

Abstract
Yes-associated protein (YAP) is a transcription co-regulator downstream of the Hippo pathway, and plays a critical role in cancer. Although YAP regulation in the canonical Hippo pathway is well established, the Hippo-independent regulation of YAP is not well explored. Here, we showed the possible new mechanism of YAP regulation by the receptor tyrosine kinase Axl. Co-immunoprecipitation and Western blot analysis demonstrated the interaction between YAP and Axl, which was enhanced by Axl ligand Growth Arrest Specific 6 (GAS6) stimulation. Furthermore, we found that YAP is phosphorylated at tyrosine residues by GAS6 stimulation in vivo and Axl directly phosphorylates YAP in vitro. Axl overexpression or GAS6 stimulation increased YAP-mediated transcriptional activity, and YAP-mediated colony forming activity in soft agar was enhanced by co-expression of Axl. In EGFR tyrosine kinase inhibitor (TKI)-sensitive lung cancer cells, YAP protein was downregulated in response to TKI treatment, while overexpression of YAP attenuated TKI sensitivity, suggesting that YAP is a key determinant of TKI response. Moreover Axl overexpression reversed TKI-induced YAP downregulation and induced TKI-resistance, which was reversed by YAP knockdown, further supporting the notion that YAP functions downstream of Axl. Together, these findings suggest a novel role of YAP in Axl-mediated TKI resistance.
Keywords: YAP, Axl, lung cancer, tyrosine kinase inhibitor, resistance
Introduction
Yes-associated protein (YAP) is an oncogenic transcriptional co-regulator and the downstream effector of the Hippo signaling pathway. The Hippo pathway is an evolutionally conserved pathway that plays an important role in organ size control in normal tissues, stem cell regulation, and cancer development and progression [1,2]. YAP is directly phosphorylated by Lats1/2 at multiple serine/threonine residues and phosphorylated YAP interacts with 14-3-3 and is sequestered in the cytoplasm or undergoes degradation through β-TrCP-mediated ubiquitination, resulting in its inactivation [3]. In contrast, unphosphorylated YAP forms a complex with TEAD transcription factors in the nucleus and induces its target genes such as CCN1, CCN2, and Axl [4]. YAP is frequently upregulated in various types of cancer, and known to promote anchorage-independent growth, cancer cell survival, proliferation, and metastasis [4]. Moreover, YAP is also shown to be involved in the immune evasion through various mechanisms [5-7]. Therefore, YAP is considered as a critical oncogenic factor that regulates various aspects of cancer.
Axl is the receptor tyrosine kinase that belongs to TAM family kinase [8]. Axl is activated by its ligand Growth Arrest Specific 6 (GAS6) or the formation of heterodimer with other receptors such as EGFR and HER2, resulting in the activation of downstream PI3 kinase-AKT and MAP kinase pathways. Axl and its ligand GAS6 are overexpressed in various cancers, and contributes to tumor progression [8,9]. Moreover, Axl is known to induce epithelial-to-mesenchymal transition (EMT) and cancer stem cells [10-12]. Therefore, Axl is a potential drug target for various cancers, and several Axl inhibitors have been investigated in clinical trials. In particular, because Axl is known to be involved in the resistance to EGFR tyrosine kinase inhibitor (TKI) in non-small cell lung cancer (NSCLC) [13,14], the combination of EGFR TKI and Axl inhibitors would be a potential therapeutic strategy to overcome the resistance to EGFR TKI [14]. Moreover, it has been reported that Axl facilitates the immune suppressive tumor microenvironment by downregulating MHC-I molecules and promoting cytokine release [15].
In addition to the canonical Hippo pathway, YAP has been shown directly to be regulated by several kinases. For example, AMPK directly phosphorylates YAP at serine 94 and inhibits the YAP-TEAD interaction [16]. CDK1 regulates YAP activity by phosphorylating at multiple serine residues during the G2/M phase of the cell cycle [17]. Nemo-like kinase (NLK) phosphorylates YAP at serine 128, which blocks the interaction between YAP and 14-3-3, leading to YAP activation [18]. To study the regulatory mechanism of YAP other than the canonical Hippo signaling pathway, we previously screened YAP interacting proteins by tandem affinity purification and mass spectrometry [19]. We particularly focused on the enzymes that may regulate YAP activity and stability. We identified several protein kinases that interacts with YAP, and have demonstrated that Aurora kinase interacts with and phosphorylates YAP at serine 397, thereby regulating YAP transcriptional activity [19]. In this study, we attempted to study other YAP regulators and focused on Axl, which is in the list of YAP binding proteins in our previous study [19]. Because Axl is known to be a target of YAP, we hypothesized that YAP may function to amplify Axl signaling through a feed-forward mechanism. In this study, we suggested that Axl plays a critical role in the regulation of YAP activity. Moreover, we also showed that Axl interacts with and phosphorylates YAP in vitro, suggesting the direct regulation of YAP by Axl.
