

Abstract
Increasing evidences demonstrate that miRNAs play an important role in development and progression of hepatocellular carcinoma (HCC). Recent studies indicate that miR-3196 regulates tumorigenesis in breast and lung cancer. However, its role and regulatory mechanism remains unknown in hepatocellular carcinoma. Here, we found that miR-3196 was downregulated in HCC tissues and decreased miR-3196 was correlated with tumor size (P=0.0297) and TNM stage (P=0.034). Forced miR-3196 suppressed HCC cell growth and chemoresistance in vivo and in vitro. Further mechanistic studies revealed that the tumor suppressor p53 transcriptionally upregulated miR-3196 expression by binding to its promoter region in HCC cells. Additional, we also found that FOXP4 was a downstream target of miR-3196 and increased miR-3196 inhibited FOXP4 expression which led to HCC growth suppression and cell apoptosis increase. Collectively, our data shed a new role of miR-3196 in HCC and indicates that p53-dependent, miR-3196-medicated FOXP4 pathway inhibits the tumorigenesis of HCC.
Keywords: miR-3196, hepatocellular carcinoma, p53, cell apoptosis
Introduction
Hepatocellular carcinoma (HCC) is the sixth most frequent cancer and the fourth most common cause of cancer-related death worldwide in 2018 [1]. Despite remarkable advances in the diagnosis and therapy of this disease, including novel chemotherapeutic interventions and target therapy, the long-term survival of HCC patients is still dismal due to the high rates of metastasis and chemoresistance resulting in tumor recurrence [2-4]. Therefore, understanding the molecular mechanisms underlying the progression and metastasis in HCC is essential for the development of effective therapeutic strategies for the cancer treatment.
MicroRNAs (miRNAs), a class of evolutionary conserved small non-coding RNAs, act as post-transcriptional regulator of gene expression in tumor initiation, development and progression by binding to complementary sequences within the 3-untranslated region (UTR) of target genes which led to the degradation of their mRNAs or the repression of their translation [5-7]. Accumulating evidences indicated that aberrantly expressed miRNAs play critical roles in HCC progresses including cell proliferation, apoptosis, drug-resistance, metastasis and stem cell renewal [8,9]. For example, miR-122 was reported to decrease in HCC cells and repressed proliferation but induced apoptosis by targeting pyruvate kinase muscle 2 (PKM2) and oncogenic distal-less 4 (DLX4) [10,11]. Overexpression of miR-25 could facilitate EMT formation by inhibiting Rho GDP dissociation inhibitor alpha (RhoGDI1) in HCC [12]. Overexpression of miR-135a promoted HCC cells migration and invasion by targeting forkhead box O1 (FOXO1) [13].
Recent studies indicated that miR-3196 plays an important role in tumorigenesis. In breast cancer, miR-3196 was reported to suppress cell proliferation and induces cell apoptosis through targeting ERBB3 [14]. MiR-3196 also inhibited orthodenticle homeobox 1 (OTX1) expression and long noncoding RNA ADPGK-AS1 promoted cell proliferation, migration and EMT via modulating miR-3196/OTX1 axis in breast cancer [15]. In lung cancer, miR-3196 was suggested to suppress cell apoptosis via targeting PUMA expression [16]. Although miR-3196 acted as a tumor suppressor or oncogen in different tumors, its role and regulatory mechanism remain unknown in hepatocellular carcinoma.
In this study, we found that miR-3196 was downregulated in HCC tissues and decreased miR-3196 was associated with tumor size increase. Overexpression of miR-3196 suppressed HCC cell proliferation and promoted chemotherapy drug-induced apoptosis in vivo and in vitro. Subsequently, our data indicated that p53 transcriptionally upregulated miR-3196 expression via binding to its promoter region in HCC cells. Additional, we also found that FOXP4 was a downstream target of miR-3196. Overall, our data suggest that p53-miR-3196-FOXP4 axis plays an important role in HCC suppression.
Materials and methods
Cell culture and reagents
The human HCC cell lines HepG2, Huh-7, SMMC-7721, SNU449 and BEL7402 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HepG2, Huh-7 and BEL7402 cells were grown in DMEM supplemented with 10% foetal bovine serum, and 1% penicillin, streptomycin. SNU449 and SMMC-7721 cells were grown in 1640 supplemented with 10% foetal bovine serum, and 1% penicillin, streptomycin. The cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. The following antibodies and reagents were used: the antibody to GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA; SC-25778, 1:1000), PARP1 (Santa Cruz Biotechnology, SC-8007, 1:1000), p53 (Santa Cruz Biotechnology, SC-126, 1:10 for ChIP, 1:1000 for WB), p21 (Preoteintech, 10355-1-AP, 1:500) and FOXP4 (Preoteintech, 16772-1-AP, 1:500).
RNA interference
RNA interference was performed as previously described [17,18]. The targeting sequences of p53 and FOXP4 were shown as following: p53 No.1: CGGCGCACAGAGGAAGAGAA; FOXP4: No.1 CCAGGGAACAATGACAGCAAA; No.2: CCAGTTTATCAAACACCTCAA.
