

Abstract
Desensitization of hepatocellular carcinoma (HCC) to paclitaxel chemotherapy is a major deterrent to successful treatment of the cancer. Abnormal activation of the PI3K/Akt/mTOR, pathway is a common outcome of chemotherapy for HCC. Therefore, we investigated whether BEZ235, a dual PI3K and mTOR inhibitor, could increase the sensitivity of HCC to paclitaxel. In vitro results showed that paclitaxel, combined with BEZ235, inhibited HCC cell proliferation and migration, arrested the cell cycle in the G2/M phase, and promoted cell apoptosis by decreasing PI3K/Akt/mTOR activity. In vivo experiments confirmed that BEZ235 enhances the anti-tumor effect of paclitaxel by reducing PI3K/Akt/mTOR activity. Immunohistochemical staining showed that paclitaxel combined with BEZ235 reduced the numbers of Ki-67- and GPC3-positive HepG2 cells in tumor tissues. We conclude that BEZ235 enhanced the sensitivity of HCC to paclitaxel, and inhibition of PI3K/Akt/mTOR signaling might be a therapeutic strategy against paclitaxel-resistant HCC.
Keywords: Paclitaxel, BEZ235, hepatocellular carcinoma, PI3K/Akt/mTOR
Introduction
Hepatocellular carcinoma (HCC) is the most common primary malignant liver cancer [1,2], with nearly 600,000 new cases diagnosed worldwide each year [3]. In 2018, the global mortality rate was about 8.2%, ranking it fourth among all types of cancer mortality [4]. The treatment of HCC is still a global health challenge [5].
Paclitaxel is a commonly used chemotherapy drug for the treatment of HCC. It promotes polymerization of cell microtubules, inhibits microtubule depolymerization, and leads to apoptosis of HCC cells [6,7]. However, HCC is usually desensitized to paclitaxel after successive treatment, which limits its clinical effectiveness. Paclitaxel often causes abnormal activation of the intracellular phosphoinositide-3-kinase/threonine-protein kinase/mammalian rapamycin (PI3K/Akt/mTOR) pathway [8-10]. Abnormal activation of Akt and mTOR can reduce the effectiveness of anti-tumor drugs through promoting cellular proliferation, cell-cycle progression, and anti-apoptosis [11-13]. Activated Akt (phosphorylated Akt, p-Akt) regulates cell function by phosphorylating downstream factors, such as enzymes, kinases, and transcription factors, and it inhibits the expression of mitochondrial pro-apoptotic-related proteins and prevents initiation of the pro-apoptotic caspase pathway, thus promoting cell survival [14,15]. mTOR, a mammalian target of rapamycin and an important serine-threonine protein kinase downstream of PI3K/Akt, phosphorylates the eukaryotic promoter 4E binding protein 1 (eIF4EBP1) and ribosomal S6 protein kinase (S6K) to promote protein synthesis and survival [16,17]. Consequently, the PI3K/Akt/mTOR signaling pathway is an important target in anti-tumor therapy, especially in the treatment of HCC. Dactolisib (BEZ235), a dual ATP-competitive PI3K and mTOR inhibitor, blocks PI3K and mTOR kinase activity, primarily through the ATP-binding groove that binds to these enzymes, an action that reduces the phosphorylation level of mTOR and mTOR downstream S6K and eIf4EBP1, thus inhibiting the activation of p110α/β/γ and p85 subunits of PI3K [18,19].
In this study, we investigated the effect of BEZ235 on paclitaxel to HCC. BEZ235 enhanced the chemotherapeutic effect of paclitaxel on HCC cells in vitro and in vivo. Thus, we believe that paclitaxel, combined with BEZ235, may be beneficial in HCC treatment and is the basis for further development of targeted therapeutic strategies for HCC.
Materials and methods
Cell lines and cell culture
HepG2 cells were cultured in RPMI-1640 (Hyclone; Salt Lake City UT, USA), supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel) and 1% penicillin-streptomycin (Invitrogen Corporation, CA, USA) and incubated at 37°C in a humidified atmosphere with 5% CO2.
Reagents and antibodies
Paclitaxel, 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-benzimidazolyl carbocyanine iodide (JC-1), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) and Annexin V-FITC/PI Kit were purchased from Sigma-Aldrich (St. Louis, MO, USA); BEZ235 was obtained from MedChem Express, Dactolisib, USA; PI3K Antibody Kit (9655#), p-Akt Antibody Kit (9916#), mTOR Antibody Kit (9964#), Bcl-2 Family Antibody Kit (9942#), Apoptosis Antibody Kit (9915#) and secondary goat anti-rabbit and anti-mouse antibodies were purchased from Cell Signal Technology (Danvers, MA, USA); Cyclin B1 and Cyclin-dependent-kinase (CDK1) were purchased from Abcam Biological Technology (USA); and Phospho Explorer Antibody chip CSP100 was obtained from Full Moon BioSystems Inc. (Sunnyvale, CA, USA). Human HepG2 cell line was bought from the American Type Culture Collection (Manassas, VA, USA).
Phospho-protein microarray analysis
Protein from cell lysates was biotinylated with Antibody Array Assay Kit (Shanghai Biochip Co., Ltd., Shanghai, China). The antibody microarray slides were first blocked with blocking solution for 30 min. The slides were then incubated with the biotinylated cell lysate in the coupling solution for 2 h, and bound biotinylated proteins were detected with Cy3-conjugated streptavidin. There are two replicates per molecule, and positive and negative controls were established. The slides were scanned on a GenePix 4000 scanner, and the images were analyzed with GenePix Pro 6.0 (Molecular Devices, Sunnyvale, CA).
MTT assay
HepG2 cell (4×103 cells/well) was plated overnight in 96-well plates. Cells of each treatment group were treated for 12 h, 24 h or 48 h. Twenty microliters of MTT (5 mg/ml) were added to each well and incubated for 4 h at 37°C. One hundred microliters of dimethyl sulfoxide were added and shaken for 10 min to completely dissolve the reaction product formazan. The optical density at wavelength 492 nm was measured with a microplate reader, and the cell proliferation inhibition rate was calculated with Graphpad Prism Version 5.0 software.
Cell scratch healing test
HepG2 cell (5×105 cells/well) was plated overnight in 6-well plates. The sterile 10-μL tip to create a cell-free zone on the monolayer cell, continuous cultured in serum-free medium. The treatment groups were photographed at 0 h, 24 h and 48 h to record scratch-healing. Cell-migration ability of the treatment groups was evaluated according to the scratch-healing size.
Transwell assay
HepG2 cell (5×104 cells/well) was inoculated with 200 µl cell suspension (FBS-free medium) in transwell chambers (24-well plates); 500 µl of 10% fetal bovine serum medium were added to the lower chamber, cultured for 24 h, taken out the transwell chamber, fixed with 4% paraformaldehyde for 20 min, and stained with 0.1% crystal violet for 5 min. Cells in 5 randomly chosen fields of each group were counted for determining cell migration.