Materials and methods
Antibodies, chemicals, and drug
Antibodies against YAP, p-YAP, Axl, Akt, p-AKT, ERK, p-ERK, EGFR, and pEGFR were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-tubulin and Flag antibodies were purchased from Sigma-Aldrich (St Louis, MO, USA). ATP-γ-S and anti-Thiophosphate ester antibody were purchased from Abcam (Cambridge, UK). Recombinant GAS6 was purchased from R & D Systems (Minneapolis, MN, USA). Erlotinib was obtained from Selleck Chemicals (Houston, TX, USA).
Cell culture, plasmid, transfection, and lentivirus infection
All the Cell lines were obtained from ATCC (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium with 10% fetal bovine serum and antibiotics. Plasmids that express YAP or shRNA against YAP were described previously [19]. Myc-His-tagged Axl was constructed on pCDH-EF1-MCS-IRES-Neo vector (#CD533A-2; System Biosciences, Palo Alto, CA, USA). 8xGTIIC-lucifease plasmid was obtained from Addgene (Cambridge, MA, USA). Lentivirus packaging and infection was performed as described previously [19]. 293T cells were co-transfected with lentivirus expression plasmid together with packaging vectors using calcium phosphate method. For luciferase assay, cells were transfected using electroporation as described previously [20].
Co-immunoprecipitation and Western blot
Co-immunoprecipitation was performed as described previously [21]. In brief, 1 mg of cells lysates were incubated with 2 μg of antibodies or control IgG in 500 μl of NP-40 lysis buffer at 4°C for overnight. Then, Protein G or A beads (15 μl) were added to the lysates, and the samples were further incubated at 4°C for 2 hours. The beads were then washed three times with NP-40 lysis buffer, and subjected to Western blot analysis. Western blot was conducted according to standard procedures. Protein samples in in SDS sample buffer were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to PVDF membranes using the mini-Protein system from Bio-Rad (Hercules, CA, USA). The membranes were blocked with 3% milk, and incubated with primary antibodies overnight, followed by one hour incubation with secondary antibodies conjugated with HRP. Signals were detected with Clarity™ Western ECL Blotting Substrates (Bio-Rad) and captured on X-ray films.
Soft agar assay and cell viability assay
Cells (5 × 103) were mixed with 0.3% agarose/DMEM/F12 at 42°C and were then laid on top of 0.5% solidified agar/DMEM/F12 in 12-well plates. After the top layer solidified, 1 ml of medium was added to each well. The cells were cultured for 14 days and medium was changed every 5 days. To visualize the colonies, 100 μl of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; 5 mg/ml) solution was added and incubated the cells for several hours. The numbers of colonies were counted under microscope. The effects of EGFR TKI on cell viability were determined as described previously [22]. In brief, the cells were seeded in 6-well plates at about 20% confluency and treated with 1 μM of erlotinb. The medium was changed 4 to 5 days later and further cultured for 10 days, and the living cells were then stained with crystal violet.
Kinase assay
His-tagged YAP protein was purified as described previously [19]. Purified active Axl protein was purchased from Enzo Life Science (Farmingdale, NY, USA). The in vitro kinase assay was performed at 30°C for 20 min by mixing 25 ng of Axl kinase with 750 ng of YAP protein in kinase buffer (50 mM HEPES-7.3; 15 mM MgCl2; 20 mM KCl; 2 mM EGTA; 100 μM ATP-γ-S). Reactions were quenched by heating at 95°C for 5 min in the presence of SDS-loading buffer. The phosphorylation signals were detected by Western blot analysis with anti-Thiophosphate ester antibody.
Luciferase assay
H1299 cells were co-transfected with 8xGTIIC-lucifease plasmid and β-actin promoter-Renilla luciferase plasmid together with YAP and/or Axl expression plasmids. 48 hours later, cells were collected and subjected to luciferase assay. For the GAS6 stimulation, cells were serum-starved for overnight before GAS6 stimulation. The luciferase assay was performed by using the dual-luciferase system, according to the manufacturers’ protocol (Promega, Madison, WI, USA).
Results
Axl interacts with YAP, which is enhanced by its ligand
To verify YAP-Axl interaction, which was identified in our previous screening [19], we performed co-immunoprecipitation and Western blot analysis. We ectopically expressed Flag-tagged YAP in BT547 cells, which express relatively low levels of YAP [19], and performed immunoprecipitation using Flag antibody. We found that Axl was pulled down only in the cells expressing Flag-YAP (Figure 1A). Alternatively, we ectopically expressed His-tagged Axl in MCF7 cells, which express low endogenous Axl protein, and performed immunoprecipitation using His antibody. Similar to the result from Flag-YAP expressed cells, we found that YAP was pulled down only in the cells expressing His-Axl (Figure 1B). To further verify the interaction, we performed immunoprecipitation of endogenous YAP protein in H1299 cells. Moreover, to study the role of Axl activity in the interaction, we serum-starved cells overnight and stimulated them with GAS6, which is the ligand of Axl. We found that endogenous Axl interacted with endogenous YAP, and the interaction was enhanced by GAS6 stimulation. Together, these results suggest that Axl is not only a YAP target, but also a YAP interacting protein.
