

Abstract
Cisplatin, as one of the front-line chemotherapeutic drugs, is employed for the treatment of esophageal squamous cell carcinoma (ESCC). However, the occurrence of cisplatin resistance and metastasis remain as challenges in clinical therapy. To investigate the mechanism involved in cisplatin resistance, in this study, we established cisplatin resistant cell lines (Res) from Eca109 and TE-1 parental cells (Par), and we observed that fibronectin (FN)-mediated cell migration and spreading abilities are significantly increased in Res cells when compared to Par cells. Furthermore, we found that the integrin α5 expression is remarkably upregulated in Res cells, and inhibition of α5 results in more apoptosis and endows the Res cells resensitize to cisplatin in vitro and in vivo. In a mechanistic manner, we identified the expression of BARD1 is significantly increased in Res cells, and silencing of BARD1 reverse the effects of α5 on cisplatin resistance. Moreover, we found that the α5/FAK/PI3K/AKT signal axis is activated in Res cells, which mediates the increased expression of BARD1, as well as the cisplatin resistance and cell survival. Thus, our results demonstrate that α5 is required for cisplatin resistance through the promotion of FAK/PI3K/AKT/BARD1 signaling to prevent cells from apoptosis and enhance the DNA damage repair ability. Taken together, our study provides plausible mechanisms of α5-mediated cisplatin resistance in ESCC cells, highlighting that inhibition of α5 may be a potential target for improving efficacy in cisplatin-based chemotherapy.
Keywords: Integrin α5, PI3K/AKT, BARD1, apoptosis, cisplatin resistance, esophageal cancer
Introduction
Esophageal cancer (EC) is the sixth most common aggressive cancer with a high mortality rate worldwide [1]. The pathological forms of EC have been categorized into two major subtypes, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) [2]. In China, the incidence of ESCC is more than 90%, which is much higher than that of EAC [3]. Although significant advances have been made in diagnostic and treatment strategies, the cancer-related mortality caused by metastasis still ranks fourth and 5-year survival rate was less than 10% [4,5]. Cisplatin-based chemotherapies, as the front-line treatment for ESCC, often gain initial responses and play vital roles in patients with postoperative recurrence and metastasis. However, cisplatin treatment eventually leads to the development of chemoresistance, resulting in therapeutic failure [6,7]. Therefore, elucidating the potential mechanisms of cisplatin resistance is critical for reversing the chemotherapy resistance in ESCC.
The prominent anticancer effects of cisplatin include induction of DNA damage and mitochondrial apoptosis [8,9]. Therefore, a prevailing hypothesis is that the development of cisplatin resistance can be attributed to the activation of DNA damage repair and anti-apoptosis ability. Homologous recombination (HR) and non-homologous end joining (NHEJ) are two major repair pathways in cisplatin-induced DNA damage response [10]. Aberrant expression of major components involved in HR and NHEJ pathways were responsible for the drug resistance. For instance, BRCA1, as a critical gene of the HR system, regulates DNA damage repair and cell proliferation by forming heterodimer with BARD1 (BRCA1-associated ring domain 1). BRCA1/BARD1 deficient was associated with suppressed DNA damage repair, and cancer cells restored sensitivity to platinum compounds or DNA damaging chemotherapeutic drugs in several cancer types including ovarian cancer, lung cancer and breast cancer [11-13]. It was also reported that activation of the PI3K/AKT pathway can enhance DNA damage repair by increasing expression of BRCA1 and BARD1 [13,14]. Additionally, the PI3K/AKT pathway plays an important role in cell apoptosis and survival [15]. Thus, the PI3K/AKT pathway is a hot target to overcome chemoresistance. In spite of the efforts that have been devoted to the mechanisms of cisplatin resistance, there is still a lack of an effective target to reverse cisplatin resistance and to improve the response to cisplatin-based therapy.
Integrins are a large family of heterodimeric membrane receptors that function as adhesion molecules and contribute to diverse biological processes such as cell adhesion, migration, and apoptosis by activation of intracellular signaling triggered by the ligation of integrins with extracellular matrix (ECM) [16]. Growing evidence has highlighted its pro-survival and anti-apoptosis functions in the regulation of response to therapeutic modalities [17,18]. As examples, the interaction between integrins and ECM promotes chemoresistance via protecting cells from drug-induced apoptosis in several cancer types including small lung cancer, myeloma and ESCC [19-21]. Focal adhesion kinase (FAK), a key signal transduction component downstream of integrins, can further activate PI3K/AKT signaling cascades which is pivotal in cancer cell survival and chemoresistance [22]. Considering the importance of the FAK/PI3K/AKT pathway in chemoresistance and also the integrin subunits can serve as an independent prognostic factor in ESCC [23-25], we hypothesized that certain integrin may participate in the cisplatin resistance in ESCC.
In the present study, we treated the Eca109 and TE-1 parent ESCC cell lines (Par) with gradually increasing concentrations of cisplatin to establish the resistant cells (Res) and found fibronectin (FN)-induced cell adhesion and migration abilities were significantly enhanced in Res cells. Furthermore, we found integrin α5 was remarkably increased in Res cells. Mechanistically, α5 modulates cisplatin-induced DNA damage and apoptosis via the upregulation of BARD1 which was induced by the FAK/PI3K/AKT signaling pathway. Moreover, blocking α5 expression restored the sensitivity to cisplatin by decreasing the efficiency of DNA damage repair and promoting apoptosis. The overall results indicated the importance of α5 in chemoresistance, which may provide new insights into the molecular mechanisms for reversing the resistance of cisplatin-based therapy in ESCC.
Material and methods
Antibodies and reagents
The experiments were performed using the following antibodies: Mouse monoclonal antibodies (mAbs) against α5 integrin, β1 integrin, FAK, and p-FAK were from BD Biosciences, β1 subunit (P5D2) was from Abcam, BARD1 obtained from Santa Cruze Biotechnology (Santa Cruz, CA, USA), Src was purchased from upstate biotechnology, α5 integrin inhibitory antibody (MAB-1956) and α5β1 integrin was from Millipore. Rabbit mAbs against AKT, p-AKT, p-Src, and γH2AX were from Cell Signaling Technology, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Santa Cruz. The peroxidase-conjugated goat antibodies against mouse and rabbit were obtained from Promega and Cell Signaling Technology, respectively. Alexa Fluor® 488 goat anti-mouse IgG and Alexa Fluor 546-phalloidin were obtained from Invitrogen (Thermo Fisher Scientific, Waltham, MA), and the control mouse IgG antibody was obtained from Millipore. Cisplatin was from Sigma-Aldrich, St Louis, MO, USA. The cell adhesion kit (ECM Array) was from Cell Biolabs. BKM120 was from Selleckchem. iScript complementary DNA (cDNA) Synthesis Kit and iTaq Universal SYBR Green Kit were from Bio-Rad. TRIzol reagent was obtained from Invitrogen.