Introduction of microRNA mimics and inhibitors
Mimics and inhibitors of miRNA-3196 were synthesized by the GenePharma Company (Shanghai, People’s Republic of China). For each transfection in a six-well plate, 100 nM miRNA mimics, scramble or inhibitor, or scramble were used. The transfection of HCC cells by Oligofectamine (Invitrogen) was performed according to the manufacturer’s instructions. The lentivirus expressing miR-3196 was purchased from GenePharma Company. The cells with stably expressing miR-3196 were obtained as previous described [19].
MTS proliferation and cell viability assay
The proliferation of cells was evaluated using the MTS Cell Proliferation Assay Kit (Colorimetric) (No.197010; Abcam, UK) following the manufacturer’s instructions. Briefly, HepG2, BEL7402 and SNU449 cells seeded in 96-well microtitre plates were cultured for approximately 22 or 46 hours and then mixed with MTS reagent (20 μL/well) and incubated for 2 hours at 37°C. Cell proliferation was finally determined by measuring the OD490 using a plate reader. At least 3 independent repeats were performed for statistical analysis.
The cell viability was determined using a CCK8 kit (Cell Counting Kit-8) following the manufacturer’s instructions.
Annexin V-FITC staining and FACS
The staining protocol was performed following the manufacturer’s instructions (BD). Generally, liver cancer cells (5×105) treated as indicated were harvested by a 5 min centrifugation at 1000 g and resuspended in 195 μL binding buffer, followed by a 10 min incubation with 5 μL Annexin V-FITC at room temperature avoiding any light. After an additional centrifugation, the cells were resuspended in 190 μL binding buffer and 10 μL PI stain was added with slight shaking. FACS (BD) analysis was employed for detecting cell apoptotic events.
Quantitative real-time polymerase chain reaction assay (q-RT-PCR)
Total RNA was isolated using TRIzol (Invitrogen). One microgram of total RNA was used to synthesize cDNA using the PrimeScriptTM RT reagent kit (Takara, RR047A) according to the manufacturer’s instructions. The primers for miR-3196 were purchased from MyBioSource.
Promoter reporters and dual-luciferase assay
The promoter of miR-3196 was constructed into the pGL3-basic vector. Luciferase activity was measured in a 1.5-ml Eppendorf tube with the Promega Dual-Luciferases Reporter Assay kit (Promega E1980) according to manufacturer’s protocols after transfection. Relative Renilla luciferase activity was normalized to firefly luciferase activity. The assay was performed as previously described [20,21].
Colony formation assays
HepG2, SNU449 and BEL7402 cells with the treatment as indicated (1×103 cells per well) were plated into 6 well plates and cultured at 37°C equipped with 5% CO2. Cells were fed with fresh growth medium every 3 days. Colonies were allowed to form for 2 weeks and were fixed with 4% paraformaldehyde, stained with crystal violet, washed with water to remove excess stain, and counted using Image J software. Each experiment was repeated three times.
Animal experiments
Animal studies were carried out in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals, with the approval of the Animal Research Committee of Dalian Medical University. Male nude mice (4-6 weeks old, 18-20 g) were obtained from the SPF Laboratory Animal Centre of Dalian Medical University (Dalian, China). The mice were used for experiments after they had been acclimatized for 1 week. Stable SNU449 cells (1×107) that were suspended in 200 μl of PBS were subcutaneously inoculated in mice. Five mice (n=5) was used in each of the experiments. After five weeks, all animals were killed by cervical decapitation, the tumour weights were measured and the tumour tissues were excised aseptically. The protocol was approved by the Animal Care and Ethics Committee of Dalian Medical University.
Statistical analysis
All results are shown as the mean ± S.D. of multiple independent experiments, not technical replicates. Detailed P values for each panel in the figures are stated in the corresponding legends. A Student’s t-test, a Mann-Whitney test (for two group comparisons) was used for statistical analyses. All statistical analyses were performed with GraphPad Prism 5 and SPSS 19.0 software. All statistical tests were two-sided, and P values <0.05 were considered to be statistically significant.
Results
MiR-3196 is a putative tumor suppressor for HCC
To investigate the role of miR-3196 in HCC, the expression levels of miR-3196 were first analyzed. Compared with the adjacent tissues, miR-3196 was significantly downregulated in HCC tissues (Figure 1A). Subsequently, the correlations between miR-3196 expression and pathological features of the patients were also assessed. As shown in Table 1, miR-3196 expression was negative correlation with tumour size (P=0.0297) and TNM stage (P=0.034).
Figure 1.

miR-3196 inhibits HCC cell growth. (A) The expression levels of miR-3196 were analyzed by q-RT-PCR in HCC tissues (n=30) and the adjacent tissues (n=30). (B-D) miR-3196 was transfected into HepG2, SNU449 and BEL7402 cells and the cell proliferation was examined by MTS assay. Data represent the mean ± SD of three independent experiments. ***P<0.001 vs. control. (E, F) The cell growth was examined by a colony formation assay. Data represent the mean ± SD of three independent experiments. **P<0.01 and ***P<0.001 vs. control. (G-K) The tumour-forming abilities of SNU449 cells with or without miR-3196 overexpression were measured in vivo (G). The tumor weight was assessed (H). The Caspase 3 activity was examined (I) and cleaved PARP1 were analyzed by western blotting (J and K).
