

Abstract
The importance and role of the estrogen receptor (ER) pathway have been well-recognized in both breast cancer development and progression. The treatment of choice in women with estrogen receptor-positive metastatic breast cancer (ER+ mBC) is classically divided into a variety of endocrine therapies, with three of the most common being: selective estrogen receptor modulators (SERM), aromatase inhibitors (AI), and selective estrogen receptor degraders (SERD). However, resistance develops in 30-50% of patients treated with these endocrine therapies due to a sophisticated and at times redundant interference at the molecular level between the ER, growth factors, and downstream cell-signaling pathways. Tumor response is heightened with adjunctive therapy that includes an mTORC1 inhibitor (everolimus), CDK4/6 inhibitors (palbociclib/ribociclib/abemaciclib), and an α isoform-specific PI3K inhibitor (alpelisib). Each of these inhibitors elicits potent anti-proliferative benefits; however, they fail to induce tumor cell death. Consequently, disease progression almost invariably occurs. Evasion of apoptosis is a hallmark of cancer. The p53 and BCL2 represent two important nodes of the apoptosis signaling pathway. Venetoclax, a potent and selective BCL2 inhibitor, synergizes with hormonal therapy in ER+ breast cancer models and is active in clinical trials. Similarly, an MDM2 inhibitor, AMG-232, which induces p53 is active in early clinical trials of both liquid and advanced solid tumor patients. In our ER+ BC cohort (Avera Cancer Institute, Sioux Falls, SD), we observed more than 70% of wild type TP53 and over 10% amplification of MDM2 and MDM4 as comparable with the TCGA data set. We summarized current treatment options, the molecular mechanisms that predispose to endocrine resistance, and a future pro-apoptotic treatment strategy for ER+ mBC patients. Our review presents critical analyses of the therapeutic options for the clinical management of ER+ Metastatic Breast Cancer in the light of a hypothesis targeting the induction of apoptosis in p53 wild type tumors. We reviewed not only the FDA approved current treatment approaches but also presented a discourse addressing the possibilities for novel combination strategy that can induce tumor cell apoptosis, a critical cellular mechanism delaying/denying tumor progression. Our review is unique as it presents patient data in support of our hypothesis.
Keywords: ER+ metastatic breast cancer, PI3K pathway, BCL2, MDM2, apoptosis
Prologue
Breast cancer and its classification
Breast cancer is a malignant clonal proliferation of benign breast tissue that continues to pose a significant threat to women across the globe. As of 2013, American women run a 1 in the 8-lifetime risk of breast cancer, up from 1 in 11 during the 1970s [1]. Fortunately, however, we have seen several advancements in the screening, prevention, and treatment of breast cancer throughout the last half-century due to an immense multi-disciplinary effort encompassing the disciplines of researchers, clinicians, pharmaceutical companies, technology, and many others. Of these advancements, breast cancer treatment has debatably seen the most innovation and is a direct consequence of our greater understanding of breast cancer’s pathophysiological foundations.
Breast cancer most frequently arises from benign ductal or lobular tissues by undergoing gradual, cumulative genetic alterations until one cell obtains enough “driver” alterations to proliferate clonally and uncontrollably along a spectrum of localized to metastatic disease [2]. This spectrum, commonly staged using the TNM system, classifies cancers clinically or pathologically using “T” for tumor size, “N” for number and location of lymph nodes containing cancer and “M” for the presence of cancer in distance sites throughout the body to help guide therapeutic decision-making and prognosis. For example, a T1N1M0 tumor is less than 2 cm in size, confined to 1-3 axillary sentinel lymph nodes and is absent of any observable metastases. In breast cancer, these “driver” alterations genetically manifest themselves molecularly through altered receptor expression that is predictively responsive to hormonal and/or molecular targeted drug therapies. Our current, clinically-relevant classification of breast tumor receptor status distinguishes breast cancers into three main subtypes: hormone receptor-positive (HR+), human epidermal growth factor receptor-2 amplified/overexpressed (HER2+) and triple-negative (TN). Hormone receptor-positive tumors include estrogen receptor-positive (ER+) and/or progesterone receptor-positive (PR+) breast cancers. HER2+ breast cancers represent an amplified ERBB2 gene that gets translated to overexpression of HER2 receptor protein. Whereas TN breast cancer is negative for all three (ER, PR, HER2) predominant receptor expressions [3,4].
Over 99% of breast cancers are diagnosed in females, and approximately 75% are HR+, with the most significance placed on ER positivity for reasons beyond the scope of this paper [5-7]. Within this population, several molecular subpopulations exist, including approximately 34% with altered PIK3CA expression, 3% altered mTOR expression, 28% CCND1 amplification, 25% TP53 mutations, 30% ESR1 mutations, 85% BCL2 overexpression, and 8-12% amplified/overexpressed MDM2 [8]. Male BC (though quite similar in expression profile to female BC) does appear to demonstrate unique expression demographics via a greater (96%) ER+ expression and a lesser (3%) p53 expression as compared to female BC [7]. Potentially important for all breast cancers in the future, these molecular aberrations currently play a crucial role in guiding translational research and treatment in advanced, metastatic ER+ breast cancer (ER+ mBC).
At the time of diagnosis, approximately 90% of breast cancers are not metastatic [3]; however, in addition to the 10% metastatic at diagnosis, approximately 10-60% of localized breast cancers develop systemic relapse [9]. Furthermore, the prognosis for ER+ mBC is a median five-year survival rate of 27%, suggesting the need for new therapies that significantly impact progression-free and overall survival in this population [4,10]. In this article, we aim to briefly describe the history of ER+ mBC treatment, current translational research in development and suggest a theoretically promising molecular therapy combination for future clinical study in ER+ mBC.