Colony formation assay
HepG2 cell (1×103 cells/well) was plated overnight in 6-well plates, and the cells of each treatment group were cultured for 2-3 weeks until clones were clearly visible and counted. The culture solution was discarded, the cells were fixed in 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet for 5 min. Cell cloning ability was expressed as the percentage of the number of clones formed in the experimental group relative to the control group.
Cell cycle assay
HepG2 cell (1×105 cells/well) was plated overnight in 6-well plates. Cells were digested and harvested after being treated with drugs for 24 h. The cells were fixed with ethanol and treated with 50 μg/ml propidium iodide (PI) and 100 μg/ml RNase A in the dark for 30 min at room temperature followed by flow cytometry analysis (BD FACSCalibur, USA). Cell cycles were analyzed with ModiFit version 3.0 software (Verity Software House, Topsham, ME).
Mitochondrial membrane potential (ΔΨm) measurement
JC-1, which is a cationic dye that exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from red (~590 nm) to green (~525 nm), can be used as a dual-emission potential-sensitive probe for measuring ΔΨm. HepG2 cells were cultured in 24-well plates overnight, treated with drugs for 24 h, counterstained with DAPI and JC-1, and examined with fluorescence microscope; HepG2 cells treated with drug were stained with 10 µg/ml JC-1 for 30 min at room temperature and analyzed with flow cytometry for changes in ΔΨm.
Apoptosis test
Cells (1×103 cells/well) were incubated overnight in 6-well plates containing cell slides. Each group of cells was treated with drug for 24 h. The cells were stained with Annexin-V FITC/PI in the dark for 15 min at room temperature, and apoptosis rate was analyzed with a fluorescence microscope.
Western blot (WB)
A mixture of protease and phosphatase inhibitor (Biyuntian Biotechnology Co., Ltd., Shanghai, China) was added in Radio Immunoprecipitation Assay lysis buffer (Biyuntian Biotechnology Co., Ltd., Shanghai, China) to lyse cells. The BCA protein assay kit (Biosharp, Hefei, China) was used for measuring protein concentration. Various concentrations of SDS-polyacrylamide gel were used to separate each sample containing approximately 20 μg of protein, and the soluble lysate was transferred to a polyvinylidene fluoride membrane (Millipore Sigma, USA). After blocking with 5% skim milk, primary antibody was incubated overnight at 4°C, and secondary antibody was applied for 1 h at room temperature. Color was developed with an ECL luminescent solution (Thermo Fisher Scientific Waltham, MA, USA). Protein was detected with a gel imager Bio-Rad (Hercules, CA, USA) and quantified with Image J Version 1.48 software (NIH, Bethesda, MD). Results were expressed as a percentage of each target molecule compared to that of the control group. The protein content of β-actin was used as a loading control.
Animal experiments
All animal experiments were approved by the Animal Experimental Ethics Committee of Anhui University of Science and Technology and were carried out in accordance with appropriate procedures. Animal experiments were carried out in the SPF-level animal room of the Central Laboratory of Medical School Anhui University of Science and Technology (NO: AUST201810088). One hundred microliters of HepG2 cell suspension (2×107/ml) were injected subcutaneously into the dorsal right side of 5-week-old female BALB/c nude mice (Vital River Laboratories, Beijing, China). When tumor volume reached 100 mm3, mice were randomized into four groups: control group: 100 μl saline daily, ip; paclitaxel (PTX) group: 10 mg/kg weekly, ip; BEZ235 group: 45 mg/kg daily, oral treatment. (The dose of BEZ235 used in vivo was based on the specifications of Selleck Chemicals); BEZ235-PTX group: PTX (10 mg/kg weekly, ip) plus BEZ235 (45 mg/kg daily, oral treatment). Each group of rats received treatment for 4 weeks, and tumor size and body weight were measured every 3 days. Tumor volume was calculated according to the formula: V=L×W2×1/2 (V, volume; L, length of tumor; W, width of tumor). Mice were sacrificed after the treatment; tumor tissues were isolated; protein was extracted for WB detection or formalin-fixed; and GPC3 and Ki67 positive cells were detected by immunohistochemistry (IHC).
Statistical analysis
All analyses were performed with SPSS version 18.0 (SPSS Inc., Chicago, IL, USA). All experiments were performed at least 3 times in duplicates. One-way ANOVA or two-tailed unpaired Student t test were used to measure significant differences between the means. Data are expressed as mean ± SD. P<0.05 was taken as statistically significant.
Results
BEZ235 significantly inhibits paclitaxel-induced activation of PI3K/Akt/mTOR pathway in HCC cells
Abnormal activation of signaling pathways may lead to acquired resistance of liver cancer to paclitaxel and treatment failure [21]. Therefore, we tested paclitaxel with a phosphorylated molecular chip (CSP100) and performed a differential analysis. The results showed that several signaling pathways have different degrees of activation, in which the activation of PI3K/Akt/mTOR pathway was the most significant (Figure 1A and 1B). To verify this result, we treated HepG2 cells with paclitaxel (5 μM) for various times (0 h, 12 h, 24 h, 36 h, 48 h and 72 h) and measured the phosphorylation levels of PI3K, mTOR, Akt, eIF4EBP1 and S6K in HepG2 cells. WB results also showed that p-PI3Kp85, p-mTOR (Ser2481), p-Akt (Ser473), p-eIF4EBP1 and p-S6K1 were progressively elevated in paclitaxel-treated HepG2 cells; after reaching the peak expression at 24 hours, the levels of these molecules declined and stabilized at higher levels than in the control group (P<0.001) (Figure 1C and 1C1), which suggested that paclitaxel induced stress activation of the PI3K/mTOR signaling pathway in HepG2 cells. Next, we evaluated the inhibitory effect of BEZ235 (0, 0.0625, 0.125, 0.25, 0.5, 1 μM) on the PI3K/Akt/mTOR signaling pathway in HepG2 cell. The results shown that the BEZ235 inhibited phosphorylation of mTOR (Ser2481), Akt (Ser473) and PI3Kp85 in a concentration-dependent manner (Figure 1D and 1D1). Thus, we selected PI3K/mTOR dual inhibitor (BEZ235) to further determined the effect of paclitaxel (5 μM) combined with PI3K/mTOR dual inhibitor (BEZ235) (0.25, 0.5, 1 μM) on the PI3K/Akt/mTOR pathway. WB results confirmed that paclitaxel-activated PI3K/AKT/mTOR pathway was inhibited by BEZ235 (P<0.05) (Figure 1E, 1E1-E5). Therefore, we believe that BEZ235 can inhibit the stress activation of the PI3K/Akt/mTOR pathway in paclitaxel-treated HCC cells.
