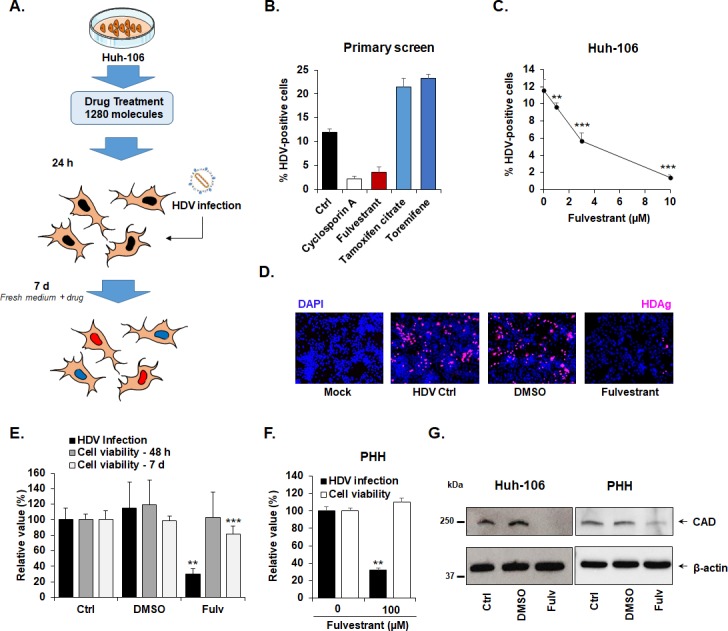
ESR1 inhibitor fulvestrant inhibits HDV replication by suppression of CAD expression. (A) Small molecule screen flow chart. Huh-106 cells were treated with individual compounds belonging to the Prestwick chemical library (10 µM) 1 day prior to infection with HDV. Infection was assessed after 7 days by immunofluorescence. Every compound was tested in triplicate. (B). ESR1 modulators affect HDV infection. From the primary screen, fulvestrant (an ESR1 inhibitor) from the one hand, and tamoxifen and toremifen from the other hand (two agonists/modulators of ESR1) exhibited antiviral or proviral activity against HDV, respectively. Results are presented as means ± SD % HDV infected cells (n=3). (C). Fulvestrant dose-dependently inhibits HDV infection. Huh-106 cells were treated with fulvestrant at the indicated concentrations and then infected with HDV for 7 days. Results are expressed as means ± SD % HDV infected cells from three independent experiments (n=9). (D) Short fulvestrant treatment for 48 hours following virus inoculation inhibits HDV infection. Huh-106 cells were infected with HDV and then treated with fulvestrant (10 µM) or dimethyl sulfoxide (DMSO) for 48 hours. Cells were then cultured in absence of drug and infection was assessed after 7 days by IF. One representative experiment is shown. (E) Fulvestrant antiviral activity is associated with slight cytotoxicity. Cell viability was assessed after 48 hours and after 7 days by Presto blue. HDV infection was assessed after 7 days by qRT-PCR. Results are expressed as means ± SD % HDV infection (HDV RNA) or cell viability compared with untreated HDV-infected cells (Ctrl, set at 100%) from three independent experiments (n=9). (F) Fulvestrant antiviral activity in PHH. PHH were infected with HDV, and then treated with fulvestrant for 48 hours at the indicated concentrations. Results are expressed as means ± SD % HDV infection (HDV RNA) or cell viability compared with untreated HDV-infected PHH (Ctrl, set at 100%) from two independent experiments (n=6). (G) Fulvestrant inhibits CAD expression. Huh-106 cells and PHH were treated with fulvestrant (Fulv, 10 µM) or DMSO for 72 hours. CAD expression was assessed by western blot. CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase; HDV, hepatitis D virus; ESR1, estrogen receptor 1; PHH, primary human hepatocyte.
gutjnl-2018-317065supp004.pdf (715.3KB, pdf)
Given our discovery of ESR1 as HDV host-dependency factor, we next focused on ESR1-targeting agents. The library includes several agonists and antagonists of ESR1, as well as characterised targets for endocrine therapy in breast cancer.33 Interestingly, the ESR1 antagonist fulvestrant exhibited a marked, significant and dose-dependent antiviral effect against HDV, whereas tamoxifen and toremifen, two ESR1 modulators with oestrogenic effects in the liver34 increased HDV infection, validating the importance of ESR1 in HDV infection (figure 6B-C). Interestingly, a 48-hour treatment started after virus inoculation induced a marked decrease in HDV-infected cells (figure 5D), suggesting an effect on HDV replication.
