

Abstract
SNAI1, an epithelial-mesenchymal transition (EMT)-inducing transcription factor, promotes tumor metastasis and resistance to apoptosis and chemotherapy. SNAI1 protein levels are tightly regulated by proteolytic ubiquitination. Here, we identified USP37 as a SNAI1 deubiquitinase that removes the polyubiquitination chain from SNAI1 and prevents its proteasomal degradation. USP37 directly binds, deubiquitinates, and stabilizes SNAI1. Overexpression of wild-type USP37, but not its catalytically inactive mutant C350S, promotes cancer cell migration. Importantly, depletion of USP37 downregulates endogenous SNAI1 protein and suppresses cell migration, which can be reversed by re-expression of SNAI1. Taken together, our findings suggest that USP37 is a SNAI1 deubiquitinase and a potential therapeutic target to inhibit tumor metastasis.
Keywords: SNAI1, USP37, EMT, deubiquitinase, degradation
Introduction
More than 90% of cancer-related deaths occur in patients with metastasis [1], a multistep process in which cancer cells in the primary tumor invade extracellular matrix and stromal cell layers, intravasate into the blood-circulating and lymphatic systems, extravasate through distant capillaries, and proliferate into secondary tumors at distant anatomic sites [2,3]. Epithelial-to-mesenchymal transition (EMT), a developmental phenomenon thought to be hijacked by pathological processes, is characterized by loss of cell-cell adhesion, repression of epithelial marker expression (e.g., E-cadherin), acquisition of mesenchymal markers (e.g., vimentin), and increased cell motility and invasiveness [4]. EMT was initially identified and characterized in early embryonic morphogenesis [5,6], which enables embryonic cells to move and form various organs [7]. The EMT program is usually inactive in adult tissues but can be reactivated during chronic inflammation or injury [7]. Notably, cancer cells that have undergone EMT become migratory and invasive, which empowers them to disseminate and metastasize [8,9].
SNAI1, an EMT-inducing zinc-finger transcription factor, binds to two E2-box elements proximal to the transcriptional start site of the CDH1 gene, leading to repression of E-cadherin expression [10,11]. As a convergence point in EMT induction [12], SNAI1 has been implicated in multiple EMT-dependent and EMT-independent functions contributing to tumor growth and metastasis [13,14]. For instance, SNAI1-induced EMT not only enhances the migratory capability of cancer cells but also suppresses host immune surveillance to promote melanoma metastasis [15]. In cultured cells and developing embryos, SNAI1 slows down cell cycle progression by repressing cyclin D2 transcription in a context-dependent manner and confers resistance to cell death by activating survival signaling, such as MEK-ERK signaling and PI3K-AKT signaling [16]. In basal-like breast cancer, SNAI1 interacts with the H3K9 methyltransferase G9a and DNA methyltransferase Dnmt1 to induce promoter hypermethylation and epigenetic silencing of fructose-1,6-biphosphatase (FBP1), thereby leading to increased glucose uptake, macromolecule biosynthesis, and maintenance of ATP production under hypoxic conditions [17]. Importantly, higher SNAI1 protein levels correlate with higher tumor grade, metastasis, and poor clinical outcome [10,18,19]. Thus, a better understanding of SNAI1 regulation will provide important insights into prevention of tumor progression and metastasis.
The expression of SNAI1 is regulated at the transcriptional level by multiple signaling pathways, such as transforming growth factor β, epidermal growth factor, and Notch pathways [9]. In addition, the activity of SNAI1 protein is regulated by its subcellular localization, which is governed by at least two kinases, GSK3β and PAK1, and by the zinc-finger transporter LIV1 [12]. SNAI1 is a labile protein with a very short half-life, due to its constant ubiquitination and proteasomal degradation. Several ubiquitin E3 ligases, including β-TrCP, FBXL14, FBXO11, and FBW7, have been shown to promote SNAI1 ubiquitination and degradation [20-23]. On the other hand, three deubiquitinating enzymes (DUBs, also called deubiquitinases), DUB3, PSMD14, and OTUB1, were found to stabilize SNAI1 through deubiquitination [24-27]. In this study, we identified USP37 as another SNAI1 deubiquitinase that directly deubiquitinates SNAI1 and promotes cancer cell migration by stabilizing SNAI1 protein.
Materials and methods
Cell lines and chemicals
The HEK293T and HCT116 cell lines were from American Type Culture Collection (ATCC) and cultured under conditions specified by the manufacturer: base medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. The SUM159 cell line was from Stephen P. Ethier (Medical University of South Carolina) and was cultured in Ham’s F12 medium supplemented with 5% FBS, 5 μg ml-1 insulin, 1 μg ml-1 hydrocortisone, and 1% penicillin and streptomycin. The chemicals used for treating cells were MG132 (Santa Cruz Biotechnology, sc-201270) and cycloheximide (Sigma, C7698). Short tandem repeat profiling and mycoplasma tests were done by ATCC or MD Anderson’s Characterized Cell Line Core Facility.