Figure 1.

BEZ235 significantly inhibited paclitaxel-induced activation of PI3K/Akt/mTOR pathway in HCC cells. A and B. The distribution of differential proteins on the PI3K/Akt and mTOR pathways in the paclitaxel-treated HepG2 signal map. C and C1. HepG2 cell was incubated with paclitaxel (5 μmol/L) for different time points, respectively. The cell lysates were gathered and the designated proteins (p-PI3Kp85, p-mTOR (Ser2481), p-Akt (Ser473), p-eIF4EBP1 and p-S6K1) were detected by WB, compare to the 0 h group. D and D1. HepG2 cell was incubated with various concentrations of BEZ235 for 24 h. The cell lysates were gathered and the designated proteins were detected by WB, compare to the 0 µM group. E, E1-E5. HepG2 cell was incubated under various concentrations of BEZ235 with a stable concertation of paclitaxel for 24 h. The cell lysates were gathered and the designated proteins (p-PI3Kp85, p-mTOR (Ser2481), p-Akt (Ser473), p-eIF4EBP1 and p-S6K1) were detected by WB, compared to that of the control and single drug groups. BEZ235-PTX: paclitaxel combined with BEZ235. Data expressed as mean ± SD, n=3 (*P<0.05; **P<0.01; ***P<0.001).
BEZ235 enhances the inhibitory effect of paclitaxel on proliferation of HCC cells
As mTOR plays an important role in cell growth, proliferation and metabolic regulation, we performed MTT and colony formation tests to investigate whether BEZ235 enhances the inhibitory effect of paclitaxel on proliferation of HCC cells. IC50s of paclitaxel and BEZ235 single treatment on HepG2 cells were analyzed (Figure 2A and 2B). When BEZ235 was co-treated with paclitaxel, the inhibition rate of paclitaxel on HepG2 cell was significantly increased. Like single paclitaxel and BEZ235 treatment, the inhibition effect of HepG2 cells both showed time-dose double dependence (Figure 2C-E). Moreover, the colony formation efficiency of the BEZ235-PTX treatment was significantly lower than that of the control and any single-drug group (P<0.001), while the paclitaxel (1 nM) or BEZ235 group (1 µM) was not significantly different than the control group (Figure 2F and 2G). Thus, BEZ235 enhanced the inhibitory effect of paclitaxel on proliferation of HCC cells.
Figure 2.
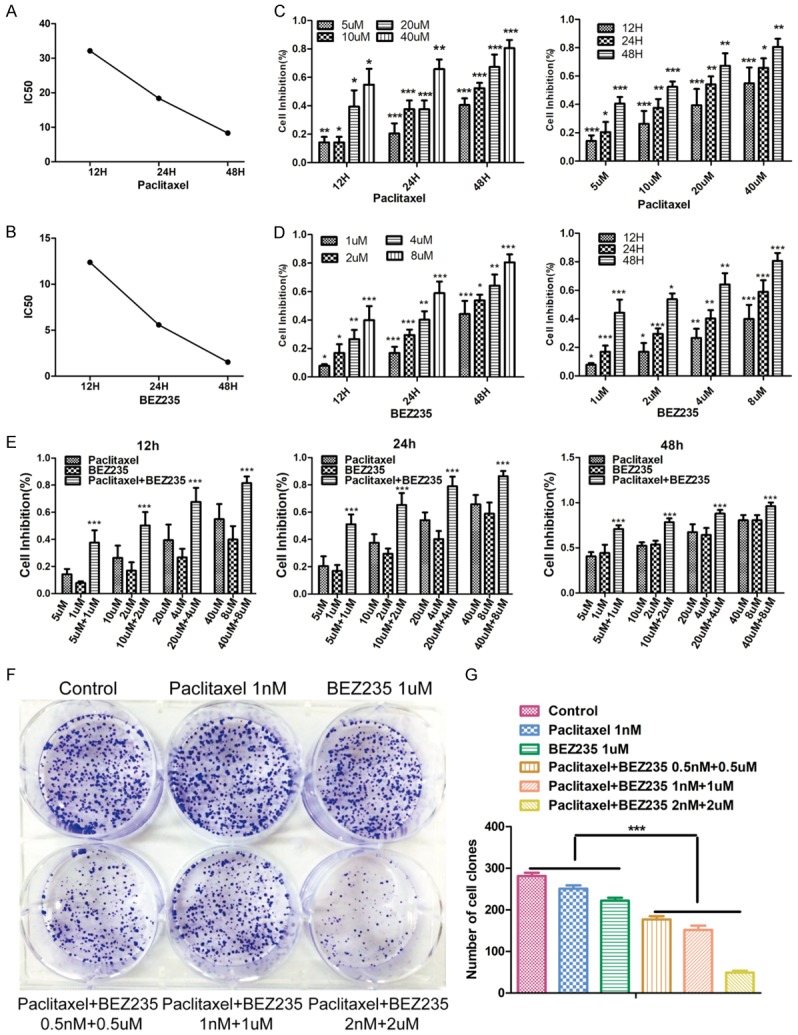
Effects of BEZ235 combined with PTX on HCC cell proliferation. A and B. The IC50 of HepG2 cell (paclitaxelIC50: 12 h: 25.959 µM; 24 h: 18.400 µM; 48 h: 8.311 µM. BEZ235IC50: 12 h: 12.387 µM; 24 h: 5.583 µM; 48 h: 1.535 µM). C-E. The inhibition rate of proliferation on HepG2 cells in each group at different concentrations (paclitaxel: 5 µM, 10 µM, 20 µM, 40 µM; BEZ235: 1 µM, 2 µM, 4 µM, 8 µM; BEZ235-PTX: 5 µM + 1 µM, 10 µM + 2 µM, 20 µM + 4 µM, 40 µM + 8 µM) and different time (12 h, 24 h, 48 h). F and G. Colony formation of HepG2 cell in different treatment (control; paclitaxel: 1 nM; BEZ235: 1 µM; paclitaxel + BEZ235: 0.5 nM + 0.5 µM; paclitaxel + BEZ235: 1 nM + 1 µM; paclitaxel + BEZ235: 2 nM + 2 µM). BEZ235-PTX: paclitaxel combined with BEZ235. Data expressed as mean ± SD, n=3 (*P<0.05; **P<0.01; ***P<0.001).
