

Abstract
Aldo-keto reductase 1B10 (AKR1B10), a member of aldo-keto reductase superfamily, contributes to detoxification of xenobiotics and metabolization of physiological substrates. Although increased expression of AKR1B10 was found in hepatocellular carcinoma (HCC), the role of AKR1B10 in the development of HCC remains unclear. This study aims to illustrate the role of AKR1B10 in hepatocarcinogenesis based on its intrinsic oxidoreduction abilities. HCC cell lines with AKR1B10 overexpression or knockdown were treated with doxorubicin or hydrogen peroxide to determinate the influence of aberrant AKR1B10 expression on cells’ response to oxidative stress. Using Akr1b8 (the ortholog of human AKR1B10) knockout mice, diethylnitrosamine (DEN) induced liver injury, chronic inflammation and hepatocarcinogenesis were explored. Clinically, the pattern of serum AKR1B10 relevant to disease progression was investigated in a patient cohort with chronic hepatitis B (n=30), liver cirrhosis (n=30) and HCC (n=40). AKR1B10 expression in HCC tissues was analyzed using both the TCGA database (n=371) and our collected HCC samples (n=67). AKR1B10 overexpression reduced hepatocyte injury while AKR1B10 knockdown augmented reactive oxygen species (ROS) accumulation and apoptotic cell death. Consistently, Akr1b8 deficiency in mice promoted DEN-induced hepatocyte damage and liver inflammation characterized by increased phospho-H2AX, serum alanine aminotransferase, interleukin-6 and tumor necrosis factor alpha level, myeloid cell infiltration and led to more severe hepatocarcinogenesis and metastasis compared with wild type mice due to significant alteration on detoxification and oxidoreduction. AKR1B10 was compensatory expressed and gradually upregulated in the process of liver disease progression in HCC and increased oxidative stress upregulated AKR1B10 through NRF2. Our results here suggested that through oxidoreduction and detoxification, AKR1B10 played an important role in protecting hepatocytes from damage induced by ROS. Deficiency of AKR1B10 might accelerate hepatotoxin and inflammation-associated hepatocarcinogenesis. AKR1B10 expression elevation in HCC could be a result of compensatory upregulation, rather than a driver of malignant transformation during the development of HCC.
Keywords: Hepatocarcinogenesis, AKR1B10, ROS, oxidoreduction
Introduction
Aldo-keto reductase 1B10 (AKR1B10) is a member of the human aldo-keto reductase (AKR) superfamily which catalyzes the NAD(P)H-dependent oxidoreduction of various carbonyl compounds [1]. The expression of AKR1B10 leads to detoxification of xenobiotics from lipid peroxidation and metabolization of physiological substrates such as farnesal, retinal, and a variety of aromatic and aliphatic aldehydes or dicarbonyl compounds. Growing evidences have suggested that the abnormal expression of AKR1B10 was associated with many kinds of cancers including colorectal carcinoma [2], non-small cell lung carcinoma (NSCLC) [2], esophageal carcinoma [4], breast cancer [5], uterine cancer [6], pancreatic cancer [7], gastric cancer [8] and particularly, the preliminary liver cancer [9]. However, the detailed mechanisms of AKR1B10 in hepatocarcinogenesis remain elusive.
Hepatocellular carcinoma (HCC) is one of the most common human cancers worldwide which accounts for the third most likely cause of cancer related mortality [10,11]. It is well known that development of HCC is a complex and multistep process, with one or more risk factors including hepatitis B or C virus infection, alcohol abuse, non-alcoholic steatohepatitis (NASH), dietary exposure to aflatoxin B1 (AFB1) from fungal contaminant, or exposure to other chemical carcinogens [12,13]. Though the detailed molecular pathogenesis of HCC has not been fully elucidated, the misregulation of oxidative stress and lesions caused by chemical carcinogens exposure and/or inflammatory damage are involved [13-15].
It is well known that hypoxia and abundant reactive oxygen species (ROS) accumulation is a prevalent tumor microenvironment and oxidative stress is an important factor in driving malignancy transformation and cancer progression [14,16,17]. ROS consisted by of free radicals or reactive nonradical species, such as superoxide anion, singlet oxygen, hydrogen peroxide (H2O2), perhydroxyl radical and hydroxyl radical from unsaturated carbonyls and aldehydes [18]. Previous studies demonstrated that AKR1B10 could efficiently catalyze and eliminate acrolein, crotonaldehyde, HNE and other unsaturated carbonyls and aldehydes [19-21], suggested that AKR1B10 might participate in the redox and elimination of ROS. AKR1B10 has been found predominantly expressed in the human digested organs like colon, small intestine, liver and the stomach where they will act directly with xenobiotics from oral intake [1,8,22]. Liver is the primary organ to detoxify various metabolites with ROS producing or xenobiotics. Indeed, early studies have reported the aberrant elevation of AKR1B10 in HCC tumor tissues and suggested that AKR1B10 might act as a oncogene to drive hepatocarcinogenesis [23,24]. In the cancer, however, excessive ROS would also cause DNA damage which in turn induced tumor cell apoptotic death. Therefore, to neutralize the extra ROS produced during fast cell proliferation and growing, tumor cells also evolved an antioxidant system to balance the double-edged sword of ROS. Since AKR1B10 functions as an enzymatic antioxidant that can neutralize the extra ROS, it is reasonable to assume that elevated AKR1B10 in tumor may play an important role in protecting hepatocytes from both endogenous and exogenous stress. However, the exact function of AKR1B10 in liver and the underlying mechanism of its upregulation in the process of hepatocarcinogenesis is still elusive.
In this study, the detoxification role of AKR1B10 in oxidative stress was analyzed in different HCC cell lines, either with AKR1B10 ectopic overexpression or knockdown. To clarify the role of AKR1B10 in hepatocarcinogenesis, the susceptibility to diethylnitrosamine (DEN) induced liver cancer were explored in Akr1b8 (the ortholog of human AKR1B10 [25]) knockout mice. In addition, the underlying mechanism relevant to AKR1B10 upregulation were investigated both in the progression of hepatocarcinogenesis in vivo and in cultured HCC cells.
Materials and methods
Patient samples
To assess the level deference of circulated AKR1B10 associated with chronic liver disease progression from CHB to cirrhosis and to hepatocellular carcinoma, serum specimens from patients with CHB (n=30), liver cirrhosis (LC, n=30) and HCC (n=40) were collected in 2010 from Beijing You’an Hospital, Capital Medical University. The clinical-pathological characteristics of the patients were shown in Table 1. All the patients had laboratory evidence(s) of chronic HBV infection by serum HBsAg and/or HBV DNA detection positive, and the inclusion criteria for the patients followed the Guideline of Prevention and Treatment for Chronic Hepatitis B (2015 Update) [26].
Table 1.
Clinicopathological parameters of patients with chronic hepatitis B (CHB, n=30), liver cirrhosis (LC, n=30) and HCC (n=40)
| Characteristics | Variables | CHB | LC | HCC |
|---|---|---|---|---|
| Cases n (%) | Cases n (%) | Cases n (%) | ||
| n=30 | n=30 | n=40 | ||
| Age (years) | Median (IQR) | 29.50 (25.50, 39.75) | 56.50 (47.00, 66.25) | 56.50 (50.00, 62.50) |
| Gender | Male | 24 (80%) | 21 (70%) | 29 (72.5%) |
| Female | 6 (20%) | 9 (30%) | 11 (27.5%) | |
| ALT (U/L) | Median (IQR) | 810.40 (176.25, 1084.60) | 54.15 (33.88, 100.25) | 43.85 (26.30, 67.38) |
| AST (U/L) | Median (IQR) | 251.40 (111.80, 574.65) | 87.00 (52.05, 158.63) | 62.70 (35.35, 127.68) |
IQR, Inter Quartile Range.