The past: history of ER+ metastatic breast cancer treatment
Initial approaches to ER+ mBC cancer treatment focused on cytotoxic effects demonstrated in early leukemic cancer therapies. It was thought that these cytotoxic chemotherapies would broadly kill the cancerous tumor cells, sacrifice a few normal cells in the process, but ultimately lead to cancer cure. Unfortunately, this strategy eventually failed and opened the door for more specific hormonal-based therapies in the latter half of the 20th century [11]. The first of these, Tamoxifen, was a selective estrogen receptor modulator (SERM) which targeted the blockade of estrogen receptors in breast tissues while activating/inhibiting other estrogen-responsive tissues. Since its FDA approval in 1985, Tamoxifen has demonstrated significant clinical success [12]; however, its activation of ER in the uterus prompted the desire for further hormonal therapies containing both greater safety and efficacy.
Over the last few decades, new classes of hormonal cancer treatments achieved FDA approval in various settings of ER+ BC, including the selective estrogen receptor degraders (SERD; Fulvestrant) which target the ER for degradation and multiple generations of aromatase inhibitors (AI) which target the final enzymatic step in estrogen’s biosynthesis from 5-hydroxytestosterone [11]. Of these, both steroid (exemestane) and non-steroid (letrozole and anastrozole) third-generation AI’s demonstrated the most promise through a large meta-analysis published in 2006 by Mauri et al. In their analysis, these third-generation AI’s demonstrated superior survival to tamoxifen in advanced breast cancer patients [13].
After demonstrating the efficacy of multiple hormonal monotherapies with distinct mechanisms of action, the next step included analyzing these therapies in different combinations and sequences. Unfortunately, conclusions of clinical efficacy from the FACT (median time to progression [mTTP] 10.8 months [high-dose fulvestrant plus anastrozole] vs. 10.2 months [anastrozole alone]; HR: 0.99; 95% CI, 0.81-1.20; P=0.91), SWOG (median progression free survival [mPFS] 15.0 months [fulvestrant plus anastrozole] vs. 13.5 months [anastrozole alone]; HR: 0.80; 95% CI, 0.68-0.94; P=0.007), and SoFEA (mPFS 4.4 months [fulvestrant plus anastrozole] vs. 4.8 months [fulvestrant plus placebo]; HR: 1.00; 95% CI, 0.83-1.21; P=0.98; or mPFS of fulvestrant plus placebo vs. 3.4 months [exemestane]; HR: 0.95; 0.79-1.14; P=0.56) studies were inconclusive in this regard [14-17]. Before further clarification could be established, the age of molecular cancer therapy had arrived, and the focus became molecular-hormonal rather than hormonal-hormonal cancer therapy combinations.
The present: addressing hormone resistance via molecular therapies
Since their FDA approval in the latter 20th century, modern hormonal therapies have demonstrated significant PFS for ER+ mBC patients [12]. However, post-diagnosis approximately 30-50% ER+ breast cancer patients on these hormonal therapies acquire resistance, requiring additional or substitutive treatment for the further clinical benefit [5,18]. Recent research suggests this resistance to occur via the PI3K-AKT-mTOR, CCND1-CDK4/6-RB, BCL2-p53-MDM2, ESR1 and other cell-signaling pathways, demonstrating the potential efficacy of molecular-based therapies in advanced breast cancer.
Initially, resistance/relapse was suspected of lying primarily downstream of intracellular and membrane-bound ER’s, within the PI3K-AKT-mTOR pathway, due in part to > 70% ER+ BC having mutations involved in this pathway [8,19]. Subsequent studies by Ellis et al. and Creighton et al. provided further support via their conclusions of ER+ BC sensitivity to both ER inhibition and PIK3CA activation, respectively [20,21]. Normal cellular signaling of this pathway relies upon estrogen binding to the intracellular or membrane-bound ER’s with differing downstream effects. For the intracellular ER, the ligand-ER complex translocates into the nucleus and activates growth-promoting gene transcription. Alternatively, cross-talk between membrane-bound ER and other tyrosine kinase receptors (EGFR, FGFR, etc.) initiate the growth-promoting PI3K-AKT-mTOR cell-signaling cascade. ER+ BC achieves resistance to this critical signaling via an estimated 28-47% PI3K over-expression, 2.6-3.8% AKT activation, and/or 3% mTOR upregulation, prompting the development of several therapies targeting these vital components [8,22].
Everolimus, a mTORC1 inhibitor, was a frontier drug targeting this pathway, showing (in combination with hormonal therapies) significant improvement in PFS for patients previously treated with AI’s alone [23]. In the BOLERO-2 trial, Baselga and group found a median PFS of 10.6 months with everolimus plus exemestane vs. 4.1 months with placebo plus exemestane (HR: 0.36; 95% CI, 0.27-0.47; P<0.001) in the postmenopausal, ER+/HER2- mBC population; however, its short-lived efficacy in combination with AI’s limited its utilization [23,24]. Genomic analysis of the BOLERO-2 cohort tumors by Hortobagyi et al. found the limited PFS benefit experienced by everolimus therapy was potentially due to differences in the type of PI3K mutation (exon 9 vs. exon 20), FGFR alteration, or degree of chromosomal instability (CIN) [25]. Furthermore, the TAMRAD study supported these results with a median PFS of 10 months with the everolimus plus tamoxifen and 5.5 months with tamoxifen alone (HR: 0.59; 95% CI, 0.33-1.07) in a similar population as the BOLERO-2 trial [26]. Despite their successes, the above-mentioned limitation of everolimus further propelled investigation into other alternative PI3K pathway inhibition therapies to overcome endocrine resistance. Of the multiple types of PI3K pathway therapies (AKT inhibitors, other mTOR inhibitors, PI3K inhibitors) under investigation, only the PI3K inhibitors have reached significance clinically.
