

Abstract
Dysregulation of the ubiquitin-proteasome pathway is closely associated with cancer initiation and progression. SPOP is an adapter protein of the CUL3-based E3 ubiquitin ligase complexes. Several whole genome/exome sequencing studies on endometrial cancers (ECs) revealed that the SPOP gene is frequently mutated. However, how SPOP mutations contribute to EC remains poorly understood. In this study, transcription factor ZBTB3 was identified as a proteolytic substrate for the SPOP-CUL3-RBX1 E3 ubiquitin ligase complex. SPOP specifically recognizes two Ser/Thr (S/T)-rich degrons located in ZBTB3 and triggers the degradation of ZBTB3 via the ubiquitin-proteasome pathway. By contrast, EC-associated SPOP mutants are defective in regulating ZBTB3 stability. SPOP inactivation promotes endometrial cell proliferation, migration, and invasion partly through ZBTB3 accumulation. Sonic hedgehog (SHH) was found to be a transcriptional target of ZBTB3. SPOP inactivation leads to ZBTB3-dependent SHH upregulation in EC cells. RUSKI-43, a small molecule inhibitor of SHH, suppresses cell proliferation, migration, and invasion in SPOP-depleted or EC-associated SPOP mutant-overexpressed EC cells. Our data indicate that pharmacological inhibition of SHH represents a possible treatment strategy for SPOP-mutated ECs.
Keywords: SPOP, ZBTB3, endometrial cancer, ubiquitination, sonic hedgehog
Introduction
Endometrial cancer is the most common gynecologic malignancy in developed countries [1]. Most patients present with low-grade, early-stage disease. However, patients with aggressive high-grade tumors that spread beyond the uterus will usually progress within one year [2]. Thus, identifying the genetic alterations that initiate EC and contribute to its progression for effective cancer prevention and treatment is necessary. The mutational landscape of ECs has been reported by several groups including the Cancer Genome Atlas (TCGA) [3-6]. Among these investigations, speckle-type POZ protein (SPOP) was identified as one of the most frequently altered genes by somatic point mutations in ECs. Nonetheless, how SPOP mutations contribute to the pathogenesis and progression of EC remains poorly understood.
The CRL3 E3 ubiquitin ligase complex is composed of the scaffold CUL3, the RING protein RBX1, and a BTB domain-containing protein that acts as an adaptor for substrate binding [7]. SPOP is a structurally well-characterized BTB protein that interacts with substrates via the MATH domain at its N terminus and binds CUL3 through the BTB domain at its C terminus [8]. SPOP has been linked to the ubiquitination-dependent degradation of multiple oncoproteins, such as SRC-3, AR, ERα, EGLN2, HDAC6, Cyclin E, DEK, TRIM24, ERG, PD-L1 and BETs [9-22]. SPOP also promotes the non-degradative ubiquitination of INF2 and MacroH2A to regulate mitochondrial dynamics and epigenetic silencing, respectively [23,24]. All EC-associated SPOP mutations identified to date affect the evolutionarily conserved residues in the MATH domain. This fact suggests that the mutations may alter the interaction between SPOP and its substrates. In addition to EC, SPOP is frequently mutated (4.6%-14.4%) in of patients with prostate cancer across different ethnic and demographic backgrounds [25]. SPOP mutations also occur in other tumor types, albeit at low frequency. The identification of additional SPOP substrates may help elucidate the underlying molecular mechanisms of SPOP-mutated cancers.
Zinc finger (ZF) and BTB/POZ domain-containing (ZBTB) proteins are an emerging family of transcription factors commonly containing a DNA-binding ZF and a transcription-repressing BTB/POZ domain [26]. The BTB domain directly interacts with transcriptional co-repressors, such as NCOR, BCOR, CTBP1 or mSin3A, and thus mediates chromatin remodeling and gene silencing/activation. By contrast, the ZF domain determines the transcriptional specificity of ZBTB proteins through binding to the sequence-specific DNA elements [26]. Human ZBTB3 plays positive roles in cancer cell growth via the regulation of the ROS detoxification pathway genes [27]. Mouse Zbtb3 was highly expressed in mouse embryonic stem cells (ESCs). Zbtb3 potentiates the ESC self-renewal in a Nanog-dependent manner, and Zbtb3 deficiency impairs the early embryonic development [28]. Therefore, ZBTB3 may regulate diverse biological processes by controlling the transcription of its targets.
In this study, we demonstrated that SPOP forms a functional CUL3-SPOP-RBX1 E3 ubiquitin-ligase complex, which targets ZBTB3 for ubiquitination and proteasomal degradation to facilitate the neoplastic transformation of EC cells. This effect is also abrogated by the EC-associated SPOP mutations. Our results showed that oncogenic deregulation of SPOP-ZBTB3-SHH axis in SPOP-mutated ECs.
Materials and methods
Cell culture and transfection
The 293T cells and EC cell lines (ECC-1 and HEC-1-A) were obtained from the American Type Culture Collection (ATCC). The 293T cells were maintained in DMEM with 10% (v/v) fetal bovine serum (FBS). The ECC-1 and HEC-1A cells were maintained in RPMI1640 with 10% (v/v) FBS. All cells were grown at 37°C with 5% CO2. The cells were transiently transfected with plasmids or siRNAs using Lipofectamine 3000 (Thermo, USA) according to the manufacturer’s instructions.
Plasmid constructions
The expression vectors for SPOP-WT or mutants were described previously [11]. The ZBTB3 cDNA was kindly provided by Dr. Jiahui Han (Xiamen University, China) and subcloned into pCMV-FLAG and pCMV-Myc expression vectors. The ZBTB3 mutants were generated by KOD-Plus Mutagenesis Kit (Toyobo, Japan) following the manufacturer’s instructions. For lentiviral knockdown, the shRNA sequence targeting SPOP or ZBTB3 were subcloned intro pLKO.3G GFP-shRNA vectors. The shRNA sequence is shown in Supplementary Table 1. For lentiviral overexpression, the SPOP or ZBTB3 cDNA were subcloned into pCDH vectors. All the constructs were verified by DNA sequencing.
Protein complex purification
The epitope-tagging strategy to isolate SPOP-containing protein complexes from human cells was performed. In brief, to obtain a FLAG-HA-SPOP expressing cell line, 293T cells were transfected with pCIN4-FLAG-HA-SPOP constructs and selected for 2 weeks in 1 mg/ml G418. The tagged SPOP protein levels were detected by WB analyses. The stable cell lines were chosen to expand for protein complex purification. For purification, the cells were lysed in BC100 buffer (20 mM Tris-Cl, pH 7.9, 100 mM NaCl, 0.2 mM EDTA, 20% glycerol) containing 0.2% Triton X-100 and fresh protease inhibitor on ice for 2 hr. The homogenate was centrifuged for 30 min at 12000 rpm at 4°C. Cleared lysates were filtered through 0.45 μM spin filters (Millipore, USA) and immunoprecipitated by anti-FLAG antibody-conjugated M2 agarose (Sigma, USA). The bound polypeptides eluted with the FLAG peptide (Sigma, USA) were further affinity purified by anti-HA antibody-conjugated agarose (Sigma, USA). The final elutes from the HA-beads with HA peptides were resolved by SDS-PAGE on a 4%-20% gradient gel (Bio-Rad) for Coomassie Blue staining. Gel bands were cut out from the gel and subjected to mass-spectrometric sequencing.