Primary HCC and the adjacent non-tumor tissues (n=67) were collected from patients who underwent routine curative surgery from 2009 to 2013 at Henan Oncology Hospital in Zhengzhou, Henan Province of China. All HCC patients were confirmed by pathological diagnosis, and none of them received any chemotherapy or radiotherapy before surgery. The clinical-pathological characteristics of the HCC patients were shown in Table 2. The normal liver tissues (n=9) were obtained from liver donors in the same hospital.
Table 2.
Clinicopathological parameters of 67 patients with HCC
| Characteristics | Variables | Cases n (%) |
|---|---|---|
| n=67 | ||
| Age (years) | Median (IQR) | 54 (46, 63) |
| Gender | Male | 56 (83.6%) |
| Female | 11 (16.4%) | |
| Liver cirrhosis | Yes | 61 (91.0%) |
| No | 6 (9.0%) | |
| Infection background | HBV | 52 (77.6%) |
| HCV | 3 (4.5%) | |
| Without HBV or HCV | 12 (17.9%) | |
| Portal vein tumor thrombosis | Present | 55 (82.1%) |
| Absent | 12 (17.9%) | |
| Tumor size | ≥5 cm | 55 (82.1%) |
| <5 cm | 12 (17.9%) | |
| BCLC stage | A | 39 (58.2%) |
| B | 7 (10.4%) | |
| C | 20 (29.9%) | |
| D | 0 | |
| N/A | 1 (1.5%) |
IQR, Inter Quartile Range. N/A, Not available; HBV, hepatitis B virus; HCV, hepatitis C virus; BCLC, Barcelona Clinic Liver Cancer; A, early stage; B, intermediate stage; C, advanced stage; D, terminal stage.
This study was approved by the Ethics Committee of Peking University Health Science Center and the informed consents were obtained from all patients before the start of study. For patients who were children, written informed consents were obtained from their guardians.
Cell lines
HepG2 and 293T cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) while Huh7, Huh1, SNU387 cells were maintained as a laboratory stock and were recently authenticated by short tandem repeat profiling. All cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) at 37°C in a 5% CO2 atmosphere.
AKR1B10 knockdown and overexpression
For AKR1B10 knock down, small-hairpin RNA inhibition plasmids were constructed by annealing two oligonucleotides and subsequently ligated into the TRC2B vector after digestion with BamHI and EcoRI. The oligonucleotide sequences used for shRNA construction were listed in Supplementary Table 1. For stable expression, 3×105 HepG2 cells were seeded in 6-well plates with 2 ml of medium without antibiotics. Following overnight incubation, the media was replaced with 1 mL of medium containing lentivirus in 10 μg/mL polybrene. Twenty-four hours later, the media was replaced with 2 mL fresh media. Forty-eight hours later, media was removed and fresh media containing 2 μg/mL puromycin was added to each well and replaced every 2 to 3 days for stable AKR1B10-sh expressed cells selection. AKR1B10 overexpression vector was constructed as previously described [27] and was used for transient transfection in Huh7 cells using lipofectamine 2000 (Life Technologies, Carlsbad, CA) according to the manufacturer’s instruction. For the doxorubicin or H2O2 administration under AKR1B10 overexpression, doxorubicin or hydrogen peroxide were added 36 hours after the transfection and were administrated for 36 hours before the cells were harvested.
Animal experiments
Akr1b8 knock out (KO) mice were generated by Professor Deliang Cao as previously described [2] and were used to hybridize with wild type (WT) C57BL/6 mice bought from Peking University Health Science Center to breed Akr1b8 homozygous KO mice. All mice were maintained under specific pathogen free conditions in the Department of Laboratory Animal Science of Peking University Health Science Center. The experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the animal ethics committee of Peking University Health Science Center (Beijing, China). For DEN administration, two-week-old WT or Akr1b8-KO male mice were intraperitoneally injected with a single DEN dose of 25 mg/kg. The animals were monitored thereafter by body weight and sacrificed by cervical dislocation at 6 (n=8), 9 (n=11) and 12 (n=4) months post DEN treatment for serum and liver isolation. For DEN-induced hepatocyte injury study, 7 male WT and Akr1b8-KO mice after DEN treatment for 1 month were used for serum detection.
Compounds
Hydrogen peroxide solution (Amresco, Houston, TX) was used to induce oxidative stress and chemotherapeutic agent doxorubicin (Cell signaling technology, Danvers, MA) was dissolved in sterile PBS to generate a 4 mg/μL stock solution and stored at -20°C. Diethylnitrosamine (Sigma-Aldrich, St. Louis, MO) was dissolved in saline to generate 0.01 g/mL DEN solution and stored at 4°C.
Western blot
Proteins were extracted using Radio Immunoprecipitation Assay (RIPA) protein lysis buffer, boiled for 10 min at 95°C. The denatured proteins were separated on 12% sodium dodecyl sulfate-polyacrylamide gels and then blotted onto nitrocellulose membranes (Amersham Biosciences, Uppsala, Sweden) using Mini-Protean II transfer apparatus (Bio-Rad, CA). The antibody (NRF2, Proteintech Group, Inc, IL; AKR1B10 and Bach1, Santa Cruz, CA; p-H2AX, CTBP and PARP, Abcam, Cambridge, UK; Beta-actin, ABGENT, CA; Gapdh, Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) were used for immunoprobing as previously described [28].
Immunocytofluorescence
AKR1B10-konckdown HepG2 Cells and control HepG2 cells were seeded in a 24-well plate at 1×105 cells per well. Twenty-four hours later, cells were treated with doxorubicin (0.5 μg/ml) for 24 hours and then fixed in 4% paraformaldehyde in phosphate-buffered saline for 20 min, permeabilized with 0.3% Triton X-100 for 10 min and subsequently incubated with p-H2AX antibody, followed by a FITC-conjugated secondary antibody. Finally, the cells were counterstained with Hoechst and examined under a Leica inverted fluorescence microscope (DM130CCB; Leica, Solms, Germany).
Biochemical markers detection
ROS Detection Kit and Cell Malondialdehyde (MDA) Detection Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangshu, China) were used to detect the cell ROS and MDA level respectively under the manufacturer’s instructions. Mice serum Alanine aminotransferase (ALT), IL-6 and TNF-α levels were determined using the commercial Mouse ELISA Kits (DingSheng, Beijing, China) according to the manufacturer’s instructions.
Luciferase reporter assay
To construct the AKR1B10-luc vector, the promoter sequences -976 nt to +278 nt of AKR1B10 (NCBI ID: NC_000007.14) were PCR amplified from HepG2 genomic DNA and inserted into the luciferase reporter vector pGL3-basic (Promega, Madison, Wis) after digestion with KpnI and XhoI. The mutant AKR1B10-luc-M vector was generated from AKR1B10-luc using AKR1B10-luc-M-F and AKR1B10-luc-M-R by Quik Change site-directed mutagenesis kit (Stratagene, La Jolla, CA). To construct NRF2 overexpression vector, the NRF2 cDNA was cloned from HepG2 cDNA and ligased into pcDNA3.1 plasmid after digestion with KpnI and XhoI. The luciferase reporter assays were carried out as described previously [29]. The primer sequences used for plasmid construction were listed in Supplementary Table 2.
Cell apoptosis assay
Apoptosis was evaluated by flow cytometry using cellular Annexin V-FITC PI Apoptosis Assay Kit (Bestbio, Shanghai, China) according to the manufacturer’s instructions and analyzed using a BD flow cytometer (BD BioSciences, San Jose, CA). Cells in early apoptosis are Annexin V positive and PI negative, while cells in late apoptosis are both Annexin V and PI positive. Annexin V and PI double-negative cells were considered viable.
Cell viability assay
Cell viability assays were carried out by CCK-8 kit (DojindoLaboratories, Rockville, MA) according to the manufacturer’s instruction. Briefly, 3×104 HepG2 cells were seeded in 96-well plates followed by Hydrogen peroxide (5 μM) or doxorubicin (0.3 μg/mL) treatment 12 hours later. Then the cell viability was detected every 24 hours.