The BELLE-2 and BELLE-3 trials were the first of these PI3K inhibitor trials to find efficacy in hormone-resistant ER+ mBC population. The BELLE-2 study analyzed a pan-PI3K inhibitor, buparlisib, plus fulvestrant vs. placebo plus fulvestrant with a median PFS of 6.9 months (95% CI 6.8-7.8) and 5.0 months (4.0-5.2), respectively and a HR 0.78 (95% CI, 0.67-0.89; one-sided P=0.00021) in the total population [27]. In the PI3K pathway activated patients, the median PFS was 6.8 months in the buparlisib group vs. 4 months in the placebo group (HR: 0.76; 95% CI, 0.60-0.97; one-sided P=0.014). Furthermore, findings were clinically meaningful for 200 patients who were confirmed carriers of PIK3CA mutations as detected by ctDNA with mPFS of 7.0 vs. 3.2 months in the buparlisib and placebo groups, respectively (HR: 0.58; 95% CI, 0.41-0.82; one-sided P=0.001) [28]. Despite 70% of all buparlisib treated patients requiring dose reduction or treatment discontinuation as a result of adverse events, their data show the PIK3CA gene to be a targetable biomarker. In parallel with the BELLE-2 trial, the BELLE-3 trial focused on buparlisib plus fulvestrant in the setting of ER+ mBC resistant to hormonal and mTOR therapies. The authors found a significantly longer median PFS in the buparlisib plus fulvestrant group (3.9 months; 95% CI, 2.8-4.2) vs. the placebo plus fulvestrant group (1.8 months; 95% CI, 1.5-2.8) with a HR 0.67 (95% CI, 0.53-0.84; one-sided P=0.00030) [29]. Despite its efficacy, the toxicity profile prevented further research using this combination of therapy and suggested the need for isoform-specific PI3K inhibitors with a more desirable toxicity profile.
Despite early successes in molecular targeting of the PI3K-AKT-mTOR pathway, others pursued the molecular targeting of other frequently deregulated pathways in ER+ mBC. The CCND1-CDK4/6-RB pathway, innately vital to cell cycle control, regulates whether a cell advances or arrests at the G1-S phase of the cell cycle. Furthermore, it was estimated that 35% HR+ breast cancers demonstrated amplification in the CCND1, the gene that encodes cyclin-D1, and 16% demonstrated amplification in the gene encoding CDK4, suggesting the theoretical utility of their inhibitors in ER+ mBC therapeutics [8,30].
This pathway derives from cell-cycle progression and the oncogenic hallmark of cancer cells. ER+ BC (like many other cancers) has demonstrated the importance of the G1-S checkpoint to the oncogenic potential of cancer. At this checkpoint, cyclin-D (D1, D2, D3) binds with cyclin-dependent kinases (CDK4/6) to promote cell cycle progression via inhibition of the tumor suppressor RB. Through CCND1 amplification and cyclin-D1-CDK4/6 complexes, uncontrolled cell cycle progression and tumor growth can proceed, leaving an excellent potential for molecular targets.
Early trials of CDK4/6 inhibitors with hormonal therapies demonstrated synergistic efficacy via significant improvement in PFS alongside a tolerable safety profile [31-33]. The first of these trials, the phase 2 PALOMA-1 trial, studied the safety and efficacy of palbociclib plus letrozole in postmenopausal patients with advanced, ER+/HER2- breast cancer. The authors reported a mPFS of 20.2 months (95% CI, 13.8-27.5) for the palbociclib plus letrozole group vs. 10.2 months (95% CI, 5.7-12.6) for the letrozole group (HR 0.488; 95% CI, 0.319-0.748; one-sided P=0.0004), demonstrating significantly improved efficacy alongside a tolerable safety profile. Of note, the most common adverse events were grade 3-4 neutropenia (54% in palbociclib group vs. 1% in the letrozole-only group) and leukopenia (19% of the palbociclib group vs. 0% in the letrozole-only group) with similar findings observed in subsequent CDK4/6 inhibitor trials [34]. The follow-up phase 3 PALOMA-2 trial extended their earlier findings in the PALOMA-1 trial, showing a median PFS of 24.8 months (95% CI, 22.1-not estimable) in the palbociclib-letrozole group, as compared with 14.5 months (95% CI, 12.9-17.1) in the placebo-letrozole group (HR 0.58; 95% CI, 0.46-0.72; P<0.001) [32].