Cell lines and cell culture
HEK293T and Human ESCC cell lines (Eca109 and TE-1) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Eca109 and TE-1 cells were maintained at 37°C in RPMI 1640 medium with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, under a humidified atmosphere containing 5% CO2. Eca109- and TE-1-Res cells were established from Par cells using continuous treatment with step-wise concentrations of cisplatin. The resistant cell lines were cultured in a complete medium containing 4 μM cisplatin. Res cells were cultured without cisplatin for 3 days before certain experiments. The degree of cisplatin resistance of each cell lines was evaluated before each experiment. The stable cell lines used in this study were established as mentioned below.
Cell viability assay
Eca109 and TE-1 -Par or -Res cells (5 × 103) were seeded into 96-well plates in triplicates overnight and then treated with interest drugs (0-160 μM cisplatin, 0.5 μM BKM120) for 48 h. Then the viability of indicated cells was estimated by the MTS ([3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]) cell proliferation assay using the CellTiter-96-Aqueous One Solution Cell Proliferation Assay reagent (Promega, Fitchburg, WI, USA), according to the manufacturer’s recommendations.
Overexpression and knockdown vectors of Integrin α5 and BARD1
The cDNA of human Integrin α5 (a generous gift from Dr. Jianguo Gu, Tohoku Medical and Pharmaceutical University, Japan) was inserted into a cloning entry vector (pDONR201, Invitrogen) use the GatewayTM cloning system kit. The cDNA of the human BARD1 was amplified by PCR from the reverse-transcribed product of Eca109 cell total RNA to yield the fragment flanked by the NheI and XhoI sites for 5’ and just before the stop codon using a mutagenic PCR primer. This complete cDNA of the BARD1 was cloned into a cloning vector (pGEMT-Easy; Promega, Madison, WI) and then subcloned into the pENTR1A vector. The resultant cDNAs were confirmed by sequence. We used the GatewayTM cloning system kit to acquire all of the expression vectors. Briefly, the LR clonase enzyme (Invitrogen) was used to transfer the cDNAs of integrin α5 and BARD1 from the entry vectors into pLenti-CMV-Hygro DEST (w117-1) (Addgene, Plasmid #17454) or pLenti-CMV-Blast DEST (706-1) (Addgene, Plasmid #17451) vectors.
For expressing short hairpin RNA, we used the pLKO.1-puro lentiviral vector. Inserted oligonucleotide sequences were listed as follows: shRNA1 against ITGA5, 5’-CCGGCCATGATGAGTTTGGCCGATTCTCGAGAATCGGCCAAACTCATCATGGTTTTT-3’; shRNA2 against ITGA5, 5’-CCGGCTCCTATATGTGACCAGAGTTCTCGAGAACTCTGGTCACATATAGGAGTTTTT-3’; and shRNA1 against BARD1, 5’-CCGGTGGTTTAGCCCTCGAAGTAAGCTCGAGCTTACTTCGAGGGCTAAACCATTTTTG-3’; shRNA2 against BARD1, 5’-CCGGTGAAAGTATGAAATCGCTATTCTCGAGAATAGCGATTTCATACTTTCATTTTTG-3’.
Virus production and infection
Virus production and infection were performed as described previously [26,27]. In brief, the lentivirus vectors were cotransfected with pCAG-HIVgp and pCMV-VSV-G-RSV-Rev into 293T cells. After transfection for 48 h, the lentivirus supernatants were collected. The indicated cells were infected with the resultant viral supernatant for 72 h, cells were selected by puromycin (pLKO.1-puro), hygromycin (pLenti-CMV-Hygro), or blasticidin (pLenti-CMV-Blast) to get resistant cells against these antibiotics. Stable cell lines were used in subsequent studies.
Western blot (WB)
Cells were seeded in 60-mm dishes overnight, then washed with ice-cold PBS and lysed in RIPA lysis buffer (Beyotime, China) with protease and phosphatase inhibitors (Beyotime, China) for 30 min. After centrifugation, the supernatants were collected and protein concentrations were determined using a BCA protein assay kit (Beyotime, China). The protein lysates were subjected to 10% SDS-PAGE. After electrophoresis, the proteins were transferred to a PVDF membrane (Millipore). The membrane was incubated with the indicated primary and secondary antibodies, and the proteins were visualized by an enhanced ECL kit (Beyotime, China). The intensity of each band was quantified with ImageJ software.
Flow cytometric analysis
For detecting integrin surface expressions, cells were grown to about 90% confluency, detached using trypsin containing 1 mM EDTA, washed with PBS and stained with indicated primary antibodies for 1 h on ice, followed by incubation with Alexa Fluor 647 goat anti-mouse IgG (Invitrogen) for 30 min on ice. Finally, the cells were washed three times with PBS and analyzed by flow cytometry (BD Biosciences).
For the apoptosis assay, indicated cells were treated with different concentrations of cisplatin. After 48 h treatment, the cells were harvested and stained with AnnexinV-FITC/PI (Multisciences, China) and then analyzed by flow cytometry.
Immunofluorescence staining
Cells were cultured on micro coverslips for 48 h and fixed with ice-cold methanol and permeabilized with 0.2% Triton-X-100. Antibodies against pFAK were used, followed by incubation with anti-mouse Alexa Fluor 488 secondary antibody (Invitrogen) and Alexa Fluor 546 phalloidin (Invitrogen). The confocal images were observed by confocal microscopy.
Cell migration assay
For the wound-healing assay, a confluent monolayer of indicated cells seeded in a 6-well plate and a “scratch” with a p200 pipet tip was made through the cell layer. After washing with PBS, RPMI 1640 medium containing 10% FBS was added in each well. Wounded areas were photographed under a light microscope (10 × objective) at 0 h and 48 h. All experiments were repeated three times.
For transwell migration assay, cells were starved in serum-free medium overnight, trypsinized, and suspended in RPMI 1640 medium containing 10% FBS. The suspended cells were centrifuged, and the supernatants were removed. Cells were resuspended with serum-free medium and diluted to 1.4 × 105 cells/ml. Each transwell (Corning transwell cell culture inserts, 8.0-mm inserts; Corning), 500 µl aliquots of the cell suspension were added; then the cells were incubated at 37°C for the appropriate time. After incubation, cells on the upper side were removed by scraping with a cotton swab. The membranes of each transwell were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet for 2 h. Cells that had migrated to the lower side were counted using a phase-contrast microscope.
Cell adhesion and spreading assays
The difference between Par and Res cells on cell adhesion was evaluated using the CytoSelect cell adhesion assay (Cell Biolabs, San Diego, USA). Indicated cell suspensions were allowed to attach to an ECM-coated 48-well plate for 40 min at 37°C. Adherent cells were stained and extracted according to the manufacturer’s protocols. Extracted samples were measured at 560 nm wavelength in a plate reader. Triplicate wells were used for each group, and the average of results was calculated.