Table 1.
miR-3196 expression and tumor index correlation analysis
| Variables | N | MiR-3196 expression | P value | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Low | High | % | ||||
| Age | ≥65 | 18 | 6 | 12 | 60 | 0.5243 |
| <65 | 12 | 3 | 9 | 40 | ||
| Gender | Male | 17 | 5 | 12 | 56.7 | 0.5156 |
| Female | 13 | 4 | 9 | 43.3 | ||
| Size | ≤5 cm | 19 | 2 | 17 | 63.3 | 0.0297 |
| >5 cm | 11 | 7 | 4 | 36.7 | ||
| Metastasis | M0 | 17 | 4 | 13 | 56.7 | 0.241 |
| M1 | 13 | 5 | 8 | 43.3 | ||
| TNM | TNM1-TNM2 | 21 | 4 | 17 | 70 | 0.034 |
| TNM3-TNM4 | 9 | 5 | 4 | 30 | ||
To further identify the effect of miR-3196 on HCC cell proliferation and tumorigenesis, we first transfected miR-3196 into HCC cells. The cell proliferation and cell viability was analyzed by MTS and a colony formation assay. As shown in Figure 1B-F, we found that overexpression of miR-3196 inhibited HCC cell growth and cell viability. Except that we also stably overexpressed miR-3196 in SNU449 cells and then the cells were subcutaneously injected into nude mice. Compared with control cells, the overexpression of miR-3196 decreased tumor growth and increased cell apoptosis (Figure 1G-K).
Increased miR-3196 promotes chemotherapy drug-induced apoptosis in HCC cells
Chemoresistance has been recognized as one of the main causes of tumor recurrence. Therefore, we want to know whether miR-3196 affected chemoresistance of HCC. To this end, we first treated p53 wild type HepG2 and SMMC-7721 cells with doxorubicin (Dox), a cornerstone of chemotherapy for HCC, which induces apoptosis via the p53 pathway. Compared with the control cells, we found that miR-3196 was augmented with increasing times and doses of doxorubicin treatment (Figure 2A-D). Similarly, miR-3196 was also upregulated in HepG2 and SMMC-7721 cells under cisplatin (CDDP) treatment, another cornerstone of chemotherapy for HCC (Figure S1A, S1B). To further evaluate function of miR-3196 in HCC chemoresistance, we used the anti-miR-3196, a synthesized inhibitor oligo, to silence miR-3196 expression in HepG2 and SMMC-7721. Then, these cells were treated with doxorubicin. We found that inhibition of miR-3196 attenuated doxorubicin-induced apoptosis and increased cell viability in HepG2 and SMMC-7721 (Figure 2E-H).
Figure 2.

miR-3196 inhibits chemoresistance of HCC cells. A, B. HepG2 and SMMC-7721 cells were treated with 2 μM Dox for the indicated times. The expression levels of miR-3196 were analyzed by q-RT-PCR. The data represent the means ± SD of three independent experiments; *P<0.05, **P<0.01, ***P<0.001 vs. control. C, D. HepG2 and SMMC-7721 cells were treated with Dox with the indicated concentration for 36 h. The expression levels of miR-3196 were analyzed by q-RT-PCR. The data represent the means ± SD of three independent experiments; *P<0.05, **P<0.01, ***P<0.001 vs. control. E-H. miR-3196 inhibitor was transfected into HepG2 and SMMC-7721 cells. The cells were then treated with 2 μM Dox for the indicated times. Cell apoptosis and cell viability were detected by western blotting and CCK8 assays. The data represent the means ± SD of three independent experiments; ***P<0.001 vs. control. I, J. SNU449 and Huh-7 cells were treated with 2 μM Dox for the indicated times. The expression levels of miR-3196 were analyzed by q-RT-PCR. The data represent the means ± SD of three independent experiments. K-N. miR-3196 was transfected into SNU449 and Huh-7 cells. The cells were then treated with 2 μM Dox for the indicated times. Cell apoptosis and cell viability were detected by western blotting and CCK8 assays. The data represent the means ± SD of three independent experiments; ***P<0.001 vs. control.
To confirm it, we then analyzed miR-3196 expression in other HCC cell lines SNU449 and Huh-7 which contain the mutant p53. The cells were treated with 2 μM doxorubicin as indicated times. Interestingly, we found that the increase of miR-3196 by doxorubicin was disappeared in SNU449 and Huh-7 cells and the protein levels of mutant p53 were not increased under doxorubicin treatment (Figures 2H, 2I and S1C, S1D). Thus, to prove the suppressive role of miR-3196 in chemoresistance, we overexpressed miR-3196 into SNU449 and Huh-7 cells. Compared with control group, we found that forced miR-3196 expression promoted doxorubicin-induced apoptosis and decreased cell viability in SUN449 and Huh-7 cells (Figure 2K-N). Taken together, our data suggest that miR-3196 facilitated HCC cell chemosensitivity.
The tumor suppressor p53 transcriptionally upregulates miR-3196 expression in HCC cells
Based on the data that miR-3196 was increased in p53 wild type HCC cells under doxorubicin treatment, we thus supposed that p53 may involve in regulating miR-3196 expression. To prove it, we first analyzed the miR-3196 and p53 expression in five HCC cell lines and found that miR-3196 expression was significantly decreased in p53 low expressed and mutant cells (Figure 3A, 3B). To further investigate the relationship between p53 and miR-3196, we established BEL7402 cell lines stably transfected wild type p53 or the mutants R175H and R273H, in which the expression of p53 and the mutants were under control of a Tet-On swith. As shown in Figure 3C, the expression p53 or the mutants were efficiently induced with doxycycline, where p53 downstream target p21 is used as the positive control. Following induction, wild type p53 not the mutants elevated miR-3196 expression (Figure 3D). Whereas, knockdown of p53 suppressed miR-3196 expression in HepG2 and SMMC-7721 cells (Figure 3E-H).