Mass-spectrometric sequencing
Nano-LC MS/MS experiment was performed on an HPLC system composed by two LC-20AD nano-flow LC pumps, an SIL-20 AC auto-sampler and an LC-20AB micro-flow LC pump (Shimadzu) connected to an LTQ-Orbitrap mass spectrometer (Thermo Fisher). Sample was loaded onto a CAPTRAP column (0.5 × 2 mm, MICHROM Bioresources) in 5 min at a flow rate of 10 μl/min. The sample was subsequently separated by a C18 reverse-phase column (0.075 × 150 mm, packed with 3 μm Aeris C18 particles, Phenomenex) at a flow rate of 300 nl/min. The mobile phases were 0.1% formic acid as the loading phase and 4% acetonitrile in 0.1% formic acid (phase A) and 96% acetonitrile with 0.1% formic acid (phase B). To achieve proper separation, a 60-min linear gradient from 5 to 40% phase B was employed. The separated sample was introduced into the mass spectrometer via nanoelectrospray. The spray voltage was set at 2.3 kV and the heated capillary at 180°C. The mass spectrometer was operated in data-dependent mode and each cycle of duty consisted one full-MS survey scan at the mass range 300~1600 Da with resolution power of 100,000 using the Orbitrap section, followed by MS/MS experiments for 10 strongest peaks using the LTQ section. The AGC expectation during full-MS and MS/MS were 1000000 and 10000, respectively. Peptides were fragmented in the LTQ section using collision-induced dissociation with helium and the normalized collision energy value set at 35%. Only 2+ and 3+ peaks were selected for MS/MS run and previously fragmented peptides were excluded for 60 s. Protein searches were performed with the Mascot 2.3.02 software (MatrixScience) against the SWISSPROT human protein database (release 2014_07) with the following criteria: 2 possible missed cleavage sites with enzymeset to trypsin, peptide mass tolerance of 25 ppm, fragment mass tolerance of 0.80 Da, Acetylated protein N-term and oxidized Met were considered as variable modifications. The acceptance criterion for peptide identifications was the rate of false positive identification less than 1%.
RNA interference
Nonspecific control siRNA and siRNAs for human CUL3 and RBX1 were purchased from GenePharma (Shanghai, China). The siRNA transfection of cells was performed following the manufacturer’s instructions. The siRNA sequence information is provided in Supplementary Table 1.
In vivo ubiquitination assay
The 293T cells were transfected with HA-ubiquitin and the indicated constructs. 36 h after transfection, the cells were treated with 30 μM MG132 for 6 h and then lysed in 1% buffer (Tris [pH 7.5], 0.5 mM EDTA, 1 mM DTT) and boiled for 10 min. For immunoprecipitation, the cell lysates were diluted 10-fold in Tris-HCl buffer and incubated with anti-FLAG M2 agarose beads (Sigma) for 4 h at 4°C. The bound beads were then washed four times with BC100 buffer (20 mM Tris-Cl, pH 7.9, 100 mM NaCl, 0.2 mM EDTA, and 20% glycerol) containing 0.2% Triton X-100. The protein was eluted with 3X FLAG peptide for 2 h at 4°C. The ubiquitinated form of ZBTB3 was detected by Western blot using anti-HA antibody.
Transcriptional reporter assay
Luciferase assay (Promega) was conducted in cells that were transiently transfected with the reporter constructs (SHH-promoter (1-2000 bp)-Luc), pTK-galactosidase, and indicated plasmids. The luciferase activity in cell lysates was measured using the luciferase assay system in a Berthold Lumat LB 9507 luminometer (Promega, USA). Luciferase activity was normalized to galactosidase activity as an internal control. Each assay was performed in triplicate, and the results were confirmed by at least three individual repeating experiments.
Real-time reverse transcription PCR (qRT-PCR)
Total RNA was isolated from ECC-1 and HEC-1-A cells by using the TRIzol reagent (Tiangen, China), and cDNA was reversed-transcribed by employing the Superscript RT kit (Toyobo, Japan) following the manufacturer’s instructions. PCR amplification was performed using the SYBR Green PCR Master Mix Kit (Toyobo, Japan). All quantitations were normalized to the level of endogenous control GAPDH. The sequences of primers for qRT-PCR were listed in Supplementary Table 1.
Cell proliferation assay
The cell proliferation rate was determined using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan) according to the manufacturer’s protocol. Briefly, the cells were seeded onto 96-well plates at a density of 1,000 cells per well. During a culture period of 2-8 days, 10 μl of the CCK-8 solution was added to the cell culture and incubated for 2 h. The resulting color was assayed at 450 nm using a microplate absorbance reader (Bio-Rad, USA). Each assay was carried out in triplicate.
Colony formation assay
EC cell lines (ECC-1 and HEC-1A) were seeded in 6-well plates containing 1 × 103 individual cells per well in triplicate. After incubating for 2 weeks, the cell lines were fixed with 100% methanol for 5 min followed by staining with Giemsa dye for 20 min (Solarbio, China).
Migration and invasion assays
Cell migration and invasion were determined by Transwell (Corning, USA) migration and invasion assays. The ECC-1 and HEC-1-A cells were precultured in serum-free medium for 48 h. For migration assay, the 2 × 104 cells were seeded in serum-free medium in the upper chamber, and the lower chamber was filled with RPMI1640 containing 5% FBS. After 48 h, we carefully removed the non-migrating cells on the upper chambers with a cotton swab and stained and counted the migrated cells underside of the filter in nine different fields. Matrigel invasion assays were performed using Transwell inserts (Costar) coated with Matrigel/fibronectin (BD Biosciences, USA).
Statistical analysis
The statistical calculations were performed using GraphPad Prism software. All data are shown as mean values ± SD for experiments performed with at least three replicates. The difference between 2 groups was analyzed using paired Student’s t-test unless otherwise specified. * represents P < 0.05; ** represents P < 0.01; *** represents P < 0.001.
Results
Identification of ZBTB3 as a novel SPOP interacting protein
To identify the molecular mediators of the tumor suppressive function of SPOP, we isolated the SPOP protein complex from the 293T cells stably expressing FLAG-HA-SPOP through the TAP methods and determined the proteins present in the complex by using mass spectrometry. As verification of the efficiency of this approach, the peptides of known SPOP substrates, such as Caprin1, INF2, GLI3, SETD2, DAXX, and TRIM24, were detected in the complex. In addition to the known binding partners, some potential novel SPOP interactors (PPP1R12A, ZBTB3, HIPK2, RXRB and SRRM1) involved in diverse biological processes were identified (Table 1). Given that the interaction between SPOP and ZBTB3 has not been previously reported, we examined the potential functional relationship between them.
Table 1.