Figure 1.
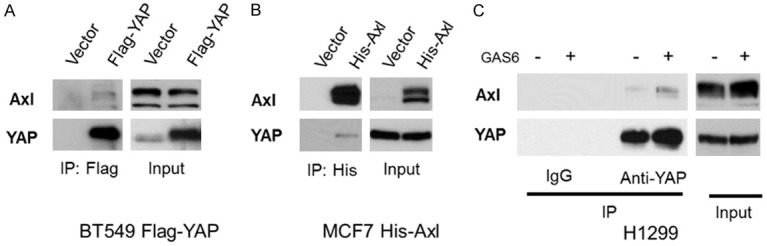
YAP interacts with Axl and the interaction is enhanced by GAS6. A. BT549 cells were stably expressed with vector control or Flag-YAP, and the cell lysates were subjected to immunoprecipitation with anti-Flag antibody, followed by Western blot with anti-YAP or anti-Axl antibody. B. MCF7 cells were stably expressed with vector control or His-tagged Axl, and the cell lysates were subjected to immunoprecipitation with anti-His-tag antibody, followed by Western blot with anti-YAP or anti-Axl antibody. C. H1299 cells were serum-deprived overnight and then treated or untreated with GAS6 (200 ng/ml) for 30 min. The cell lysates were then subjected to immunoprecipitation with anti-YAP or control IgG, followed by Western blot analysis with anti-YAP or anti-Axl antibody.
YAP tyrosine phosphorylation is enhanced by Axl activation
Because Axl is a receptor tyrosine kinase, we next investigated the tyrosine phosphorylation of YAP after Axl activation. H1299 cells were serum-starved overnight and stimulated with GAS6. Then, we performed immunoprecipitation with the anti-phosphotyrosine 4G10 antibody or control mouse IgG, followed by Western blot analysis with anti-YAP antibody (Figure 2A). Alternatively, we performed immunoprecipitation with the anti-YAP antibody, followed by Western blot analysis with the anti-phosphotyrosine 4G10 antibody (Figure 2B). Axl activation was verified by evaluating phosphorylation of AKT and ERK, which are the downstream molecules of the Axl-mediated signaling pathway (Figure 2C). To examine if YAP is directly phosphorylated by Axl, we performed in vitro kinase assay with the purified YAP protein, Axl protein, and ATP-γ-S as a kinase substrate, and the phosphorylation was detected by Western blot analysis with the anti-Thiophosphate ester antibody. We identified that the YAP protein from amino acid (aa) 271 to aa504 was phosphorylated but not the one from aa1 to 270 (Figure 2D). By using in silico analysis, we found the potential phosphorylation sites at tyrosine (Y) 391 and 407. Thus, we substituted Y391 and/or Y407 to phenylalanine (F), and performed in vitro kinase assay. Both Y391F and Y407F single mutants showed reduced phosphorylation but the phosphorylation was completely abrogated in the Y391/407F double mutant, indicating that Axl can phosphorylate Y391 and Y407 in YAP in vitro.
Figure 2.
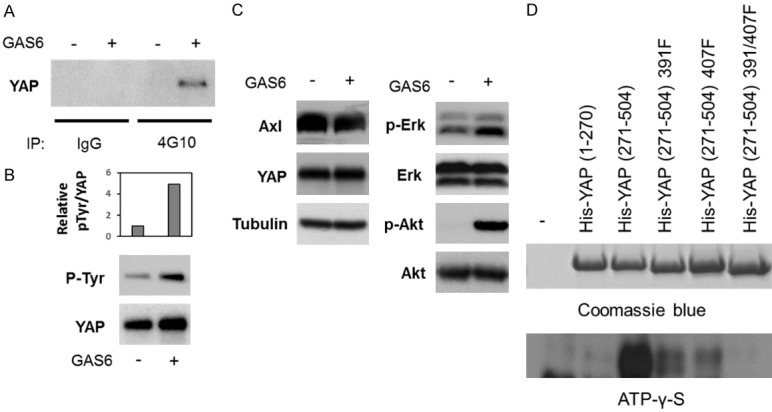
Tyrosine phosphorylation in YAP is enhanced by Gas6 treatment. A-C. H1299 cells were serum-starved overnight and treated or untreated with 200 ng/ml of GAS6 for 30 min. A. The cell lysates were subjected to immunoprecipitation with anti-phosphotyrosine (4G10) antibody or control mouse IgG, followed by Western blot analysis with anti-YAP antibody. B. The cell lysates were subjected to immunoprecipitation with anti-YAP antibody, followed by Western blot analysis with 4G10 antibody (p-Tyr) or anti-YAP antibody. The intensity of the band signals were analyzed by using ImageJ, and the relative ratio of the signals from p-Tyr and YAP was shown in the upper graph. C. The cell lysates were subjected to Western blot analysis with the indicated antibodies. D. His-tagged YAP recombinant proteins were subjected to in vitro kinase assay with purified Axl protein and ATP-γ-S, followed by Western blot analysis with anti-Thiophosphate ester antibody that recognizes ATP-γ-S.