Plasmids and siRNA
pRK5-HA-ubiquitin and the lysine-specific mutant plasmids (K6, K11, K27, K29, K33, K48, and K63) were from Addgene (plasmid number: 17608, 22900, 22901, 22902, 22903, 17607, 17605, and 17606). Sixty-eight human DUB open reading frames were obtained from the Dana-Farber/Harvard Cancer Center DNA Resource Core or MD Anderson’s Functional Genomics Core and individually subcloned into the pBabe-SFB vector using the Gateway system (Invitrogen). The pBabe-puro-SNAI1 plasmid was from Robert A. Weinberg (Whitehead Institute for Biomedical Research). Full-length human SNAI1 was subcloned into the pcDNA3.1-MYC vector. pLOC-USP37 and pGIPZ-USP37 shRNA #1 (V2LHS_200776) and #7 (V3LHS_317043) were from MD Anderson’s Functional Genomics Core. The SFB-USP37C350S and pLOC-USP37C350S mutants were generated using a QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer’s protocol. USP37 siRNA oligonucleotides were synthesized by Sigma and the sequences are as follows: USP37 siRNA #1 (5’-GAUUUGACAGAAUGAGCGAdTdT-3’), USP37 siRNA #2 (5’-GAAUAAAGUCAGCCUAGUAdTdT-3’), and USP37 siRNA #3 (5’-CCAAGGAUAUUUCAGCUAAdTdT-3’). Cells were transfected with the indicated oligonucleotide (100 nM) using the Oligofectamine reagent (Invitrogen). Forty-eight hours after siRNA transfection, cells were used for functional assays or collected for Western blot analysis.
Lentiviral transduction
For the generation of stable USP37-knockdown cells, virus-containing supernatant was collected 48 hours and 72 hours after co-transfection of pCMV-VSV-G, pCMV Δ8.2, and the pGIPZ-USP37 shRNA vector into HEK293T cells, and was then added to the target cells. Forty-eight hours later, the infected cells were selected with 1 μg ml-1 puromycin (Gibco, A11138-03). For the generation of stable USP37-overexpressing cells, virus-containing supernatant was collected 48 hours and 72 hours after co-transfection of pCMV-VSV-G, pCMV Δ8.2, and the pLOC-USP37 or pLOC-USP37C350S vector into HEK293T cells, and was then added to the target cells. Forty-eight hours later, the infected cells were selected with 10 μg ml-1 blasticidin (Life Technologies, R21001).
Immunoblotting
Western blot analysis was performed with precast gradient gels (Bio-Rad) using standard methods. Briefly, cultured cells were lysed in RIPA buffer (Millipore, 20-188) containing protease inhibitors (Roche). Proteins were separated by SDS-PAGE and blotted onto a nitrocellulose membrane (Bio-Rad), followed by incubation with the specific primary antibodies and peroxidase-conjugated secondary antibodies. Protein bands were visualized by chemiluminescence (Denville Scientific). The following antibodies were used: antibodies against β-actin (1:1,000, Santa Cruz Biotechnology, sc-47778), FLAG (1:5,000, Sigma, F3165, clone M2; or 1:2,000, Sigma, F7425), HA (1:2,000, Santa Cruz Biotechnology, sc-7392), MYC (1:2,000, Santa Cruz Biotechnology, sc-40, clone 9E10), USP37 (1:2,000, Abcam, ab190184), SNAI1 (1:700, Cell Signaling Technology, 3879, clone C15D3), and V5 (1:2,000, Sigma, V8012, clone V5-10). The ImageJ program was used for densitometric analysis of Western blots, and the quantification results were normalized to an internal control.
Purification of SFB-tagged proteins
HEK293T cells were transfected with SFB-tagged GFP, USP29, USP37, or USP37C350S proteins. Forty-eight hours later, cells were collected and lysed in NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing protease inhibitors (Roche), followed by pulldown with Streptavidin Sepharose beads (Sigma-Aldrich, GE17-5113-01) at 4°C for 2 hours. The beads were washed with NETN buffer three times, 10 minutes each time. The beads were then incubated in Biotin (Sigma) solution (1-2 mg ml-1, dissolved in NETN lysis buffer) at 4°C for 2 hours to elute SFB-tagged proteins.
Immunoprecipitation and pulldown assays
Cells were lysed in NETN buffer or CHAPS buffer (25 mM Tris-HCl, pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.33% CHAPS) containing protease inhibitors. For immunoprecipitation of tagged proteins, cell extracts were incubated with anti-MYC agarose beads (ThermoFisher Scientific, 20168) overnight at 4°C. For the pulldown of SFB-tagged proteins, cell extracts were incubated with S-protein beads (Millipore, 69704) at 4°C for 2 hours.
In vitro binding assay
Purified His-SNAI1 (Abcam, ab134870) was incubated with mammalian purified SFB-DUBs, followed by pulldown with S-protein beads at 4°C for 2 hours. After the pulldown assay, the nickel magnetic agarose beads or S-protein beads were washed with NETN buffer and the bound proteins were eluted by boiling in 1× Laemmli buffer.