The number of total/unique peptides identified by mass spectrometry analysis
| Group | Gene Name | FLAG-HA-SPOP | |
|---|---|---|---|
|
| |||
| Peptide count | Unique pep count | ||
| Novel | PPP1R12A | 12 | 10 |
| ZBTB3 | 10 | 6 | |
| HIPK2 | 8 | 4 | |
| RXRB | 7 | 4 | |
| SRRM1 | 5 | 2 | |
| Known | Caprin1 | 11 | 6 |
| INF2 | 7 | 4 | |
| GLI3 | 6 | 2 | |
| SETD2 | 4 | 4 | |
| DAXX | 4 | 2 | |
| TRIM24 | 3 | 2 | |
To verify that ZBTB3 is a bona fide SPOP interacting protein, we first examined whether SPOP could interact with ZBTB3 in cells. Co-immunoprecipitation (co-IP) analysis showed that Myc-SPOP was co-immunoprecipitated by FLAG-ZBTB3 (Figure 1A), and FLAG-SPOP was able to co-immunoprecipitate Myc-ZBTB3 (Figure 1B), suggesting an interaction between the two exogenously expressed proteins. FLAG-SPOP was able to immunoprecipitate endogenous ZBTB3 and a known SPOP substrate INF2 in ECC-1 cells (Figure 1C). The potential binding between the endogenous SPOP and ZBTB3 was investigated next. We performed immunoprecipitation by using the anti-ZBTB3 antibody in cell lysates prepared from ECC-1 cells. As shown in Figure 1D, endogenous SPOP was efficiently co-immunoprecipitated by the ZBTB3, suggesting an endogenous interaction between these two proteins.
Figure 1.
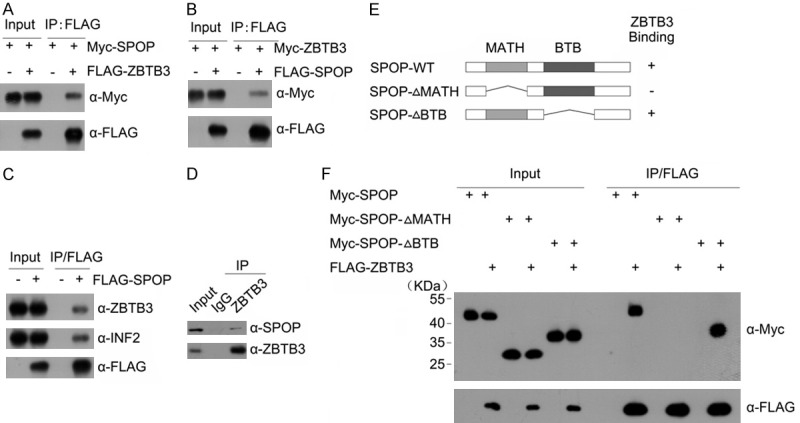
Identification of ZBTB3 as a novel SPOP Interactor. Western blot of whole cell lysates (WCLs) and co-IP samples of anti-FLAG antibody obtained from 293 T cells transfected with indicated plasmids (A, B). (C) Western blot of WCLs and co-IP samples of anti-FLAG antibody obtained from ECC-1 cells infected with lentivirus expressing FLAG-SPOP or control. The cells were treated with 20 μM MG132 for 8 h before harvesting. (D) Western blot of co-IP samples of IgG or anti-ZBTB3 antibodies obtained from cell lysates of ECC-1 cells. The cells were treated with 20 μM MG132 for 8 h before harvesting. (E) Schematic representation of SPOP deletion mutants. Binding capacity of SPOP to ZBTB3 is indicated with the symbol. (F) Western blot of WCLs and co-IP samples of anti-FLAG antibody obtained from 293 T cells transfected with indicated plasmids.
SPOP contains two structural domains: a substrate-binding MATH domain at the N-terminus and a CUL3-binding BTB domain at the C-terminus. To determine the domain that mediates its interaction with ZBTB3, two deletion mutants of SPOP corresponding to the deletion of these two domains were generated, namely, SPOP-ΔBTB and ΔMATH (Figure 1E). Co-IP assay was then performed to test the ability of the overexpressed ZBTB3 to bind the two deletion mutants. As shown in Figure 1F, the interaction was abolished between SPOP-ΔMATH and ZBTB3 while full-length SPOP or SPOP-ΔBTB interacted with ZBTB3. Overall, these findings demonstrate that SPOP interacts with ZBTB3 in vivo through the MATH domain.
ZBTB3 is a bona fide substrate of SPOP-CUL3-RBX1 E3 ubiquitin-ligase complex
We next explored whether the SPOP-CUL3-RBX1 E3 ubiquitin-ligase complex could promote the ubiquitination and degradation of ZBTB3. As shown in Figure 2A, SPOP decreased the protein level of ectopically co-expressed ZBTB3 in a dose-dependent manner. This effect was completely blocked when cells were treated with the proteasome inhibitors MG132 or Bortezomib (Figure 2A). By contrast, the lysosome inhibitor Chloroquine had no effect on SPOP-mediated ZBTB3 degradation (Figure 2A). These results indicated that SPOP downregulates ZBTB3 protein via the proteasomal-but not the lysosomal-degradation pathway. Moreover, SPOP-ΔMATH and SPOP-ΔBTB mutant didn’t promote the degradation of overexpressed or endogenous ZBTB3 (Figure 2B, 2C), indicating that the MATH and BTB domains are both required for SPOP-mediated ZBTB3 degradation. The endogenous SPOP in endometrial cells was also depleted using two SPOP-specific shRNAs, and an increase was observed in the ZBTB3 protein level (Figure 2D, 2E) but not the mRNA level (Figure 2F). This result indicated that the effect of SPOP on ZBTB3 is not mediated through the regulation of ZBTB mRNA expression. SPOP knockdown also prolonged the half-life of endogenous ZBTB protein (Figure 2G, 2H), further suggesting that SPOP regulates ZBTB3 at the protein level.
Figure 2.

ZBTB3 is a bona fide substrate of the SPOP-CUL3-RBX1 E3 ubiquitin ligase complex. (A) Western blot of WCLs from 293 T cells transfected with the indicated plasmids. and treated with MG132 (20 μM), Bortezomib (200 nM), Chloroquine (100 mM) or DMSO for 8 h. (B) Western blot of WCLs of 293 T cells transfected with indicated plasmids. (C) Western blot of WCLs of ECC-1 cells infected with empty vector (EV) or lentivirus expressing wild-type or mutant SPOP. (D) Western blot of the WCLs of ECC-1 cells infected with control or lentivirus expressing SPOP-specific shRNAs (shSPOP#1,2). (E) Western blot of the WCLs of HEC-1-A cells infected with control or lentivirus expressing SPOP-specific shRNAs (shSPOP#1,2). (F) Quantitative RT-PCR measurement of SPOP and ZBTB3 mRNA levels in SPOP-depleted ECC-1 cells. GAPDH mRNA levels were used for normalization. Data are shown as means ± SD (n=3). *P < 0.01. (G) Western blot of WCLs of ECC-1 cells infected with control or lentivirus expressing SPOP-specific shRNAs and then treated with 50 μg/ml cycloheximide (CHX) and harvested at different time points. (H) At each time point, the intensity of each ZBTB3 protein was normalized to the intensity of actin and then to the value at 0 h. Western blot of the WCLs of ECC-1 cells transfected with control siRNAs or siRNAs towards Cul3 (I) or RBX1 (J). (K) Western blots of the products of in vivo ubiquitination assays performed using cell lysate from 293 T cells transfected with the indicated plasmids and treated with 20 μM MG132 for 8 h.