Figure 3.
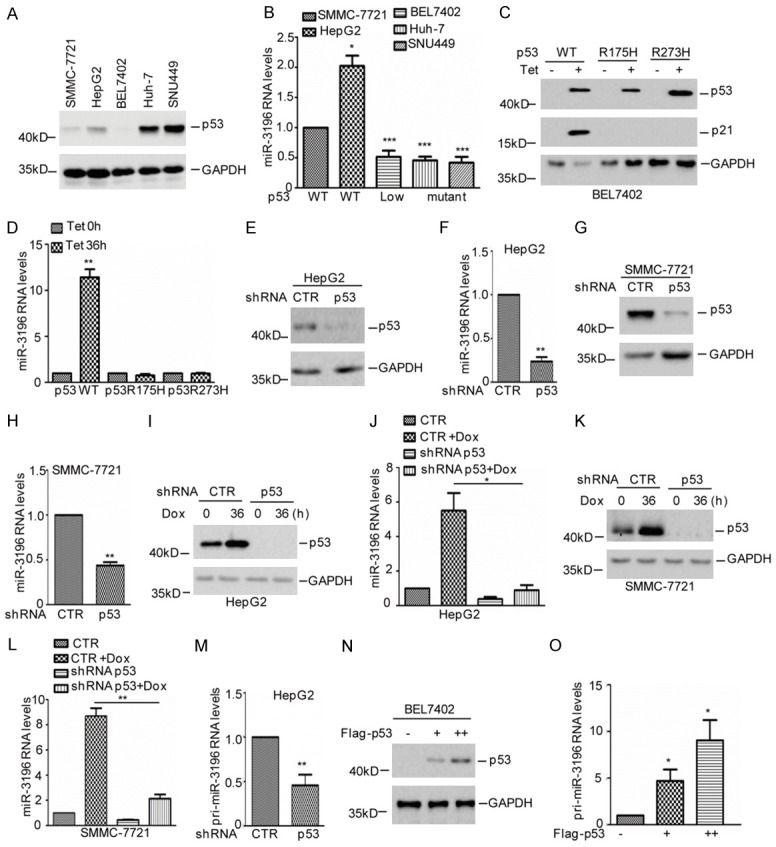
p53 upregulates miR-3196 expression. A, B. The expression levels of p53 and miR-3196 were analyzed in HCC cell lines. The data were obtained from three times independent experiments. *P<0.05 and ***P<0.001 vs. control. C, D. BEL7402 cells with doxycycline-inducible (Tet) expression of either wide type p53 or mutant p53 were incubated with doxcycline for 24 h. Cell lysates were subjected to western blot analysis with the indicated antibodies. The expression levels of miR-3196 were examined by q-RT-PCR. The data were obtained from three times independent experiments. **P<0.01 vs. control. E-H. p53 was knocked down in HepG2 and SMMC-7721 cells. Cell lysates were subjected to western blot analysis with the indicated antibodies. The expression levels of miR-3196 were examined by q-RT-PCR. The data were obtained from three times independent experiments. **P<0.01 vs. control. I-L. HepG2 and SMMC-7721 cells with or without p53 knockdown were treated with 2 μM Dox for 36 h. Cell lysates were subjected to western blot analysis with the indicated antibody. The expression levels of miR-3196 were examined by q-RT-PCR. The data were obtained from three times independent experiments. *P<0.05, **P<0.01. M. p53 was knocked down in HepG2 cells and the expression levels of pri-miR-3196 was analyzed by q-RT-PCR. The data were obtained from three times independent experiments. **P<0.01 vs. control. N, O. p53 was overexpressed in BEL7402 cells and the expression levels of pri-miR-3196 was analyzed by q-RT-PCR. The data were obtained from three times independent experiments. *P<0.05 vs. control.
Additionally, HepG2 and SMMC-7721 with or without p53 knockdown were treated with or without 2 μM doxorubicin. The expression levels of miR-3196 were analyzed by q-RT-PCR. As shown in Figure 3I-L, inhibition of p53 decreased miR-3196 expression and abolished the doxorubicin-induced miR-3196 increase. Accumulating evidences indicate that p53 regulates miRNA expression by both transcription dependent and independent means [22-24]. We thus wondered whether p53 could transcriptionally upregulated miR-3196 expression in HCC cells. To test this hypothesis, the expression levels of pri-miR-3196 were measured using q-RT-PCR analysis in HepG2 cells with or without p53 knockdown. As shown in Figure 3M, knockdown of p53 suppressed pri-miR-3196 expression in HCC cells. Whereas, overexpression of p53 promoted pri-miR-3196 expression (Figure 3N, 3O). Taken together, these data indicate that p53 transciptionally upregulates miR-3196 expression in HCC cells.
p53 binds to the promoter of miR-3196
To determine the p53 binding regions on the miR-3196 promoter, we first cloned the upstream sequences of miR-3196 (-1837 to 0 bp) and different truncations by PCR. Then, we inserted them into the pGL3-based luciferase reporter plasmids named P1-P4 (Figure 4A). After that, we transfected them into 293T cells with or without p53 overexpression. Compared with the control cells, the luciferase activities of P1 and P2 were increased in p53 overexpressing cells (Figure 4B). To further confirm it, these plasmids were transfected into HepG2 cells with or without p53 knockdown and the luciferase activities were measured. As shown in Figure 4C, we found that the knockdown of p53 decreased the luciferase activities of P1 and P2. Additionally, we also found that the mutant p53 lost its effect on luciferase activities of P2 (Figure 4D). Thus, these data indicated that the region (-1337 to -837 bp) was a key region for the promotion of miR-3196 by p53.