BEZ235 enhanced paclitaxel inhibition of HepG2 cellular migration
To explore whether BEZ235 can enhance the inhibitory effect of paclitaxel on HCC cell migration ability, scratch healing and transwell experiments were performed. The scratch healing assay showed that paclitaxel and BEZ235 monotherapy moderately inhibited cell migration (paclitaxel, 24 h: 66.5%, 48 h: 49.8%; BEZ235, 24 h: 77.2%, 48 h: 65.0%). When the two drugs were combined, the decrease in cell migration (24 h: 96.1%, 48 h: 93.6%) was greater than with either drug alone (P<0.05) (Figure 3A and 3C). Similarly, the transwell assay showed that the migration inhibition rate of paclitaxel was reduced to 38.8%; BEZ235 reduced this number to 40.0%, and co-treatment with the two drugs reduced it to 70.9% (Figure 3B and 3D). All these data revealed that BEZ235 enhanced paclitaxel inhibition of HepG2 cellular migration.
Figure 3.
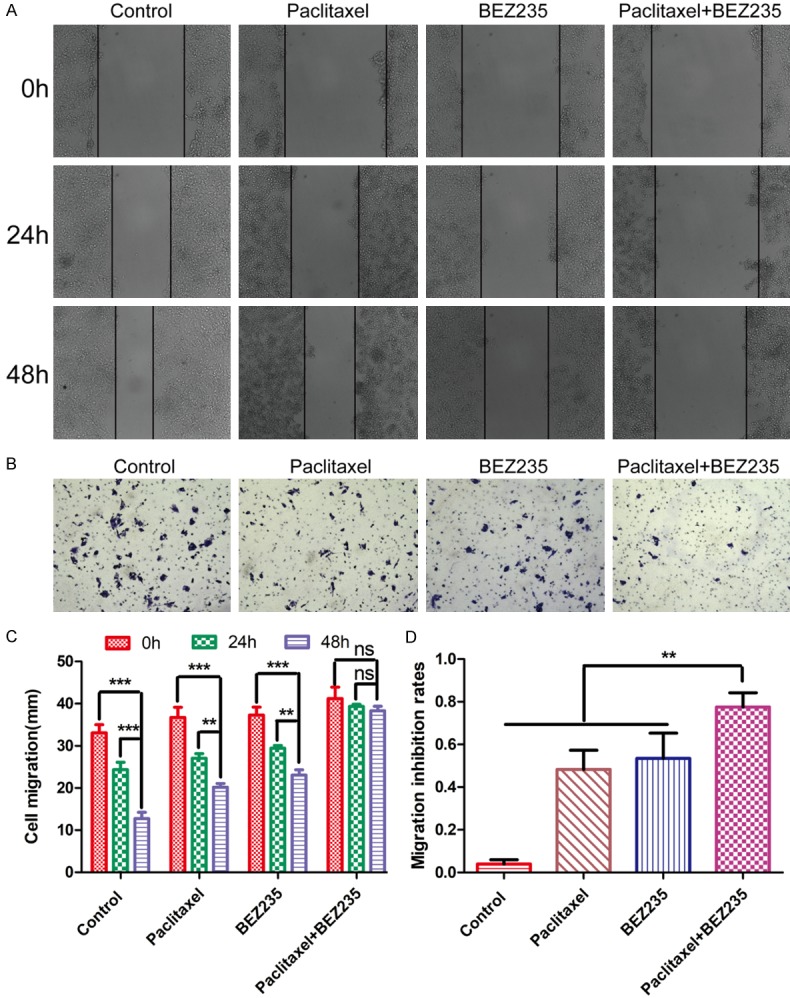
Effects of BEZ235 combined with paclitaxel on HCC cell migration. A and C. Degree of scratch healing at 0 h, 24 h and 48 h in each group (control; paclitaxel: 5 µM; BEZ235: 1 µM; paclitaxel + BEZ235: 5 µM + 1 µM). ×100 magnification. B and D. Cell migration ability at 24 h in different groups (control; pacliaxel: 1 nM; BEZ235: 1 µM. Paclitaxel + BEZ235: 1 nM + 1 µM). ×100 magnification. BEZ235-PTX: paclitaxel combined with BEZ235. Paclitaxel (5 µM), BEZ235 (1 µM). Data expressed as mean ± SD, n=3. ns = not significant (*P<0.05; **P<0.01; ***P<0.001).
BEZ235 combined with paclitaxel treatment enhanced accumulation of HepG2 cells in the G2/M phase
The effect of paclitaxel combined with BEZ235 on HepG2 cell-cycle progression was investigated by flow cytometric analysis. After treatment of HepG2 cells for 24 h with drugs, the proportions of cells in G2/M phase after BEZ235 treatment (6.13%) or paclitaxel treatment (16.18%) were higher than in the control group (2.62%) (P<0.05). With paclitaxel combined with BEZ235, the proportions of cells in G2/M phase (33.39%) were significantly higher than in the control or with any single drug treatment (P<0.001). Especially, the proportion of sub-G1 DNA content was significantly increased with BEZ235-paclitaxel treatment (P<0.01), which revealed that paclitaxel combined with BEZ235 promoted apoptosis of HepG2 cells (Figure 4A and 4B). Endogenous cyclin B1 and p-CDK1 control G2/M phase progression; thus, we evaluated the expression of cyclin B1 and p-CDK1. In the HepG2 cell line treated with BEZ235 plus paclitaxel, cyclin B1 and p-CDK1 were down-regulated. These results indicate that BEZ235-PTX blocks cell-cycle progression at the G2/M phase and implies that it might promote apoptosis (Figure 4C and 4D).
Figure 4.

Cell-cycle analysis of HepG2 cell treated with paclitaxel and BEZ235. A and B. HepG2 cell stained with propidium iodide after treatment with paclitaxel (16 nM), BEZ235 (1 μM) and BEZ235-PTX (16 nM + 1 uM) for 24 h. Cell cycle distribution was analyzed by flow cytometry. C and D. Total protein was extracted from HepG2 cell after treatment with paclitaxel (16 nM), BEZ235 (1 μM) and BEZ235-PTX (16 nM + 1 uM) for 24 h. BEZ235-PTX: paclitaxel combined with BEZ235. Data expressed as mean ± SD, n=3. ns = not significant (*P<0.05, **P<0.01, ***P<0.001).
BEZ235 increased paclitaxel-induced apoptosis in HepG2 cells
To investigate the effect of apoptosis induced by paclitaxel combined with BEZ235 in HepG2 cells, JC-1 staining and flow cytometry analysis for mitochondrial membrane potential (ΔΨm) were performed. In these experiments, the intensity ratio of green/red fluorescence of BEZ235-PTX (25.63%) was significantly higher than that of paclitaxel (13.10%), BEZ235 (10.21%) and control groups (6.03%) (P<0.01) (Figure 5A). In flow cytometry analysis, the green/red fluorescence intensity ratio of BEZ235-PTX (0.33%) was significantly higher than in the control (0.09%), paclitaxel (0.15%) or BEZ235 groups (0.04%) (P<0.05) (Figure 5C). Moreover, Annexin V/PI staining revealed that the apoptosis rate of BEZ235-PTX (76.34%) was higher than that of other groups (paclitaxel: 35.07%; BEZ235: 24.11%; control: 9.12%) (P<0.001) (Figure 5B).