The cell spreading assay was performed as described previously [28,29]. Briefly, 6-well plates were coated with FN (10 µg/ml) in PBS at 4°C overnight and then blocked with 1% bovine serum albumin (BSA) in RPMI 1640 medium for 1 h at 37°C. The indicated cells were detached and suspended in serum-free RPMI 1640 medium with 0.1% BSA at 6 × 104 cells/ml. After 40 min incubation on FN-coated plates, non-adherent cells were gently removed by PBS, and the attached cells were fixed with 4% paraformaldehyde (PFA) in PBS, and images were then taken by phase-contrast microscopy.
RNA isolation and qPCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Then reverse transcribed with an iScript complementary DNA (cDNA) Synthesis Kit. The resulting cDNA was used for real-time PCR with the iTaq Universal SYBR Green Kit. Primers are listed in Table 1. GAPDH was used as a control. Real-time PCR and data collection were performed on the ABI PRISM 7700 Sequence Detector (Applied Biosystems).
Table 1.
Primer sequences for qRT-PCR
| Target | Forward primer (5’-3’) | Reverse primer (5’-3’) |
|---|---|---|
| ITGA4 | GCTTCTCAGATCTGCTCGTG | GTCACTTCCAACGAGGTTTG |
| ITGA5 | TGCAGTGTGAGGCTGTGTACA | GTGGCCACCTGACGCTCT |
| ITGA8 | ACATTCTGGTGGACTGTGG | AATCCCTTGTTGTTGCGTTC |
| ITGAV | AATCTTCCAATTGAGGATATCAC | AAAACAGCCAGTAGCAACAAT |
| ITGB1 | GAAGGGTTGCCCTCCAGA | GCTTGAGCTTCTCTGCTGTT |
| ITGB3 | CCGTGACGAGATTGAGTCA | AGGATGGACTTTCCACTAGAA |
| ITGB6 | TCAGCGTGACTGTGAATATCC | GTGACATTTGGAGCTGTTCAC |
| ITGB7 | TGCAGCTCATCATGGATGCTTA | CCGTCTTCTCAGGACCCTTACA |
| 53BP1 | GCCTGATCAATGGACCCTACTGGAAGTCAGG | CCGCTCGAGTTAGTGAGAAACATAATCGTGTTT |
| MDC1 | TGCTCTTCACAGGAGTGGTG | GGGCACACAGGAACTTGACT |
| BRCA1 | CTGAAGACTGCTCAGGGCTATC | AGGGTAGCTGTTAGAAGGCTGG |
| BARD1 | AGCGTAGGGATGGACCTCTT | CCATTGAGAATCCCAAGCAT |
| CHK1 | ATATGAAGCGTGCCGTAGACT | TGCCTATGTCTGGCTCTATTCTG |
| CHK2 | AAGAAGTTGTTGGTAGTG | TTCCTCAGACAGAAGATC |
| LIG4 | TGCTGCTGAGTTGCATAATGT | AGCAGCTAGCATTGGTTTTGA |
| RAD50 | TCCACGATAGGTACTTCGCC | TGAGGACAACAGAACTTGTGAAC |
| RAD51 | GGTCTGGTGGTCTGTGTTGA | GGTGAAGGAAAGGCCATGTA |
| RAD52 | CTGGCACTGTCCAAAGCATA | TAGATCGAGCTCCCTGTGTG |
| NHEJ1 | TGCAGATTCATGACAAAGGG | ACTACCAGGAGAGTGGGGCT |
| XRCC3 | CGTCTTCCGTGCAGATGTAG | CATCACTGAGCTGGCCG |
| XRCC4 | TTTCAGCTGAGATGTGCTCC | AGGAGACAGCGAATGCAAAG |
| XRCC5 | GAAGGCTCGGATGCAGTCTA | CCTGCTGAAAACTTCCGTGT |
| XRCC6 | TGGTTCATTTGTTTCCCGAT | AGACCAGGAAGCGAGCACT |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
Xenograft assay
The Eca109-Con or Eca109-α5-KD2 cells (1 × 107, suspended in 0.1 ml PBS) were injected into the subcutaneous tissue of 5 weeks old BALB/c-nude mice. Tumor growth was monitored every 5 days. Once the tumor size reached ~ 100 mm3, the Con and α5-KD2 group mice were randomly divided into two groups (5 mice/group): PBS group and cisplatin group (ip, 4 mg/kg, twice a week). The tumor tissues were harvested after 30 days and their volumes and weights were measured. All experiments were conducted according to the protocols approved by the Institutional Animal Care and Use Committee of Yangzhou University.
Statistical analysis
Statistical analyses were performed via a Student’s t test using GraphPad Prism5. The results are presented as the means ± standard derivation. Statistical significance was defined as P < 0.05 (not signifcant (n.s.); *P < 0.05; **P < 0.01; ***P < 0.001).
Results
Cell migration ability is enhanced in cisplatin resistant ESCC cells
To explore the mechanism of chemoresistance to cisplatin in ESCC cells, cisplatin resistant (Res) cell lines were established from parental (Par) Eca109 and TE-1 cells via a continuous treatment with gradually increasing concentrations of cisplatin (Cis). Cell viability assay was performed to examine the sensitivity of Par and Res cells to cisplatin via MTS reagents. As shown in Figure 1A (upper panel), Res cells exhibited significant higher MTS activity compared with that in Par cells after treatment with the indicated concentration of cisplatin for 48 h. The curves also indicated that the IC50 value of Par and Res cells were 5.676 μM and 31.46 μM in Eca109 cells, 4.329 μM and 28.58 μM in TE-1 cells, respectively, which means the Res cells showed about 6-folds increase in resistance to cisplatin compared with Par cells. Consistently, exposure to cisplatin for 48 h can induce the expression level of γH2AX, a DNA damage marker [30], in both Par and Res cells, however, the response of Res cells was remarkably attenuated, indicating less cytotoxic effects were induced in Res cells (Figure 1A, lower panel). Then the cell behaviors, such as proliferation and migration of both cells were compared. As shown in Figure 1B, there was no significant difference between Par and Res cells in cell growth. Interestingly, the Res cells exhibited an increased cell migration ability when compared to Par cells, as showed by wound healing assay (Figure 1C) and boyden chamber analysis (Figure 1D).
Figure 1.