Figure 4.
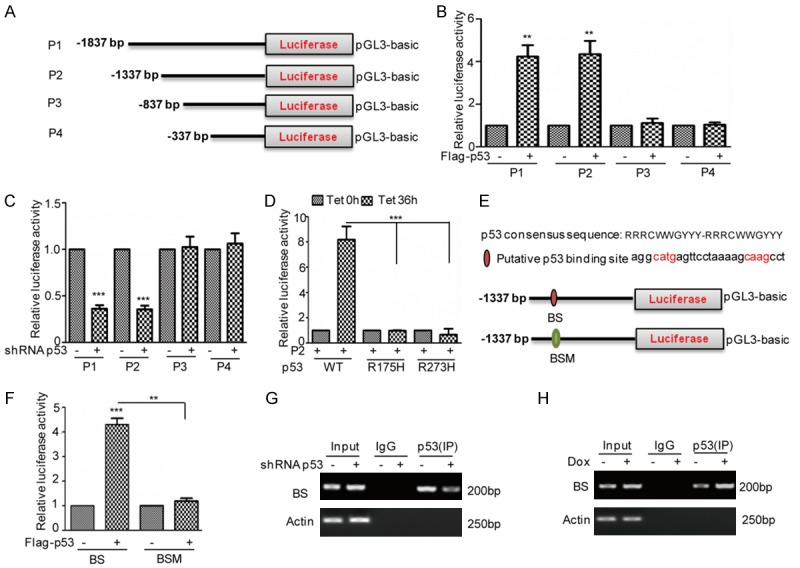
p53 binds to the promoter of miR-3196. (A) Schematic illustration of pGL3-based reported constructs that were used in luciferase assays to examine the transcriptional activity of miR-3196. (B) The promoters of miR-3196, named P1, P2, P3, and P4, were individually transfected into 293T cells with or without p53 overexpression. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; **P<0.01 vs. control. (C) The promoters of miR-3196, named P1, P2, P3, and P4, were individually transfected into HepG2 cells with or without p53 knockdown. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; ***P<0.001 vs. control. (D) P2 was transfected into BEL7402 cells with or without p53 or mutants expression. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; ***P<0.001 vs. control. (E, F) A schematic illustration of p53 wild type binding site (WT) and the matching mutant (Mut) that were used in the luciferase assays is shown. p53 wild type binding site (BS) and the matching mutant (BSM) were individually transfected into 293T cells with or without p53 overexpression. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; **P<0.01, ***P<0.001 vs. control. (G, H) ChIP analysis showed the binding of p53 to the promoter of miR-3196 in HepG2 cells with p53 knockdown (G) or 2 μM Dox treatment (H). An isotype-matched IgG was used as a negative control.
To determine the p53 binding sites, we next examined the genomic DNA region (-1337 to -837 bp) of miR-3196 and identified a putative p53-binding site on miR-3196 promoter (Figure 4E). To verify that the potential binding site was indeed responsive to p53, a series of pGL3-based luciferase reporter plasmids named wild type binding site (BS) and the binding site mutant (BSM) were generated (Figure 4E). The two plasmids were individually transfected into 293T cells with or without p53 overexpression. We found that the luciferase activity of BS, not BSM was significantly increased in p53 expressed cells (Figure 4F). In addition, the subsequent chromatin immunoprecipitation (ChIP) assays showed that the chromatin fragments, corresponding to the putative p53-binding sites, were specifically present in anti-p53 immunoprecipitates from HepG2 cells, and the bond was decreased when p53 was knocked down (Figure 4G). Whereas, the bond was increased under doxorubicin treatment (Figure 4H). Thus, these data indicate the wild type p53 could bind to the promoter of miR-3196 and contribute to its increase in HCC cells.