YAP activity is enhanced by Axl activation
Next, we studied whether Axl affects YAP function. We transiently transfected H1299 cells with the luciferase plasmid containing TEAD-binding sites (8xGTIIC-lucifease plasmid) together with the empty vector, YAP, Axl, or YAP plus Axl expression plasmid (Figure 3A). Alternatively, we transiently transfected H1299 cells with the 8xGTIIC-luciferase plasmid together with the empty vector or YAP expression plasmid, followed by stimulation with GAS6 (Figure 3B). We observed that YAP-mediated transcriptional activity was further enhanced by Axl overexpression (Figure 3A) and Axl activation (Figure 3B). In our previous study, we showed that YAP promotes anchorage-independent cell proliferation [19]. Thus, we overexpressed YAP together with Axl in BT549 cells and cultured them in soft agar. Consistent with the results from the luciferase assay (Figure 3A, 3B), we observed that YAP-mediated anchorage-independent proliferation was further enhanced by overexpression of Axl (Figure 3C). Together, these results suggest that Axl regulates YAP function.
Figure 3.
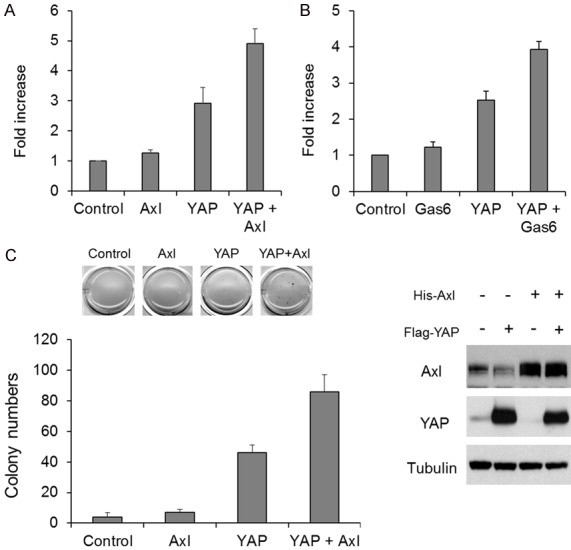
Axl enhances YAP activity. A. H1299 cells were transiently transfected with TEAD/YAP-responsive luciferase plasmid (8xGTIIC-luc) and YAP expression plasmid and/or Axl expression plasmid. 48 hours after transfection, luciferase activity was determined. B. H1299 cells were transfected with TEAD/YAP-responsive luciferase plasmid (8xGTIIC-luc) with/without YAP expression plasmid. 48 hours after transfection, the cells were treated or untreated with GAS6 for 1 hour, and subjected to luciferase assay. C. Flag-YAP and/or His-tagged Axl were stably expressed in BT549 cells, and they were cultured in soft agar for 3 weeks. The numbers of colonies were counted and shown as the bar graph (n = 3, the left graph). Protein expression was confirmed by Western blot analysis (the right panels).
YAP plays a role in Axl-mediated TKI resistance
Some studies suggest that YAP is also involved in EGFR inhibitor resistance [23-27]. To verify the YAP-mediated resistance to EGFR TKI, we overexpressed YAP in PC9 cells, which express mutant EGFR and sensitive to EGFR TKI, and investigated the TKI sensitivity. Consistent with previous studies [23-27], YAP conferred resistance to EGFR TKI erlotinib in PC9 cells (Figure 4A). It has been demonstrated that Axl overexpression is associated with resistance to EGFR TKI in lung cancer [14]. Because YAP is also involved in TKI resistance and we found that YAP is activated by Axl, we next investigated the role of YAP in Axl-mediated TKI resistance. We overexpressed Axl in PC9 cells and confirmed that Axl overexpression attenuated the sensitivity to erlotinib (Figure 4B). However, when YAP was knocked down in Axl-overexpressing cells, the effect of Axl on TKI resistance was completely abrogated (Figure 4B). There results suggest that YAP functions as a downstream effector of Axl.
Figure 4.
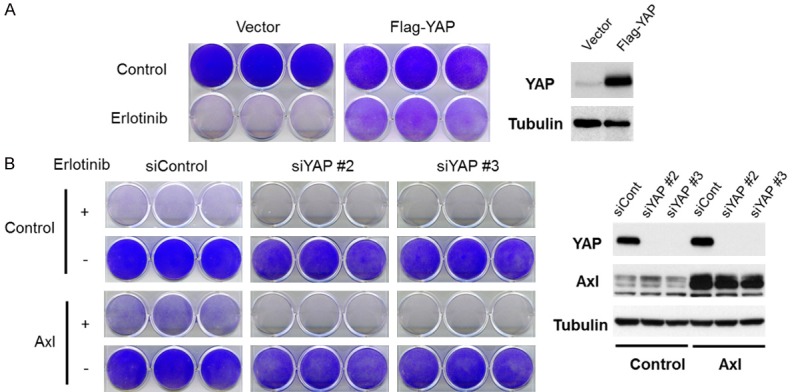
YAP is involved in Axl-mediated resistance to an EGFR inhibitor in EGFR mutant lung cancer cells. A. YAP was stably expressed in PC9 cells, and the YAP-overexpressing cells and control cells were cultured with1 μM of erlotinib for 10 days. The cells were then fixed and stained with crystal violet. Expression of YAP was confirmed by Western blot analysis (the right panels). B. YAP was knocked down in control or Axl-overexpressing PC9 cells. The cells were then cultured with 1 μM of erlotinib for 10 days. The cells were then fixed and stained with crystal violet. Expression of YAP and Axl was confirmed by Western blot analysis (the right panels).