Comparison of cell proliferation and migration ability in Par and Res ESCC cells. A. The viability curve of Eca109- and TE-1-Par, Res cells under different concentrations of cisplatin treatment (0, 2.5, 5, 10, 20, 40, 80, 160 μM for Eca109 cells and 0, 1.875, 3.75, 7.5, 15, 30, 60, 120 μM for TE-1 cells) for 48 h (upper panel). Data were represented from three independent experiments. Cell lysates from indicated cells treated with or without cisplatin (Cis) were immunoblotted by anti-γH2AX and anti-H2AX antibodies (lower panel). B. The growth of indicated cells was measured by the MTS proliferation assay. Relative MTS activities were normalized to those at 0 h (n=3 individual experiments). C. The indicated cells were cultured until confluence. A scratch was made with a p200 pipet in each well, and photographs were taken at 0 h and 48 h (left panel; Scale bar, 250 µm). Quantitative data were from three independent experiments (right panel). D. The migration ability was analyzed by transwell assay. The representative images were recorded by phase-contrast microscopy (left panel; Scale bar, 350 µm). The quantitative number of migrated cells was obtained from three independent experiments (right panel). The p values were determined by a two-tail unpaired t-test (n.s., P > 0.05; **, P < 0.01).
Cisplatin resistant cells exhibit increased FN-induced cell-matrix adhesion
Since cell-matrix adhesion plays essential roles in tumor cell migration and invasive potentials [31], we detected the ECM binding profiles of Par and Res cells. As shown in Figure 2A, Res cells attached strongly to fibronectin (FN) compared with other ECM proteins, indicating that the increased migration ability of Res cells may be related to the inducement of the adhesiveness to FN. This phenomenon was further confirmed via cell spreading assay on FN-coated condition, the Res cells exhibit enhanced spreading ability compared with Par cells (Figure 2B). It is well known that FAK is involved in focal adhesion formation via tyrosine phosphorylation during the cell adhesion process, which can facilitate intracellular signaling events [32]. To investigate whether the FN-mediated FAK signaling was aberrantly activated in Res cells, the phosphorylation level of FAK was detected using cell lysates collected after adhesion to FN at indicated times. As shown in Figure 2C, the response of the FN-induced activation of FAK was attenuated in Par cells, compared with Res cells. Consistently, immunofluorescence staining showed that a significant increase in both the size and intensity of p-FAK in Res cells as compared to that in Par cells (Figure 2D, upper panel). Additionally, the formation of actin stress fibers was also more abundant in Res cells, as detected by phalloidin staining (Figure 2D, lower panel). Taken together, these observations indicate that cell-FN adhesion for migration is upregulated in cisplatin resistant cells.
Figure 2.

Detecting the FN-induced cell adhesion, FAK signaling and actin filament formation in Par and Res cells. A. Adhesion ability of Eca109-Par and Res cells upon various ECM proteins. Cell suspensions were planted on the ECM-coated plate for 40 min at 37°C. Attached cells were stained and checked by colorimetric detection. The quantitative data were presented as the means ± standard derivation from three independent experiments (***, P < 0.001 by two-tail unpaired t-test). B. Eca109-Par and Res cells were detached, suspended in serum-free RPMI 1640 for 1 h, and then plated on the FN-precoated (10 µg/ml) dishes. After incubation for 40 min, spread cells were fixed with PFA, and the representative photos were taken (left panel; Scale bar, 50 µm). The percentages of adherent cells were statistically analyzed as the means ± standard derivation of three independent experiments (***, P < 0.001 by two-tail unpaired t-test). C. Eca109-Par and Res cells were plated on the FN-precoated (10 µg/ml) dishes for indicated times. Cell lysates were collected and then immunoblotted by anti-pFAK and anti-FAK antibodies. D. Immunofluorescence staining of p-FAK (top) and phalloidin (bottom) in Eca109-Par and Res cells. Cells were cultured on FN-coated coverslips, then cells were fixed, permeabilized, and visualized with p-FAK and phalloidin-Alexa Fluor 549 (actin), respectively. The images were taken by confocal microscopy. Scale bar, 20 µm.
Upregulated integrin α5 affects the sensitivity of cisplatin in Res cells
FN is one of the extracellular matrix glycoproteins which binds to specific integrins and plays an important role in cell adhesion [33]. To identify which integrin subunit was responsible for the enhanced adhesion and migration of Res cells, we analyzed the expression of FN-associated integrins via qPCR analysis and found that integrin α5 was significantly upregulated more than 10-folds in Eca109-Res cells compared to Par cells (Figure 3A). Western blot and FACS analysis were performed to further confirm the upregulated integrin α5 in the Eca109-Res cells (Figure 3B). Importantly, a similar result was observed in TE-1-Par and Res cells (Figure 3B). Thus, these data indicated that integrin α5 might be a key regulator involved in cisplatin resistance of ESCC cells. To test this hypothesis, we established integrin α5-knockdown (KD) Res cells and then restored α5 expression in the KD cells (Rescue) (Figure 3C). As expected, α5-KD cells showed a significant decrease in cell migration as compared with the Con cells, and restoration of α5 largely rescued this phenotype in both Eca109 and TE-1 cells (Figure 3D). To further study whether the cisplatin resistance is induced by the increased expression of α5, the sensitivity to cisplatin was measured. As shown in Figure 3E, silencing of α5 remarkably resensitize Res cells to cisplatin (Eca109, IC50=4.214/3.336 μM for α5-KD1/KD2; TE-1, IC50=4.37/2.696 μM for α5-KD1/KD2) compared with Con group (Eca109, IC50=33.71 μM; TE-1, IC50=27.47 μM), and this effect can be largely abolished by restoration of α5 (Eca109, IC50=21.97 μM; TE-1, IC50=16.33 μM). Furthermore, FACS analysis also demonstrated that cisplatin induced more apoptosis in Res cells upon α5 knockdown (Figure 3F). Importantly, the restoration of α5 in the KD cells largely abolished the sensitivity. Together, these results suggest that α5 mediates the cisplatin resistance in ESCC cells.
Figure 3.