miR-3196 inhibits FOXP4 expression in HCC
Having identified p53 as the upstream transcriptional factor of miR-3196 in HCC cells, we next sought to identify the downstream targets of miR-3196. To this end, we searched the target-predicting algorithms (TargetScan and miRWalk) to seek potential target genes. Of the 18 genes that overlapped among these algorithms, we found that FOXP4 was an important oncogene among these potential targets (Figure 5A). To confirm a direct relationship between miR-3196 and FOXP4, we introduced miR-3196 mimics into Huh-7 and SNU449 cells. Then the expression levels of FOXP4 were detected by western blotting and we found that overexpression of miR-3196 suppressed endogenous FOXP4 protein levels (Figure 5B). Whereas, introduction of miR-3196 inhibitor into HepG2 and SMMC-7721 cells increased FOXP4 expression (Figure 5C). Similar results were also obtained in BEL7402 cells (Figure S1E, S1F). Additional, we also found that the 3’ UTR of FOXP4 contained four potential regions that matched perfectly to the miR-3196 “seed” region. To further identify the potential binding sites of miR-3196, we firstly cloned the 3’ UTR of FOXP4 into a dual-luciferase vector which were named BS1-BS4 (Figure 5D). We then transfected them into 293T cells together with miR-3196. Notably, we found that the luciferase activity of BS3 not the BS1, BS2 and BS4 was suppressed by miR-3196 (Figure 5E). To further confirm it, the wild type and the mutant of BS3 were constructed (Figure 5F). These plasmids were then introduced into SNU449 cell with or without the miR-3196 overexpression. Compared with the control group, we found that the luciferease activity of wild type BS3 not the mutant was decreased by miR-3196 (Figure 5G). Whereas, inhibition of miR-3196 increased the luciferease activity of wild type BS3 not the mutant in HepG2 cells (Figure 5H). Taken together, our data suggest that FOXP4 is a real target of miR-3196.
Figure 5.

miR-3196 suppresses FOXP4 expression. (A) Potential target genes of the miR-765 as predicted by TargetScan, miRWalk. (B) miR-3196 was transfected into Huh-7 and SNU449 cells. Cell lysates were subjected to western blot analysis with the indicated antibody. (C) miR-3196 inhibitor was transfected into HepG2 and SMMC-7721 cells. Cell lysates were subjected to western blot analysis with the indicated antibody. (D, E) Sequences of the potential miR-3196 binding site at the 3’ UTR of FOXP4 were inserted into pSICHEK2 vector named BS1, BS2, BS3 and BS4. These plasmids were individually transfected into 293T cells with or without miR-3196 overexpression. The luciferase activity was measured. The data represent the means ± SD of three independent experiments; **P<0.01 vs. control. (F) Sequences of the potential miR-3196 binding site at the 3’ UTR of FOXP4 (BS3), and nucleotides mutated, the red indicated the mutated region. (G) The wild type 3’ UTR (WT) and mutated 3’ UTR (MUT) of FOXP4 were transfected into SNU449 cell with miR-3196 overexpression (G) or HepG2 cell with miR-3196 inhibition (H). The luciferase activities were measured. The data represent the mean ± SD of three independent experiments. **P<0.01 vs. CTR.
p53-miR-3196-FOXP4 signaling axis plays a critical role in HCC
Considering that p53 facilitated miR-3196 expression, we want to know whether p53 could suppress FOXP4 expression via miR-3196. To investigate it, we overexpressed miR-3196 into HepG2 cells with or without p53 knockdown. We found that p53 knockdown increased FOXP4 protein levels. However, the increase was abolished when miR-3196 was overexpressed (Figure 6A, 6B). Whereas, the decrease of FOXP4 by p53 was also reversed by miR-3196 inhibitor (Figure 6C, 6D).
Figure 6.

p53-miR-3196-FOXP4 axis plays a key role in HCC suppression. A, B. miR-3196 together with 3’ UTR of FOXP4 was transfected into HepG2 cells with or without p53 knockdown. The protein levels of FOXP4 and the luciferase activity were measured. Data represent the mean ± SD of three independent experiments. **P<0.01 vs. control. C, D. miR-3196 inhibitor together with 3’ UTR of FOXP4 was transfected into BEL7402 cells with or without p53 overexpression. The protein levels of FOXP4 and the luciferase activity were measured. Data represent the mean ± SD of three independent experiments. **P<0.01 vs. control. E, F. p53 was knocked down in HepG2 cells together with miR-3196 overexpression or FOXP4 knockdown. The cell proliferation was examined by a colony formation assay. Data represent the mean ± SD of three independent experiments. ***P<0.001 vs. control. G, H. miR-3196 was overexpressed in HepG2 cells together with p53 knockdown. The cells were then treated with 2 μM Dox for 36 h and cell apoptosis was analyzed by western blotting and Flow Cytometer. I, J. FOXP4 was knocked down in HepG2 cells together with p53 knockdown. The cells were then treated with 2 μM Dox for 36 h and cell apoptosis was analyzed by western blotting and Flow Cytometer. K. miR-3196 inhibitor was transfected into BEL7402 cells with or without p53 overexpression. The cells were then treated with 2 μM Dox for 36 h and cell apoptosis was analyzed by western blotting.
Subsequently, the effect of p53-miR-3196-FOXOP4 pathway on HCC suppression was investigated. We found that elevations of cell viability and chemoresistance by p53 knockdown were also reversed by miR-3196 overexpression or FOXP4 depletion in HepG2 cells (Figure 6E-J). Otherwise, the increase of cell apoptosis by p53 was abolished when miR-3196 was inhibited in BEL7402 cells (Figure 6K). Taken together, our data indicate p53-miR-3196-FOXP4 axis plays an important role in HCC suppression.
Discussion
A lot of studies have demonstrated that dysregulation of miRNAs play an important role in the HCC initiation, development and progression [8]. Several miRNAs have been identified as novel prognostic biomarkers and effective therapeutic targets of HCC [25,26]. In this study, we found that miR-3196 was significantly downregulation in HCC tissues and decreased miR-3196 promoted HCC cells growth and chemoresistance. For the first time, our data provide a more comprehensive understanding of the tumor suppressor role of miR-3196 in HCC.