Next, we aimed investigate the mechanism of action of the antiviral activity of fulvestrant by investigating a functional link of ESR1 and CAD suggested by protein/protein interaction studies.27 To address this question, CAD expression was analysed in Huh-106 cells and PHH in the presence or absence of the ESR1 antagonist (figure 6E-G). As shown in figure 6G, fulvestrant treatment resulted in a loss of CAD protein expression in both Huh-106 cell line and PHH, suggesting that the antiviral activity of ESR1-inhibitor fulvestrant is mediated by ESR1-mediated downregulation of CAD expression, which in turn is required for HDV replication, confirming the previous observations with PALA.
Taken together, our dual screening approach uncovers the pyrimidine pathway and its associated and regulatory proteins as host factors for HDV infection and targets for antiviral therapy.
Discussion
Chronic hepatitis D is the most severe form of viral hepatitis, and at present time, no treatment allows robust viral clearance. Using a dual screening approach, we identified ESR1 and CAD as host factors and antiviral targets for HDV infection. The functional impact of the pyrimidine biosynthesis for HDV replication is corroborated by: (1) a marked decrease in HDV infection after CAD and ESR1 silencing; (2) a significant antiviral effect of CAD and ESR1 inhibitors PALA and fulvestrant; and (3) kinetic experiments mapping viral replication as the step of the viral life cycle mediated by these host factors and targeted by the antiviral (4) validation studies in primary human hepatocytes.
Whereas the molecular virology of HDV is well described, the molecular interactions between the virus and liver host factors are still largely unknown.5 In mammalian cells, the pyrimidine biosynthesis pathway leads to the production of de novo nucleotides and plays a key role in RNA and DNA production, as well as protein glycosylation or cell membrane assembly.25 Moreover, uridine nucleotides and derivatives regulate key physiological processes, such as lipid metabolism, regulation of normal central nervous system activity or modulation of reproduction,35 making this pathway a major actor of important metabolic processes. As a source of nucleotides for HDV RNA, this pathway is likely to be required for optimal virus replication. Moreover, as a key regulator of viral glycosylation,25 an alteration of this pathway may alter post-translational modification of host or viral proteins, disrupting viral infection.
CAD is a multifunctional enzyme exhibiting carbamoylphosphate synthetase, aspartate transcarbamoylase and dihydroorotase activities catalysing the three first steps of the pyrimidine biosynthesis pathway,36 playing a key role in HDV infection as such. The observed antiviral activity of the CAD-specific inhibitor PALA validates the importance of this factor and pathway in HDV infection. It is of interest to note that another enzyme of the pyrimidine biosynthesis pathway DHODH, which catalyses the production of orotate from DHO, has been described as an antiviral target by small molecule GSK983.37 Here, we confirmed that the CAD-mediated DHO and uridine starvation is responsible for the observed antiviral effect (figures 4 and 5). Interestingly, HSPG synthesis has been shown to be dependent on the pyrimidine synthesis pathway in a CAD-defective animal model.38 However, we did not observe any effect of CAD inhibition on either HBV infection or HDV entry (figure 5E–H). While recent studies have suggested that the inhibition of pyrimidine biosynthesis can stimulate innate antiviral responses,29 no induction of IFN expression was observed in our model (figure 4E). Thus, it is likely that the modulation of CAD activity or expression affects HDV infection through its classical enzymatic activity linked to pyrimidine metabolism, mainly affecting viral replication through uridine starvation, affecting both genomic and antigenomic HDV RNAs. In this context, our study suggests that the first steps of replication are particularly susceptible to PALA. These results are consistent with the timing of HDV RNA amplification, much more pronounced during the early phase of replication, before L-HDAg-mediated slowdown of replication.5
Among the genes regulating the pyrimidine pathway, we identified ESR1 as a HDV host-dependency factor and antiviral target. ESR1 is a nuclear hormone receptor expressed in the mammary gland and female reproductive track and also in lung and liver.39 In our mechanistic analyses, we show that ESR1 inhibition by fulvestrant led to a decrease in CAD protein expression, which most likely explains the antiviral effect of the molecule.