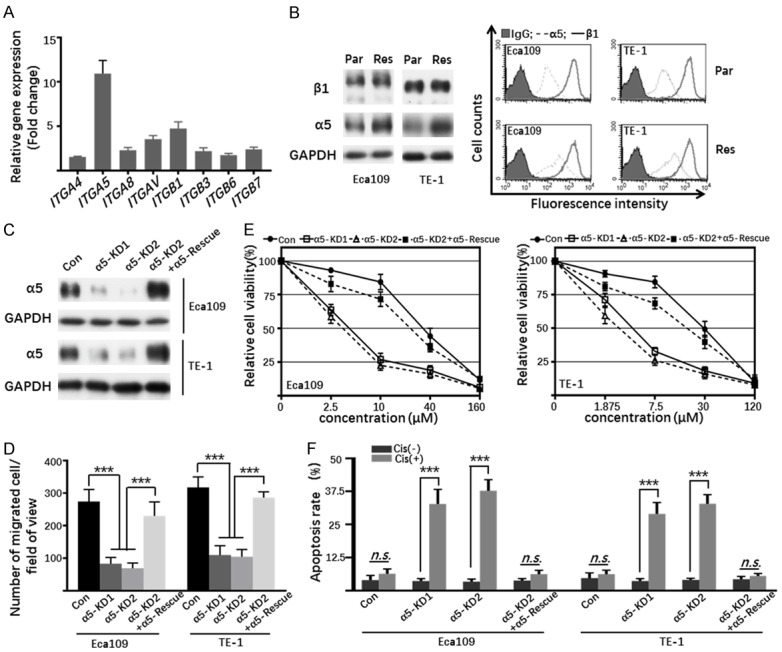
The effect of upregulated α5 on the sensitivity of chemotherapy in Res cells. A. The mRNA levels of ITGA4, ITGA5, ITGA8, ITGAV, ITGB1, ITGB3, ITGB6 and ITGB7 in Eca109-Par and Res cells were determined by qPCR. The columns represented the fold increase of mRNA in Res cells as compared to Par cells. The quantitative data were obtained from three independent experiments. B. Cell lysates from indicated cells were immunoblotted by anti-β1 and anti-α5 antibodies, GAPDH was used as a loading control (left panel). Indicated cells were collected and incubated with anti-β1 (bold line) and anti-α5 (dotted line) or with anti-IgG antibody (grey shadow), followed by incubation with Alexa Fluor 647 goat anti-mouse IgG subjected to FACS analysis (right panel). C. Cell lysates from Con, α5-KD1, α5-KD2, α5-KD2+α5-Rescue Eca109 and TE-1 Res cells were immunoblotted with anti-α5 antibody, GAPDH was used as a loading control. D. The migration of indicated cells treated with or without cisplatin was determined by transwell assay. Representative photos were taken and then the migrated cells were counted. The quantitative data were obtained from three independent experiments (***, P < 0.001, by two-tail unpaired t-test). E. The viability curve of Con, α5-KD1, α5-KD2, α5-KD2+α5-Rescue Eca109 (left panel), and TE-1 (right panel) Res cells under different concentrations of cisplatin treatment. Data were represented as the means ± standard derivation (n=3). F. The apoptosis of Con, α5-KD1, α5-KD2, α5-KD2+α5-Rescue Eca109 and TE-1 Res cells treated with or without cisplatin for 48 h were detected by FACS analysis through AnnexinV-FITC/PI staining. The percentages of apoptotic cells were statistically analyzed as the means ± standard derivation of three independent experiments (n.s., P > 0.05; ***, P < 0.001, by two-tail unpaired t-test).
Integrin α5 regulates cisplatin-induced DNA damage through BARD1
Considering that the induction of DNA damage was one of the major prominent anticancer effects upon cisplatin treatment [9], we hypothesized the effect of α5 on cisplatin resistance was mediated by DNA damage repair. We found that silencing of α5 endowed Res cells more sensitive to cisplatin-induced DNA damage, and restored α5 resensitized the cells to cisplatin, as reflected by the expression of γH2AX (Figure 4A). This means increased α5 contributes to DNA damage repair and further results in cisplatin resistant in ESCC cells. HR and NHEJ were two major pathways responsible for DNA repair, we then investigated the genes involved in these pathways. Subsequent qPCR results showed the mRNA levels of several repair-related genes, especially BARD1, were much higher in Res cells than that in Par cells (Figure 4B). Additionally, among these genes, only BARD1 and BRCA1 were significantly downregulated in α5-KD cells in presence of cisplatin, when compared to Res cells (Figure 4B), indicating that α5 induces BARD1/BRCA1 expression. Therefore, we hypothesized BARD1 was the major downstream effector of α5 to regulate the cisplatin response. To test it, the effect of BARD1 on cisplatin resistance was assessed by cell viability assay, as shown in Figure 4C, overexpression of BARD1 in α5-KD2 cells enable the Res cells to recapitulate the ability of resistance to cisplatin. Furthermore, the silencing of BARD1 reversed α5-induced resistance in Res cells, and this effect can be largely abolished by restoration of BARD1 (Figure 4D). Collectively, these results indicate that α5 mediated-cisplatin resistance is at least partly through BARD1.
Figure 4.

α5-mediated cisplatin resistant was through BARD1. A. Cell lysates of Con, α5-KD1, α5-KD2 and α5-KD2+α5-rescue Eca109-Res cells treated with or without cisplatin for 48 h, then were immunoblotted by anti-γH2AX and anti-H2AX antibodies. B. The mRNA levels of 53BP1, MDC1, BRCA1, BARD1, CHK1, CHK2, LIG4, RAD50, RAD51, RAD52, NHEJ1, XRCC3, XRCC4, XRCC5 and XRCC6 in Eca109-Par, Res, and Res-α5-KD2 cells treated with or without cisplatin were determined by qPCR. Graphic representation of the fold increases of mRNA in Res cells as compared to Par cells (gray column), and the effect of cisplatin in α5-KD2 Res cells compared to Res cells (dark column), respectively. The quantitative data were statistically analyzed as the means ± standard derivation (n=3, ***, P < 0.001, by two-tail unpaired t-test). C. Immunoblotting of α5 and BARD1 in Con, α5-KD2, and α5-KD2+BARD1-OE Eca109-Res cells, GAPDH used as a loading control (left panel). The cell viability under different concentrations of cisplatin treatment (0, 5, 20, 80 μM) for 48 h. Data were represented as the means ± standard derivation of three independent experiments (right panel). D. Immunoblotting of α5, BARD1 in Con, α5-OE, α5-OE+BARD1-KD1, α5-OE+BARD1-KD2, and α5-OE+BARD1-KD1+BARD1-Rescue Eca109-Res cells, GAPDH was used as a loading control (left panel). The cell viability under different concentrations of cisplatin treatment (0, 10, 40, 160 μM) for 48 h. Data were represented as the means ± standard derivation of three independent experiments (right panel).
The fundamental roles of α5 in PI3K/AKT/BARD1-induced DNA damage repair and α5 inhibition restores the sensitivity to cisplatin in vitro and in vivo
To elucidate the potential mechanisms involved in the upregulation of BARD1 induced by α5 in Res cells, we compared the downstream signaling of integrin α5 in Par and Res cells. As shown in Figure 5A, the phosphorylated levels of FAK, AKT, and Src were remarkably increased in Res cells, compared with Par cells, suggesting that the PI3K pathway was activated in Res cells. Given the evidence that PI3K/AKT participates in chemoresistance via regulating the cell cycle, inhibiting apoptosis and upregulating DNA damage repair associated genes, including BRCA1, BRCA2 and BARD1 [13,34], we hypothesized that knockdown α5 reversed cisplatin resistance through the down-regulation of BARD1 might be due to the suppression of PI3K pathway. To test this, anti-α5 blocking antibody and BKM120 (a pan PI3K inhibitor) were used to check whether they can overcome the cisplatin resistance in ESCC cells. As shown in Figure 5B, both blocking α5 and inhibition of PI3K led to decreased BARD1 expression and increased γH2AX expression, which indicated the deficiency of DNA damage repair in Res cells. Moreover, BKM120 can significantly inhibit not only the expression of pAKT, but also the levels of BARD1 (Figure 5B), confirming that the PI3K pathway is responsible for the upregulated BARD1 in Res cells. Furthermore, although both α5 and PI3K inhibition led the Res cells susceptible to cisplatin as reflected by cell viabilities and apoptosis, the effects of α5 inhibition group were more obvious (Figure 5C and 5D), suggesting that α5 plays fundamental roles in DNA damage repair and re-sensitizing the Res cells to cisplatin. Moreover, we evaluate the impact of α5 on cisplatin resistance in vivo. As the results shown in Figure 5E-G, cisplatin treatment has no obvious effect on Res cells, however, knockdown of α5 rendered tumor xenografts significantly more sensitive to cisplatin, indicating that inhibition of α5 can also resensitize Eca109-Res cells to cisplatin in vivo. Taken together, our results highlight the vital roles of α5/PI3K/AKT/BARD1 in cisplatin resistance in ESCC cells.