Figure 5.

Effects of BEZ235-PTX on apoptosis of HepG2 cells. A. HepG2 cells were treated with each treatment group for 24 h, then counterstained with JC-1 and DAPI and observed with fluorescence microscopy. ×400 magnification. B. HepG2 cells were treated with each treatment group for 24 h, counterstained with DAPI/Annexin V-FITC/PI and observed with fluorescence microscopy. ×400 magnification. C. HepG2 cells were treated with each treatment group for 24 h, the ΔΨm were dectected by flow cytometry. D. The expression levels of mitochondria-associated pro-apoptotic proteins (Bad, Bax, Bak, Bim, Bid, Bik, puma) and caspase proteins (caspase-9, -3, -7) in each treatment group were analyzed by WB. E. Statistical graph of the ratio of apoptosis-related proteins to β-actin. F. Related apoptotic molecule regulation pattern chart. Treatment groups: control; paclitaxel: 25 µM; BEZ235: 1 µM; paclitaxel + BEZ235: 25 µM + 1 µM. Data expressed as mean ± SD, n=3 (*P<0.05; **P<0.01; ***P<0.001).
To further clarify the mechanisms of BEZ235-PTX induction of apoptosis, we evaluated the expression of the mitochondria apoptosis-related proteins (Bad, Bik, Bid, Bim, Bax, Bak and puma) by WB. The results revealed that the expression levels of apoptosis-related proteins after BEZ235-PTX treatment were higher than in control or with BEZ235 and paclitaxel single treatment (P<0.05) (Figure 5D and 5E). The expression level of caspase-3, -7, -9 and poly ADP ribose polymerase (PARP) proteins were further analyzed to evaluate the effect of BEZ235 on paclitaxel-induced apoptosis. WB results showed that the expression levels of caspase-3, -7, -9 and PARP in BEZ235 group and paclitaxel group were slightly higher than that of the control group, while the combination of BEZ235 and paclitaxel produced an additive effect on these proteins (P<0.05) (Figure 5D and 5E). Consequently, we concluded that BEZ235 increased paclitaxel-induced apoptosis in HepG2 cells (Figure 5F).
BEZ235 significantly enhanced the anti-tumor effect of paclitaxel in vivo by PI3K/mTOR double inhibition
Anti-tumor effects of paclitaxel combined with BEZ235 in vivo were evaluated also in a murine xenograft model using HepG2 cells. Tumor tissues of each group were isolated and collected after 28-day treatment of tumor-bearing mice. The tumor growth of animals treated BEZ235-PTX was significantly inhibited compared with that in the paclitaxel, BEZ235 and control groups (P<0.01) (Figure 6A). The molecular mechanism of the anti-tumor activity of BEZ235-PTX was further examined by WB analysis of protein lysates from HepG2 xenografts. The results showed that BEZ235-PTX significantly reduced the phosphorylation levels of PI3K, Akt, mTOR, eIF4EBP1 and S6K compared with levels in the control, paclitaxel and BEZ235 groups (P<0.01) (Figure 6B). IHC staining showed that the proportion of Ki-67- and GPC3-positive HepG2 cells was significantly reduced in the BEZ235-PTX-treated animals compared with the proportion in control or each single drug groups, which indicated that cell proliferation and activity of HepG2 cells were inhibited (Figure 6C). Thus, we concluded that BEZ235 significantly enhanced the antitumor effect of paclitaxel in HCC in vivo, at least in part by inhibiting the PI3K/Akt/mTOR pathway.
Figure 6.
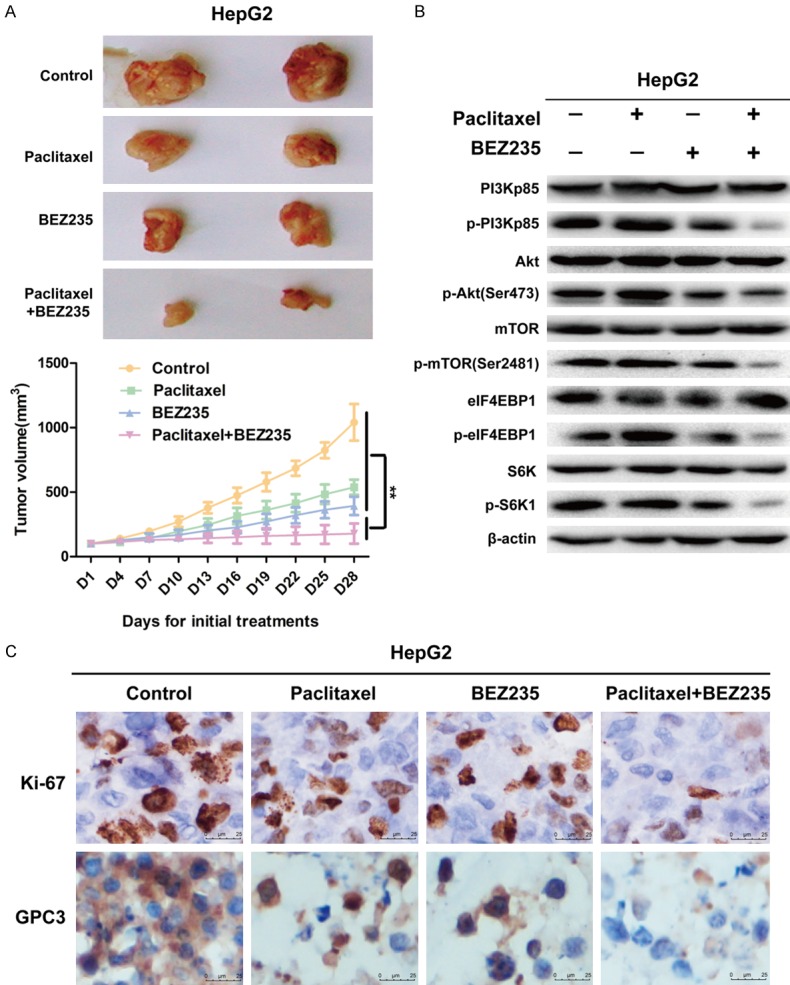
BEZ235 significantly enhanced the anti-tumor effect of paclitaxel in vivo by PI3K/mTOR double inhibition. A. tumor tissues of each group (control group: 100 μl saline daily, ip; PTX group: 10 mg/kg weekly, ip; BEZ235 group: 45 mg/kg daily, oral treatment; BEZ235-PTX group: PTX: 10 mg/kg weekly, ip and BEZ235: 45 mg/kg daily, oral treatment) were isolated and collected after 28-day-treatment of tumor-bearing mice. Tumor volume expressed as mean ± SD. n=5. B. Partial tumor tissue was lysed to extract tissue protein for WB analysis. C. Each treatment groups IHC analysis results of Ki-67 and GPC3. Data expressed as mean ± SD, n=3 (*P<0.05; **P<0.01; ***P<0.001).