Our screening strategy also identified small molecules exhibiting significant antiviral activity against HDV infection. Among them was found ribavirin, a nucleoside analogue previously used for the treatment of chronic HCV infection.40 Given its concentration in the primary screen (10 µM), HDV inhibition is most likely due to the ability of ribavirin to inhibit de novo synthesis of GTP as recently suggested as a possible mechanism for the inhibition of hepatitis E virus (HEV) replication in Huh7 cells (IC50 of 18.9 µM for GTP depletion; EC50 of 3 µM for HEV replication41). However, clinical attempts to treat hepatitis D using ribavirin were largely unsuccessful.4
Collectively, we identified previously undiscovered pathways and host factors for HDV infection and antiviral targets. Host-targeting agents are a promising approach for the development of new antiviral treatments, notably because they limit the emergence of resistant variants.8–10 42 In particular for RNA viruses, which do not integrate into the genome, host-targeting agents hold promise for viral cure.43 Indeed, in chronic HCV infection antibodies targeting a viral host entry factor claudin-1 cure viral infection in a state-of-the-art mouse model,12 and a miRNA-122 antagonist has been shown to cure chronic HCV infection in monotherapy in a subset of patients.44 Two host compounds targeting HDV factors have been developed up to clinical proof of concept: these include Myrcludex B, a small peptide-targeting HBV/HDV entry factor NTCP and the prenylation inhibitor lonafranib.4 13 14 In randomised clinical trials, both compounds demonstrated a significant and robust antiviral effect in chronically infected patients,4 13 14 supporting the validity of the concept of host-targeting antivirals for HDV infection. However, long-term safety, potential resistance in subsets of patients and data on sustained virological response remain to be determined. The discovery of novel host factors described in this study may overcome these limitations by complementary, safer and more efficient approaches. Compounds targeting essential host factors of the viral replication step may be conceptually superior to entry or prenylation inhibitors by targeting the most vulnerable step of the viral life cycle. Indeed, viral replication is the target of many effective clinically licenced antiviral therapies such as nucleoside analogues for HBV infection or polymerase/protease inhibitors for HIV and HCV infection.
A theoretical disadvantage of HTA is their putative adverse effects on physiological processes mediated by the host factors. In this context, given that an HDV treatment will occur in a context of diseased liver, assessment of safety will be a major parameter in development. In this regard, it is of interest to note that pyrimidine synthesis inhibitors, such as leflunomide or teriflunomide, are currently used for the treatment of rheumatoid arthritis and multiple sclerosis.45 46 The safety profile of these compounds suggests that pyrimidine biosynthesis is a targetable pathway for antiviral strategies. Indeed, the FDA-approved leflunomide, targeting DHODH, induces uridine starvation at concentrations similar to PALA concentrations we used in this study (0–100 µM).47 Although detailed toxicity analyses in animal model and human tissues will be required to assess the therapeutic window of PALA, the dose–response effects of leflunomide suggests that PALA doses required to inhibit HDV infection are most likely in a range that will have an acceptable clinical safety profile.
In conclusion, by uncovering key host factors for HDV infection our results significantly improve the understanding of the HDV life cycle and contribute to the development of novel antiviral strategies for HDV cure.
Material and methods
Human subjects
Human serum from patients with chronic HBV/HDV infection followed at the Strasbourg University Hospitals, Strasbourg, France, was obtained with informed consent. PHHs were obtained from liver tissue from patients undergoing liver resection for liver metastasis at the Strasbourg University Hospitals with informed consent.
Cell lines and human hepatocytes
Huh-106,7 HepAD387 and HepG2-NTCP7 cells have been described. PHHs were isolated and cultured as described.48
Key reagents
Fulvestrant (I4409), DL-dihydroorotic acid (DHO, D7003), L-glutamine (59 202C), uridine (U3003) and TFV (1643601) were purchased from Sigma-Aldrich (Merck). Sparfocid acid (L-Aspartic acid, N-(phosphonoacetyl)-, disodium salt (9 CI) or PALA, NSC: 224131) was first obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institute of Health, Bethesda, Maryland, USA. For further analyses, PALA was synthetised at the Institute of Chemistry, University of Strasbourg, as described in online supplementary material and methods. PreS1 peptide was synthetised by Bachem.7
HDV and HBV production and infection
The production of recombinant HDV and HBV infectious virus as well as the protocol for HDV infection of Huh-106 cells and PHHs and for HBV infection of HepG2-NTCP cells have been described.7 49 50 A detailed protocol is presented in online supplementary material and methods.
RNAi loss-of-function and small molecule screens
Screening was performed at the High Throughput Screening platform of the Institut de Génétique et de Biologie Moléculaire et Cellulaire in Illkirch, France. The Human ON-TARGETplus ‘Druggable Genome’ siRNA Library was used for gene expression silencing. For the small molecule screen, the Chemical Library (PCL) containing 1280 FDA-approved molecules was obtained from Prestwick. Detailed information about both screening strategies as well as the algorithm for selection of HDV-dependency candidates are presented in online supplementary material and methods.