Figure 5.
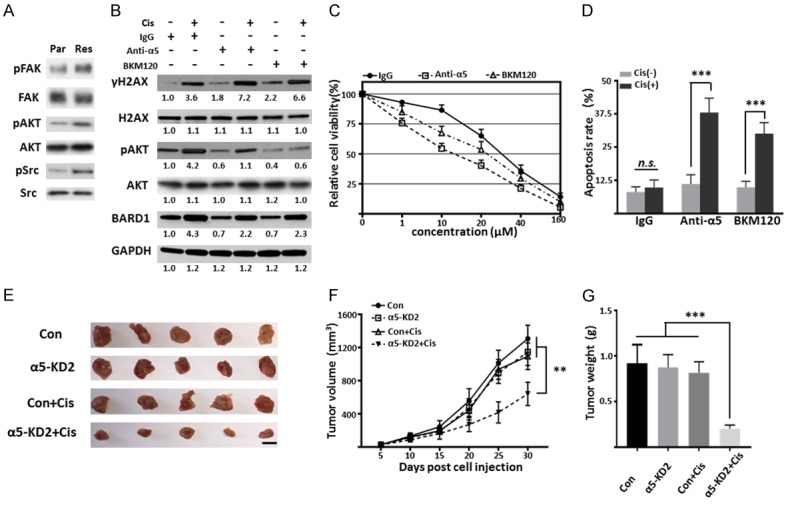
Inhibition of α5 restores the sensitivity to cisplatin in vitro and in vivo. (A) Cell lysates of Eca109-Par and Res cells were immunoblotted by anti-pFAK, anti-FAK, anti-pAKT, anti-AKT, anti-pSrc, and anti-Src antibodies. (B) Cell lysates of Eca109-Res cells treated with or without cisplatin, IgG (10 μg/ml), α5 blocking antibody (10 μg/ml), or BKM120 (0.5 μM) were immunoblotted by anti-γH2AX, anti-H2AX, anti-pAKT, anti-AKT, and anti-BARD1 antibodies, GAPDH was used as a loading control. (C) The sensitivity of Res cells treated with IgG, α5 blocking antibody, or BKM120 to different concentrations of cisplatin for 48 h. Data were represented as the means ± standard derivation of three independent experiments. (D) The percentage of apoptotic cells of Eca109-Res cells treated with IgG, α5 blocking antibody, BKM120 and then treated with or without cisplatin for 48 h. The percentages of apoptotic cells were statistically analyzed as the means ± standard derivation of three independent experiments (n.s, P > 0.05; ***, P < 0.001, by two-tail unpaired t-test). (E-G) Represent tumor images (E), tumor growth curves (F), and tumor weight (G, at day 30) of BALB/c-nude mice with subcutaneous injection of Con and α5-KD2 Eca109-Res cells with or without cisplatin treatment. Tumor volume was measured every 5 days after injection and the cisplatin (4 mg/kg) were intraperitoneally injected twice a week. Values are presented as the means ± standard derivation (n=5, **, P < 0.05, ***, P < 0.001 by two-tail unpaired t-test). Scale bar, 1 cm.
Discussion
A growing body of evidence implicated the interaction between members of the integrin family and ECM ligands induced intracellular signaling pathway was critical for cancer cell survival and chemoresistance [17,31,35]. In the present study, we found that integrin α5 and BARD1 were highly expressed in cisplatin resistant ESCC cell lines, which resulted in the activation of DNA damage repair and anti-apoptosis effects via FAK/PI3K/AKT signaling pathway. Blocking α5 attenuated signal transduction, reduced BARD1 expression and restored the sensitivity to cisplatin in vitro and in vivo.
Integrin α5β1, as a major FN receptor, was involved in the modulation of tumor progression, metastasis, and chemoresistance through the downstream signaling pathways and the cross-talk with receptor tyrosine kinases (RTKs) [17,18]. Our data showed that the expression of α5 was significantly enhanced in Res cells compared with other integrin subunits, as expected, the Res cells exhibited a significant increase in FN-induced cell adhesive and migratory properties. Although it was difficult to exclude the possible involvement of other α subunits (e.g. αV) in drug resistance, the sensitivity to cisplatin was significantly affected by knockdown or rescue α5 gene which provided evidence to some extent that α5 was of pivotal importance in this process. In line with our observations, Xie, et al. reported that upregulated α5 promoted tumor progression and was an independent prognostic factor in ESCC [24], and Martinkova, et al. demonstrated that α5β1 antagonists regulate chemotherapy-induced p53 signaling and decrease aggressiveness in glioblastoma cells [36]. These results suggested that upregulated α5 might be associated with malignant phenotype and cisplatin resistance in ESCC cells. Furthermore, our results indicated that the resistance of ESCC cells to cisplatin was mediated by α5-induced the signal axis consisting of FAK, Src and PI3K/AKT. Consistent with our observation, several studies have reported that integrins were involved in anti-apoptosis and chemotherapy via various mechanisms. For example, Aoudjit, et al. described the inhibitory effect of β1 in paclitaxel- and vincristine-induced apoptosis in breast cancer cells was mediated by activation of PI3K signaling, which prevented the downregulation of Bcl-2 [18]; Hodkinson et al. reported that α2β1, α3β1, α6β1, and αvβ1 mediated activation of PI3K signaling could override drug-induced apoptosis by preventing cell cycle arrest through downregulation of cyclins and promotion the expression of p21 and p27 [37]; Cordes et al. reported attachment to FN and laminin protected lung cancer and breast cancer cells from cytotoxic drug ukrain- and radiation-induced apoptosis [38]. Given the fact that the chemosensitivity mediated by integrins was majorly depended on PI3K signaling evens, it was necessary to identify whether these effects were targetable. Indeed, we found the resistance to cisplatin in ESCC cells can be more effectively overcome by inhibition of α5 as compared to the PI3K inhibitor, which means disruption of α5-mediated cell adhesion and signaling appears to be a key event in the restoration of cisplatin sensitivity. It was reasonable that integrin-mediated signaling events were critical for cell adhesion, invasion and survival in different death factors-induced incidences [39,40], however, detail mechanisms are required for better understanding of α5 in modulating cisplatin sensitivity.