Colony formation assay
For doxorubicin treatment, HepG2 AKR1B10 knockdown cells or control cells were seeded in 6-well plates at 1×105 cells per well, respectively. The cells were insulted with 0, 0.1, 0.3, 0.5 μg/mL doxorubicin after 4 days, followed by replacing the medium using flesh medium with doxorubicin at the same concentration every 2 days. One week later, cells were fixed and stained with methanol/crystal violet and photographed. For Hydrogen peroxide treatment, HepG2 AKR1B10 knock down Cells or control cells were seeded in 6-well plates at 3×105 cells per well, respectively. The cells were insulted with 0, 10, 50, 100 μM hydrogen peroxide after 24 hours, followed by operation same as doxorubicin treatment.
Immunohistochemistry
Liver tissues were fixed in 4% formalin overnight, embedded in paraffin, sectioned at 4 μm and stained using a standard technique as described previously [28]. The primary antibodies used for IHC including anti-Ki67 (Abcam, Cambridge, UK), anti-CD34 (Abcam, Cambridge, UK), anti-F4/80 (Abcam, Cambridge, UK). More than 3 randomly chosen magnification fields were imaged from each mouse liver with at least 3 mice per group.
Next-generation sequencing and data analysis
RNA was extracted with a standard method from mice liver tumors followed by concentration measurement using Qubit® RNA Assay Kit in Qubit® 3.0 Fluorometer (Life Technologies, CA, USA). Same amount of RNA from three Akr1b8-KO or WT mice was mixed as the KO or WT sample respectively and the transcriptome sequencing was relegated to Vazyme Biotech (Nanjing, Jiangsu, China). Briefly, the libraries were generated and sequenced on an Illumina Hiseq X Ten platform. Genes with corrected p values less than 0.05 and the absolute value of log2<1 were assigned as significantly differentially expressed. The Gene Ontology (GO) terms enrichment and pathway annotation using the Kyoto Encyclopedia of Genes and Genomes (KEGG) were analyzed and the false discovery rate (FDR) was used to test for the statistical enrichment. To improve the enrichment accuracy and reduce the enrichment background due to some big pathways with large amount of genes, the pathways with enriched genes less than 100 were considered. The gene correlation network was depicted by the STRING database and the discrete cluster was isolated by Cytoscape Mcode.
Statistical analysis
For statistical analysis, two-tailed Student’s t-test and Fisher’s exact test were performed using the Statistical Product and Service Solutions (SPSS) v21.0 (Xishu Software Company, Shanghai, China). In all cases, a P value of <0.05 was considered as statistically significant. Data was presented as the mean standard deviation (s.d.) from at least three independent experiments.
Results
AKR1B10 was involved in reducing oxidative stress and protecting hepatocytes against ROS induced injury
To investigate the function of AKR1B10 in hepatocytes under oxidative damage, AKR1B10 was detected in four HCC cell lines including HepG2, Huh1, SNU387 and Huh7 and was silenced by shRNA in HepG2 cells with higher endogenous AKR1B10 expression, or was ectopically overexpressed in Huh7 cells with lower endogenous AKR1B10 expression (Figure 1A and Supplementary Figure 1). Then cells were treated by doxorubicin (Dox) or hydrogen peroxide (H2O2) respectively. Dox belongs to the anthracyclines of antitumor agents which contains carbonyl groups and can efficiently induce generation of free radicals and intercalate with DNA to lead to apoptotic cell death [30,31]. H2O2 is a major source of ROS and is widely used to trigger oxidative damage in the cell level [18,32]. As shown in Figure 1B, the ROS assay revealed that compared with the control cells, AKR1B10 knockdown caused more severe ROS accumulation under H2O2 and Dox exposure. MDA was usually generated by peroxidation of polyunsaturated fatty acids and was widely used to assess lipid peroxidation and ROS accumulation [33]. We then tested the MDA level in AKR1B10 knockdown HepG2 cells. Consistent with the more serious oxidative stress observed in AKR1B10 knockdown cells, the MDA level was significantly higher in AKR1B10 knockdown cells than that in the control cells after H2O2 or Dox treatment (Figure 1C). When the AKR1B10 was overexpressed, the MDA level reduced significantly. These results indicated that the expression of AKR1B10 might play an important role in defending hepatocytes against oxidative stress-induced damage by reducing ROS accumulation.
Figure 1.

AKR1B10 reduced oxidative stress and protected hepatocytes against apoptotic cell death. A. Western blot analysis confirmed AKR1B10-knockdown in HepG2 cells (top) or AKR1B10-overexpression in Huh7 cells (bottom). B. ROS accumulation assayed by DCFH-DA (2,7-dichlorofuorescin diacetate) probe in AKR1B10-knockdown HepG2 cells treated either with doxorubicin (Dox, 0.5 μg/mL) or with hydrogen peroxide (H2O2, 50 µM) for 36 hours, respectively. The merge indicated combined DCFH-DA (green) and the bright filed. The scale bar is 400 μm. C. MDA levels were detected in AKR1B10 knockdown HepG2 cells (left panel) or in ectopically overexpression Huh7 cells (right panel), after Dox (0.5 μg/mL) or H2O2 (50 µM) treatment for 36 hours respectively. D. Western blot and immunofluorescent analysis of p-H2AX level in AKR1B10-overexpressed Huh7 cells (top) or AKR1B10-knockdown HepG2 cells (bottom) after Dox (0.5 μg/mL) or H2O2 (50 µM) treatment for 36 hours, respectively. The scale bar is 500 μm. E, F. Flow cytometry and western blot analysis of apoptosis in AKR1B10 knockdown HepG2 cells after Dox (0.5 μg/mL) or H2O2 (50 µM) treatment for 48 hours. G. CCK-8 assays of HepG2 cells with or without AKR1B10 knockdown after Dox (0.3 μg/mL) or H2O2 (5 µM) treatment. Cell viability were detected at indicated time points and normalized to that of control cells without treatment. H. Colony formation assays of HepG2 cells after Dox or H2O2 treatment with different dose for 7 days. Statistical significance was determined with Student t two-tailed test. *indicated P<0.05. **indicated P<0.01.
ROS are considered as carcinogenic potentials that facilitate cancer promotion and progression, while excessive amounts of ROS may induce cell oxidative damage and death [34]. To investigate the likely protective roles of AKR1B10 in hepatocytes, we detected the DNA damage and cell apoptosis induced by ROS in HCC cells with AKR1B10 overexpression or knockdown. First, expression level of phospho-H2AX (p-H2AX), a marker for DNA damage, was analyzed by western blot and immunofluorescent assays. As shown in Figure 1D, AKR1B10 overexpression significantly reduced the p-H2AX level in Huh7 cells after H2O2 or Dox administration and conversely, knockdown of AKR1B10 expression in HepG2 cells led to a significant increase of p-H2AX expression compared to the control cells. Next, apoptosis of these HCC cells was detected by flow cytometry and western blot assays. The results revealed that exposure to H2O2 or Dox caused an increased incidence of apoptosis after silencing of AKR1B10 expression (Figure 1E, 1F). Moreover, the enhanced apoptosis induced by AKR1B10 knockdown upon oxidative stress was demonstrated by increased cleaved-PARP levels. Finally, the results of CCK-8 and colony formation assays showed that in consistent with the enhanced apoptosis, AKR1B10 knockdown significantly reduced cell proliferation and colony formation ability in HepG2 cells upon oxidative stress (Figure 1G, 1H). Taken together, these data suggested that AKR1B10 could protect hepatocytes and knockdown of AKR1B10 accelerated DNA damage and apoptotic cell death under oxidative stress.