The next of these trials, the phase 3 MONALEESA-2 trial, studied the safety and efficacy of ribociclib plus letrozole in postmenopausal ER+ mBC patients previously untreated by systemic therapy. The authors found a significantly longer PFS for the ribociclib plus letrozole group over the placebo group (HR: 0.56; 95% CI, 0.43-0.72; P=3.29×10-6 for superiority) with an 18-month PFS rate of 63.0% (95% CI, 54.6-70.3) in the ribociclib group and 42.2% (95% CI, 34.8-49.5) in the placebo group [31]. Recently, it has been reported by Tripathy et al. in their phase 3 MONALEESA-2 trial that ribociclib plus AI or tamoxifen significantly improved mPFS (23.8 months) compared with placebo plus hormonal therapy (13.0 months; HR: 0.55; 95% CI, 0.44-0.69; P<0.0001) and had a manageable safety profile in patients with premenopausal ER+/HER2- advanced breast cancer [35]. The latest of these initial CDK4/6 inhibitor trials, the phase 3 MONARCH-3 trial utilized abemaciclib or placebo with a nonsteroidal AI in postmenopausal HR+/HER2- mBC. The abemaciclib arm demonstrated a significantly longer median PFS than the placebo arm (28.18 versus 14.76 months; HR: 0.540; 95% CI, 0.418-0.698; P=0.000002) and an objective response rate (ORR) of 61.0% in the abemaciclib arm vs. 45.5% in the placebo arm (measurable disease, P=0.003) [36]. Following the success of these trials, all three CDK4/6 inhibitors achieved FDA approval with anti-estrogen therapy in ER+/HER2- breast cancer patients. Despite the CDK4/6 inhibitors’ significant improvements in efficacy, resistance eventually became an issue.
One mechanism of resistance to these CDK4/6 inhibitors is believed to occur via PIK3CA mutations within the previously described PI3K-AKT-mTOR pathway. In their preclinical data, Vora et al. discovered that the combination of CDK4/6 and PI3K inhibition led to tumor regressions in PIK3CA-mutant xenografts [37]. Subsequently, trial investigators sought to study the safety and efficacy of alpelisib, a p110-α-specific PI3K inhibitor, in two initial phase 1b trials of alpelisib plus hormonal therapy for ER+ mBC patients resistant to previously exposed hormonal therapy. In its first trial, the alpelisib plus letrozole combination demonstrated a clinical benefit rate (lack of progression > 6 months) of 35% (44% in patients with PIK3CA-mutated and 20% in PIK3CA wild-type tumors; 95% CI, 17%-56%) and 31% (lack of progression > 12 months), alongside a more tolerable toxicity profile over the pan-PI3K inhibitor buparlisib [38]. The second of these trials studying alpelisib plus fulvestrant provided further support of its tolerability in combination with hormonal therapy. The combination demonstrated longer mPFS for PIK3CA-altered tumors (9.1 months; 95% CI, 6.6-14.6 months) vs. wild-type tumors (4.7 months; 95% CI, 1.9-5.6 months) and an ORR in the PIK3CA-altered tumor group of 29% (95% CI, 17%-43%), vs. none in the wild-type tumor group [39].
Most recently, the phase 3 SOLAR-1 trial (clinicaltrials.gov identifier: NCT02437318) of alpelisib plus fulvestrant in postmenopausal, advanced breast cancer patients previously treated with hormonal therapy has demonstrated a significantly longer mPFS for the combination (11.0 months) vs. placebo plus fulvestrant (5.7 months; HR: 0.65; 95% CI, 0.50-0.85; P=0.00065) while maintaining the tolerable safety profile [40]. Consequently, from these trials, alpelisib has achieved FDA approval in combination with fulvestrant for postmenopausal, ER+, HER2-, PIK3CA-mutated, advanced breast cancer patients. Additionally, the recentlypublished, multi-center phase 2 LORELEI trial showed that taselisib (a β-sparing PI3K inhibitor) plus letrozole achieved higher objective response (50%) than placebo plus letrozole (39%) with an odds ratio (OR) of 1.55 (95% CI, 1.00-2.38; P=0.049) for the overall study population and an OR 2.03 (95% CI, 1.06-3.88; P=0.033) in the PIK3CA-mutated subset of postmenopausal women with early-stage ER+/HER2- BC in the neoadjuvant setting [41].
Another preclinical discovery, this time by O’Brien et al., found that triple therapy with an anti-estrogen, PI3K inhibitor (pan or α-specific), and a CDK4/6 inhibitor showed the best synergistic long-term tumor growth inhibition over mono or dual therapy of these agents in preclinical ER+ BC mouse models. Their studies demonstrated ribociclib plus an anti-estrogen inhibited tumor growth while the addition of a PI3K inhibitor to the dual therapy led to robust tumor regression [42]. These promising preclinical results quickly reached the clinic based on the result of phase I/II study by Juric et al. with a 4-arm analysis of letrozole, ribociclib, and alpelisib in advanced ER+ BC. The most intriguing of these being the 2 arms studying the triple therapy combination of the previously mentioned drugs (NCT01872260).
ESR1 alterations are also implicated as another well-known source of endocrine resistance in ER+ mBC, especially after prior adjuvant AI therapy. Through a variety of mechanisms, including gene amplification, point-mutations, and chromosomal rearrangements, ESR1 becomes constitutively active independent of hormone presence. Furthermore, these alterations are believed to transcriptionally drive metastasis in these ER+ mBC patients [43]. The most common therapeutic approach includes fulvestrant which has demonstrated some efficacy in ESR1-altered ER+ mBC patients previously exposed to other endocrine therapies; however, more potent and selective SERD’s remain necessary to provide better clinical benefit in these patients [44].