Considering cisplatin was a DNA-damaging drug and also our data showed that knockdown α5 can promote cisplatin-induced apoptosis and inhibit the PI3K/AKT signaling, we speculated that the development of cisplatin resistance was caused by enhanced DNA damage repair and anti-apoptosis abilities. In agreement with this hypothesis, previous reports showed integrins provided survival advantage against death receptors, such as Fas- and TRAIL-mediate cell apoptosis, which was important for cancer immune escape and chemotherapy [41,42]. Our data also showed the attachment of α5 to FN induced FAK/PI3K/AKT signaling pathway which contributed to the upregulation of BARD1 and apoptosis induced by cisplatin. It is worth noting that BARD1 was significantly increased in Res cells as compared with other DNA damage repair associated genes including BRCA1. Therefore, we speculated BARD1 might be the major effector involved in the cisplatin resistance in ESCC cells. It was conceivable since BARD1 acts as a tumor suppressor in a BRCA1-dependent or independent manner. Briefly, in addition to forming a heterodimer with BRCA1 to participate in DNA damage repair [43], BARD1 can also control apoptosis and stabilize p53 by directly binding with it or other oncogenic pathway proteins including BCL3, and poly (ADP-ribose) (PAR) [44-46]. Another explanation might be the expression patterns during the cell cycle of the two proteins were different. BARD1 gradually increased in cell cycle and mostly expressed in mitosis, while the maximal levels of BRCA1 occurred during S-phase [47]. Taken together, the possibility to explain the fundamental roles of BARD1 in ESCC chemoresistance was that BARD1 exerts its DNA damage repair and pro-apoptosis functions by forming a complex with BRCA1 in S phase and in a BRCA1-independent manner in mitosis. Further evidence is needed for confirming the hypothesis.
PI3K/AKT/mTOR signaling cascade plays a central role in governing several key cellular processes, such as cell growth, survival, and apoptosis [15]. We showed that the expression level of BARD1/BRCA1 was increased and PI3K/AKT was activated in Res cells, which implicated in the cisplatin resistance in ESCC cells. Consistently, previous reports showed that activated PI3K pathway is responsible for the enhanced BARD1/BRCA1/2 via mitogen-activated protein kinase/extracellular regulated kinase (MAPK/ERK), inhibiting PI3K significantly decreased the complex expression, impaired the HR repair and thus resensitized the tumor to chemotherapy [13,14]. These results above confirmed the implication of integrin/PI3K/AKT signaling in both cell survival and chemoresistance was a general complicity phenomenon in ESCC cells.
In summary, the current study delineated the mechanisms of upregulated α5 implicated in cisplatin resistant in ESCC cells. α5 regulated chemoresistance by promoting the hyperactivation of PI3K/AKT signaling for cell survival and upregulated BARD1 to enhance DNA damage repair. Moreover, blocking α5 can re-sensitive the Res cells to cisplatin. Our results suggested that α5 might be a potential novel biomarker for predicting cisplatin response and anti-α5 therapy was worth considering to overcome cisplatin resistance in ESCC.
Acknowledgements
We gratefully acknowledge support from the National Natural Science Foundation of China (No. 31800675), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 18KJB320028), the Postdoctoral Science Foundation in Jiangsu Province (No. 2018K263C), the Key Project for Social Development in Jiangsu Province (BE2019698), Strengthening Health Care via Science and Education Project and Clinical Medical Innovation Platform Foundation of Yangzhou (2018, YXZX20184, Gastroenterology) and Major public health projects in Yangzhou: Screening projects of early gastrointestinal diseases (2018).
Disclosure of conflict of interest
None.
References
- 1.McGuire S. World cancer report 2014. geneva, switzerland: world health organization, international agency for research on cancer, who press, 2015. Adv Nutr. 2016;7:418–9. doi: 10.3945/an.116.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–2509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, Zheng R, Zhang S, Zhao P, Zeng H, Zou X. Report of cancer incidence and mortality in China, 2010. Ann Transl Med. 2014;2:61. doi: 10.3978/j.issn.2305-5839.2014.04.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Wei Z, Zhang J, Hu W, Ma Z, Liu Q. Which factors are associated with extremely short-term survival after surgery in patients with esophageal squamous cell carcinoma? Asia Pac J Clin Oncol. 2016;12:308–313. doi: 10.1111/ajco.12503. [DOI] [PubMed] [Google Scholar]
- 5.Kim T, Grobmyer SR, Smith R, Ben-David K, Ang D, Vogel SB, Hochwald SN. Esophageal cancer-the five year survivors. J Surg Oncol. 2011;103:179–183. doi: 10.1002/jso.21784. [DOI] [PubMed] [Google Scholar]
- 6.Goense L, van Rossum PS, Kandioler D, Ruurda JP, Goh KL, Luyer MD, Krasna MJ, van Hillegersberg R. Stage-directed individualized therapy in esophageal cancer. Ann N Y Acad Sci. 2016;1381:50–65. doi: 10.1111/nyas.13113. [DOI] [PubMed] [Google Scholar]
- 7.Phatak P, Byrnes KA, Mansour D, Liu L, Cao S, Li R, Rao JN, Turner DJ, Wang JY, Donahue JM. Overexpression of miR-214-3p in esophageal squamous cancer cells enhances sensitivity to cisplatin by targeting survivin directly and indirectly through CUG-BP1. Oncogene. 2016;35:2087–2097. doi: 10.1038/onc.2015.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 2018;73:e478s. doi: 10.6061/clinics/2018/e478s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol. 2001;67:93–130. doi: 10.1016/s0079-6603(01)67026-0. [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 11.Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J. Clin. Oncol. 2008;26:20–25. doi: 10.1200/JCO.2007.11.6905. [DOI] [PubMed] [Google Scholar]
- 12.Dai CH, Li J, Chen P, Jiang HG, Wu M, Chen YC. RNA interferences targeting the Fanconi anemia/BRCA pathway upstream genes reverse cisplatin resistance in drug-resistant lung cancer cells. J Biomed Sci. 2015;22:77. doi: 10.1186/s12929-015-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Zhu Y, Liu Y, Zhang C, Chu J, Wu Y, Li Y, Liu J, Li Q, Li S, Shi Q, Jin L, Zhao J, Yin D, Efroni S, Su F, Yao H, Song E, Liu Q. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat Commun. 2018;9:1595. doi: 10.1038/s41467-018-03951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, Diez O, Rubio IT, Perez J, Rodon J, Cortes J, Ellisen LW, Scaltriti M, Baselga J. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–210. doi: 10.2147/OTT.S89967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 17.Aoudjit F, Vuori K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract. 2012;2012:283181. doi: 10.1155/2012/283181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aoudjit F, Vuori K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene. 2001;20:4995–5004. doi: 10.1038/sj.onc.1204554. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Zou L, Ma G, Wu X, Huang F, Feng T, Li S, Lin Q, He X, Liu Z, Cao X. Integrin beta1 is a critical effector in promoting metastasis and chemo-resistance of esophageal squamous cell carcinoma. Am J Cancer Res. 2017;7:531–542. [PMC free article] [PubMed] [Google Scholar]