Loss of Akr1b8 (AKR1B10 ortholog gene) in mice enhanced chemical-induced liver tumorigenesis and metastasis
Since AKR1B10 could protect hepatocytes from DNA damage and apoptotic cell death under oxidative stress, its role in HCC development and progression were addressed by using Akr1b8 (the ortholog of human AKR1B10 [25]) KO C57 mice. Two-week-old male WT and Akr1b8-KO mice were intraperitoneal injected with a single dose of 25 mg/kg DEN. The mice were sacrificed at 6 (n=8), 9 (n=11) and 12 (n=4) months post DEN administration (Figure 2A). Macroscopically, nodules were found in the livers of both WT and Akr1b8-KO mice 6 months after DEN administration (Figure 2B). However, significantly higher tumor incidence (75.0% vs 12.5%, P=0.041) and tumor number (P=0.0398) were observed in the livers of Akr1b8-KO mice, as compared with WT mice (Figure 2C). Compared with WT mice under the same treatment, a more severe tumor development tendency remained in Akr1b8-KO mice 9 months after DEN treatment in both tumor incidence (90.9% VS 63.6%) and tumor weight verse liver weight (45.1% vs 32.7%). Though all mice developed liver tumor 12 months after DEN treatment, Akr1b8-KO mice still showed more advanced tumor development characterized by higher tumor weight verse liver weight than WT mice (P=0.0286) (Figure 2C). Hematoxylin and eosin (H&E) staining and IHC assays showed that nodules from the Akr1b8-KO mice retained major histological features of primary HCC with the confluent areas of cytological atypia such as huge nuclei and multinuclear hepatocytes (Figure 2D). IHC for the proliferation marker Ki67 revealed ongoing proliferation of cells in Akr1b8-KO nodules. Further IHC assays of CD34 staining showed that there was a significant angiogenesis in Akr1b8-KO tumors (Figure 2D). In addition, lung metastasis from HCC was observed in one of the Akr1b8-KO mice after 12 months of DEN administration (Figure 2E). The basic leucine zipper transcription factor, BTB and CNC homology 1 (Bach1) has been identified in metastatic signatures in variety cancers which can induce expression of metastatic genes and can also increase glycolysis to promotes metastasis [35,36]. Consistently, western blot assays further showed that the Bach1 expression was more obvious in the livers of Akr1b8-KO mice than that in WT mice at both 9 and 12 months post DEN injection (Figure 2F). Taken together, these data indicated that deficiency of Akr1b8 enhanced chemical-induced liver tumorigenesis and progression in mice.
Figure 2.

Knockout of Akr1b8 in mice promoted DEN-induced hepatocarcinogenesis and metastasis. A. Schematic of DEN intraperitoneal injection and the experimental design. B. Representative photographs of mice livers harvested post 6, 9 and 12 months DEN administration. The scale bar is 5 mm. C. Liver tumorigenesis in WT and Akr1b8-KO mice post 6, 9, and 12 months DEN injection. Liver tumors ≥0.5 mm were considered. D. Histological and pathological determination characterized by H&E staining, Ki67 and CD34 IHC staining of the liver sections in WT and Akr1b8-KO mice 6 months after DEN injection. For Ki67 and CD34 IHC staining, the representative positive staining cells were showed by arrows. The scale bar is 200 μm. E. Gross (arrowhead) and histological appearance of lung metastasis in Akr1b8-KO mice. The scale bar is 400 μm. F. Western blot analysis of Bach1 level in livers of WT and Akr1b8-KO mice 9 and 12 months after DEN injection. Data represent means ± s.d. (n≥4). Statistical significance was determined with Fisher’s exact test or Student t two-tailed test. *indicated P<0.05.
Loss of Akr1b8 promoted hepatocyte damage and liver inflammation
To investigate the underlying mechanism relevant to increased susceptibility to DEN-induced hepatocarcinogenesis observed in Akr1b8-KO mice, the makers of hepatocyte damage and inflammatory response in WT and Akr1b8-KO mice were assessed. Compared to the untreated mice, DEN treatment significantly increased the serum ALT level in 1-month-old WT and Akr1b8-KO mice, implying the hepatocyte injury induced by DEN and especially in the KO mice at the time point of 9-month post DEN treatment (Figure 3A). Consistently, western blot assays showed that in the livers of Akr1b8-KO mice, the p-H2AX activation was more obvious than that in WT mice, indicating more serious hepatocyte injury and DNA damage after DEN treatment in Akr1b8-KO mice (Figure 3B). In addition, IHC for the peripheral blood lymphocyte marker CD3 identified an extensive periductular and periportal mixed inflammatory infiltrate in the livers of 6 or 9 month DEN treated Akr1b8-KO mice (Figure 3C). To further confirm the liver inflammation, we detected serum IL-6 and TNF-α, the main pro-inflammatory cytokines that indicated inflammatory response level of the mice. The results showed that the serum IL-6 and TNF-α levels were higher in Akr1b8-KO mice compared with the WT mice, and the difference was statistically significant for IL-6 at 6 months and for TNF-α at 9 months post DEN treatment, respectively (Figure 3D). The IHC of F4/80, a widely used marker of macrophages or Kuppfer cells also showed a significant macrophage infiltration in the livers of Akr1b8-KO mice 6 months after DEN treatment (Figure 3E) and in the tumor tissues 9 months after DEN treatment (Supplementary Figure 2). Taken together, these data suggested that in Akr1b8-KO mice the hepatocytes became more sensitive to DEN-induced damage and were more prone to liver necro-inflammation, thereby enhancing HCC development in mice.
Figure 3.
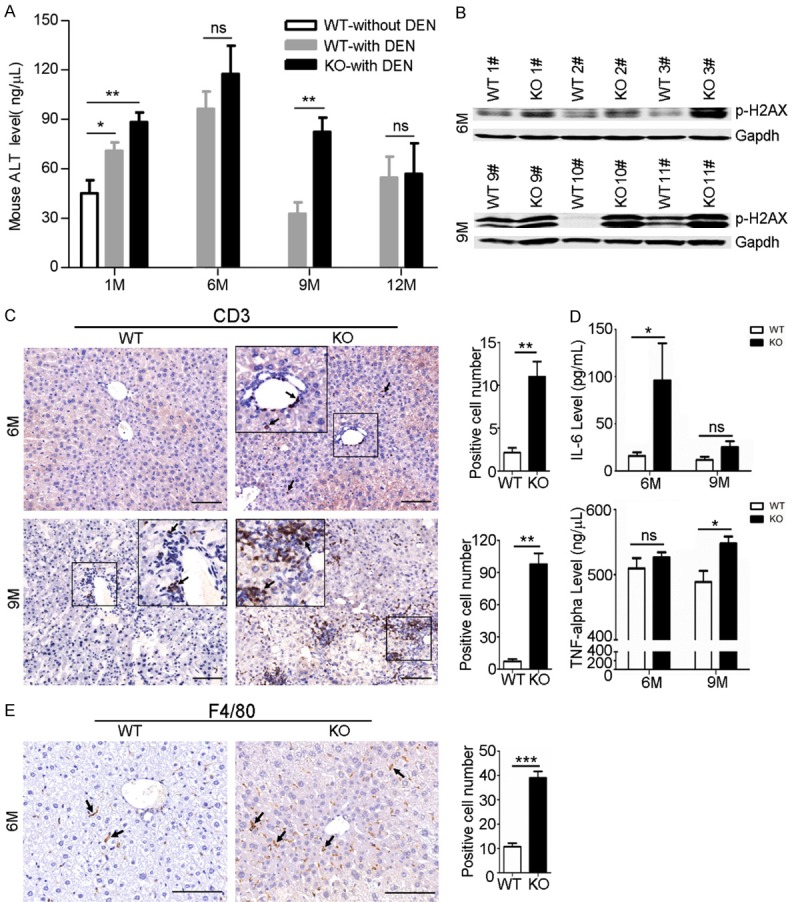
Akr1b8 Knockout in mice promoted hepatocyte damage and Liver inflammation. A. Changes of serum ALT level in WT or Akr1b8-KO mice with or without DEN administration for 1, 6, 9 and 12 months, respectively. B. Western blot analysis of p-H2AX level in livers of WT and Akr1b8-KO mice 6 and 9 months after DEN injection. C. IHC of CD3 in the none-tumor liver tissues of WT and Akr1b8-KO mice 6 and 9 months after DEN injection. The scale bar is 100 μm. Average numbers of CD3 positive stained cells in five high power fields were calculated and the data represent means ± s.d. (n=5). D. Changes of serum IL-6 and TNF-α levels in WT and Akr1b8-KO mice 6 and 9 months after DEN injection. E. F4/80 IHC staining of macrophages infiltration in the livers of WT and Akr1b8-KO mice after 6-month DEN treatment. Average numbers of F4/80 positive stained cells in five fields were calculated and the data represent means ± s.d. (n=5). The scale bar is 100 μm. The arrows indicated positive staining. Statistical significance was determined with Student t two-tailed test. *indicated P<0.05. **indicated P<0.01. ***indicated P<0.001.