Next, whether other subunits of the SPOP-CUL3-RBX1 E3 ubiquitin-ligase complex are also required for ZBTB3 degradation was evaluated. CUL3 or RBX1 was depleted through gene-specific siRNAs, and the changes in ZBTB3 protein level in ECC-1 cells were examined. As shown in Figure 2I and 2H, the knockdown of either CUL3 or RBX1 increased the ZBTB3 protein levels. This result suggested that the integrity of SPOP-CUL3-RBX1 E3 ligase complex is required for ZBTB3 degradation. ZBTB3 protein was robustly polyubiquitinated upon SPOP-WT co-expression in a dose-dependent manner. By contrast, minimal or no ZBTB3 polyubiquitination was observed when SPOP-ΔMATH or SPOP-ΔBTB was co-expressed (Figure 2K). Overall, these data demonstrate that the SPOP-CUL3-RBX1 E3 ubiquitin-ligase complex regulates ZBTB3 protein stability through ubiquitin-dependent proteasomal degradation in EC cells.
SPOP binding consensus motifs in ZBTB3 are required for SPOP-mediated ZBTB3 degradation
Previous studies reported that one or several SPOP binding consensus (SBC) motifs (Φ-π-S-S/T-S/T; Φ: nonpolar residues, π: polar residues) are present in the SPOP substrates [8]. Accordingly, we examined the amino acid sequence of ZBTB3 and found two potential SBC motifs (196-LSSTS-200 aa, 272-PSSST-276 aa) (Figure 3A). Three ZBTB3 mutants were generated (M1, M2, and M3), in which single or double SBC motifs were deleted to examine whether these two regions are required for SPOP-ZBTB3 interaction (Figure 3A). Co-IP assay demonstrated that deletion of the first or second motif in ZBTB3 moderately reduced its SPOP binding capacity compared with that of ZBTB3-WT. However, the deletion of both motifs in ZBTB3 completely abrogated its SPOP-binding capacity (Figure 3B). These data indicate that both motifs are required for SPOP-ZBTB3 interaction. Next, we decided to determine whether both motifs are required for SPOP-mediated ZBTB degradation. As shown in Figure 3C, ZBTB3-M1 and M2 mutants were still degraded by SPOP, albeit less than the ZBTB3-WT. By contrast, the ZBTB3-M3 mutant was resistant to SPOP-mediated degradation. The half-lives of ZBTB3-WT and ZBTB3-M3 mutant were likewise measured. As shown in Figure 3D and 3E, the ZBTB3-M3 mutant exhibited a significantly prolonged half-life compared with ZBTB3-WT. In vivo ubiquitination assays were performed to further determine the importance of these two motifs as degrons. The results showed that the deletion of these two motifs in ZBTB3 totally abolished SPOP-mediated ZBTB3 ubiquitination (Figure 3F). Overall, these data demonstrate that two SBC motifs function as ZBTB3 degrons, which are essential for SPOP binding and subsequent degradation through the ubiquitin-proteasome pathway.
Figure 3.

Identification of degrons in ZBTB3 are required for SPOP-mediated ZBTB3 degradation. A. Diagram showing wild-type ZBTB3 proteins and SBC motif-deleted mutants. The SBC motif is depicted in red. B. Western blot of WCLs and co-IP samples of anti-FLAG antibody obtained from 293 T cells transfected with indicated plasmids. C. Western blot of WCLs of 293 T cells transfected with indicated plasmids. D. Western blots of WCLs from 293 T cells transfected with the indicated constructs, treated with 50 μg/ml cycloheximide (CHX) and harvested at different time points. E. Quantification of the western blots carried out in. At each time point, the intensity of ZBTB3 protein was normalized to the intensity of actin and then to the value at 0 h. F. Western blots of the products of in vivo ubiquitination assays performed using cell lysate from 293 T cells transfected with the indicated plasmids and treated with 20 μM MG132 for 8 h.
EC-associated mutants of SPOP are defective in promoting ZBTB3 degradation and ubiquitination
SPOP was mutated in 5.7%-10% of patients with EC across multiple independent cohorts [11]. Notably, all the SPOP mutations found in ECs exclusively occur in the MATH domain, which is responsible for ZBTB3 binding (Figure 4A). Therefore, this study proposed that EC-associated SPOP mutations may cause dysfunction in mediating ZBTB3 destruction. We first examined the interactions between the SPOP mutants and ZBTB3. As shown in Figure 4B, the ZBTB3 binding ability of the SPOP mutants was severely impaired compared with wild-type SPOP. SPOP-mediated degradation and ubiquitination of ZBTB3 protein were also markedly attenuated for these mutants (Figure 4C, 4D). The stable overexpression of a few hotspot mutants of SPOP in ECC-1 cells failed to degrade ZBTB3 protein and instead led to elevated endogenous levels of ZBTB3, showing a dominant-negative effect exerted by the EC-associated SPOP mutants (Figure 4E). Indeed, the coexpression of SPOP mutants (G75R, P94A or M117I) was observed to reduce the interaction between wild-type SPOP and ZBTB3 (Figure 4F), and suppress the wild-type SPOP-induced ZBTB3 ubiquitination (Figure 4G). These data indicate that EC-associated SPOP mutations abrogate SPOP-mediated ZBTB3 degradation and lead to ZBTB3 stabilization in EC cells.
Figure 4.
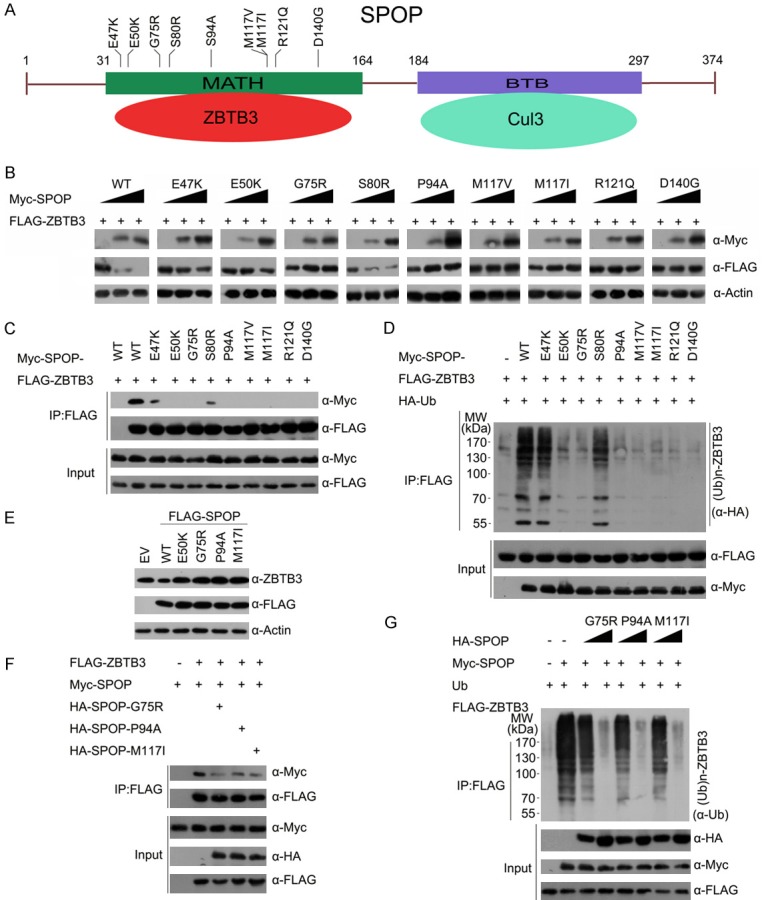
EC-associated mutants of SPOP are defective in promoting ZBTB3 degradation and ubiquitination. A. Distribution of the most common mutations in the SPOP gene found in endometrial cancer samples. B. Western blot of WCLs of 293 T cells transfected with indicated plasmids. C. Western blot of WCLs and co-IP samples of anti-FLAG antibody obtained from 293 T cells transfected with indicated plasmids. D. Western blots of the products of in vivo ubiquitination assays performed using cell lysate from 293 T cells transfected with the indicated plasmids and treated with 20 μM MG132 for 8 h. E. Western blot of the indicated proteins in ECC-1 cells infected with empty vector (EV) or lentivirus expressing wild-type or mutant SPOP. F. Western blot of WCLs and co-IP samples of anti-FLAG antibody obtained from 293 T cells transfected with indicated plasmids. G. Western blot of the in vivo ubiquitination assay in 293T cells transfected with the indicated plasmids and treated with 20 μM MG132 for 8 h.