Deubiquitination of SNAI1 in vivo and in vitro
For the in vivo deubiquitination assay, HEK293T cells were transfected with indicated plasmids and then treated with the proteasome inhibitor MG132 (10 μM) for 6 hours. Cells were lysed in RIPA buffer (Millipore, 20-188) containing protease inhibitors (Roche). For denaturing, lysates with 1% SDS were heated at 95°C for 5 minutes, followed by 10-fold dilution with lysis buffer (to 0.1% SDS) and sonication, as described previously [28]. The cell extracts were subjected to immunoprecipitation and Western blot analysis with the indicated antibodies. For the preparation of ubiquitinated SNAI1 as the substrate for the in vitro deubiquitination assay of SNAI1, HEK293T cells were co-transfected with HA-ubiquitin and MYC-SNAI1 and then treated with MG132 for 6 hours. Ubiquitinated SNAI1 was purified from the cell extracts with anti-MYC beads. After extensive wash with NETN buffer, the bound proteins were incubated with purified SFB-tagged GFP, USP37, or USP37C350S proteins in deubiquitination buffer (50 mM Tris-HCl at pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM dithiothreitol, 5% glycerol) at 37°C for 2 hours. The beads were then washed three times with deubiquitination buffer, and the bound proteins were eluted by boiling in 1× Laemmli buffer and then subjected to immunoblotting analysis with the indicated antibodies.
Cell proliferation assay
To measure cell proliferation rates, we plated equal numbers of cells in 6-well plates. Cells were trypsinized and counted on the indicated days. Cell counts were obtained from a TC10 Automated Cell Counter (Bio-Rad).
Migration assay
To measure cell motility, we plated the indicated cells in the top chamber with the non-coated membrane (24-well insert; pore size, 8 μm; BD Biosciences) in serum-free medium, and medium supplemented with serum was used as a chemoattractant in the lower chamber. After incubation for 18 hours, cells that did not migrate through the pores were removed with a cotton swab. Then cells on the lower surface of the membrane were fixed with 10% formalin, stained with 0.2% crystal violet at room temperature for 45 minutes, and then counted.
Statistical analysis
Each experiment was repeated three times or more. Data are presented as mean ± s.e.m., and a two-tailed unpaired t-test was used to compare two groups of independent samples.
Results
USP37 binds, deubiquitinates, and stabilizes SNAI1
Deubiquitinases are proteases that remove mono-ubiquitin or ubiquitin chains from substrate proteins [29]. To identify deubiquitinases that associate with SNAI1, we first screened for SNAI1-interacting DUBs by an S-protein bead pulldown assay using a panel of 68 human SFB-tagged DUBs [30-32]. We transiently co-transfected each SFB-tagged DUB with MYC-tagged SNAI1 into HEK293T cells and pulled down the DUBs with S-protein beads, and we detected the physical association of SNAI1 with 23 DUBs (Figure 1A). Because nearly one-third of the 68 DUBs interacted with SNAI1 in this pulldown assay, we performed a secondary screen to determine which DUBs modulate SNAI1 protein levels. We transfected the 23 identified DUBs individually into HEK293T cells and found that only USP29, USP36, and USP37 markedly upregulated SNAI1 protein (Figure 1B). The interactions between these three DUBs and SNAI1 were verified by a confirmatory pulldown assay (Figure 1C) and by a co-immunoprecipitation assay, in which SFB-tagged USP29, USP36, or USP37 was co-immunoprecipitated with MYC-SNAI1, but not with MYC-GFP (Figure 1D).
Figure 1.

USP37 is a direct positive regulator of SNAI1. A. Twenty-three of 68 DUBs physically associated with SNAI1. Each SFB-tagged DUB was co-transfected with MYC-SNAI1 into HEK293T cells, followed by pulldown with S-protein beads and immunoblotting with antibodies against FLAG and MYC. B. Upper panel: each SFB-tagged DUB was transfected into HEK293T cells, followed by immunoblotting with antibodies against SNAI1, FLAG, and β-actin. Lower panel: quantification of SNAI1 protein levels (normalized to β-actin). C. SFB-tagged DUBs and MYC-SNAI1 were co-transfected into HEK293T cells, followed by pulldown with S-protein beads and immunoblotting with antibodies against FLAG and MYC. D. SFB-tagged DUBs and MYC-SNAI1 or MYC-GFP were co-transfected into HEK293T cells, followed by immunoprecipitation (IP) with anti-MYC beads and immunoblotting with antibodies against FLAG and MYC. E. Left panel: HEK293T cells were co-transfected with HA-ubiquitin, MYC-SNAI1, and SFB-tagged DUBs, followed by immunoprecipitation with anti-MYC beads and immunoblotting with antibodies against HA and MYC. Cells were treated with 10 μM MG132 for 6 hours before collection. Before immunoprecipitation, lysates were heated at 95°C for 5 minutes in the presence of 1% SDS (for denaturing), followed by 10-fold dilution with lysis buffer and sonication. Right panel: immunoblotting of HA-ubiquitin, MYC-SNAI1, FLAG-DUBs, and HSP90 in the input. F. Upper panel: HEK293T cells were co-transfected with SNAI1, MYC-GFP, and SFB-tagged GFP or DUBs, treated with 100 μg ml-1 cycloheximide (CHX), harvested at different time points, and then immunoblotted with antibodies against SNAI1, MYC, and FLAG. MYC-GFP served as the control for transfection. LE: long exposure. SE: short exposure. Lower panel: quantification of SNAI1 protein levels (normalized to MYC-GFP). G. Upper panel: SFB-GFP, SFB-USP29, and SFB-USP37 were purified from HEK293T cells transfected with SFB-tagged GFP or DUBs, via pulldown with Streptavidin beads, followed by elution with biotin. Purified SFB-GFP, SFB-USP29, or SFB-USP37 was incubated with purified His-SNAI1, followed by pulldown with S-protein beads and immunoblotting with antibodies against SNAI1 and FLAG. Lower panel: purified proteins were analyzed by SDS-PAGE and Coomassie blue staining.