Tumors in Akr1b8-KO mice displayed significant alteration on detoxification and oxidoreduction
AKR1B10 participates in the intracellular oxidoreductation metabolism of α, β-unsaturated carbonyls, which are highly reactive and cytotoxic, by detoxifying the electrophilic xenobiotics to less toxic alcohol forms in a NADPH-dependent way [19]. AKR1B10 also is a key enzyme in retinoic acid (RA) metabolism through its high catalytic efficiency on all-trans-retinaldehyde, triggering a decrease in the RA biosynthesis flow, and may involve in different signaling pathways related to RA [37]. The biological functions of AKR1B10 were depicted in Figure 4A. To associate the tumorigenesis in Akr1b8-KO mice to the loss of Akr1b8 function, the global gene expression of liver tumors from WT and Akr1b8-KO mice after 12 month DEN administration was performed using transcriptome sequencing by Illumina HiSeq. The RNA-seq data identified 829 differently expressed genes between WT and Akr1b8-KO mice. The GO analysis of the differently expressed genes showed that oxidoreductation terms such as oxidoreductase activity, monooxygenase activity and carboxylic ester hydrolase activity in the molecular function process were enriched (Figure 4B). In the KEGG pathway annotation analysis, the top 10 pathways enriched in detoxification and xenobiotics metabolism pathways including chemical carcinogenesis, metabolism of xenobiotics by cytochrome P450, propanoate metabolism and drug metabolism (Figure 4C). Further gene correlation analysis among these differently expressed genes identified a strongly concentrated correlation among the genes of oxidoreductase activity in GO terms, chemical carcinogenesis and retinol metabolism in the KEGG pathways (Figure 4D and Supplementary Figure 3). Taken together, these data suggested that hepatocytes with Akr1b8 knockout might undergo tumorigenesis due to loss of efficient oxidoreduction and detoxification of Akr1b8 and the resulted excessive oxidative stress.
Figure 4.

Different oxidoreduction and detoxification profile in the DEN-induced liver tumors between WT and Akr1b8-KO mice. (A) The biological function of AKR1B10 in carbonyls and retinoic acid (RA) metabolism. R, R1 and R2 stand for various chemical groups. ADH4/7, alcohol dehydrogenase 4/7. ALDH1a1, aldehyde dehydrogenase 1 family member A1. SDR, short-chain dehydrogenase. RAR, retinoic acid receptor. RXR, retinoid X receptor. PPAR, peroxisome proliferator-activated receptor-α. (B, C) KEGG and GO analysis of the different expression genes in liver tumors between WT and Akr1b8-KO mice after 12 months DEN administration. The top 10 terms from GO analysis (B) in the molecular function process and the top 10 pathway items from KEGG analysis (C) were shown. FDR, false discovery rate. (D) Gene relative network among the differently expressed genes enriched in oxidoreductase activity in GO terms and chemical carcinogenesis and retinol metabolism in KEGG pathways. Genes enriched in oxidoreductase activity, chemical carcinogenesis and retinol metabolism were indicated by red, blue and green color, respectively. The genes with more than one color indicated they were involved in different GO terms or KEGG pathways.
AKR1B10 was compensatory upregulated in the process of liver tumorigenesis
Chronic liver diseases including viral hepatitis, necrosis, fibrosis, cirrhosis and HCC are often accompanied by high ROS production and oxidative stress in liver, irrespective of the cause of the liver dysfunction [38,39]. To investigate whether AKR1B10 expression could be upregulated accompanied with chronic liver disease progression we detected the AKR1B10 level in the serum of patients with chronic hepatitis B (CHB, n=30), liver cirrhosis (LC, n=30) and HCC (n=40). The results showed that with the progression of liver diseases, the AKR1B10 levels were sequentially increased (Figure 5A). To confirm if upregulation of AKR1B10 expression attributed to tumor development, AKR1B10 mRNA level was quantitatively determined in 67 paired HCC tumor tissues and non-tumor liver tissues, and 9 normal liver tissues by RT-qPCR assays. The results showed that AKR1B10 expression was significantly higher in liver tumors compared to the non-tumor and normal liver tissues (Figure 5B), which was further confirmed by Western blot (Figure 5C). In line with these results, analysis of data from TCGA database revealed that AKR1B10 mRNA expression was significantly higher in tumors than that in the non-tumor tissues and more than that, the higher AKR1B10 expression was significantly associated with poor survival rate in HCC patients (Figure 5D, 5E). To further conform the compensatory or adaptive upregulation of AKR1B10/Akr1b8, we detected the Akr1b8 expression in WT mice livers with (n=5) or without (n=6) DEN administration for 1 month. The results showed that upon DEN treatment, there was a significant Akr1b8 expression increase in mRNA level (P=0.0043, Supplementary Figure 4A). Concordantly, this up-regulation was further confirmed by other researchers in rats with (n=3) or without DEN (n=3) treatment through the whole transcript array assay (GEO database ID: GSE123408 [40]) which showed a dramatically Akr1b8 expression increase of about 64 times in DEN treated rat livers (Supplementary Figure 4B). The results suggested that AKR1B10 expression levels might be compensatively upregulated during the progression of chronic liver diseases to protect hepatocytes against excessive oxidative stress and maintain a relatively higher level of ROS homeostasis in the tumor microenvironment. Notably, we also found that another two members Akr1b3 and Akr1b7 in aldo-keto reductase superfamily which has the similar detoxification enzymatic function of Akr1b8 [41], were also upregulated in liver tumors or upon DEN treatment in mice (Supplementary Figure 5).
Figure 5.
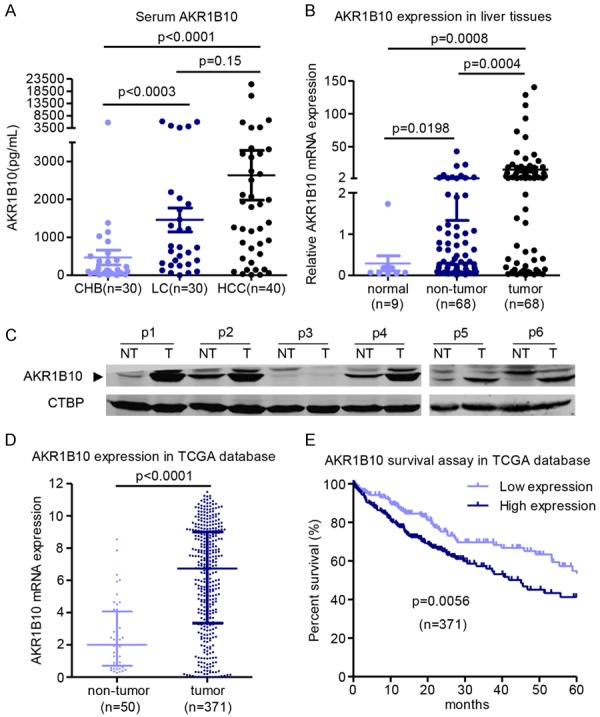
AKR1B10 was compensatory upregulated with the increased oxidative stress in liver disease progression. A. AKR1B10 concentrations in serum of patients with chronic hepatitis B (CHB), liver cirrhosis (LC) and HCC. B. AKR1B10 mRNA expression in liver tissues detected by RT-qPCR assay. C. Western blot assay about the AKR1B10 expression in HCC samples and the adjacent none-tumor tissues (n=6). p, patient. NT, non-tumor. T, tumor. D. AKR1B10 mRNA expression data collected from TCGA database. E. The overall survival of the 371 patients analyzed according to the AKR1B10 expression level in TCGA database.