Validation of CAD as a HDV host factor using individual siRNAs
Huh-106 cells were reverse-transfected with the four individual siRNAs from the siCAD pool, a pool of siRNA targeting SLC10A1 (siNTCP) or a non-targeting siRNA control (siCtrl) using lipofectamin RNAi max as described.7 50 Gene expression was assessed, 2 days after transfection, by western blot detection of CAD protein using a rabbit monoclonal anti-CAD antibody (Abcam ab40800) as previously described.7 50 β-actin expression was assessed as a loading control using a mouse monoclonal anti-β-actin antibody (Sigma, A5441). Two days after transfection, cells were infected by HDV, and infection was assessed after 7 days as described in online supplementary material and methods.
Statistical analysis
All experiments were performed at least twice in an independent manner. Statistical analyses were performed using a two-tailed Mann-Whitney U test unless otherwise stated; p<0.05 (*), p<0.01 (**) and p<0.001 (***) were considered statistically significant. Significant p values are indicated by asterisks in the individual figures and figure legends. The number of biological replicates is indicated in the figure legends (n).
Additional methodological information are available in online supplementary material.
Acknowledgments
We would like to thank J Taylor (Fox Chase Cancer Center) for the gift of the HDV expression plasmid (pSVL(D3)) and our colleagues S Durand and M Renaud for excellent technical support. We thank the INGESTEM infrastructure for access to the IGBMC high-throughput high-content screening workstation.
Footnotes
Contributors: TB initiated the study. TB and ERV designed and supervised research. ERV, AW, LB and TB set up, designed and performed the siRNA and small molecule screens. TG and PPa produced the sparfosic acid. ERV, CB, LH, VT-L and EC performed the validation experiments. CSu performed the HDV northern blots. PPe provided human hepatocytes. ERV, AW, CB, LH, EC, MBZ, CSu, CSc, LB and TB analysed the data. ERV, HES and AK performed the bioinformatical analyses of the screens. EV and TB wrote the manuscript. All the authors approved the study.
Funding: This work was supported by Inserm, the University of Strasbourg, the European Union (Infect-ERA hepBccc, ERC-2014-AdG-671231-HEPCIR and Horizon 2020 research and innovation programme under grant agreement 667273 - HEPCAR), Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS 15/1099) and the French Cancer Agency (ARC IHU201301187). This work has been published under the framework of the LabEx ANR-10-LAB-28 and benefits from a funding from the state managed by the French National Research Agency as part of the Investments for the Future (Investissements d’Avenir) programme. ERV is the recipient of an ANRS fellowship (ECTZ50121).
Competing interests: None declared.
Ethics approval: Protocols were approved by the local Ethics Committee of the Strasbourg University Hospitals (CPP) and the Ministry of Higher Education and Research of France (DC 2016 2616).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: The datasets generated in this study, including the results from both RNAi and small molecule primary screens, are available within online supplementary material files. The rest of the data are available from the corresponding authors on reasonable request.