Lastly, HER2 mutations represent an interesting phenomenon in ER+ mBC. Nayar et al. observed in a small sample of 8 metastatic ER+ BC tumor biopsies that each had HER2 mutations. However, when they analyzed biopsies from primary, treatment-naïve ER+ BC patients, 4 of the 5 biopsy samples did not demonstrate any pre-existing mutations in HER2. This suggests a mutual exclusivity to simultaneous ER and HER2 mutations and the possibility that HER2 mutations developed in resistance to prior endocrine therapy for ER+ BC disease. Through their in vitro studies, they confirmed this logic by demonstrating the resistance of HER2-mutated tumors to anti-estrogen therapies [45]. At this time, multiple entities have reported a potential for HER2-mutated BC to respond to HER2-targeted therapies, regardless of HER2 overexpression/amplification. In the ER+ BC setting, the correlation of these somatic HER2 mutations with response to endocrine therapy is already recognized, and one clinical trial combining neratinib, a pan-HER inhibitor, with fulvestrant is currently in recruiting patients (NCT01670877).
Epilogue
The promotion of a pro-apoptotic future: BCL2 and MDM2 inhibition
A novel approach to ER+ mBC treatment was recently published by Lok et al., presenting an idea to attack ER+ mBC via an apoptotic mechanism of BCL2 inhibition [46,47]. Reminiscent of the renowned Hallmarks of Cancer work by Hanahan et al., their focus promoted apoptosis as the means to maximally reduce the opportunity for further mutations and achieve maximum clinical benefit [48]. In 2000, Perillo et al. showed that BCL2 expression can be upregulated downstream effector molecule during ER stimulation which is significant knowing 30% of ER+ mBC patients possess ESR1-activating mutations [49,50]. Since then, it has been established that approximately 85% of primary ER+ BC demonstrate BCL2 overexpression [49].
The precise mechanism by which BCL2 overexpression occurs in the setting of ER+ BC remains to be fully elucidated. It is theoretically possible that this overexpression is simply a downstream response by BCL2 of the many pro-proliferative and pro-survival signaling that occurs from the upstream PI3K-AKT-mTOR pathway activation (See Figure 2). However, there is also the potential for this overexpression to occur as a form of resistance after previous anti-estrogen therapies or by some other undiscovered mechanism(s). Nonetheless, the demographic predominance of BCL2 overexpression represents another promising therapeutic target in ER+ BC. Venetoclax (ABT-199/GDC-0199), a potent and selective BCL2 inhibitor, has shown positive apoptotic results and achieved FDA approval in the CLL, SLL and AML settings [51-53]. However, BCL2 inhibition had not reached ER+ BC (or other solid tumors) until recently. Lok et al. in their recent phase 1b study utilized venetoclax in combination with tamoxifen, demonstrating a radiologic ORR of 54%, stable disease (SD) rate of 21% and clinical benefit rate (CBR) of 75% in the cohort of ER+/BCL2+ mBC patients without ESR1 mutations and an ORR 40% and CBR 70% in those with ESR1 mutations, all while maintaining a very tolerable toxicity profile [46]. Furthermore, an ongoing phase II trial (VERONICA) of fulvestrant with or without venetoclax for ER+ mBC patients who have acquired resistance to CDK4/6 inhibitors (NCT03584009) may provide more answers regarding synergistic effects of other hormonal treatments with venetoclax and yet another option in the arsenal of ER+ mBC therapies.
Figure 2.
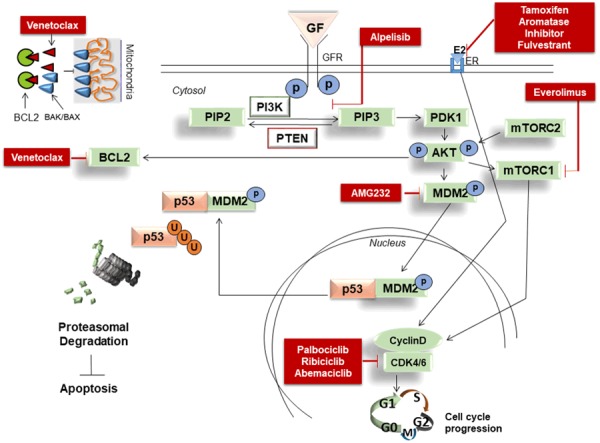
Important cellular signaling in ER+ BC, drugs either FDA approved or currently under development and their mechanisms of action. Insert shows the detailed mode of action of venetoclax.
In addition to BCL2 inhibition, recently surfaced interest in MDM2 inhibition is gaining traction clinically. Currently, there are three trials studying a potent and selective MDM2 inhibitor, AMG-232, in various non-breast cancer settings. Two are completed (NCT02016729 and NCT02110355) and one is in recruitment (NCT03041688). Preclinical evidence suggests MDM2 as another potential apoptotic mechanism for future study. Canon et al. demonstrated AMG-232 to possess cell stasis and pro-apoptotic activity in multiple distinct TP53 wild-types (WT) tumor cell lines [54]. Notably, however, AMG-232 appears to rely on WT TP53; it did not affect p53 (or its downstream effectors) activity in TP53-mutant xenografts. Acting as an inhibitor of MDM2-p53 interaction, AMG-232 binds to MDM2, prevents the ubiquitination of WT p53 by its negative regulator MDM2 and allows p53 to fulfill its important pro-apoptotic role.