- 20.Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, Chilvers ER, Dransfield I, Donnelly SC, Strieter R, Haslett C. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5:662–668. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 21.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C, You J, Fu J, Wang X, Zhang Y. Phosphatidylinositol 3-Kinase/Akt mediates integrin signaling to control RNA polymerase I transcriptional activity. Mol Cell Biol. 2016;36:1555–1568. doi: 10.1128/MCB.00004-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon J, Lee TS, Lee HW, Kang MC, Yoon HJ, Kim JH, Park JH. Integrin alpha 6: a novel therapeutic target in esophageal squamous cell carcinoma. Int J Oncol. 2013;43:1523–1530. doi: 10.3892/ijo.2013.2097. [DOI] [PubMed] [Google Scholar]
- 24.Xie JJ, Guo JC, Wu ZY, Xu XE, Wu JY, Chen B, Ran LQ, Liao LD, Li EM, Xu LY. Integrin alpha5 promotes tumor progression and is an independent unfavorable prognostic factor in esophageal squamous cell carcinoma. Hum Pathol. 2016;48:69–75. doi: 10.1016/j.humpath.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 25.Vay C, Hosch SB, Stoecklein NH, Klein CA, Vallbohmer D, Link BC, Yekebas EF, Izbicki JR, Knoefel WT, Scheunemann P. Integrin expression in esophageal squamous cell carcinoma: loss of the physiological integrin expression pattern correlates with disease progression. PLoS One. 2014;9:e109026. doi: 10.1371/journal.pone.0109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hang Q, Isaji T, Hou S, Zhou Y, Fukuda T, Gu J. N-Glycosylation of integrin alpha5 acts as a switch for EGFR-mediated complex formation of integrin alpha5beta1 to alpha6beta4. Sci Rep. 2016;6:33507. doi: 10.1038/srep33507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hang Q, Isaji T, Hou S, Im S, Fukuda T, Gu J. Integrin alpha5 suppresses the phosphorylation of epidermal growth factor receptor and its cellular signaling of cell proliferation via N-Glycosylation. J Biol Chem. 2015;290:29345–29360. doi: 10.1074/jbc.M115.682229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hang Q, Isaji T, Hou S, Wang Y, Fukuda T, Gu J. A key regulator of cell adhesion: identification and characterization of important N-Glycosylation sites on integrin alpha5 for cell migration. Mol Cell Biol. 2017;37 doi: 10.1128/MCB.00558-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou S, Hang Q, Isaji T, Lu J, Fukuda T, Gu J. Importance of membrane-proximal N-glycosylation on integrin beta1 in its activation and complex formation. FASEB J. 2016;30:4120–4131. doi: 10.1096/fj.201600665R. [DOI] [PubMed] [Google Scholar]
- 30.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz MA, Horwitz AR. Integrating adhesion, protrusion and contraction during cell migration. Cell. 2006;125:1223–1225. doi: 10.1016/j.cell.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khalili AA, Ahmad MR. A review of cell adhesion studies for biomedical and biological applications. Int J Mol Sci. 2015;16:18149–18184. doi: 10.3390/ijms160818149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010;107:7491–7496. doi: 10.1073/pnas.0914242107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang XH, Flores LM, Li Q, Zhou P, Xu F, Krop IE, Hemler ME. Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res. 2010;70:2256–2263. doi: 10.1158/0008-5472.CAN-09-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinkova E, Maglott A, Leger DY, Bonnet D, Stiborova M, Takeda K, Martin S, Dontenwill M. alpha5beta1 integrin antagonists reduce chemotherapy-induced premature senescence and facilitate apoptosis in human glioblastoma cells. Int J Cancer. 2010;127:1240–1248. doi: 10.1002/ijc.25187. [DOI] [PubMed] [Google Scholar]
- 37.Hodkinson PS, Elliott T, Wong WS, Rintoul RC, Mackinnon AC, Haslett C, Sethi T. ECM overrides DNA damage-induced cell cycle arrest and apoptosis in small-cell lung cancer cells through beta1 integrin-dependent activation of PI3-kinase. Cell Death Differ. 2006;13:1776–1788. doi: 10.1038/sj.cdd.4401849. [DOI] [PubMed] [Google Scholar]
- 38.Cordes N, Blaese MA, Plasswilm L, Rodemann HP, Van Beuningen D. Fibronectin and laminin increase resistance to ionizing radiation and the cytotoxic drug Ukrain in human tumour and normal cells in vitro. Int J Radiat Biol. 2003;79:709–720. doi: 10.1080/09553000310001610240. [DOI] [PubMed] [Google Scholar]
- 39.Stupack DG, Cheresh DA. Get a ligand, get a life: integrins, signaling and cell survival. J Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 40.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 41.Gendron S, Couture J, Aoudjit F. Integrin alpha2beta1 inhibits Fas-mediated apoptosis in T lymphocytes by protein phosphatase 2A-dependent activation of the MAPK/ERK pathway. J Biol Chem. 2003;278:48633–48643. doi: 10.1074/jbc.M305169200. [DOI] [PubMed] [Google Scholar]
- 42.Lane D, Goncharenko-Khaider N, Rancourt C, Piche A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene. 2010;29:3519–3531. doi: 10.1038/onc.2010.107. [DOI] [PubMed] [Google Scholar]
- 43.Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev. 2002;12:86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 44.Dechend R, Hirano F, Lehmann K, Heissmeyer V, Ansieau S, Wulczyn FG, Scheidereit C, Leutz A. The Bcl-3 oncoprotein acts as a bridging factor between NF-kappaB/Rel and nuclear co-regulators. Oncogene. 1999;18:3316–3323. doi: 10.1038/sj.onc.1202717. [DOI] [PubMed] [Google Scholar]
- 45.Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feki A, Jefford CE, Berardi P, Wu JY, Cartier L, Krause KH, Irminger-Finger I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene. 2005;24:3726–3736. doi: 10.1038/sj.onc.1208491. [DOI] [PubMed] [Google Scholar]
- 47.Choudhury AD, Xu H, Baer R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J Biol Chem. 2004;279:33909–33918. doi: 10.1074/jbc.M403646200. [DOI] [PubMed] [Google Scholar]