Expression of YAP is downregulated by EGFR TKI, which is reversed by Axl
We next investigated YAP protein expression in both TKI-sensitive and resistant cells after TKI treatment. We selected H1299, A549 and H460 cells as EGFR TKI resistant lines and H322, PC9, HCC827 as EGFR TKI sensitive lines. Both PC9 and HCC827 cells express mutant EGFR, while H322 cells express wild type EGFR but are somehow sensitive to EGFR TKI [28]. We observed that YAP expression was downregulated in the TKI sensitive cells but not in TKI resistant cells (Figure 5A). YAP phosphorylation was not affected by TKI treatment (Figure 5A). Because YAP protein is downregulated after TKI treatment, we next studied the stability of YAP protein after TKI treatment in PC9 cells. We treated PC9 cells with erlotinib in the presence of cycloheximide, and determined YAP expression levels by Western blot analysis. As shown in Figure 5B, the YAP expression level did not change in the absence of TKI but it was significantly downregulated in the presence of TKI, suggesting that the inhibition of EGFR destabilizes YAP protein in EGFR-TKI sensitive cells. We next studied the role of Axl in YAP protein stability. PC9 wild type and Axl-overexpressing cells were treated with erlotinib, and YAP expression was determined by Western blot analysis. As shown in Figure 5C, Axl overexpression significantly inhibited the YAP downregulation by TKI. Moreover, we investigated YAP expression in the TKI-resistant HCC827-derived GR5 cells, in which Axl expression is upregulated [29]. EGFR TKI downregulated the YAP protein expression in the parental HCC827 cells, but not in the GR5 cells. Together, these results suggest that Axl is involved in YAP protein stabilization, which may contribute to the Axl-mediated TKI resistance.
Figure 5.
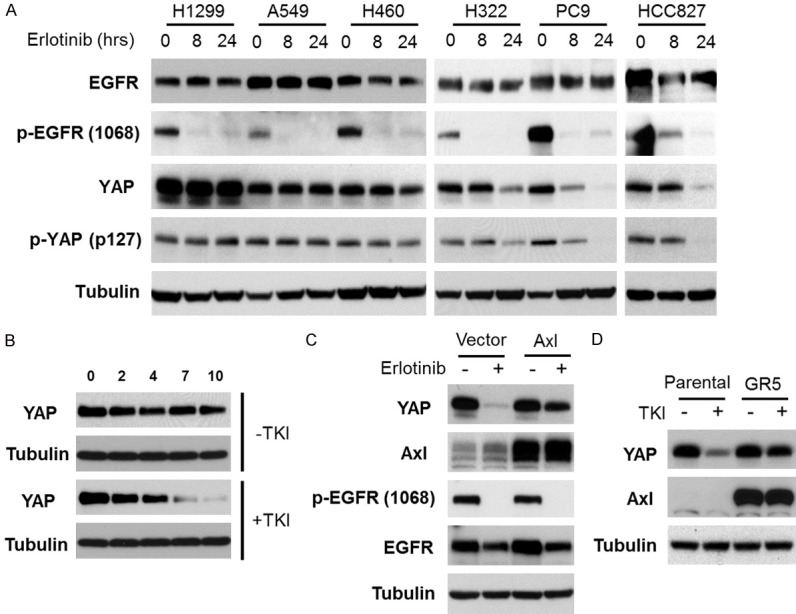
YAP is downregulated in response to EGFR inhibitors in EGFR inhibitor sensitive cells. A. Erlotinib resistant (H1299, A549 and H460) and sensitive (H322, PC9, HCC827) cells were treated with 1 μM of erlotinib for the indicated periods of time, and subjected to Western blot analysis with the indicated antibodies. B. PC9 cells were treated or untreated with 1 μM of erlotinib in the presence of 50 μg/ml of cycloheximide. Expression of YAP and tubulin was determined by Western blot analysis. C. PC9 control and Axl-overexpressing cells were treated with 1 μM of erlotinib for 24 hours, and subjected to Western blot analysis with the indicated antibodies. D. HCC827 parental and TKI-resistant (GR5) cells were treated with 1 μM of erlotinib for 24 hours, and subjected to Western blot analysis with the indicated antibodies.