To determine whether these three SNAI1-interacting DUBs promote SNAI1 deubiquitination, we co-transfected USP29, USP36, USP37, or a negative control (USP44 or GFP) with MYC-SNAI1 and HA-ubiquitin into HEK293T cells, treated the cells with MG132 to inhibit proteasomal degradation, pulled down SNAI1 by anti-MYC beads, and detected SNAI1 polyubiquitination by immunoblotting. USP29, USP36, and USP37, but not USP44 or GFP, reduced the polyubiquitination of SNAI1 (Figure 1E). To determine which of the three DUBs stabilizes SNAI1 protein, we transiently transfected HEK293T cells with pBabe-SNAI1, MYC-GFP, and SFB-tagged GFP or DUB, treated the cells with 100 μg ml-1 cycloheximide (CHX), and determined SNAI1 levels at different time points. Although all three DUBs increased the steady-state level of SNAI1 protein (Figure 1B), only USP29 and USP37 increased the half-life of ectopically expressed SNAI1 protein (Figure 1F).
Next, to determine whether USP29 and USP37 are able to interact with SNAI1 directly, we performed an in vitro binding assay using purified proteins. As shown in Figure 1G, bacterially purified His-SNAI1 protein bound to mammalian purified SFB-USP37 but not to SFB-GFP or SFB-USP29 under cell-free conditions, suggesting that USP37 may directly regulate SNAI1. Collectively, these results indicate that USP37 is a strong candidate for the SNAI1 deubiquitinase.
USP37 stabilizes SNAI1 protein through deubiquitination
To further investigate the effect of USP37 on SNAI1, we co-expressed SFB-USP37 with MYC-SNAI1 in HEK293T cells and examined endogenous SNAI1 protein levels. Wild-type USP37, but not the catalytically inactive mutant USP37C350S, markedly upregulated SNAI1 protein, which was similar to the effect of MG132 treatment (Figure 2A), suggesting that the enzymatic activity of USP37 is required for elevating SNAI1 protein levels. To determine whether USP37’s catalytic activity is required for stabilizing SNAI1 protein, we transfected HEK293T cells with MYC-SNAI1 and USP37 or USP37C350S, treated the cells with 100 μg ml-1 cycloheximide, and determined MYC-SNAI1 levels at different time points. Overexpression of wild-type USP37, but not USP37C350S, increased the half-life of SNAI1 protein (Figure 2B). Conversely, knockdown of USP37 in HEK293T cells markedly shortened the half-life of ectopically expressed MYC-SNAI1 protein (Figure 2C).
Figure 2.

USP37 stabilizes SNAI1 protein through deubiquitination. A. HEK293T cells were transfected with SFB-GFP, SFB-USP37, or SFB-USP37C350S, treated with 10 μM MG132 for 6 hours, harvested, and immunoblotted with antibodies against SNAI1, FLAG, and β-actin. B. Upper panel: HEK293T cells were co-transfected with V5-GFP and SFB-tagged GFP, USP37, or USP37C350S, treated with 100 μg ml-1 cycloheximide (CHX), harvested at different time points, and then immunoblotted with antibodies against MYC, V5, and FLAG. V5-GFP served as the control for transfection. Lower panel: quantification of SNAI1 protein levels (normalized to V5-GFP). C. Upper panel: HEK293T cells were co-transfected with MYC-SNAI1, V5-GFP and USP37 siRNA #3 or scrambled siRNA, treated with 100 μg ml-1 cycloheximide (CHX), harvested at different time points, and then immunoblotted with antibodies against MYC, V5, β-actin and FLAG. V5-GFP serves as the control for transfection. Lower panel: quantification of SNAI1 protein levels (normalized to V5-GFP). D. HEK293T cells were co-transfected with MYC-SNAI1, HA-ubiquitin (Ub), and SFB-tagged USP37 or USP37C350S, followed by immunoprecipitation with anti-MYC beads and immunoblotting with antibodies against HA and MYC. Cells were treated with 10 μM MG132 for 6 hours. Before immunoprecipitation, lysates were heated at 95°C for 5 minutes in the presence of 1% SDS (for denaturing), followed by 10-fold dilution with lysis buffer and sonication. E. SFB-GFP, SFB-USP37, and SFB-USP37C350S were purified from HEK293T cells transfected with SFB-tagged GFP or DUBs. Ubiquitinated MYC-SNAI1 was purified with anti-MYC beads from HEK293T cells co-transfected with MYC-SNAI1 and HA-Ub, and was then incubated with purified SFB-tagged GFP or DUBs. After the in vitro deubiquitination, bound proteins were eluted and immunoblotted with antibodies against HA and MYC. Purified proteins were analyzed by SDS-PAGE and Coomassie blue staining.