Discussion
HCC resistance to paclitaxel may involve multiple molecular mechanisms, including activation of the PI3K/Akt/mTOR and MAPK pathway and overexpression of P-glycoprotein (P-gp) [20-24]. The PI3K/Akt/mTOR signaling pathway is frequently activated in human tumors, such as gastric cancer, ovarian cancer and HCC [25,26]. Activation of the PI3K/Akt/mTOR pathway induces cell proliferation, migration, regulation of the cell cycle and promotes cell apoptosis [27]. Thus, we applied a novel PI3K/mTOR dual inhibitor, BEZ235, combined with paclitaxel on HCC cells. Paclitaxel treatment increased phosphorylation of PI3Kp85, mTOR, Akt, eIF4EBP1 and S6K1 in HepG2 cells, while BEZ235 significantly enhanced the anti-proliferative, anti-migration and pro-apoptotic effects of paclitaxel. In in vivo work, tumor growth was inhibited more in HepG2 xenograft models when paclitaxel combined with BEZ235 was administered than with either paclitaxel or BEZ235 alone.
The classic anti-mitotic agent paclitaxel can inhibit cell mitosis and arrest cell cycle in G2/M phase to induce cell death by binding to β-tubulin and promoting tubulin polymerization [6]. However, paclitaxel treatment often activates the PI3K/Akt/mTOR pathway in cancer cells. Akt and mTOR signaling is involved in cell survival and proliferation [28], so activation of the PI3K/Akt/mTOR pathway can impair the anti-cancer effect of paclitaxel and even induce paclitaxel resistance. In this research, CSP100 antibody microarray and WB results both revealed that the levels of p-PI3Kp85, p-Akt (Ser473), p-mTOR (Ser2481), p-S6K1 and p-eIF4EBP1 in HepG2 cells were up-regulated after 24 h treatment with paclitaxel. Activated PI3Kp85 phosphorylates Akt (p-Akt) reduces tumor apoptosis and promotes proliferation by up-regulating phosphorylation of downstream factors, such as kinases and transcription factors [29]. p-Akt (Ser473) activating the mTORC1 signaling, then activated mTORC1 further phosphorylates and activates S6K1 and eIF4EBP1, which initiates translation of the protein and promotes cell survival [30]. Our in vivo results also showed that the phosphorylation levels of Akt, mTOR, S6K and eIF4EBP1 were upregulated after paclitaxel treatment. Cyclin B1/CDK1 complex is an important enzyme that promotes the conversion to the G2/M phase. When p-Akt (Ser473) induces CDK1 phosphorylation (p-CDK1), then p-CDK1 inhibits the activity of the Cyclin B1/CDK1 complex, which cannot promote cell-cycle progression [31]. Our cell-cycle experimental results indicated that the expression levels of cyclin B1 and p-CDK1 in the paclitaxel-treated animals were higher than in controls. p-Akt (Ser473) inhibits the activity of caspase-9 hydrolase and prevents the initiation of the apoptotic cascade to exert an anti-apoptotic effect [39]. Therefore, we conjectured that the activated PI3K/Akt/mTOR pathway may be the main reason for the reduction of paclitaxel sensitivity to HCC cells (Figure 7).
Figure 7.
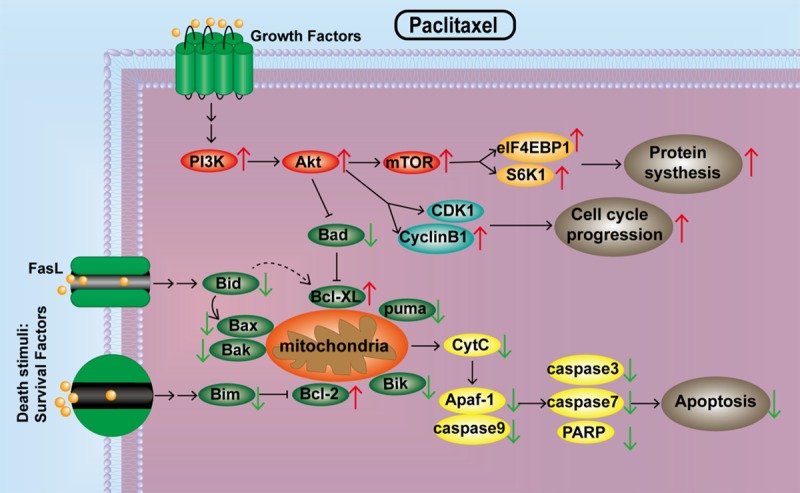
Paclitaxel abnormally activates the PI3K/Akt/mTOR pathway, leading to desensitization of HCC cells.
To inhibit the PI3K/Akt/mTOR pathway stress activated by paclitaxel, we selected mTOR and PI3K dual inhibitor (BEZ235) combined with paclitaxel on the HepG2 cell. The results revealed that the expression levels of p-PI3Kp85, p-mTOR (Ser2481), p-Akt (Ser473), p-eIF4EBP1 and p-S6K1 were significantly down-regulated. Moreover, the proportion of G2/M phase in paclitaxel combined with BEZ235 treatment was significantly higher than that of the paclitaxel treatment. The key molecules regulating G2/M phase transition are cyclin B1 and CDK1. When Akt is inhibited, it can cause CDK1 dephosphorylation, enhance cyclin B1/CDK1 activity, stimulate G2/M transformation, arrest cell cycle in the G2/M phase and inhibit cell-cycle progression [32]. Thus, these results confirmed that BEZ235 abolished the activation of PI3K/Akt/mTOR signaling by paclitaxel and inhibited the proliferation, migration and regulation of cell-cycle progression of paclitaxel-treated HepG2 cells.