YAP targets mRNA expression is associated with AXL and GAS6 mRNA expression in lung adenocarcinoma
To verify the clinical significance of the Axl-mediated YAP activation, we analyzed co-occurrence of AXL or GAS6 expression and YAP target gene expression in lung adenocarcinoma from The Cancer Genome Atlas (TCGA) database. Using cBioPortal platform [30], we analyze the mRNA expression of AXL, GAS6 as well as eight YAP target genes including CCN1, CCN2, SERPINE1, REEP1, PRSS23, FGF2, and CENPA in 568 lung adenocarcinoma (Figure 6A). Moreover, using the tool for analyzing the tendency of their expression, we found that most of the YAP target genes are significantly correlated with the expression of AXL and GAS6 (Figure 6B). These results suggest that Axl activation is associated with YAP activation in lung adenocarcinoma.
Figure 6.
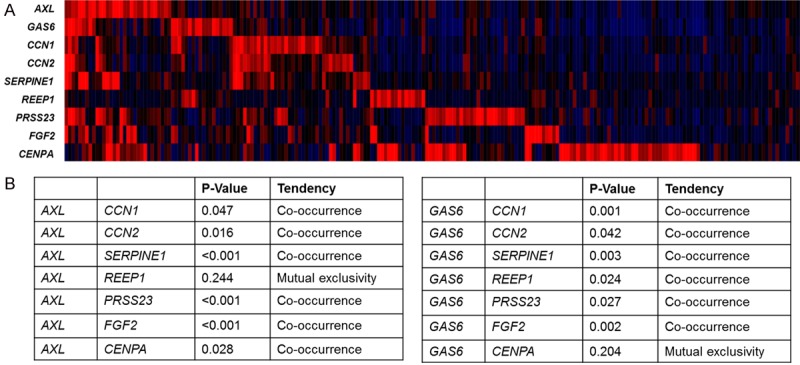
Co-expression of YAP target genes and Axl and GAS6. A. Heat map of mRNA expression of Axl, GAS6, CCN1 and CCN2 in lung adenocarcinoma from TCGA database were analyzed by using cBioPortal platform. B. The tendency of the expression between AXL or GAS6 and YAP target genes was analyzed by using cBioPortal platform.
Discussion
In the present study, we showed that Axl interacts with YAP and regulates YAP activity. Our data also showed that tyrosine phosphorylation of YAP is enhanced by Axl activation and YAP is an Axl substrate in vitro. Therefore, it is plausible that Axl interacts with YAP directly and phosphorylates it in vivo. Because YAP activity was also enhanced by Axl activation (Figure 3), the YAP tyrosine phosphorylation at Y391 and/or Y407 may be involved in enhanced YAP activation.
It has been shown that EGFR signaling regulates Yorkie (Drosophila YAP homologue) activity via the Ajuba protein in Drosophila and Yorkie plays a role in EGFR-mediated cell proliferation [31]. Also, another report has shown that EGFR regulates YAP activity via PI3K-PDK1 pathway in hepatocellular carcinoma cells, and the EGFR-mediated YAP activity is critical for cell survival rather than cell proliferation [32]. In this report, we discovered that EGFR signaling in EGFR-mutant lung cancer was essential protein stability of YAP (Figure 5B). Interestingly, Axl was able to restore the YAP expression that was downregulated by TKI treatment (Figure 5C, 5D). Therefore, it would be interesting to study whether the YAP phosphorylation by Axl contributes to YAP protein stability. Indeed, the phosphorylation of S381, 384, and 387 has been shown to be critical for the interaction between YAP and β-TrCP [33]. Because Y391 and 407 are relatively close to these sites, the phosphorylation by Axl may affect the interaction between YAP and β-TrCP, thereby inhibiting YAP degradation. It would be also important to study how the EGFR signaling pathway regulates YAP stability in EGFR mutant lung cancer cells.
We showed that YAP overexpression attenuated the sensitivity of EGFR mutant lung cancer cells to the EGFR TKI while the knockdown of YAP sensitized the cells to it (Figure 4). These results are consistent with the previous reports [23,25-27]. In the acquired resistant cells, either YAP activity or expression is upregulated, which is accompanied with Axl upregulation [25,27]. One report also showed that the knockdown of YAP attenuates Axl expression in the TKI-resistant cells, but Axl expression in the resistant cells after YAP knockdown is still higher than one in wild type cells [25]. Our results showed that YAP knockdown did not affect the endogenous Axl expression in PC9 cells (Figure 4B). Moreover, TKI-treatment, which significantly reduced the YAP protein levels, did not alter Axl protein levels (Figure 5C). Therefore, the upregulation of Axl protein in TKI resistant cells may be, at least in part, due to other mechanisms such as gene amplification, other transcription factors, and micro RNAs, as reported previously [34]. Moreover, we found that TKI resistance induced by Axl overexpression was reversed by the knockdown of YAP, further supporting the notion that YAP can function downstream of Axl. Considering that YAP overexpression reduced the TKI sensitivity (Figure 4) and that YAP is significantly downregulated only in the TKI-sensitive cells (Figure 5A), YAP may function as a key factor that controls cell survival after TKI exposure.