We reasoned that USP37 stabilizes SNAI1 through deubiquitination. First, we transiently transfected HEK293T cells with HA-ubiquitin, MYC-SNAI1, and SFB-tagged USP37 or USP37C350S, and then treated the cells with 10 μM MG132 for 6 hours. MYC-SNAI1 was immunoprecipitated by anti-MYC beads, and its polyubiquitination was detected by an antibody against HA. As expected, wild-type USP37, but not USP37C350S, substantially reduced SNAI1 polyubiquitination (Figure 2D). To further determine whether USP37 directly deubiquitinates SNAI1, we incubated purified USP37 or USP37C350S and ubiquitinated SNAI1 purified from HEK293T cells in a cell-free system. Wild-type USP37, but not USP37C350S, markedly decreased SNAI1 polyubiquitination in vitro (Figure 2E), suggesting that USP37 can directly deubiquitinate SNAI1.
USP37 promotes cancer cell migration
To validate that USP37 regulates endogenous SNAI1 protein, we individually transfected three independent USP37 siRNAs (#1, #2, and #3) or the scrambled siRNA into HEK293T cells, and we found that all three USP37 siRNAs reduced endogenous SNAI1 protein levels (Figure 3A). Conversely, overexpression of wild-type USP37, but not USP37C350S, increased endogenous SNAI1 protein levels (Figure 3B), suggesting that USP37 consistently stabilizes SNAI1 in HEK293T cells. To determine whether USP37 regulates SNAI1 in cancer cells, we transfected two independent USP37 siRNAs (#2 and #3) into two cell lines expressing high levels of endogenous SNAI1 protein: a breast cancer cell line SUM159 and a colon cancer cell line HCT116. In both cell lines, we found that both siRNAs drastically reduced SNAI1 protein levels (Figure 3C). On the other hand, overexpression of wild-type USP37 increased endogenous SNAI1 protein levels in both SUM159 and HCT116 cells, whereas overexpression of the USP37C350S mutant did not (Figure 3D).
Figure 3.
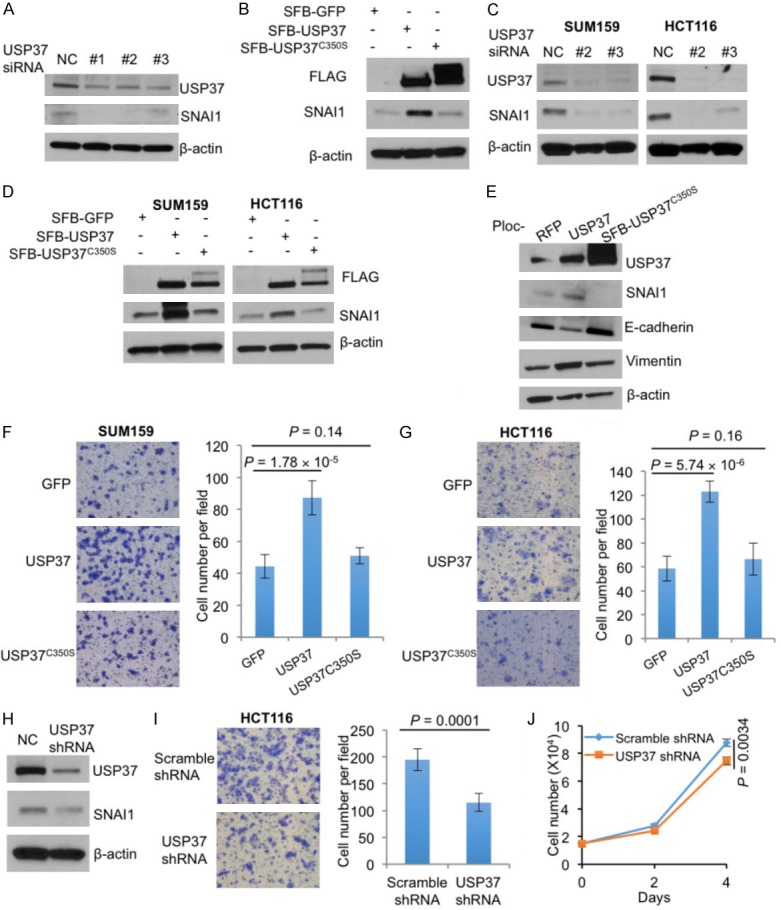
USP37 promotes cancer cell migration. (A) Immunoblotting of USP37, SNAI1, and β-actin in HEK293T cells transfected with USP37 siRNA or a scrambled control (NC). (B) Immunoblotting of FLAG-USP37, SNAI1, and β-actin in HEK293T cells transfected with SFB-tagged GFP, USP37, or USP37C350S. (C) Immunoblotting of USP37, SNAI1, and β-actin in SUM159 and HCT116 cells transfected with USP37 siRNA or a scrambled control (NC). (D) Immunoblotting of FLAG-USP37, SNAI1, and β-actin in SUM159 and HCT116 cells transfected with SFB-tagged GFP, USP37, or USP37C350S. (E) Immunoblotting of USP37, SNAI1, E-cadherin, vimentin, and β-actin in HEK293T cells transduced with RFP, USP37, or USP37C350S. (F) Transwell migration assays of SUM159 cells transfected with SFB-tagged GFP, USP37, or USP37C350S. (G) Transwell migration assays of HCT116 cells transfected with SFB-tagged GFP, USP37, or USP37C350S. (H) Immunoblotting of USP37, SNAI1, and β-actin in HCT116 cells transduced with USP37 shRNA or scramble shRNA (NC). (I) Transwell migration assays of HCT116 cells transduced with scrambled shRNA or USP37 shRNA. (J) Growth curves of HCT116 cells transduced with scrambled shRNA or USP37 shRNA. n = 3 biological replicates. Error bars in (F), (G), (I), and (J) are s.e.m. P values were calculated from a two-tailed t-test.