More importantly, mitochondrial apoptosis experiments (JC-1 staining) and Annexin-V FITC/PI staining results also showed that BEZ235 significantly increased paclitaxel-induced apoptosis rate in HepG2 cells. The levels of pro-apoptotic proteins (Bad, Bid, Bik, Bax and Bim) were much higher in BEZ235-PTX treatment than that of paclitaxel and BEZ235 single groups. The WB results further showed that BEZ235-PTX treatment significantly amplified the activity of caspase pathway. In the process of inducing apoptosis, after p-Akt (Ser473) was inhibited, the Bcl-2 family member Bad was dephosphorylated, preventing Bcl-XL from initiating anti-apoptosis [33,34]. The pro-apoptotic proteins Bad, Bid, Bik, Bax and Bim can be present in the cytoplasm and translocate into the mitochondria after death signal transduction, where they promote cytochrome C (Cyt C) release [35-38]. After DNA damage, activated p53 induces Puma transcription, and Puma interacts with Bcl-2/Bcl-XL also to induce Cyt C release. Cyt C is released from the mitochondria into the cytoplasm, which forms an apoptotic complex with caspases-9 through the multimerization of Apaf-1 factor. Caspase-9 cleaves and activates the downstream effect of caspase (including caspase-3 and -7) to achieve apoptosis [39-41]. The xenograft mice results of in vivo experiments showed that paclitaxel combined with BEZ235 inhibited PI3K/Akt/mTOR pathway activation, suppressed cell proliferative and promoted apoptotic effects in HCC. Consequently, high levels of expression of anti-apoptosis related proteins and cleaved-caspase signaling in the BEZ235-PTX co-treatment strongly confirmed that BEZ235 promoted apoptosis of paclitaxel-treated HCC HepG2 cells (Figure 8). While we admit that our study has limitations. The study of only one PI3K/Akt/mTOR pathway may not have been enough. Therefore, crosstalk between PI3K/Akt/mTOR with other pathways will be explored in our future research, as will in-depth exploration of the mechanism of paclitaxel desensitization.
Figure 8.
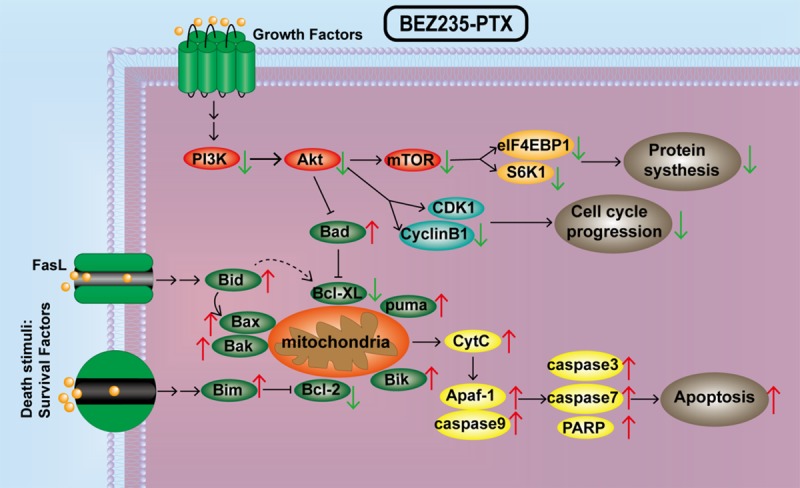
BEZ235 enhances paclitaxel sensitizes on HepG2 cells through inhibiting the PI3K/Akt/mTOR pathway. PTX = paclitaxel.
In conclusion, our results confirmed that acquired desensitization to paclitaxel in HepG2 cells involves the PI3K/mTOR signaling pathway and cell-death signaling, while BEZ235 mainly abolishes the activation of the PI3K/Akt/mTOR pathway induced by paclitaxel. Blocking PI3K/mTOR signaling inhibited cell proliferation and migration, arrested cell cycle in G2/M phase, and promoted apoptosis, which enhanced the chemotherapy sensitivity of paclitaxel on HCC. These findings may have implications for the chemotherapy of HCC in clinical practice.
Acknowledgements
The National Natural Science Fund of China (81872017, 81572431), Anhui Provincial Science and Technology program (1604a0802094), University Natural Science Research Project of Anhui Province (KJ2018ZD011, KJ2019A0305, KJ2019A0093) and Huainan Science and Technology Project (2017B41) funded this research.
Disclosure of conflict of interest
None.
References
- 1.Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H, Hall MN. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. 2017;32:807–823. e12. doi: 10.1016/j.ccell.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Dou C, Xu Q, Liu J, Wang Y, Zhou Z, Yao W, Jiang K, Cheng J, Zhang C, Tu K. SHMT1 inhibits the metastasis of HCC by repressing NOX1-mediated ROS production. J Exp Clin Cancer Res. 2019;38:70. doi: 10.1186/s13046-019-1067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allain C, Angenard G, Clément B, Coulouarn C. Integrative genomic analysis identifies the core transcriptional hallmarks of human hepatocellular carcinoma. Cancer Res. 2016;76:6374–6381. doi: 10.1158/0008-5472.CAN-16-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, Znaor A, Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Wang X, Li B, Hou Y, Yang J, Yi L. Development of a novel morphological paclitaxel-loaded PLGA microspheres for effective cancer therapy: in vitro and in vivo evaluations. Drug Deliv. 2018;25:166–177. doi: 10.1080/10717544.2017.1422296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu B, Liang Y, Tan Y, Xie C, Shen J, Zhang M, Liu X, Yang L, Zhang F, Liu L, Cai S, Huai , Zheng D, Zhang R, Zhang C, Chen K, Tang X, Sui X. Genistein-loaded nanoparticles of star-shaped diblock copolymer mannitol-core PLGA-TPGS for the treatment of liver cancer. Mater Sci Eng C Mater Biol Appl. 2016;59:792–800. doi: 10.1016/j.msec.2015.10.087. [DOI] [PubMed] [Google Scholar]
- 8.Zhu M, Li W, Lu Y, Dong X, Chen Y, Lin B, Xie X, Guo J, Li M. Alpha fetoprotein antagonizes apoptosis induced by paclitaxel in hepatoma cells in vitro. Sci Rep. 2016;6:26472. doi: 10.1038/srep26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco E, Sangai T, Wu S, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, Ferrari M. Colocalized delivery of rapamycin and paclitaxel to tumors enhances synergistic targeting of the PI3K/Akt/mTOR pathway. Mol Ther. 2014;22:1310–1319. doi: 10.1038/mt.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang SQ, Wang C, Chang LM, Zhou KR, Wang JW, Ke Y, Yang DX, Shi HG, Wang R, Shi XL, Ma LY, Liu HM. Geridonin and paclitaxel act synergistically to inhibit the proliferation of gastric cancer cells through ROS-mediated regulation of the PTEN/PI3K/Akt pathway. Oncotarget. 2016;7:72990–73002. doi: 10.18632/oncotarget.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang X, Zhou S, Tao X, Wang J, Wang F, Liang Y. Targeted delivery of docetaxel via Pi-Pi stacking stabilized dendritic polymeric micelles for enhanced therapy of liver cancer. Mater Sci Eng C Mater Biol Appl. 2017;75:1042–1048. doi: 10.1016/j.msec.2017.02.098. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Niu J, Li J, Shen X, Shen S, Straubinger RM, Qu J. Temporal effects of combined birinapant and paclitaxel on pancreatic cancer cells investigated via large-scale, ion-current-based quantitative proteomics (IonStar) Mol Cell Proteomics. 2018;17:655–671. doi: 10.1074/mcp.RA117.000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco E, Sangai T, Wu S, Hsiao A, Ruiz-Esparza GU, Gonzalez-Delgado CA, Cara FE, Granados-Principal S, Evans KW, Akcakanat A, Wang Y, Do KA, Meric-Bernstam F, Ferrari M. Colocalized delivery of rapamycin and paclitaxel to tumors enhances synergistic targeting of the PI3K/Akt/mTOR pathway. Mol Ther. 2014;22:1310–1319. doi: 10.1038/mt.