In contrast to the previous model [25], we also found that Axl activation by GAS6 or its overexpression increased YAP transcriptional activity (Figure 3A, 3B). Besides, the YAP downregulation induced by TKI was reversed by Axl overexpression (Figure 5C). To study the clinical significance of Axl-mediated YAP activation, we analyzed TCGA database (Figure 6). Because AXL is also one of YAP-target genes, co-occurrence of expression of AXL and other YAP target genes can be explained by only YAP activation. However, we also showed that GAS6, which is not a YAP target gene, also positively co-expressed with YAP target genes. Therefore, these data further support our hypothesis that Axl activation lead to YAP activation in lung adenocarcinoma.
Although YAP is a potential drug target in various cancer, there is no FDA-approved clinical drug available. If the blockage of Axl attenuates YAP activity in lung cancer, Axl TKIs may be used as an alternative strategy to target YAP. Also, the combination of EGFR TKIs and Axl TKIs may be more effective to downregulate YAP. Because both YAP and Axl are involved in immune evasion, it would be worthwhile to test the combination of immune checkpoint inhibitors and Axl/EGFR TKIs.
In summary, this study provides an entirely new concept regarding the role of YAP and Axl in the EGFR TKI-resistance (Figure 7). In the previous model, YAP facilitates TKI resistance though the upregulation of Axl. We showed the possibility that Axl also regulates YAP activity and stability, thereby amplifying the signaling through the feed-forward mechanism (Figure 7). Clearly further studies are required to solidify this model in the future.
Figure 7.
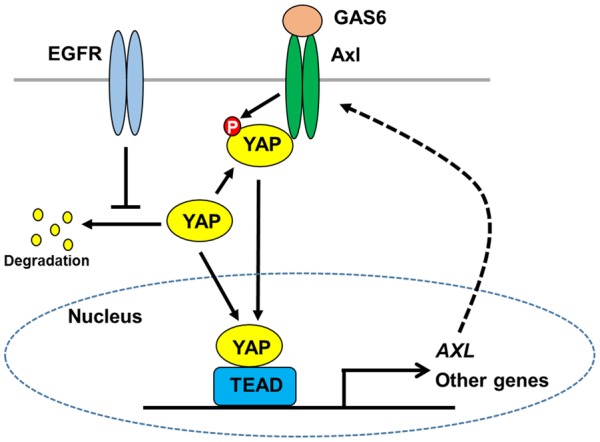
Schematic model of Axl-mediated YAP regulation in lung cancer. EGFR activity is critical for YAP protein stability in EGFR TKI-sensitive cells. Axl interacts with YAP and enhances its activity. Axl also directly phosphorylates YAP, which may be involved in YAP activation and/or protein stability. YAP induces the expression of various genes, which may contribute to cell survival. AXL is a YAP target gene and YAP-mediated transcription of AXL may contribute to the upregulation of Axl in the resistant cells, thereby amplify the signaling through the feed-forward mechanism.
Acknowledgements
This work is supported by a startup grant from QBRI, Qatar Foundation (HY); The University of Texas MD Anderson Cancer Center-China Medical University and Hospital Sister Institution Fund (MCH); The publication of this article was funded by the Qatar National Library.
Disclosure of conflict of interest
None.
References
- 1.Zhao B, Tumaneng K, Guan KL. The hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–883. doi: 10.1038/ncb2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu FX, Guan KL. The hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian hippo pathway. Semin Cell Dev Biol. 2012;23:785–793. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murakami S, Shahbazian D, Surana R, Zhang W, Chen H, Graham GT, White SM, Weiner LM, Yi C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene. 2017;36:1232–1244. doi: 10.1038/onc.2016.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, Zhang J, Li-Ning-Tapia EM, Kapoor A, Wu CJ, Patel NB, Guo Z, Ramamoorthy V, Tieu TN, Heffernan T, Zhao D, Shang X, Khadka S, Hou P, Hu B, Jin EJ, Yao W, Pan X, Ding Z, Shi Y, Li L, Chang Q, Troncoso P, Logothetis CJ, McArthur MJ, Chin L, Wang YA, DePinho RA. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016;6:80–95. doi: 10.1158/2159-8290.CD-15-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA, Park SH, Shin EC, Kim J. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol Res. 2018;6:255–266. doi: 10.1158/2326-6066.CIR-17-0320. [DOI] [PubMed] [Google Scholar]
- 8.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paccez JD, Vogelsang M, Parker MI, Zerbini LF. The receptor tyrosine kinase Axl in cancer: biological functions and therapeutic implications. Int J Cancer. 2014;134:1024–1033. doi: 10.1002/ijc.28246. [DOI] [PubMed] [Google Scholar]
- 10.Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33:1316–1324. doi: 10.1038/onc.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cichon MA, Szentpetery Z, Caley MP, Papadakis ES, Mackenzie IC, Brennan CH, O’Toole EA. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene. 2014;33:4185–4192. doi: 10.1038/onc.2013.388. [DOI] [PubMed] [Google Scholar]