MiR-3196 was reported to play an ambivalent role in tumorigenesis as either a tumor suppressor or an oncogene in a tissue-specific manner. In lung cancer, miR-3196 was indicated to act as an oncogene and suppressed cell apoptosis via targeting PUMA [16]. In breast cancer, miR-3196 acted as a tumor suppressor and inhibited tumorigenesis by regulating orthodenticle homeobox 1 (OTX1) expression [14,15]. Consistently, we found that miR-3196 was significantly downregulated in HCC tissues and decreased miR-3196 was positive associated with tumor size increase. Overexpression of miR-3196 inhibited HCC cell growth in vivo and in vitro.
Doxorubicin (Dox) is the cornerstone of chemotherapy for HCC; however, Dox resistance is an obstacle to successful treatment in patients with HCC. Dox induces apoptosis in human HCC cells via the p53 pathway. It is noteworthy that most tumors were observed overexpression of mutant p53, including HCC [27,28]. Interestingly, our data indicated that Dox induced miR-3196 increase in p53 wild type HCC cells and p53 facilitated miR-3196 expression via binding its promoter region. Increased miR-3196 by p53 elevated chemosensitivity of HCC via targeting FOXP4.
FOXP4 is a member of the FoxP subfamily and play key roles in embryonic development and oncogenesis [29]. Recent studies indicated that the expression levels of FOXP4 were upregulated in prostate cancer and Circular RNA circABCC4 facilitates prostate cancer progression by upregulating FOXP4 expression [30]. Several miRNAs have been reported to suppress FOXP4 in cancer. For example, miR-3184-5p inhibited FOXP4 expression in breast cancer [31]. In HCC cells, miR-338-3p and miR-4316 were reported to inhibit HCC cell growth and metastasis via targeting FOXP4 [32,33]. Similarly, our data uncover that miR-3196 facilitated cell proliferation and chemoresistance via regulating FOXP4 expression in HCC.
In summary, our findings shed light on a new role of miR-3196 in HCC and suggested that the p53-dependent, miR-3196-mediated FOXP4 pathway suppressed cell growth and decreased chemoresistance in HCC (Figure 7).
Figure 7.
Schematic diagram of the role of p53-miR-3196-FOXP4 axis in HCC.
Acknowledgements
This research was supported by National Nature Science Foundation of China (Nos. 81773966 and 81471755 to Deguang Sun), the Liaoning Provincial Natural Science Foundation of China (Nos. 20170540255 and L2016029 to Chengshun Gao; No.201602237 to Deguang Sun).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Poon RT, Fan ST, Lo CM, Liu CL, Wong J. Long-term survival and pattern of recurrence after resection of small hepatocellular carcinoma in patients with preserved liver function: implications for a strategy of salvage transplantation. Ann Surg. 2002;235:373–382. doi: 10.1097/00000658-200203000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Zhang D, Liu S, Li X, Hu W, Han C. KLF4 suppresses the migration of hepatocellular carcinoma by transcriptionally upregulating monoglyceride lipase. Am J Cancer Res. 2018;8:1019–1029. [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12:436. doi: 10.1038/nrclinonc.2015.121. [DOI] [PubMed] [Google Scholar]
- 5.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Jiang J, Ma B, Li X, Jin W, Han C, Wang L, Wang H. MiR-1281, a p53-responsive microRNA, impairs the survival of human osteosarcoma cells upon ER stress via targeting USP39. Am J Cancer Res. 2018;8:1764–1774. [PMC free article] [PubMed] [Google Scholar]
- 8.Xu X, Tao Y, Shan L, Chen R, Jiang H, Qian Z, Cai F, Ma L, Yu Y. The role of microRNAs in hepatocellular carcinoma. J Cancer. 2018;9:3557–3569. doi: 10.7150/jca.26350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanthaje S, Makol A, Chakraborti A. Sorafenib response in hepatocellular carcinoma: microRNAs as tuning forks. Hepatol Res. 2018;48:5–14. doi: 10.1111/hepr.12991. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Zhang M, Tu J, Pang L, Cai W, Liu X. MicroRNA-122 affects cell aggressiveness and apoptosis by targeting PKM2 in human hepatocellular carcinoma. Oncol Rep. 2015;34:2054–2064. doi: 10.3892/or.2015.4175. [DOI] [PubMed] [Google Scholar]
- 11.Xie XH, Xu XP, Sun CY, Yu ZJ. Regulation of the oncogenic function of distal-less 4 by microRNA-122 in hepatocellular carcinoma. Mol Med Rep. 2015;12:1375–1380. doi: 10.3892/mmr.2015.3554. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, Wang X, Su Z, Fei H, Liu X, Pan Q. MiR-25 promotes hepatocellular carcinoma cell growth, migration and invasion by inhibiting RhoGDI1. Oncotarget. 2015;6:36231–36244. doi: 10.18632/oncotarget.4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng YB, Liang XH, Zhang GX, Jiang N, Zhang T, Huang JY, Zhang L, Zeng XC. miRNA-135a promotes hepatocellular carcinoma cell migration and invasion by targeting forkhead box O1. Cancer Cell Int. 2016;16:63. doi: 10.1186/s12935-016-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji ZC, Han SH, Xing YF. Overexpression of miR-3196 suppresses cell proliferation and induces cell apoptosis through targeting ERBB3 in breast cancer. Eur Rev Med Pharmacol Sci. 2018;22:8383–8390. doi: 10.26355/eurrev_201812_16536. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Wu W, Wu M, Ding J. Long noncoding RNA ADPGK-AS1 promotes cell proliferation, migration, and EMT process through regulating miR-3196/OTX1 axis in breast cancer. In Vitro Cell Dev Biol Anim. 2019;55:522–532. doi: 10.1007/s11626-019-00372-1. [DOI] [PubMed] [Google Scholar]