ZBTB3 is an important mediator of SPOP inactivation-induced cell growth, migration and invasion
Previous studies reported that ZBTB3 is associated with tumorigenesis and stem cell self-renewal [27,28]. These observations led us to explore whether ZBTB3 is a molecular mediator of SPOP mutation-induced malignant transformation in EC. Indeed, ZBTB3 depletion in ECC-1 cells decreased the cell growth (Figure 5A). By contrast, SPOP depletion enhanced cell growth, and co-depletion of SPOP and ZBTB3 reduced cell growth compared with the levels with depletion of SPOP alone (Figure 5A). Similar results were obtained when we stably overexpressed SPOP-G75R mutant in ECC-1 cells simultaneously with shRNA-mediated knockdown of endogenous SPOP (Figure 5B). Colony formation assays were performed, and the results were consistent with CCK-8 cell growth assay (Figure 5C, 5D). Migration and invasion assays were also performed in ECC-1 cells after depletion of SPOP and/or ZBTB3. We observed that the increase of migration and invasion in ECC-1 cells by SPOP depletion or stable overexpression of SPOP-G75R mutant was partly diminished by ZBTB3 depletion (Figure 5E-H). Similar effects were observed in another EC cells HEC-1-A (Supplementary Figure 1). Overall, our data suggest that SPOP inactivation promotes endometrial cell proliferation, migration, and invasion, at least in part, by upregulating ZBTB3.
Figure 5.

SPOP suppresses cell proliferation, migration and invasion partially dependent on ZBTB3. (A) Western blot (left panel) and cell proliferation assay (right panel) of ECC-1 cells infected with lentivirus expressing the indicated shRNAs. Standard deviation (S.D.) of at least three independent experiments is shown to indicate statistical significance. *P < 0.05. (B) Western blot (left panel) and cell proliferation assay (right panel) of ECC-1 cells infected with empty vector or lentivirus expressing HA-SPOP-G75R in combination with control shRNA or ZBTB3-spefic shRNAs. Data are shown as means ± SD (n=3). *P < 0.05. (C) Cell colony formation assay of ECC-1 cells infected with lentivirus expressing the indicated shRNAs. All data shown are mean values ± SD from three replicates. *P < 0.05. (D) Cell colony formation assay of ECC-1 cells infected with empty vector or lentivirus expressing SPOP-G75R in combination with control shRNA or ZBTB3-spefic shRNAs. Cell migration (E) and invasion (F) assay of ECC-1 cells infected with lentivirus expressing the indicated shRNAs. Data are shown as means ± SD (n=3). *P < 0.05. (G, H) Cell migration (G) and invasion (H) assay of ECC-1 cells with lentivirus expressing FLAG-SPOP-G75R in combination with control shRNA or ZBTB3-spefic shRNAs. Data are shown as means ± SD (n=3). *P < 0.05.
ZBTB3-regulated gene expression in EC cells
ZBTB3 is a transcription factor, but the transcriptional targets were poorly understood. RNA-sequencing (RNA-seq) was performed to identify the differentially expressed genes in SPOP-depleted ECC-1 cells and further explore the molecular mechanism by which ZBTB3 affects the endometrial cell phenotypes. A total of 114 genes were identified, wherein the expression was significantly altered by ZBTB3 depletion (Figure 6A). Among the 114 genes, 39 genes were downregulated and 75 genes were upregulated above 1.5-fold (Figure 6B). For validation, we verified that the mRNAs of SHH and IL22RAI were downregulated, whereas BIK was upregulated in ZBTB3-depleted ECC-1 or HEC-1A cells by RT-qPCR methods (Figure 6C). Conversely, the mRNA of SHH and IL22RAI were upregulated, whereas BIK was downregulated in ZBTB3 (WT or M3 mutant) stably overexpressed ECC-1 or HEC-1A cells (Figure 6D). Importantly, SPOP depletion led to the upregulation of SHH, IL22RAI and downregulation of BIK, but these effects were reversed by co-depletion of ZBTB3 (Figure 6E). Taken together, these results suggest that a SPOP-ZBTB3 regulatory axis controls the mRNA expression of a subset genes in EC cells.
Figure 6.
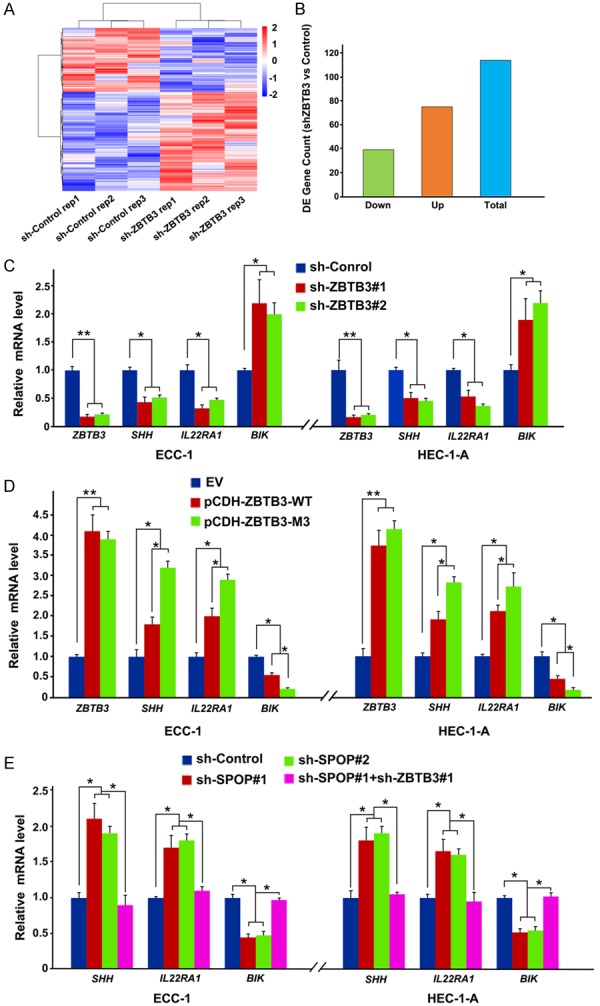
ZBTB3 regulates the mRNA expression of a subset genes. (A) Hierarchical clustering of the differentially expressed genes in sh-control and ZBTB3-depleted ECC-1 cells. (B) Quantitative data of up- and down-regulated genes shown in (A). (C) RT-qPCR measurement of the mRNA expression of differentially expressed genes in sh-control and ZBTB3-depleted ECC-1 or HEC-1-A cells. Data are shown as means ± SD (n=3). *P < 0.05; **P < 0.01. (D) RT-qPCR measurement of the mRNA expression of differentially expressed genes in EV, SPOP-WT or SPOP-M3 mutant overexpressed ECC-1 or HEC-1-A cells. Data are shown as means ± SD (n=3). *P < 0.05; **P < 0.01. (E) RT-qPCR measurement of the mRNA expression of differentially expressed genes in sh-control, SPOP/ZBTB3-depleted ECC-1 or HEC-1-A cells. Data are shown as means ± SD (n=3). *P < 0.05.