In our ER+ BC cohort (Avera Cancer Institute, Sioux Falls, SD), we observed more than 70% of wild type TP53 and over 10% amplification of MDM2 and MDM4 as comparable with the TCGA data set. Approximately 8-12% of ER+ BC are MDM2-amplified/overexpressed and 75% were TP53 WT within the Cancer Genome Atlas (TCGA) cohort, and cohorts from the Avera Cancer Center (See Figure 1A and 1B) [8]. Recently, Lu et al. reported preclinical evidence of in vivo anti-tumor activity by another MDM2 inhibitor, MI-77301, in both endocrine-sensitive and resistant tumor cell lines derived from poor-prognosis, treatment-resistant, breast cancer patients without demonstrating significant evidence of toxicity in mice [55]. Preclinical evidence from our laboratory demonstrated a robustly increased expression of pro-apoptotic markers (cleaved-CASPASE3/7 and PARP-1) and pro-apoptotic/cell cycle inhibitor transcripts (p21 and PUMA) and decreased transcription of pro-survival transcripts (survivin/stathmin mRNA expression) in ER+/TP53 WT BC cell lines with using a combination of venetoclax-AMG-232 (unpublished data).
Figure 1.

A. Alterations of TP53, MDM2, and MDM4 genes in the 129 patients with ER+ BC in the Avera cohort: Y-axis represents the absolute number of alterations while the numbers on the individual bars represent the percentage of alterations in an individual gene. B. Alterations in TP53, MDM2, and MDM4 in ER+ breast invasive carcinomas: Oncoprint presents genetic alterations of TP53, MDM2 and MDM4 in ER+ breast invasive carcinomas (TCGA, 2015) in 594 patients/samples (cBioPortal). The types of alterations are presented in the figure. We acknowledge the cBioPortal for Cancer Genomics site (http://cbioportal.org) which provides a Web resource for exploring, visualizing, and analyzing multi-dimensional cancer genomics data. The portal reduces molecular profiling data from cancer tissues and cell lines into readily understandable genetic, epigenetic, gene expression and proteomic events (Gao et al., 2013, Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal, Sci. Signal., 2 April, Vol. 6, Issue 269, p. pl1 [DOI: 10.1126/scisignal.2004088]). We acknowledge the TCGA Research Network for generating TCGA datasets.
Consequently, we believe further study is warranted regarding MDM2 inhibitors in the ER+/TP53 WT mBC setting. We hypothesize that a new clinical trial utilizing triple therapy of a BCL2 inhibitor (venetoclax), an anti-estrogen (tamoxifen/AI/fulvestrant) and an MDM2 inhibitor (AMG-232/MI-77301) will show promising results for this population. Our hypothesis relies on the previously demonstrated clinical safety and efficacy of the venetoclax-tamoxifen dual therapy alongside the theoretically synergistic potential of AMG-232 or MI-77301 to mechanistically inhibit MDM2, upregulate TP53 and promote maximal apoptotic signaling. Furthermore, BCL2/MDM2 inhibitor combination has demonstrated efficacy in relapsed/refractory AML patients. In a recent phase 1b trial, 35.9% of patients treated with the combination responded according to results presented at the December 2018 American Society of Hematology Annual Meeting in San Diego, CA. A focus on the cellular signaling pathways for the induction of apoptosis is vital to achieve maximum clinical benefit for patients. We propose that future trials involving triple therapies would elucidate the clinical importance of our hypothesis.
Acknowledgements
The authors acknowledge the Avera Cancer Institute, Sioux Falls, SD for their support. The authors also acknowledge Foundation Medicine for DNA sequencing. We acknowledge the TCGA Research Network for generating TCGA datasets used in this study. We acknowledge the cBioPortal for Cancer Genomics site (http://cbioportal.org) which provides a Web resource for exploring, visualizing, and analyzing multi-dimensional cancer genomics data.
Disclosure of conflict of interest
None.
References
- 1.Desantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 2.Hayes DF, Lippman ME. Breast Cancer. In: Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J, editors. Harrison’s Principles of Internal Medicine. 20e. New York, NY: McGraw-Hill Education; 2018. [Google Scholar]
- 3.Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321:288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 4.Karuturi M, Valero V, Chavez-MacGregor M. Metastatic breast cancer. In: Kantarjian HM, Wolff RA, editors. The MD anderson manual of medical oncology. 3e. New York, NY: McGraw-Hill Medical; 2016. [Google Scholar]
- 5.Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123:21–27. doi: 10.1309/4wv79n2ghj3x1841. [DOI] [PubMed] [Google Scholar]
- 6.Lange CA, Yee D. Progesterone and breast cancer. Womens Health (Lond) 2008;4:151–162. doi: 10.2217/17455057.4.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moelans CB, de Ligt J, van der Groep P, Prins P, Besselink N, Hoogstraat M, ter Hoeve N, Lacle M, Kornegoor R, van der Pol C, de Leng W, Barbe E, van der Vegt B, Martens J, Bult P, Smits VT, Koudijs M, Nijman I, Voest E, Selenica P, Weigelt B, Reis-Filho J, van der Wall E, Cuppen E, van Diest PJ. The molecular genetic make-up of male breast cancer. Endocr Relat Cancer. 2019 doi: 10.1530/ERC-19-0278. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howlader N, Noone A, Krapcho M. National cancer institute. SEER cancer statistics review: 1975-2011. 2015 [Google Scholar]