AKR1B10 was upregulated via NRF2 upon oxidative stress in hepatocytes
It is known that persistent inflammation caused by oxidative stress and hepatocyte injury is the major reason to drive and promote tumorigenesis in liver and hypoxia and abundant ROS accumulation is a prevalent tumor microenvironment [14,16]. To investigate the potential mechanism of AKR1B10 upregulation in HCC, Dox was used to induce oxidative stress and cellular toxicity in Huh7 cells. The ROS assay revealed that doxorubicin could cause evident ROS production (Figure 6A). RT-qPCR and western blot assays showed that Dox treatment could increase AKR1B10 expression both in mRNA and protein level (Figure 6B, 6C). Nuclear factor related factor 2 (NRF2) is a critical transcription factor for regulating expression of genes involved in decreasing intracellular ROS level by binding to antioxidant response element (ARE) [42] and the western blot assay showed that the doxorubicin treatment induced significant NRF2 expression (Figure 6C). Since ARE sequence was identified in AKR1B10 promoter located from -228 to -214 by the JASPAR database [43] (Figure 6D), we then detected the possible regulating role of NRF2 on AKR1B10 expression in hepatocytes. The luciferase reporter assays demonstrated that overexpression of NRF2 could increase the transcriptional activity of AKR1B10 promoter, while had no effect on the ARE-mutated AKR1B10 promoter (Figure 6E). Consistently, the western blot assay demonstrated that AKR1B10 expression level was increased in NRF2-overexressed Huh7 cells, compared with cells expressing the vector control (Figure 6F). Additionally, the luciferase reporter assays also showed that the AKR1B10 promoter activity was increased by Dox and H2O2 treatment in a dose-dependent manner, implying endogenous expression of NRF2 induced by ROS could modulate AKR1B10 expression (Supplementary Figure 6). Taken together, these data suggested that the upregulation of AKR1B10 expression might be mediated by NRF2 through the key motif in AKR1B10 promoter in the cellular response to ROS in hepatocytes.
Figure 6.
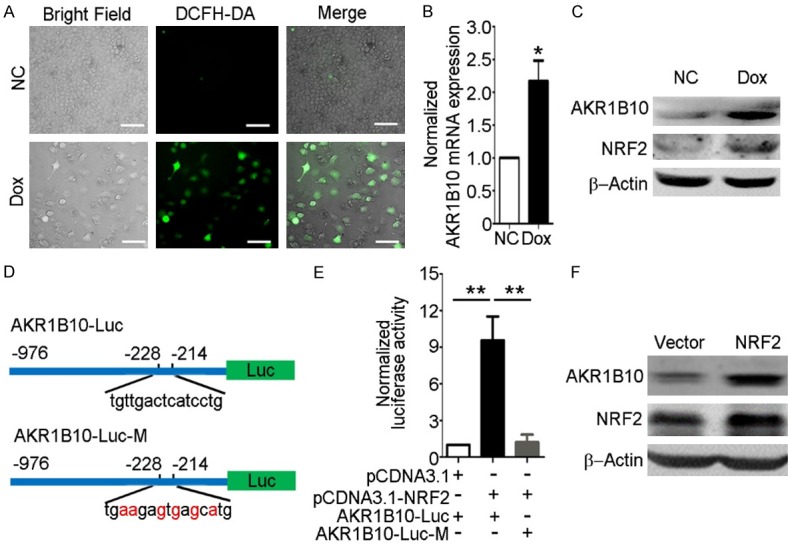
AKR1B10 was upregulated via NRF2 upon oxidative stress. A. ROS assay in Huh7 cells with Dox (0.5 μg/mL) treatment for 36 hours. DCFH-DA was the probe targeting the ROS. The scale bar is 200 μm. B, C. RT-qPCR and western blot assays of AKR1B10 in Huh7 cells after treatment with Dox (0.5 μg/mL) for 16 hours and 36 hours, respectively. D. Schematic of NRF2 transcriptional regulation element and the site-directed ARE mutation located in AKR1B10 promoter. The nucleotides in red indicated the mutant sites compared to the wild type AKR1B10 promoter sequence. E. Luciferase activity assay detected in Huh7 cells with AKR1B10 promoter and NRF2 expression plasmids transfection. F. Western blot assay of AKR1B10 protein level with NRF2 overexpression. Data showed the mean ± s.d. of three independent experiments (*P<0.05, **P<0.01, Student’s t-test, two-tailed).
Discussion
As a cytosolic NADPH-dependent reductase, AKR1B10 plays a critical role in detoxifying intracellular cytotoxic carbonyls and mediating oxidative stress decrease. Elevated expression of AKR1B10 has been observed in several kinds of tumors including HCC, but it is still unclear whether AKR1B10 contributes to the pathogenesis of the disease. Our data here demonstrated that AKR1B10 could protect hepatocytes against ROS-induced injury and the upregulation of AKR1B10 was not a driver in hepatocarcinogenesis, but a compensatory or adaptive response to the malignant cell microenvironment.
It is well known that oxidative stress induced by ROS is a major contributing factor to the tumorigenesis in liver regardless of etiology [38]. The balance between ROS production and antioxidant defenses determines the degree of oxidative stress. With the important role of AKR1B10 in oxidoreduction and detoxification of xenobiotics, it is logical to presume that AKR1B10 might protect hepatocytes from oxidative stress induced from ROS. In this study, we demonstrated that overexpression of AKR1B10 reduced the ROS-induced DNA damage, while knockdown of AKR1B10 increased the DNA damage, and the later accompanied with elevated cell apoptosis. These findings suggested that the AKR1B10 upregulation was responsible for the resistance to ROS-induced hepatocyte damage through the oxidoreduction and detoxification activities of AKR1B10. Consistent with these results from in vitro experiments, deficiency of AKR1B10 ortholog gene (Akr1b8) in mice enhanced chemical-induced liver tumorigenesis due to significant alteration on detoxification and oxidoreduction. More Importantly, Akr1b8-KO mice were much more predisposed to severe hepatocyte damage and liver inflammation, suggested that loss of Akr1b8-dependent oxidoreduction and detoxification led to the enhanced hepatocarcinogenesis. Similar to our findings, Shen et al. demonstrated that Akr1b8 loss also aggravated the oxidative and carbonyl-induced DNA damage in the colonic mucosa under dextran sulfate sodium (DSS) treatment [2]. Therefore, AKR1B10 functions as a critical protein in protecting hepatocytes and colonic epithelium cells from oxidative stress and carbonyl damage.