SNAI1-induced EMT promotes cell migration. To determine whether USP37 induces EMT, we stably transduced USP37 or USP37C350S into HEK293T cells. As expected, overexpression of USP37 downregulated the epithelial marker E-cadherin and upregulated SNAI1 and the mesenchymal marker vimentin (Figure 3E), suggesting that USP37 may be implicated in EMT. Notably, overexpression of the deubiquitinase-dead mutant USP37C350S, which did not upregulate SNAI1, did not affect the expression of EMT markers (Figure 3E).
To determine the biological effect of USP37 in cancer cells, we transfected wild-type USP37 or USP37C350S into SUM159 cells and performed Transwell migration assays. Wild-type USP37, but not USP37C350S, markedly increased cell migration (Figure 3D and 3F). A similar effect was observed in HCT116 cells (Figure 3D and 3G). Conversely, shRNA-mediated stable knockdown of USP37 in HCT116 cells downregulated SNAI1 protein (Figure 3H) and reduced cell migration (Figure 3I) with a minor effect on cell proliferation (Figure 3J).
To determine whether SNAI1 functionally mediates the effect of USP37 on cell migration, we transiently transfected MYC-GFP or MYC-SNAI1 into HCT116 cells transduced with USP37 shRNA or the control shRNA and then performed Transwell migration assays. Ectopic expression of SNAI1 in USP37-knockdown HCT116 cells fully reversed USP37 shRNA-induced inhibition of cell migration (Figure 4A and 4B). These results indicate that USP37 promotes cancer cell migration and that this function is mediated, at least in part, by its substrate SNAI1.
Figure 4.
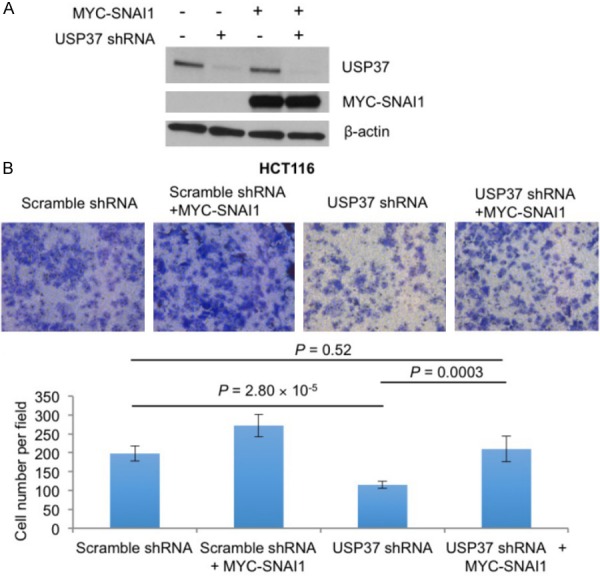
SNAI1 mediates USP37-induced cell migration. A. Immunoblotting of USP37, SNAI1, and β-actin in HCT116 cells transduced with USP37 shRNA or a scramble control with or without MYC-SNAI1 overexpression. B. Transwell migration assays of HCT116 cells transduced with USP37 shRNA or a scramble control with or without MYC-SNAI1 overexpression. Error bars are s.e.m. P values were calculated from a two-tailed t-test.
Discussion
In summary, our findings suggest that USP37 deubiquitinates and stabilizes SNAI1 and promotes cell migration. In cancer, SNAI1 regulates tumor recurrence, metastasis, immune evasion, cell cycle progression, cell survival, and cancer metabolism [15-18]. Because SNAI1 protein is a transcription factor localized predominantly in the nucleus, it is difficult to directly target SNAI1 by small-molecule inhibitors. Considering the important roles of SNAI1 in tumor progression, identifying targets upstream of SNAI1 that are amenable to therapeutic intervention could be a promising strategy to suppress cancer metastasis.