- 10.Noone AM, Howlader N, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975-2015. 2018. p. 2019. [Google Scholar]
- 11.Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72:7–25. doi: 10.1016/j.steroids.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Litherland S, Jackson IM. Antioestrogens in the management of hormone-dependent cancer. Cancer Treat Rev. 1988;15:183–194. doi: 10.1016/0305-7372(88)90002-3. [DOI] [PubMed] [Google Scholar]
- 13.Mauri D, Pavlidis N, Polyzos NP, Ioannidis JP. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst. 2006;98:1285–1291. doi: 10.1093/jnci/djj357. [DOI] [PubMed] [Google Scholar]
- 14.Reinert T, Barrios CH. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther Adv Med Oncol. 2015;7:304–320. doi: 10.1177/1758834015608993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergh J, Jönsson PE, Lidbrink EK, Trudeau M, Eiermann W, Brattström D, Lindemann JP, Wiklund F, Henriksson R. FACT: an open-label randomized phase iii study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer. J. Clin. Oncol. 2012;30:1919–1925. doi: 10.1200/JCO.2011.38.1095. [DOI] [PubMed] [Google Scholar]
- 16.Mehta RS, Barlow WE, Albain KS, Vandenberg TA, Dakhil SR, Tirumali NR, Lew DL, Hayes DF, Gralow JR, Hortobagyi GN, Livingston RB. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med. 2012;367:435–444. doi: 10.1056/NEJMoa1201622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston SR, Kilburn LS, Ellis P, Dodwell D, Cameron D, Hayward L, Im YH, Braybrooke JP, Brunt AM, Cheung KL, Jyothirmayi R, Robinson A, Wardley AM, Wheatley D, Howell A, Coombes G, Sergenson N, Sin HJ, Folkerd E, Dowsett M, Bliss JM SoFEA investigators. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): a composite, multicentr. Lancet Oncol. 2013;14:989–998. doi: 10.1016/S1470-2045(13)70322-X. [DOI] [PubMed] [Google Scholar]
- 18.Chia S, Bryce C, Gelmon K. The 2000 EBCTCG overview: a widening gap. Lancet. 2005;365:1665–1666. doi: 10.1016/S0140-6736(05)66524-5. [DOI] [PubMed] [Google Scholar]
- 19.LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J. Clin. Oncol. 2016;34:3803–3815. doi: 10.1200/JCO.2014.59.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellis MJ, Lin L, Crowder R, Tao Y, Hoog J, Snider J, Davies S, Deschryver K, Evans DB, Steinseifer J, Bandaru R, Liu W, Gardner H, Semiglazov V, Watson M, Hunt K, Olson J, Baselga J. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res Treat. 2010;119:379–390. doi: 10.1007/s10549-009-0575-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, Lluch A, Gray JW, Brown PH, Hilsenbeck SG, Osborne CK, Mills GB, Lee AV, Schiff R. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12:R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13:224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beaver JA, Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol. 2012;8:651–657. doi: 10.2217/fon.12.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, Anderson I, Robinson DM, He W, Garg A, McDonald ER 3rd, Bitter H, Huang A, Taran T, Bachelot T, Lebrun F, Lebwohl D, Baselga J. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J. Clin. Oncol. 2016;34:419–426. doi: 10.1200/JCO.2014.60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Treilleux I, Arnedos M, Cropet C, Ferrero JM, Lacourtoisie SA, Spaeth D, Levy C, Legouffe E, Pujade-Lauraine E, Wang Q, Bachelot T. Predictive markers of everolimus efficacy in hormone receptor positive (HR+) metastatic breast cancer (MBC): final results of the TAMRAD trial translational study. J. Clin. Oncol. 2013;31:510–510. [Google Scholar]
- 27.Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng LM, Hurvitz S, Masuda N, Takahashi M, Vuylsteke P, Hachemi S, Dharan B, Di Tomaso E, Urban P, Massacesi C, Campone M. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–916. doi: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chopra N, Turner NC. Targeting PIK3CA-mutant advanced breast cancer in the clinical setting. Lancet Oncol. 2017;18:842–843. doi: 10.1016/S1470-2045(17)30430-8. [DOI] [PubMed] [Google Scholar]
- 29.Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, O’Regan R, Mouret-Reynier MA, Kalev D, Egle D, Csőszi T, Bordonaro R, Decker T, Tjan-Heijnen VCG, Blau S, Schirone A, Weber D, El-Hashimy M, Dharan B, Sellami D, Bachelot T. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19:87–100. doi: 10.1016/S1470-2045(17)30688-5. [DOI] [PubMed] [Google Scholar]
- 30.Geradts J, Wilson PA. High frequency of aberrant p16(INK4A) expression in human breast cancer. Am J Pathol. 1996;149:15–20. [PMC free article] [PubMed] [Google Scholar]
- 31.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, Janni W, Verma S, Conte P, Arteaga CL, Cameron DA, Petrakova K, Hart LL, Villanueva C, Chan A, Jakobsen E, Nusch A, Burdaeva O, Grischke EM, Alba E, Wist E, Marschner N, Favret AM, Yardley D, Bachelot T, Tseng LM, Blau S, Xuan F, Souami F, Miller M, Germa C, Hirawat S, O’Shaughnessy J. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 32.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–1936. doi: 10.1056/NEJMoa1607303. [DOI] [PubMed] [Google Scholar]
- 33.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen SC, Manso L, Freedman OC, Garnica Jaliffe G, Forrester T, Frenzel M, Barriga S, Smith IC, Bourayou N, Di Leo A. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017;35:3638–3646. doi: 10.1200/JCO.2017.75.6155. [DOI] [PubMed] [Google Scholar]