Pharmaceutical inhibition of hedgehog pathway overcomes spop mutation-caused malignant transformation of EC cells
The secreted protein sonic hedgehog (SHH) is the ligand of the hedgehog signaling pathway, which plays critical roles in a range of physiological (e.g., embryonic development and stem cell maintenance) and pathological (e.g., cancer) events [29]. SHH protein expression was very weak in the normal endometrium but increased in endometrial hyperplasia and carcinoma [30]. Given that the mRNA level of SHH was regulated by SPOP-ZBTB3 axis (Figure 6E), we investigated whether SPOP plays a tumor suppressor role in EC partly through modulating ZBTB3-SHH signaling. First, the SHH protein was decreased in ZBTB3-depleted ECC-1 or HEC-1-A cells (Figure 7A). SPOP depletion led to the upregulation of SHH protein level, but this effect was reversed by ZBTB3 co-depletion (Figure 7B). These WB results were consistent with mRNA level changes examined by RT-qPCR methods (Figure 6E). Moreover, we found that stable overexpression of an EC-associated SPOP-G75R mutant also led to the upregulation of SHH protein level (Figure 7C) and mRNA level (Figure 7D).
Figure 7.
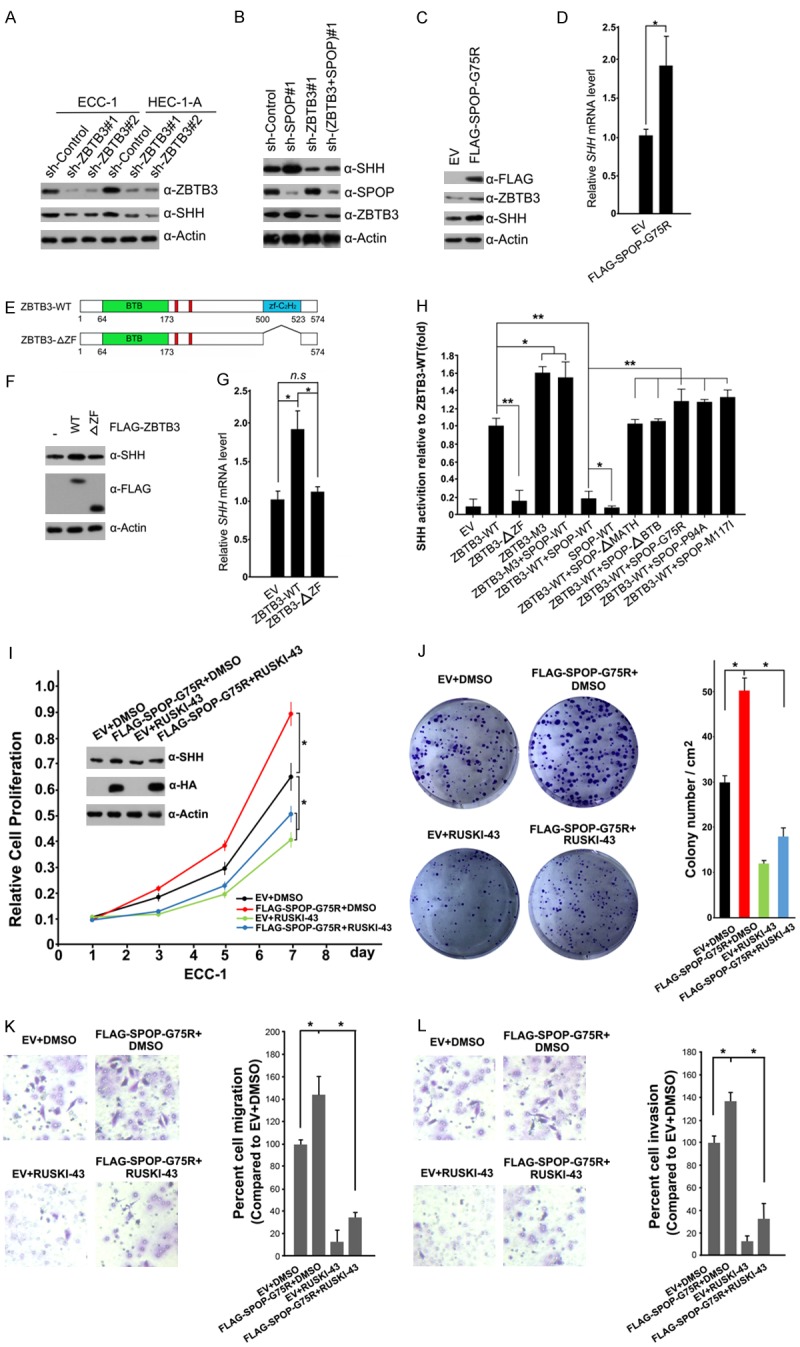
SPOP suppresses ZBTB3-SHH signaling. (A) Western blot of the indicated proteins in WCLs from ECC-1 or HEC-1-A cells infected with indicated shRNAs. (B) Western blot of the indicated proteins in WCLs from ECC-1 infected with the indicated shRNAs. (C) Western Blot of the indicated proteins in WCLs from of EV and FLAG-SPOP-G75R-overexpressing ECC-1 cells. (D) Quantitative RT-qPCR measurement of mRNA expression of SHH from of EV and FLAG-SPOP-G75R-overexpressing ECC-1 cells. Data are shown as means ± SD (n=3). *P < 0.05. (E) Schematic representation of ZBTB3 deletion mutants. (F) Western blot of the indicated proteins in WCLs obtained from ECC-1 cells transfected with indicated plasmids. (G) Quantitative RT-qPCR measurement of mRNA expression of SHH in ECC-1 cells infected with the indicated plasmids. Data are shown as means ± SD (n=3). *P < 0.05. (H) 293T cells were transfected with the SHH-luc reporter, pTKgalactosidase (internal control), and indicated plasmids. After 24 hr, the luciferase activities were measured by luminometer. Data are shown as means ± SD (n=3). *P < 0.05; **P < 0.01. (I) Western blot and cell proliferation assay of ECC-1 cells infected with lentivirus expressing EV or FLAG-SPOP-G75R, and treated with DMSO or RUSKI-43 (10 μM). Data are shown as means ± SD (n=3). *P < 0.05; **P < 0.01. (J) Cell colony formation assay of ECC-1 cells infected with lentivirus expressing EV or FLAG-SPOP-G75R, and treated with DMSO or RUSKI-43 (10 μM). Data are shown as means ± SD (n=3). *P < 0.05. Cell migration (K) and invasion (L) assay of ECC-1 cells infected with lentivirus expressing EV or FLAG-SPOP-G75R, and treated with DMSO or RUSKI-43 (10 μM). Data are shown as means ± SD (n=3). *P < 0.05.