DUB3, PSMD14, and OTUB1 have recently been shown to deubiquitinate SNAI1 and promote tumor metastasis in breast cancer or esophageal squamous cell carcinoma [24-27]. DUB3 did not stand out as a SNAI1-interacting DUB in our initial screening, indicating that the interaction between DUB3 and SNAI1 may not be strong enough in HEK293T cells, and that screening methods other than our interaction screen may identify additional weak-interacting SNAI1 DUBs. It should be noted that we previously used the same approach to identify the DUBs for PTEN [30], β-catenin [32], EZH2 [33], and YAP [34], demonstrating the value of this method. In our screen, we did find that OTUB1 interacted with SNAI1 (Figure 1A); however, OTUB1 did not increase SNAI1 protein levels as much as USP29, USP36, or USP37 did (Figure 1B), indicating that OTUB1 may not play a major role in stabilizing SNAI1 protein in our system.
Notably, USP37 is highly expressed in breast cancer and positively correlated with mortality rates [35]. It has been reported that USP37 can interact with and stabilize the Hedgehog pathway component Gli-1 and regulate EMT, cell invasion, and stemness via the Hedgehog pathway [35]. Several other proteins have also been identified as USP37 substrates. For example, USP37 regulates the stability of oncogenic proteins 14-3-3γ [36], PLZF/RARA [37], and c-Myc [38]. In addition, USP37 plays an important role in regulating DNA replication by deubiquitinating Cdt1 [39]. Taken together with our findings, these results suggest that USP37 may emerge as a potential therapeutic target.
Acknowledgements
We thank Dr. Fan Yao for technical advice and scientific discussion. We thank MD Anderson’s Functional Genomics Core and Characterized Cell Line Core Facility for reagents and technical assistance. We also thank Erica Goodoff for critical reading and editing. L.M. is supported by US National Institutes of Health (NIH) grants R01CA166051 and R01CA181029, a Cancer Prevention and Research Institute of Texas (CPRIT) grant RP190029, and a Stand Up To Cancer Innovative Research Grant (award number: 403235). Z.X. was supported by a Rosalie B. Hite Graduate Fellowship. All work was performed at MD Anderson Cancer Center.
Disclosure of conflict of interest
None.
References
- 1.Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18:43–73. doi: 10.1615/critrevoncog.v18.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 3.Sun Y, Ma L. The emerging molecular machinery and therapeutic targets of metastasis. Trends Pharmacol Sci. 2015;36:349–359. doi: 10.1016/j.tips.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber CE, Li NY, Wai PY, Kuo PC. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J Burn Care Res. 2012;33:311–318. doi: 10.1097/BCR.0b013e318240541e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamburger V. Introduction: johannes holtfreter, pioneer in experimental embryology. Dev Dyn. 1996;205:214–216. doi: 10.1002/(SICI)1097-0177(199603)205:3<214::AID-AJA2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 6.Holtfreter J. Tissue affinity, a means of embryonic morphogenesis. Arch Exp Zellforsch. 1939;23:169–209. [Google Scholar]
- 7.Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–1808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brabletz T. To differentiate or not-routes towards metastasis. Nat Rev Cancer. 2012;12:425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- 9.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The role of snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13:963–972. doi: 10.2174/15680096113136660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 12.Barrallo-Gimeno A, Nieto MA. The snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 13.Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007;67:11721–11731. doi: 10.1158/0008-5472.CAN-07-2318. [DOI] [PubMed] [Google Scholar]
- 14.Davidson NE, Sukumar S. Of snail, mice, and women. Cancer Cell. 2005;8:173–174. doi: 10.1016/j.ccr.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 16.Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung MC, Evers BM, Zhou BP. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Chen WJ, Wang H, Tang Y, Liu CL, Li HL, Li WT. Multidrug resistance in breast cancer cells during epithelial-mesenchymal transition is modulated by breast cancer resistant protein. Chin J Cancer. 2010;29:151–157. doi: 10.5732/cjc.009.10447. [DOI] [PubMed] [Google Scholar]