References
- 1. Taylor JM. Virology of hepatitis D virus. Semin Liver Dis 2012;32:195–200. 10.1055/s-0032-1323623 [DOI] [PubMed] [Google Scholar]
- 2. Fattovich G, Giustina G, Christensen E, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European concerted action on viral hepatitis (Eurohep). Gut 2000;46:420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. The Lancet 2011;378:73–85. 10.1016/S0140-6736(10)61931-9 [DOI] [PubMed] [Google Scholar]
- 4. Rizzetto M. Investigational drugs in development for Hepatitis D. Expert Opin Investig Drugs 2017;26:999–1005. 10.1080/13543784.2017.1357695 [DOI] [PubMed] [Google Scholar]
- 5. Sureau C, Negro F. The hepatitis delta virus: replication and pathogenesis. J Hepatol 2016;64:S102–16. 10.1016/j.jhep.2016.02.013 [DOI] [PubMed] [Google Scholar]
- 6. Webb CT, Lupták A. Kinetic parameters of trans scission by extended hdv-like ribozymes and the prospect for the discovery of genomic trans-cleaving RNAs. Biochemistry 2018;57:1440–50. 10.1021/acs.biochem.7b00789 [DOI] [PubMed] [Google Scholar]
- 7. Verrier ER, Colpitts CC, Bach C, et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016;63:35–48. 10.1002/hep.28013 [DOI] [PubMed] [Google Scholar]
- 8. Verrier ER, Colpitts CC, Sureau C, et al. Hepatitis B virus receptors and molecular drug targets. Hepatol Int 2016;10:567–73. 10.1007/s12072-016-9718-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumert TF, Verrier ER, Nassal M, et al. Host-targeting agents for treatment of hepatitis B virus infection. Curr Opin Virol 2015;14:41–6. 10.1016/j.coviro.2015.07.009 [DOI] [PubMed] [Google Scholar]
- 10. Colpitts CC, Verrier ER, Baumert TF. Targeting viral entry for treatment of Hepatitis B and C virus infections. ACS Infect Dis 2015;1:420–7. 10.1021/acsinfecdis.5b00039 [DOI] [PubMed] [Google Scholar]
- 11. Harak C, Meyrath M, Romero-Brey I, et al. Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat Microbiol 2016;2:16247 10.1038/nmicrobiol.2016.247 [DOI] [PubMed] [Google Scholar]
- 12. Mailly L, Xiao F, Lupberger J, et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat Biotechnol 2015;33:549–54. 10.1038/nbt.3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koh C, Canini L, Dahari H, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis 2015;15:1167–74. 10.1016/S1473-3099(15)00074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol 2016;65:490–8. 10.1016/j.jhep.2016.04.016 [DOI] [PubMed] [Google Scholar]
- 15. Li Q, Brass AL, Ng A, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 2009;106:16410–5. 10.1073/pnas.0907439106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brass AL, Dykxhoorn DM, Benita Y, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008;319:921–6. 10.1126/science.1152725 [DOI] [PubMed] [Google Scholar]
- 17. Busse-Wicher M, Wicher KB, Kusche-Gullberg M. The exostosin family: proteins with many functions. Matrix Biol 2014;35:25–33. 10.1016/j.matbio.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 18. Sharma N, Kumari R. Rpb4 and Rpb7: multifunctional subunits of RNA polymerase II. Crit Rev Microbiol 2013;39:362–72. 10.3109/1040841X.2012.711742 [DOI] [PubMed] [Google Scholar]
- 19. Hull MW, McKune K, Woychik NA. RNA polymerase II subunit RPB9 is required for accurate start site selection. Genes Dev 1995;9:481–90. 10.1101/gad.9.4.481 [DOI] [PubMed] [Google Scholar]
- 20. Kraus RJ, Yu X, Cordes BA, et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog 2017;13:e1006404 10.1371/journal.ppat.1006404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morinet F, Parent M, Pillet S, et al. Hypoxia inducible factor one alpha and human viral pathogens. Curr Res Transl Med 2017;65:7–9. 10.1016/j.retram.2016.11.002 [DOI] [PubMed] [Google Scholar]
- 22. Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun 2017;8:517 10.1038/s41467-017-00530-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo D, Wang Z, Wu J, et al. The Role of Hypoxia Inducible Factor-1 in Hepatocellular Carcinoma. Biomed Res Int 2014;2014:1–11. 10.1155/2014/409272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol 2015;21:12171–8. 10.3748/wjg.v21.i42.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem 2004;279:33035–8. 10.1074/jbc.R400007200 [DOI] [PubMed] [Google Scholar]
- 26. Löffler M, Fairbanks LD, Zameitat E, et al. Pyrimidine pathways in health and disease. Trends Mol Med 2005;11:430–7. 10.1016/j.molmed.2005.07.003 [DOI] [PubMed] [Google Scholar]
- 27. Wang Y, Wang Z, Liu T, et al. Effects of flanking regions on HDV cotranscriptional folding kinetics. RNA 2018;24:1229–40. 10.1261/rna.065961.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swyryd EA, Seaver SS, Stark GR. N-(phosphonacetyl)-L-aspartate, a potent transition state analog inhibitor of aspartate transcarbamylase, blocks proliferation of mammalian cells in culture. J Biol Chem 1974;249:6945–50. [PubMed] [Google Scholar]