The expression of AKR1B10 varies greatly by cancer type and tissue of origin. AKR1B10 is constitutively and typically limited expression to the small intestine, stomach, colon and liver in humans, contributing to detoxification of xenobiotics and metabolizes physiological substrates [1,19,44]. Decreased expression of AKR1B10 has been discovered in colon and gastric cancer [2,8,45], which are organs contacting to the xenobiotics or dicarbonyl compounds directly from the daily diet and exogenous intake. Thus, it is reasonable to assume that AKR1B10 low-expression results in decreased detoxification function to the cytotoxic compounds and leads to cell injury, inflammation, compensatory proliferation and tumorigenesis. Up-regulation of AKR1B10 has been observed in non-small cell lung carcinoma (NSCLC), esophageal carcinoma, breast, uterine, pancreatic cancers and liver cancers [3-7]. Since knockdown of AKR1B10 in HCC cells inhibited cell proliferation, most studies assumed that AKR1B10 might function as an oncogenic gene in the development of HCC [24,46]. It is well known that inflammation and injury of hepatocytes caused by viral infection or chemical carcinogen could induce high ROS production and oxidative stress in the liver, which is becoming recognized as a key factor in the progression of HCC [39]. Based on the antioxidant defense ability of AKR1B10, it is reasonable to assume that AKR1B10 would be upregulated accompanied with chronic liver disease progression and tumorigenesis. Here we demonstrated that AKR1B10 expression was gradually upregulated in the process of chronic liver disease progression, and HCC tissues showed the highest AKR1B10 expression. In addition, higher AKR1B10 expression was significantly associated with poor survival rate in HCC patients. Recently, Ye et al. also found that the serum AKR1B10 level was gradually increased from CHB, LC to HCC patients [47]. Commonly, alcoholic and non-alcoholic fatty liver, HBV or HCV infection or chemical-induced hepatic toxicity are the origination of liver disease from persistent wound-healing response activated by hepatic injury, liver cell death and the resulting inflammatory cascade which leads to HCC development at last [14]. Starmann et al. found that AKR1B10 was striking highly expressed in steatohepatitis compared to normal liver [48] and Qi et al. found expression of AKR1B10 was increased significantly in the liver tumors with HBV infection and/or AFB1 exposure [46]. These results suggested that the expression of AKR1B10 might increase compensatively during chronic liver disease and HCC development due to the increasing ROS production and oxidative stress. This upregulation of AKR1B10 expression in HCC reduced ROS accumulation and protected malignant hepatocytes from suffering ROS-induced injury and apoptosis, rather than promoted cellular proliferation and tumorigenesis. The higher expression of AKR1B10 in HCC might reflect more serious malignancy and poor prognosis of liver tumor as our calculated results from TCGA database. Therefore, AKR1B10 deficiency in normal tissues may play an initiating role in development of tumor such as colon cancer and HCC, while when the tumor happens, compensatory or adaptive response induced AKR1B10 increase may help the transformed cell survival and play a promoting role in tumor progression.
NRF2 is an essential transcription factor that regulates an array of detoxifying and antioxidant defense genes expression in the liver [43]. NRF2 is activated in response to ROS accumulation and oxidative stress in hepatic inflammation, fibrosis, hepatocarcinogenesis [42,49]. Thus, NRF2 plays a pivotal role as a protector through target gene induction [43]. We observed that NRF2 increased AKR1B10 promoter activity and enhanced AKR1B10 expression under oxidative stress, suggesting the compensative upregulation of AKR1B10 in the process of chronic liver disease progression and HCC was mediated, at least partially by NRF2-induced transcriptional activation with increased oxidative stress.
In summary, our findings indicated that AKR1B10 could protect hepatocytes against ROS-induced injury through reducing oxidative stress. Loss of AKR1B10 ortholog gene (Akr1b8) in mice enhanced chemical-induced liver tumorigenesis with more serious liver damage and inflammation due to significant alteration on detoxification and oxidoreduction. AKR1B10 was compensatory unregulated during the process of liver tumorigenesis. Therefore, our results here provided a novel insight into the mechanism of AKR1B10-mediated hepatocarcinogenesis via reducing ROS-induced hepatocyte damage, which suggest the potential of AKR1B10 as a therapeutic target to prevent and treat HCC.
Acknowledgements
This work was supported by National Natural Science Foundation of China (grant numbers 81572366, 81372603), Beijing Natural Science Foundation (7182079), the interdisciplinary medicine Seed Fund of Peking University (grant number BMU2017MX015) and the National Key R&D Program of China (grant number 2017YFC0908100). We thank Professor Guangde Zhou from department of Pathology and Hepatology, Beijing 302 Hospital, Beijing, China for his professional histopathology analysis and consultant.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem. 1998;273:11429–11435. doi: 10.1074/jbc.273.19.11429. [DOI] [PubMed] [Google Scholar]
- 2.Shen Y, Ma J, Yan R, Ling H, Li X, Yang W, Gao J, Huang C, Bu Y, Cao Y, He Y, Wan L, Zu X, Liu J, Huang MC, Stenson WF, Liao DF, Cao D. Impaired self-renewal and increased colitis and dysplastic lesions in colonic mucosa of AKR1B8-deficient mice. Clin Cancer Res. 2015;21:1466–1476. doi: 10.1158/1078-0432.CCR-14-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, Taniguchi H, Ishikawa S, Ito H, Yamamoto S, Iwanari H, Hironaka M, Ishikawa Y, Niki T, Sohara Y, Kodama T, Nishimura M, Fukayama M, Dosaka-Akita H, Aburatani H. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res. 2005;11:1776–1785. doi: 10.1158/1078-0432.CCR-04-1238. [DOI] [PubMed] [Google Scholar]
- 4.Breton J, Gage MC, Hay AW, Keen JN, Wild CP, Donnellan C, Findlay JB, Hardie LJ. Proteomic screening of a cell line model of esophageal carcinogenesis identifies cathepsin D and aldo-keto reductase 1C2 and 1B10 dysregulation in Barrett’s esophagus and esophageal adenocarcinoma. J Proteome Res. 2008;7:1953–1962. doi: 10.1021/pr7007835. [DOI] [PubMed] [Google Scholar]
- 5.Ma J, Luo DX, Huang C, Shen Y, Bu Y, Markwell S, Gao J, Liu J, Zu X, Cao Z, Gao Z, Lu F, Liao DF, Cao D. AKR1B10 overexpression in breast cancer: association with tumor size, lymph node metastasis and patient survival and its potential as a novel serum marker. Int J Cancer. 2012;131:E862–71. doi: 10.1002/ijc.27618. [DOI] [PubMed] [Google Scholar]
- 6.Yoshitake H, Takahashi M, Ishikawa H, Nojima M, Iwanari H, Watanabe A, Aburatani H, Yoshida K, Ishi K, Takamori K, Ogawa H, Hamakubo T, Kodama T, Araki Y. Aldo-keto reductase family 1, member B10 in uterine carcinomas: a potential risk factor of recurrence after surgical therapy in cervical cancer. Int J Gynecol Cancer. 2007;17:1300–6. doi: 10.1111/j.1525-1438.2007.00932.x. [DOI] [PubMed] [Google Scholar]
- 7.Chung YT, Matkowskyj KA, Li H, Bai H, Zhang W, Tsao MS, Liao J, Yang GY. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol. 2012;25:758–766. doi: 10.1038/modpathol.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao HB, Xu Y, Chen LG, Guan TP, Ma YY, He XJ, Xia YJ, Tao HQ, Shao QS. AKR1B10, a good prognostic indicator in gastric cancer. Eur J Surg Oncol. 2014;40:318–324. doi: 10.1016/j.ejso.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Heringlake S, Hofdmann M, Fiebeler A, Manns MP, Schmiegel W, Tannapfel A. Identification and expression analysis of the aldo-ketoreductase1-B10 gene in primary malignant liver tumours. J Hepatol. 2010;52:220–227. doi: 10.1016/j.jhep.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 11.Sartorius K, Sartorius B, Aldous C, Govender PS, Madiba TE. Global and country underestimation of hepatocellular carcinoma (HCC) in 2012 and its implications. Cancer Epidemiol. 2015;39:284–290. doi: 10.1016/j.canep.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. Biomed Res Int. 2013;2013:810461. doi: 10.1155/2013/810461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erkekoglu P, Oral D, Chao MW, Kocer-Gumusel B. Hepatocellular carcinoma and possible chemical and biological causes: a review. J Environ Pathol Toxicol Oncol. 2017;36:171–190. doi: 10.1615/JEnvironPatholToxicolOncol.2017020927. [DOI] [PubMed] [Google Scholar]