- 20.Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinas-Castells R, Beltran M, Valls G, Gomez I, Garcia JM, Montserrat-Sentis B, Baulida J, Bonilla F, de Herreros AG, Diaz VM. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J Biol Chem. 2010;285:3794–3805. doi: 10.1074/jbc.M109.065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Zhang X, Ye M, Jing P, Xiong J, Han Z, Kong J, Li M, Lai X, Chang N, Zhang J, Zhang J. FBW7 loss promotes epithelial-to-mesenchymal transition in non-small cell lung cancer through the stabilization of snail protein. Cancer Lett. 2018;419:75–83. doi: 10.1016/j.canlet.2018.01.047. [DOI] [PubMed] [Google Scholar]
- 24.Liu T, Yu J, Deng M, Yin Y, Zhang H, Luo K, Qin B, Li Y, Wu C, Ren T, Han Y, Yin P, Kim J, Lee S, Lin J, Zhang L, Zhang J, Nowsheen S, Wang L, Boughey J, Goetz MP, Yuan J, Lou Z. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat Commun. 2017;8:13923. doi: 10.1038/ncomms13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Y, Wang Y, Lin Y, Liu Y, Wang Y, Jia J, Singh P, Chi YI, Wang C, Dong C, Li W, Tao M, Napier D, Shi Q, Deng J, Evers BM, Zhou BP. Dub3 inhibition suppresses breast cancer invasion and metastasis by promoting Snail1 degradation. Nat Commun. 2017;8:14228. doi: 10.1038/ncomms14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu R, Liu Y, Zhou H, Li L, Li Y, Ding F, Cao X, Liu Z. Deubiquitinating enzyme PSMD14 promotes tumor metastasis through stabilizing SNAIL in human esophageal squamous cell carcinoma. Cancer Lett. 2018;418:125–134. doi: 10.1016/j.canlet.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 27.Zhou H, Liu Y, Zhu R, Ding F, Cao X, Lin D, Liu Z. OTUB1 promotes esophageal squamous cell carcinoma metastasis through modulating snail stability. Oncogene. 2018;37:3356–3368. doi: 10.1038/s41388-018-0224-1. [DOI] [PubMed] [Google Scholar]
- 28.Zhong B, Liu X, Wang X, Chang SH, Liu X, Wang A, Reynolds JM, Dong C. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol. 2012;13:1110–1117. doi: 10.1038/ni.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Z, Zhang P, Ma L. The role of deubiquitinases in breast cancer. Cancer Metastasis Rev. 2016;35:589–600. doi: 10.1007/s10555-016-9640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, Wang M, Chen D, Sun Y, Hung MC, Chen J, Ma L. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15:1486–1494. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z, Zhang P, Hu X, Kim J, Yao F, Xiao Z, Zeng L, Chang L, Sun Y, Ma L. USP51 promotes deubiquitination and stabilization of ZEB1. Am J Cancer Res. 2017;7:2020–2031. [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Alavi Naini F, Sun Y, Ma L. Ubiquitin-specific peptidase 2a (USP2a) deubiquitinates and stabilizes beta-catenin. Am J Cancer Res. 2018;8:1823–1836. [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Xiao Z, Wang S, Zhang M, Wei Y, Hang Q, Kim J, Yao F, Rodriguez-Aguayo C, Ton BN, Lee M, Wang Y, Zhou Z, Zeng L, Hu X, Lawhon SE, Siverly AN, Su X, Li J, Xie X, Cheng X, Liu LC, Chang HW, Chiang SF, Lopez-Berestein G, Sood AK, Chen J, You MJ, Sun SC, Liang H, Huang Y, Yang X, Sun D, Sun Y, Hung MC, Ma L. ZRANB1 is an EZH2 deubiquitinase and a potential therapeutic target in breast cancer. Cell Rep. 2018;23:823–837. doi: 10.1016/j.celrep.2018.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao F, Zhou Z, Kim J, Hang Q, Xiao Z, Ton BN, Chang L, Liu N, Zeng L, Wang W, Wang Y, Zhang P, Hu X, Su X, Liang H, Sun Y, Ma L. SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nat Commun. 2018;9:2269. doi: 10.1038/s41467-018-04620-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin T, Li B, Feng X, Fan S, Liu L, Liu D, Mao J, Lu Y, Yang J, Yu X, Zhang Q, Zhang J, Song B, Li M, Li L. Abnormally elevated USP37 expression in breast cancer stem cells regulates stemness, epithelial-mesenchymal transition and cisplatin sensitivity. J Exp Clin Cancer Res. 2018;37:287. doi: 10.1186/s13046-018-0934-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JO, Kim SR, Lim KH, Kim JH, Ajjappala B, Lee HJ, Choi JI, Baek KH. Deubiquitinating enzyme USP37 regulating oncogenic function of 14-3-3gamma. Oncotarget. 2015;6:36551–36576. doi: 10.18632/oncotarget.5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang WC, Shih HM. The deubiquitinating enzyme USP37 regulates the oncogenic fusion protein PLZF/RARA stability. Oncogene. 2013;32:5167–5175. doi: 10.1038/onc.2012.537. [DOI] [PubMed] [Google Scholar]
- 38.Pan J, Deng Q, Jiang C, Wang X, Niu T, Li H, Chen T, Jin J, Pan W, Cai X, Yang X, Lu M, Xiao J, Wang P. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene. 2015;34:3957–3967. doi: 10.1038/onc.2014.327. [DOI] [PubMed] [Google Scholar]
- 39.Hernandez-Perez S, Cabrera E, Amoedo H, Rodriguez-Acebes S, Koundrioukoff S, Debatisse M, Mendez J, Freire R. USP37 deubiquitinates Cdt1 and contributes to regulate DNA replication. Mol Oncol. 2016;10:1196–1206. doi: 10.1016/j.molonc.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]