- 16.Xu C, Zhang L, Duan L, Lu C. MicroRNA-3196 is inhibited by H2AX phosphorylation and attenuates lung cancer cell apoptosis by downregulating PUMA. Oncotarget. 2016;7:77764–77776. doi: 10.18632/oncotarget.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du T, Li H, Fan Y, Yuan L, Guo X, Zhu Q, Yao Y, Li X, Liu C, Yu X, Liu Z, Cui CP, Han C, Zhang L. The deubiquitylase OTUD3 stabilizes GRP78 and promotes lung tumorigenesis. Nat Commun. 2019;10:2914. doi: 10.1038/s41467-019-10824-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D, Lin J, Chao Y, Zhang L, Jin L, Li N, He R, Ma B, Zhao W, Han C. Regulation of the adaptation to ER stress by KLF4 facilitates melanoma cell metastasis via upregulating NUCB2 expression. J Exp Clin Cancer Res. 2018;37:176. doi: 10.1186/s13046-018-0842-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin J, Zhang D, Fan Y, Chao Y, Chang J, Li N, Han L, Han C. Regulation of cancer stem cell self-renewal by HOXB9 antagonizes endoplasmic reticulum stress-induced melanoma cell apoptosis via the miR-765-FOXA2 axis. J Invest Dermatol. 2018;138:1609–1619. doi: 10.1016/j.jid.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Ma B, Yuan Z, Zhang L, Lv P, Yang T, Gao J, Pan N, Wu Q, Lou J, Han C, Zhang B. Long non-coding RNA AC023115.3 suppresses chemoresistance of glioblastoma by reducing autophagy. Biochim Biophys Acta Mol Cell Res. 2017;1864:1393–1404. doi: 10.1016/j.bbamcr.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Wang Y, Li X, Xia X, Li N, He R, He H, Han C, Zhao W. ZBTB7A enhances osteosarcoma chemoresistance by transcriptionally repressing lncRNALINC00473-IL24 activity. Neoplasia. 2017;19:908–918. doi: 10.1016/j.neo.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob JJ, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 23.Gurtner A, Falcone E, Garibaldi F, Piaggio G. Dysregulation of microRNA biogenesis in cancer: the impact of mutant p53 on Drosha complex activity. J Exp Clin Cancer Res. 2016;35:45. doi: 10.1186/s13046-016-0319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Järvelin AI, Noerenberg M, Davis I, Castello A. The new (dis)order in RNA regulation. Cell Commun Signal. 2016;14:9. doi: 10.1186/s12964-016-0132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding B, Lou W, Liu J, Li R, Chen J, Fan W. In silico analysis excavates potential biomarkers by constructing miRNA-mRNA networks between non-cirrhotic HCC and cirrhotic HCC. Cancer Cell Int. 2019;19:186. doi: 10.1186/s12935-019-0901-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Z, He R, Luo H, Lu C, Ning Z, Wu Y, Han C, Tan G, Wang Z. Integrin-beta5, a miR-185-targeted gene, promotes hepatocellular carcinoma tumorigenesis by regulating beta-catenin stability. J Exp Clin Cancer Res. 2018;37:17. doi: 10.1186/s13046-018-0691-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Teufel A, Wong EA, Mukhopadhyay M, Malik N, Westphal H. FoxP4, a novel forkhead transcription factor. Biochim Biophys Acta. 2003;1627:147–152. doi: 10.1016/s0167-4781(03)00074-5. [DOI] [PubMed] [Google Scholar]
- 30.Huang C, Deng H, Wang Y, Jiang H, Xu R, Zhu X, Huang Z, Zhao X. Circular RNA circABCC4 as the ceRNA of miR-1182 facilitates prostate cancer progression by promoting FOXP4 expression. J Cell Mol Med. 2019;23:6112–61119. doi: 10.1111/jcmm.14477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X, Xiao Y, Zhou Y, Zhou Z, Yan W. LncRNA FOXP4-AS1 is activated by PAX5 and promotes the growth of prostate cancer by sequestering miR-3184-5p to upregulate FOXP4. Cell Death Dis. 2019;10:472. doi: 10.1038/s41419-019-1699-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang G, Sun Y, He Y, Ji C, Hu B, Sun Y. MicroRNA-338-3p inhibits cell proliferation in hepatocellular carcinoma by target forkhead box P4 (FOXP4) Int J Clin Exp Pathol. 2015;8:337–344. [PMC free article] [PubMed] [Google Scholar]
- 33.E C, Yang J, Li H, Li C. LncRNA LOC105372579 promotes proliferation and epithelial-mesenchymal transition in hepatocellular carcinoma via activating miR-4316/FOXP4 signaling. Cancer Manag Res. 2019;11:2871–2879. doi: 10.2147/CMAR.S197979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.