- 14.Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, Liang X, Ma C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut. 2015;64:1593–1604. doi: 10.1136/gutjnl-2014-307671. [DOI] [PubMed] [Google Scholar]
- 15.Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14. doi: 10.1186/1477-3163-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 17.Kangari P, Zarnoosheh Farahany T, Golchin A, Ebadollahzadeh S, Salmaninejad A, Mahboob SA, Nourazarian A. Enzymatic antioxidant and lipid peroxidation evaluation in the newly diagnosed breast cancer patients in Iran. Asian Pac J Cancer Prev. 2018;19:3511–3515. doi: 10.31557/APJCP.2018.19.12.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu JM, Lin PH, Yao Q, Chen C. Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. J Cell Mol Med. 2010;14:840–860. doi: 10.1111/j.1582-4934.2009.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong L, Liu Z, Yan R, Johnson S, Zhao Y, Fang X, Cao D. Aldo-keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem Biophys Res Commun. 2009;387:245–250. doi: 10.1016/j.bbrc.2009.06.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem. 2009;284:26742–26748. doi: 10.1074/jbc.M109.022897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin HJ, Maser E. Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem Biol Interact. 2009;178:145–150. doi: 10.1016/j.cbi.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Hyndman DJ, Flynn TG. Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim Biophys Acta. 1998;1399:198–202. doi: 10.1016/s0167-4781(98)00109-2. [DOI] [PubMed] [Google Scholar]
- 23.Liu TA, Jan YJ, Ko BS, Wu YJ, Lu YJ, Liang SM, Liu CC, Chen SC, Wang J, Shyue SK, Liou JY. Regulation of aldo-keto-reductase family 1 B10 by 14-3-3epsilon and their prognostic impact of hepatocellular carcinoma. Oncotarget. 2015;6:38967–38982. doi: 10.18632/oncotarget.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matkowskyj KA, Bai H, Liao J, Zhang W, Li H, Rao S, Omary R, Yang GY. Aldoketoreductase family 1B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Hum Pathol. 2014;45:834–843. doi: 10.1016/j.humpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi A, Rajput S, Wang C, Ma J, Cao D. Murine aldo-keto reductase family 1 subfamily B: identification of AKR1B8 as an ortholog of human AKR1B10. Biol Chem. 2010;391:1371–1378. doi: 10.1515/BC.2010.144. [DOI] [PubMed] [Google Scholar]
- 26.Hou J, Wang G, Wang F, Cheng J, Ren H, Zhuang H, Sun J, Li L, Li J, Meng Q, Zhao J, Duan Z, Jia J, Tang H, Sheng J, Peng J, Lu F, Xie Q, Wei L Chinese Society of Hepatology, Chinese Medical Association; Chinese Society of Infectious Diseases, Chinese Medical Association. Guideline of prevention and treatment for chronic hepatitis B (2015 update) J Clin Transl Hepatol. 2017;5:297–318. doi: 10.14218/JCTH.2016.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zu X, Yan R, Robbins S, Krishack PA, Liao DF, Cao D. Reduced 293T cell susceptibility to acrolein due to aldose reductase-like-1 protein expression. Toxicol Sci. 2007;97:562–568. doi: 10.1093/toxsci/kfm033. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Qi X, Zeng Z, Wang L, Wang J, Zhang T, Xu Q, Shen C, Zhou G, Yang S, Chen X, Lu F. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci Rep. 2017;7:2796. doi: 10.1038/s41598-017-03070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Q, Chen X, Lu F, Zhang T, Hao M, Wang Y, Zhao J, McCrae MA, Zhuang H. Aberrant expression of microRNA 155 may accelerate cell proliferation by targeting sex-determining region Y box 6 in hepatocellular carcinoma. Cancer. 2012;118:2431–2442. doi: 10.1002/cncr.26566. [DOI] [PubMed] [Google Scholar]
- 30.Muggia FM, Green MD. New anthracycline antitumor antibiotics. Crit Rev Oncol Hematol. 1991;11:43–64. doi: 10.1016/1040-8428(91)90017-7. [DOI] [PubMed] [Google Scholar]
- 31.Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40:553–624. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol. 2004;287:C895–C902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 33.Esterbauer H, Cheeseman KH. Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990;186:407–421. doi: 10.1016/0076-6879(90)86134-h. [DOI] [PubMed] [Google Scholar]
- 34.Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. doi: 10.1016/j.gene.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 35.Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, Ziegler DV, Xu X, Ghosh T, Mondal T, Kanduri C, Lindahl P, Sayin VI, Bergo MO. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell. 2019;178:330–345. e22. doi: 10.1016/j.cell.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Wu H, Lei R, Chong RA, Wei Y, Lu X, Tagkopoulos I, Kung SY, Yang Q, Hu G, Kang Y. Transcriptional network analysis identifies BACH1 as a master regulator of breast cancer bone metastasis. J Biol Chem. 2012;287:33533–33544. doi: 10.1074/jbc.M112.392332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruiz FX, Porté S, Parés X, Farrés J. Biological role of aldo-keto reductases in retinoic acid biosynthesis and signaling. Front Pharmacol. 2012;3:58. doi: 10.3389/fphar.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma-On C, Sanpavat A, Whongsiri P, Suwannasin S, Hirankarn N, Tangkijvanich P, Boonla C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med Oncol. 2017;34:57. doi: 10.1007/s12032-017-0914-5. [DOI] [PubMed] [Google Scholar]
- 39.Ivanov AV, Valuev-Elliston VT, Tyurina DA, Ivanova ON, Kochetkov SN, Bartosch B, Isaguliants MG. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget. 2017;8:3895–3932. doi: 10.18632/oncotarget.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito Y, Nakajima K, Masubuchi Y, Kikuchi S, Saito F, Akahori Y, Jin M, Yoshida T, Shibutani M. Expression characteristics of genes hypermethylated and downregulated in rat liver specific to nongenotoxic hepatocarcinogens. Toxicol Sci. 2019;169:122–136. doi: 10.1093/toxsci/kfz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert-Langlais S, Pointud JC, Lefrancois-Martinez AM, Volat F, Manin M, Coudoré F, Val P, Sahut-Barnola I, Ragazzon B, Louiset E, Delarue C, Lefebvre H, Urade Y, Martinez A. Aldo keto reductase 1B7 and prostaglandin F2alpha are regulators of adrenal endocrine functions. PLoS One. 2009;4:e7309. doi: 10.1371/journal.pone.0007309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, Baranasic D, Arenillas DJ, Sandelin A, Vandepoele K, Lenhard B, Ballester B, Wasserman WW, Parcy F, Mathelier A. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46:D260–D266. doi: 10.1093/nar/gkx1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103:1007–1013. doi: 10.1172/JCI4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohashi T, Idogawa M, Sasaki Y, Suzuki H, Tokino T. AKR1B10, a transcriptional target of p53, is downregulated in colorectal cancers associated with poor prognosis. Mol Cancer Res. 2013;11:1554–1563. doi: 10.1158/1541-7786.MCR-13-0330-T. [DOI] [PubMed] [Google Scholar]
- 46.Qi LN, Li LQ, Chen YY, Chen ZH, Bai T, Xiang BD, Qin X, Xiao KY, Peng MH, Liu ZM, Liu TW, Qin X, Li S, Han ZG, Mo ZN, Santella RM, Winkler CA, O’Brien SJ, Peng T. Genome-wide and differential proteomic analysis of hepatitis B virus and aflatoxin B1 related hepatocellular carcinoma in Guangxi, China. PLoS One. 2013;8:e83465. doi: 10.1371/journal.pone.0083465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye X, Li C, Zu X, Lin M, Liu Q, Liu J, Xu G, Chen Z, Xu Y, Liu L, Luo D, Cao Z, Shi G, Feng Z, Deng H, Liao Q, Cai C, Liao DF, Wang J, Jin J, Cao D. A large-scale multicenter study validates aldo-keto reductase family 1 member b10 as a prevalent serum marker for detection of hepatocellular carcinoma. Hepatology. 2019;69:2489–2501. doi: 10.1002/hep.30519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starmann J, Falth M, Spindelbock W, Lanz KL, Lackner C, Zatloukal K, Trauner M, Sultmann H. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS One. 2012;7:e46584. doi: 10.1371/journal.pone.0046584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.