- 29. Lucas-Hourani M, Dauzonne D, Jorda P, et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog 2013;9:e1003678 10.1371/journal.ppat.1003678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reyes GR. Ribavirin: recent insights into antiviral mechanisms of action. Curr Opin Drug Discov Devel 2001;4:651–6. [PubMed] [Google Scholar]
- 31. Sulkowski MS, Cooper C, Hunyady B, et al. Management of adverse effects of Peg-IFN and ribavirin therapy for hepatitis C. Nat Rev Gastroenterol Hepatol 2011;8:212–23. 10.1038/nrgastro.2011.21 [DOI] [PubMed] [Google Scholar]
- 32. Perry CM, Frampton JE, McCormack PL, et al. Nelfinavir: a review of its use in the management of HIV infection. Drugs 2005;65:2209–44. 10.2165/00003495-200565150-00015 [DOI] [PubMed] [Google Scholar]
- 33. Jeselsohn R, Buchwalter G, De Angelis C, et al. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 2015;12:573–83. 10.1038/nrclinonc.2015.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellmén J, Hakulinen P, Partanen A, et al. Estrogenic effects of toremifene and tamoxifen in postmenopausal breast cancer patients. Breast Cancer Res Treat 2003;82:103–11. 10.1023/B:BREA.0000003957.54851.11 [DOI] [PubMed] [Google Scholar]
- 35. Connolly GP, Duley JA. Uridine and its nucleotides: biological actions, therapeutic potentials. Trends Pharmacol Sci 1999;20:218–25. 10.1016/S0165-6147(99)01298-5 [DOI] [PubMed] [Google Scholar]
- 36. Lee L, Kelly RE, Pastra-Landis SC, et al. Oligomeric structure of the multifunctional protein CAD that initiates pyrimidine biosynthesis in mammalian cells. Proc Natl Acad Sci U S A 1985;82:6802–6. 10.1073/pnas.82.20.6802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deans RM, Morgens DW, Ökesli A, et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol 2016;12:361–6. 10.1038/nchembio.2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franks DM, Izumikawa T, Kitagawa H, et al. C. elegans pharyngeal morphogenesis requires both de novo synthesis of pyrimidines and synthesis of heparan sulfate proteoglycans. Dev Biol 2006;296:409–20. 10.1016/j.ydbio.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 39. Droog M, Mensink M, Zwart W. The estrogen receptor α-cistrome beyond breast cancer. Mol Endocrinol 2016;30:1046–58. 10.1210/me.2016-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loustaud-Ratti V, Debette-Gratien M, Jacques J, et al. Ribavirin: past, present and future. World J Hepatol 2016;8:123–30. 10.4254/wjh.v8.i2.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Debing Y, Emerson SU, Wang Y, et al. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob Agents Chemother 2014;58:267–73. 10.1128/AAC.01795-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Levrero M, Testoni B, Zoulim F. HBV cure: why, how, when? Curr Opin Virol 2016;18:135–43. 10.1016/j.coviro.2016.06.003 [DOI] [PubMed] [Google Scholar]
- 43. Zeisel MB, Baumert TF. Clinical development of hepatitis C virus host-targeting agents. Lancet 2017;389:674–5. 10.1016/S0140-6736(17)30043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van der Ree MH, de Vree JM, Stelma F, et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet 2017;389:709–17. 10.1016/S0140-6736(16)31715-9 [DOI] [PubMed] [Google Scholar]
- 45. Zhang C, Chu M. Leflunomide: a promising drug with good antitumor potential. Biochem Biophys Res Commun 2018;496:726–30. 10.1016/j.bbrc.2018.01.107 [DOI] [PubMed] [Google Scholar]
- 46. Bar-Or A, Pachner A, Menguy-Vacheron F, et al. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014;74:659–74. 10.1007/s40265-014-0212-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baumann P, Mandl-Weber S, Volkl A, et al. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther 2009;8:366–75. 10.1158/1535-7163.MCT-08-0664 [DOI] [PubMed] [Google Scholar]
- 48. Lupberger J, Zeisel MB, Xiao F, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 2011;17:589–95. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verrier ER, Yim SA, Heydmann L, et al. Hepatitis B virus evasion from cyclic guanosine monophosphate-adenosine monophosphate synthase sensing in human Hepatocytes. Hepatology 2018;68:1695–709. 10.1002/hep.30054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verrier ER, Colpitts CC, Bach C, et al. Solute Carrier NTCP Regulates innate antiviral immune responses targeting Hepatitis C virus infection of hepatocytes. Cell Rep 2016;17:1357–68. 10.1016/j.celrep.2016.09.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
gutjnl-2018-317065supp001.pdf (3.8MB, pdf)
gutjnl-2018-317065supp003.pdf (104.2KB, pdf)
gutjnl-2018-317065supp002.pdf (104.2KB, pdf)
gutjnl-2018-317065supp004.pdf (715.3KB, pdf)