- 12.Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT, Micklem DR, Akslen LA, Glackin C, Lorens JB. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124–1129. doi: 10.1073/pnas.0909333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, Wistuba II, Coombes KR, Minna JD, Heymach JV. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguilera TA, Rafat M, Castellini L, Shehade H, Kariolis MS, Hui AB, Stehr H, von Eyben R, Jiang D, Ellies LG, Koong AC, Diehn M, Rankin EB, Graves EE, Giaccia AJ. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016;7:13898. doi: 10.1038/ncomms13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–510. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013;73:6722–6733. doi: 10.1158/0008-5472.CAN-13-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon S, Kim W, Kim S, Kim Y, Song Y, Bilousov O, Kim J, Lee T, Cha B, Kim M, Kim H, Katanaev VL, Jho EH. Phosphorylation by NLK inhibits YAP-14-3-3-interactions and induces its nuclear localization. EMBO Rep. 2017;18:61–71. doi: 10.15252/embr.201642683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang SS, Yamaguchi H, Xia W, Lim SO, Khotskaya Y, Wu Y, Chang WC, Liu Q, Hung MC. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene. 2017;36:1265–1275. doi: 10.1038/onc.2016.292. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi H, Chen CT, Chou CK, Pal A, Bornmann W, Hortobagyi GN, Hung MC. Adenovirus 5 E1A enhances histone deacetylase inhibitors-induced apoptosis through Egr-1-mediated Bim upregulation. Oncogene. 2010;29:5619–5629. doi: 10.1038/onc.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL, Yao J, Wei Y, Nie L, Jiao S, Chang WC, Chen CH, Yu Y, Hortobagyi GN, Hung MC. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene. 2018;37:208–217. doi: 10.1038/onc.2017.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, Du Y, Ko HW, Herbst R, Hung MC. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin Cancer Res. 2013;19:845–854. doi: 10.1158/1078-0432.CCR-12-2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee TF, Tseng YC, Nguyen PA, Li YC, Ho CC, Wu CW. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci Rep. 2018;8:271. doi: 10.1038/s41598-017-18527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu BS, Xia HW, Zhou S, Liu Q, Tang QL, Bi NX, Zhou JT, Gong QY, Nie YZ, Bi F. Inhibition of YAP reverses primary resistance to EGFR inhibitors in colorectal cancer cells. Oncol Rep. 2018;40:2171–2182. doi: 10.3892/or.2018.6630. [DOI] [PubMed] [Google Scholar]
- 25.Ghiso E, Migliore C, Ciciriello V, Morando E, Petrelli A, Corso S, De Luca E, Gatti G, Volante M, Giordano S. YAP-dependent AXL overexpression mediates resistance to EGFR Inhibitors in NSCLC. Neoplasia. 2017;19:1012–1021. doi: 10.1016/j.neo.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu PC, You B, Yang YL, Zhang WQ, Wang YC, Xu Z, Dai Y, Liu S, Yang CT, Li H, Hu B, Jablons DM, You L. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget. 2016;7:51922–51933. doi: 10.18632/oncotarget.10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGowan M, Kleinberg L, Halvorsen AR, Helland A, Brustugun OT. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer. 2017;8:497–504. doi: 10.18632/genesandcancer.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Schaeybroeck S, Kyula J, Kelly DM, Karaiskou-McCaul A, Stokesberry SA, Van Cutsem E, Longley DB, Johnston PG. Chemotherapy-induced epidermal growth factor receptor activation determines response to combined gefitinib/chemotherapy treatment in non-small cell lung cancer cells. Mol Cancer Ther. 2006;5:1154–1165. doi: 10.1158/1535-7163.MCT-05-0446. [DOI] [PubMed] [Google Scholar]
- 29.Lee PC, Fang YF, Yamaguchi H, Wang WJ, Chen TC, Hong X, Ke B, Xia W, Wei Y, Zha Z, Wang Y, Kuo HP, Wang CW, Tu CY, Chen CH, Huang WC, Chiang SF, Nie L, Hou J, Chen CT, Huo L, Yang WH, Deng R, Nakai K, Hsu YH, Chang SS, Chiu TJ, Tang J, Zhang R, Wang L, Fang B, Chen T, Wong KK, Hsu JL, Hung MC. Targeting PKCdelta as a therapeutic strategy against heterogeneous mechanisms of EGFR inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell. 2018;34:954–969. e954. doi: 10.1016/j.ccell.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy BV, Irvine KD. Regulation of hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev Cell. 2013;24:459–471. doi: 10.1016/j.devcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia H, Dai X, Yu H, Zhou S, Fan Z, Wei G, Tang Q, Gong Q, Bi F. EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 2018;9:269. doi: 10.1038/s41419-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levin PA, Brekken RA, Byers LA, Heymach JV, Gerber DE. Axl receptor axis: a new therapeutic target in lung cancer. J Thorac Oncol. 2016;11:1357–1362. doi: 10.1016/j.jtho.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]