ZBTB3 harbors a ZF domain, which is responsible for DNA binding. A ZBTB3-ΔZF mutant was generated, in which the ZF domain of ZBTB3 was deleted, to determine whether this domain was essential to the transcription activation function of ZBTB3 (Figure 7E). Compared with ZBTB3-WT, the ZBTB3-ΔZF mutant was unable to elevate the protein and mRNA levels of SHH (Figure 7F, 7G). ZBTB3-WT, but not the ZBTB3-ΔZF mutant, activated the SHH promotor luc-reporter activity. SPOP-binding defective mutant ZBTB3-M3 activated the SHH reporter more potently than did ZBTB3-WT.
Co-expression of wild-type SPOP suppressed ZBTB3-mediated activation of SHH luc-reporter but had no obvious effect on ZBTB3-M3 mutant-mediated activation of SHH luc-reporter. Moreover, co-expression of the EC-associated SPOP mutants (G75R, P94A or M117I) or domain deletion mutants (ΔMATH, ΔBTB) had no obvious effect on ZBTB3-mediated activation of SHH luc-reporter (Figure 7H). These data suggest that ZBTB3 stabilization caused by SPOP inactivation led to upregulation of its transcriptional target SHH in EC cells.
Next, we investigated whether the pharmaceutical inhibition of Hedgehog pathway will overcome SPOP mutation-caused malignant transformation of EC cells. RUSKI-43 is a potent and specific inhibitor of SHH palmitoylation, thereby blocking autocrine and paracrine Hedgehog signaling [31]. It is proposed to have therapeutic potential for Hedgehog-dependent cancers as well. Indeed, the treatment of ECC-1 cells with RUSKI-43 significantly reduced SPOP-G75R mutant overexpression-enhanced cell growth (Figure 7I, 7J), migration (Figure 7K), and invasion (Figure 7L). Similar results were also observed in HEC-1-A cells (Supplementary Figure 2).
Taken together, these data suggest that SPOP may suppress EC cell proliferation, migration, and invasion, at least in part, by inhibiting ZBTB3-mediated Hedgehog pathway activation.
Discussion
Recurrent SPOP mutations in ECs have been confirmed by several independent cancer exome or genome sequencing studies [3-6]. Although frequent mutations of SPOP in ECs have been identified, the functional impact of these mutations on ECs remains poorly unknown. A study that used a mouse model, in which SPOP expression was ablated in uterine cells, showed that SPOP was required for normal uterine function by regulating the homeostasis of key signaling cues required for embryo implantation and endometrial decidualization [32]. Our previous study revealed that SPOP targets estrogen receptor-α (ERα) for ubiquitination and proteasomal degradation in EC cells. However, this effect is abrogated by the EC-associated SPOP mutations [11]. In this study, we demonstrated that ZBTB3 is a bona fide substrate for the SPOP-CUL3-RBX1 E3 ubiquitin-ligase complex. SPOP recognizes two SBC motifs in ZBTB3 and promotes ZBTB3 ubiquitination and proteasomal degradation. EC-associated SPOP mutants are defective in promoting ZBTB3 degradation and ubiquitination. ZBTB3, as a transcription factor, also promoted endometrial cell proliferation, migration, and invasion partly through promoting the SHH gene transcription (Figure 8). These findings support that ZBTB3 acts as an oncogene in EC, and its expression is dysregulated in SPOP-mutated ECs.
Figure 8.
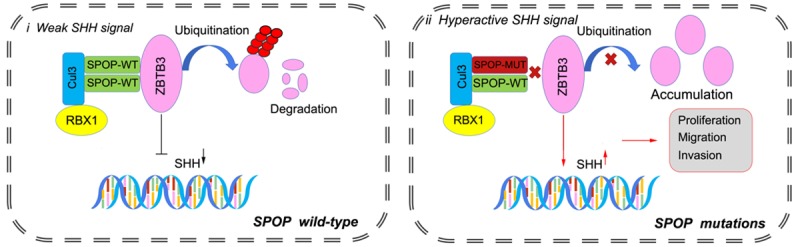
Schematic of the proposed mechanism through which SPOP mutants enhance ZBTB3-SHH axis-induced malignant transformation in SPOP-mutated endometrial cancer.
Accumulating evidence supports the notion that Hedgehog pathway plays a critical role during EC initiation and progression [30,33]. Constitutive activation of the Hedgehog pathway has been implicated in tumorigenesis and may be involved in the early events of endometrial carcinogenesis [33]. Gli proteins are downstream transcription factors of the Hedgehog pathway. Interestingly, previous reports demonstrated that SPOP targeted GLI2 and GLI3 for proteasomal degradation to suppress Hedgehog pathway activation [34,35]. More importantly, this process is evolutionary conserved from worms to humans [36]. A recent study showed Sufu; Spop double knockout (DKO) mice develop highly aggressive, earlyonset Medulloblastoma which is dependent on constitutive Hedgehog pathway activation [37]. Our studies showed that the Hedgehog pathway ligand SHH was also controlled by SPOP, suggesting SPOP can regulate Hedgehog pathway at multiple levels. These findings suggest that the inhibition of Hedgehog pathway by small molecular inhibitors, such as RUSKI-43, may represent a promising therapeutic strategy for targeting SPOP-mutated ECs.
Acknowledgements
This work was supported by The Natural Science Foundation of Zhejiang Province (LY20C070001 to X.J.), The National Natural Science Foundation of China (81972396, 81672558 and 81201533 to C.W.; 31801165 to X.J.; 91954106, 81872109 to K.G), The Natural Science Foundation of Shanghai (18ZR1430100 to K.G.), The Natural Science Foundation of Ningbo (2018A610213 to X.J.), the Program of “Xinmiao” (Potenial) Talents in Zhejiang Province (2019R405061 to J.W., and 2019R405011 to Q.L.), the Student Research and Innovation Program of Ningbo University (2018SRIP1925 to Q.L., 2019SRIP1907 to J.W.) and the K.C. Wong Magna Fund in Ningbo University.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Lortet-Tieulent J, Ferlay J, Bray F, Jemal A. International patterns and trends in endometrial cancer incidence, 1978-2013. J Natl Cancer Inst. 2018;110:354–361. doi: 10.1093/jnci/djx214. [DOI] [PubMed] [Google Scholar]
- 2.Sutton G, Axelrod JH, Bundy BN, Roy T, Homesley HD, Malfetano JH, Mychalczak BR, King ME. Whole abdominal radiotherapy in the adjuvant treatment of patients with stage III and IV endometrial cancer: a gynecologic oncology group study. Gynecol Oncol. 2005;97:755–763. doi: 10.1016/j.ygyno.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC NIH Intramural Sequencing Center (NISC) Comparative Sequencing Program. Hieter P, Mullikin JC, Merino MJ, Bell DW. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, Bortolomai I, Buza N, Hui P, Abu-Khalaf M, Ravaggi A, Bignotti E, Bandiera E, Romani C, Todeschini P, Tassi R, Zanotti L, Carrara L, Pecorelli S, Silasi DA, Ratner E, Azodi M, Schwartz PE, Rutherford TJ, Stiegler AL, Mane S, Boggon TJ, Schlessinger J, Lifton RP, Santin AD. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhn E, Wu RC, Guan B, Wu G, Zhang J, Wang Y, Song L, Yuan X, Wei L, Roden RB, Kuo KT, Nakayama K, Clarke B, Shaw P, Olvera N, Kurman RJ, Levine DA, Wang TL, Shih IeM. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst. 2012;104:1503–1513. doi: 10.1093/jnci/djs345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307–2320. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, Hoggard T, Harper JW, White KP, Schulman BA. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36:39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, Bond R, He B, Coarfa C, Mitsiades N. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–5643. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–669. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, Dong Y, Huang H, Yu L, Wang C. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell Death Dis. 2015;6:e1687. doi: 10.1038/cddis.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Peng S, Dai X, Gan W, Nie X, Wei W, Hu G, Guo J. Tumor suppressor SPOP ubiquitinates and degrades EglN2 to compromise growth of prostate cancer cells. Cancer Lett. 2017;390:11–20. doi: 10.1016/j.canlet.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan Y, Ci Y, Dai X, Wu F, Guo J, Liu D, North BJ, Huo J, Zhang J. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget. 2017;8:47890–47901. doi: 10.18632/oncotarget.18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ju LG, Zhu Y, Long QY, Li XJ, Lin X, Tang SB, Yin L, Xiao Y, Wang XH, Li L, Zhang L, Wu M. SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 2019;26:1156–1168. doi: 10.1038/s41418-018-0198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, Moch H, Prive GG, Carr SA, Garraway LA. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–89. doi: 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, Cui Y, Wang L, Pan Y, Zhang X, Zhu Y, Wang C, Halling GC, Cheng L, Sukov WR, Karnes RJ, Vasmatzis G, Zhang Q, Zhang J, Cheville JC, Yan J, Sun Y, Huang H. Truncated ERG oncoproteins from TMPRSS2-ERG fusions are resistant to SPOP-mediated proteasome degradation. Mol Cell. 2015;59:904–916. doi: 10.1016/j.molcel.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 17.Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, Asara JM, Beck AH, Huang J, Pandolfi PP, Wei W. SPOP promotes ubiquitination and degradation of the ERG oncoprotein to suppress prostate cancer progression. Mol Cell. 2015;59:917–930. doi: 10.1016/j.molcel.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, Guo J, Huang YH, Fan C, Ren S, Sun Y, Freeman GJ, Sicinski P, Wei W. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, Chen T, Shimizu K, Beca F, Blattner M, Vasudevan D, Buckley DL, Qi J, Buser L, Liu P, Inuzuka H, Beck AH, Wang L, Wild PJ, Garraway LA, Rubin MA, Barbieri CE, Wong KK, Muthuswamy SK, Huang J, Chen Y, Bradner JE, Wei W. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063–1071. doi: 10.1038/nm.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, Bielski CM, Kryukov GV, Cascione L, Napoli S, Enchev RI, Mutch DG, Carney ME, Berchuck A, Winterhoff BJN, Broaddus RR, Schraml P, Moch H, Bertoni F, Catapano CV, Peter M, Carr SA, Garraway LA, Wild PJ, Theurillat JP. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23:1046–1054. doi: 10.1038/nm.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, Yang Y, Wu D, He Y, Zhang J, Lu D, Liu X, Yu L, Zhao S, Li Y, Lin D, Wang Y, Wang L, Chen Y, Sun Y, Wang C, Huang H. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med. 2017;23:1055–1062. doi: 10.1038/nm.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, Houtman R, Cato ACB, Tschopp P, Gu L, Corsinotti A, Zhong Q, Fankhauser C, Fritz C, Poyet C, Wagner U, Guo T, Aebersold R, Garraway LA, Wild PJ, Theurillat JP, Brown M. TRIM24 is an oncogenic transcriptional activator in prostate cancer. Cancer Cell. 2016;29:846–858. doi: 10.1016/j.ccell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin X, Wang J, Gao K, Zhang P, Yao L, Tang Y, Tang L, Ma J, Xiao J, Zhang E, Zhu J, Zhang B, Zhao SM, Li Y, Ren S, Huang H, Yu L, Wang C. Dysregulation of INF2-mediated mitochondrial fission in SPOP-mutated prostate cancer. PLoS Genet. 2017;13:e1006748. doi: 10.1371/journal.pgen.1006748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernández-Muñoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y, van Lohuizen M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci U S A. 2005;102:7635–7640. doi: 10.1073/pnas.0408918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattner M, Lee DJ, O’Reilly C, Park K, MacDonald TY, Khani F, Turner KR, Chiu YL, Wild PJ, Dolgalev I, Heguy A, Sboner A, Ramazangolu S, Hieronymus H, Sawyers C, Tewari AK, Moch H, Yoon GS, Known YC, Andren O, Fall K, Demichelis F, Mosquera JM, Robinson BD, Barbieri CE, Rubin MA. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costoya JA. Functional analysis of the role of POK transcriptional repressors. Brief Funct Genomic Proteomic. 2007;6:8–18. doi: 10.1093/bfgp/elm002. [DOI] [PubMed] [Google Scholar]
- 27.Lim JH. Zinc finger and BTB domain-containing protein 3 is essential for the growth of cancer cells. BMB Rep. 2014;47:405–410. doi: 10.5483/BMBRep.2014.47.7.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye B, Liu B, Yang L, Zhu X, Zhang D, Wu W, Zhu P, Wang Y, Wang S, Xia P, Du Y, Meng S, Huang G, Wu J, Chen R, Tian Y, Fan Z. LncKdm2b controls self-renewal of embryonic stem cells via activating expression of transcription factor Zbtb3. EMBO J. 2018;37 doi: 10.15252/embj.201797174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 30.Feng YZ, Shiozawa T, Miyamoto T, Kashima H, Kurai M, Suzuki A, Ying-Song J, Konishi I. Overexpression of hedgehog signaling molecules and its involvement in the proliferation of endometrial carcinoma cells. Clin Cancer Res. 2007;13:1389–1398. doi: 10.1158/1078-0432.CCR-06-1407. [DOI] [PubMed] [Google Scholar]
- 31.Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, Resh MD. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat Chem Biol. 2013;9:247–249. doi: 10.1038/nchembio.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hai L, Szwarc MM, He B, Lonard DM, Kommagani R, DeMayo FJ, Lydon JP. Uterine function in the mouse requires speckle-type poz protein. Biol Reprod. 2018;98:856–869. doi: 10.1093/biolre/ioy060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao X, Siu MK, Au CW, Chan QK, Chan HY, Wong ES, Ip PP, Ngan HY, Cheung AN. Aberrant activation of hedgehog signaling pathway contributes to endometrial carcinogenesis through beta-catenin. Mod Pathol. 2009;22:839–847. doi: 10.1038/modpathol.2009.45. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137:2001–2009. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai H, Liu A. Spop promotes skeletal development and homeostasis by positively regulating Ihh signaling. Proc Natl Acad Sci U S A. 2016;113:14751–14756. doi: 10.1073/pnas.1612520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J. A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell. 2006;10:719–729. doi: 10.1016/j.devcel.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Yin WC, Satkunendran T, Mo R, Morrissy S, Zhang X, Huang ES, Uuskula-Reimand L, Hou H, Son JE, Liu W, Liu YC, Zhang J, Parker J, Wang X, Farooq H, Selvadurai H, Chen X, Sau-Wai Ngan E, Cheng SY, Dirks PB, Angers S, Wilson MD, Taylor MD, Hui CC. Dual regulatory functions of SUFU and targetome of GLI2 in SHH subgroup medulloblastoma. Dev Cell. 2019;48:167–183. e165. doi: 10.1016/j.devcel.2018.11.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.