- 34.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 35.Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, Hurvitz SA, Chow L, Sohn J, Lee KS, Campos-Gomez S, Villanueva Vazquez R, Jung KH, Babu KG, Wheatley-Price P, De Laurentiis M, Im YH, Kuemmel S, El-Saghir N, Liu MC, Carlson G, Hughes G, Diaz-Padilla I, Germa C, Hirawat S, Lu YS. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018;19:904–915. doi: 10.1016/S1470-2045(18)30292-4. [DOI] [PubMed] [Google Scholar]
- 36.Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, Frenzel M, Hardebeck MC, Cox J, Barriga S, Toi M, Iwata H, Goetz MP. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer. 2019;5:5. doi: 10.1038/s41523-018-0097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-Ortiz M, Kim S, Schlegel R, Huang A, Engelman JA. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, Li Y, Cantley LC, Winer E, Arteaga CL. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2-metastatic breast cancer. Clin Cancer Res. 2017;23:26–34. doi: 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juric D, Janku F, Rodón J, Burris HA, Mayer IA, Schuler M, Seggewiss-Bernhardt R, Gil-Martin M, Middleton MR, Baselga J, Bootle D, Demanse D, Blumenstein L, Schumacher K, Huang A, Quadt C, Rugo HS. Alpelisib plus Fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 2019;5:e184475. doi: 10.1001/jamaoncol.2018.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, Yamashita T, Lu YS, Inoue K, Takahashi M, Pápai Z, Longin AS, Mills D, Wilke C, Hirawat S, Juric D. LBA3_PRAlpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase III SOLAR-1 trial. Ann Oncol. 2018;29 [Google Scholar]
- 41.Saura C, Hlauschek D, Oliveira M, Zardavas D, Jallitsch-Halper A, de la Peña L, Nuciforo P, Ballestrero A, Dubsky P, Lombard JM, Vuylsteke P, Castaneda CA, Colleoni M, Santos Borges G, Ciruelos E, Fornier M, Boer K, Bardia A, Wilson TR, Stout TJ, Hsu JY, Shi Y, Piccart M, Gnant M, Baselga J, de Azambuja E. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2-negative, early-stage breast cancer (LORELEI): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2019;20:1226–1238. doi: 10.1016/S1470-2045(19)30334-1. [DOI] [PubMed] [Google Scholar]
- 42.O’Brien NA, Tomaso ED, Ayala R, Tong L, Issakhanian S, Linnartz R, Finn RS, Hirawat S, Slamon DJ. In vivo efficacy of combined targeting of CDK4/6, ER and PI3K signaling in ER+ breast cancer. American Association for Cancer Research Annual Meeting; April 5-9, 2014; San Diego, CA. Abstract 4756. [Google Scholar]
- 43.Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat. 2019:5. doi: 10.20517/2394-4722.2019.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dustin D, Gu G, Fuqua SAW. ESR1 mutations in breast cancer. Cancer. 2019;125:3714–3728. doi: 10.1002/cncr.32345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA, Painter C, Freeman S, Persky NS, Marini L, Helvie K, Oliver N, Rozenblatt-Rosen O, Ma CX, Regev A, Winer EP, Lin NU, Wagle N. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet. 2019;51:207–216. doi: 10.1038/s41588-018-0287-5. [DOI] [PubMed] [Google Scholar]
- 46.Lok SW, Whittle JR, Vaillant F, Teh CE, Lo LL, Policheni AN, Bergin AR, Desai J, Ftouni S, Gandolfo LC, Liew D, Liu HK, Mann GB, Moodie K, Murugasu A, Pal B, Roberts AW, Rosenthal MA, Shackleton K, Silva MJ, Siow ZR, Smyth GK, Taylor L, Travers A, Yeo B, Yeung MM, Bujak AZ, Dawson SJ, Gray DHD, Visvader JE, Lindeman GJ. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2-positive metastatic breast cancer. Cancer Discov. 2019;9:354–369. doi: 10.1158/2159-8290.CD-18-1151. [DOI] [PubMed] [Google Scholar]
- 47.Drago JZ, Chandarlapaty S, Jhaveri K. Targeting apoptosis: a new paradigm for the treatment of estrogen receptor-positive breast cancer. Cancer Discov. 2019;9:323–325. doi: 10.1158/2159-8290.CD-19-0050. [DOI] [PubMed] [Google Scholar]
- 48.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 49.Merino D, Lok SW, Visvader JE, Lindeman GJ. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene. 2016;35:1877–87. doi: 10.1038/onc.2015.287. [DOI] [PubMed] [Google Scholar]
- 50.Perillo B, Sasso A, Abbondanza C, Palumbo G. 17β-Estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20:2890–2901. doi: 10.1128/mcb.20.8.2890-2901.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, Konopleva M, Andreeff M. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell. 2017;32:748–760. e6. doi: 10.1016/j.ccell.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D’Rozario J, Assouline S, Owen C, Gerecitano J, Robak T, De la Serna J, Jaeger U, Cartron G, Montillo M, Humerickhouse R, Punnoose EA, Li Y, Boyer M, Humphrey K, Mobasher M, Kater AP. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107–1120. doi: 10.1056/NEJMoa1713976. [DOI] [PubMed] [Google Scholar]
- 54.Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther. 2015;14:649–658. doi: 10.1158/1535-7163.MCT-14-0710. [DOI] [PubMed] [Google Scholar]
- 55.Lu J, McEachern D, Li S, Ellis MJ, Wang S. Reactivation of p53 by MDM2 inhibitor MI-77301 for the treatment of endocrine-resistant breast cancer. Mol Cancer Ther. 2016;15:2887–2893. doi: 10.1158/1535-7163.MCT-16-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]