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang X, Lyu Y, Li A, Liang Y, Zheng D. Therapeutic effect of sorafenib-loaded TPGS-b-PCL nanoparticles on liver cancer. J Biomed Nanotechnol. 2018;14:396–403. doi: 10.1166/jbn.2018.2529. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer. 2014;13:98. doi: 10.1186/1476-4598-13-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenerson HL, Yeh MM, Kazami M, Jiang X, Riehle KJ, McIntyre RL, Park JO, Kwon S, Campbell JS, Yeung RS. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology. 2013;144:1055–65. doi: 10.1053/j.gastro.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, Liu Y, Gui B, Sini M, Smith JW, Dombrowski F, Calvisi DF, Evert M, Chen X. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice. Hepatology. 2015;61:200–13. doi: 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018;13:76. doi: 10.1186/s13014-018-1020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aung W, Tsuji AB, Sudo H, Sugyo A, Ukai Y, Koudav K, Kurosawa Y, Furukawa T, Saga T, Higashi T. Combined treatment of pancreatic cancer xenograft with 90Y-ITGA6B4-mediated radioimmunotherapy and PI3K/mTOR inhibitor. World J Gastroenterol. 2017;232:7551–7562. doi: 10.3748/wjg.v23.i42.7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Shao J, Li X, Cui L, Tan Z. Docetaxel promotes cell apoptosis and decreases SOX2 expression in CD133-expressing hepatocellular carcinoma stem cells by suppressing the PI3K/AKT signaling pathway. Oncol Rep. 2019;41:1067–1074. doi: 10.3892/or.2018.6891. [DOI] [PubMed] [Google Scholar]
- 21.Villaruz LC, Socinski MA. Is there a role of nab-paclitaxel in the treatment of advanced non-small cell lung cancer? The data suggest yes. Eur J Cancer. 2016;56:162–171. doi: 10.1016/j.ejca.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barve A, Jin W, Cheng K. Prostate cancer relevant antigens and enzymes for targeted drug delivery. J Control Release. 2014;187:118–132. doi: 10.1016/j.jconrel.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang X, Chen L, Li A, Cai S, Zhang Y, Liu X, Jiang Z, Liu X, Liang Y, Ma D. Anti-GPC3 antibody modified sorafenib loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Deliv. 2018;25:1484–1494. doi: 10.1080/10717544.2018.1477859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Cheng Q, Owusu-Ansah KG, Chen J, Song G, Xie H, Zhou L, Xu X, Jiang D, Zheng S. Cabazitaxel, a novel chemotherapeutic alternative for drug-resistant hepatocellular carcinoma. Am J Cancer Res. 2018;8:1297–1306. [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HJ, Venkatarame Gowda Saralamma V, Kim SM, Ha SE, Raha S, Lee WS, Kim EH, Lee SJ, Heo JD, Kim GS. Pectolinarigenin induced cell cycle arrest, autophagy, and apoptosis in gastric cancer cell via PI3K/AKT/mTOR signaling pathway. Nutrients. 2018;10 doi: 10.3390/nu10081043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aziz AUR, Farid S, Qin K, Wang H, Liu B. PIM kinases and their relevance to the PI3K/AKT/mTOR pathway in the regulation of ovarian cancer. Biomolecules. 2018;8 doi: 10.3390/biom8010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing S, Yu W, Zhang X, Luo Y, Lei Z, Huang D, Lin J, Huang Y, Huang S, Nong F, Zhou C, Wei G. Isoviolanthin extracted from dendrobium officinale reverses TGF-β1-Mediated epithelial-mesenchymal transition in hepatocellular carcinoma cells via deactivating the TGF-β/Smad and PI3K/Akt/mTOR signaling pathways. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19061556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Xie C, Li A, Liu X, Xing Y, Shen J, Huo Z, Zhou S, Liu X, Xie Y, Cao W, Ma Y, Xu R, Cai S, Tang X, Ma D. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11:5134–5149. [PMC free article] [PubMed] [Google Scholar]
- 29.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Zhu M, Wang Q, Hou Y, Li L, Weng H, Zhao Y, Chen D, Ding H, Guo J, Li M. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018;9:1027. doi: 10.1038/s41419-018-1036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakayama S, Torikoshi Y, Takahashi T, Yoshida T, Sudo T, Matsushima T, Kawasaki Y, Katayama A, Gohda K, Hortobagyi GN, Noguchi S, Sakai T, Ishihara H, Ueno NT. Prediction of paclitaxel sensitivity by CDK1 and CDK2 activity in human breast cancer cells. Breast Cancer Res. 2009;11:R12. doi: 10.1186/bcr2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Fan M, Candas D, Zhang T, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, Duru N, He F, Chen M, Finkel T, Weinstein LS, Li JJ. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014;29:217–32. doi: 10.1016/j.devcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–75. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szlyk B, Braun CR, Ljubicic S, Patton E, Bird GH, Osundiji MA, Matschinsky FM, Walensky LD, Danial NN. A phospho-BAD BH3 helix activates glucokinase by a mechanism distinct from that of allosteric activators. Nat Struct Mol Biol. 2013;21:36–42. doi: 10.1038/nsmb.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bao L, Zhang M, Han S, Zhan Y, Guo W, Teng F, Liu F, Guo M, Zhang L, Ding G, Wang Q. MicroRNA-500a promotes the progression of hepatocellular carcinoma by post-transcriptionally targeting BID. Cell Physiol Biochem. 2018;47:2046–2055. doi: 10.1159/000491472. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Oesterlund EJ, Chi X, Pogmore J, Leber B, Andrews DW. Bim escapes displacement by BH3-mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. Elife. 2017;8 doi: 10.7554/eLife.37689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan H, Chen L, Sui X, Wu B, Zhou S, Li A, Zhang Y, Liu X, Wang D, Cai S, Liu X, Liang Y, Tang X. Enhanced delivery of sorafenib with anti-GPC3 antibody-conjugated TPGS-b-PCL/Pluronic P123 polymeric nanoparticles for targeted therapy of hepatocellular carcinoma. Mater Sci Eng C Mater Biol Appl. 2018;91:395–403. doi: 10.1016/j.msec.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 38.Gu Z, Serradj N, Ueno M, Liang M, Li J, Baccei ML, Martin JH, Yoshida Y. Skilled movements require non-apoptotic Bax/Bak pathway-mediated corticospinal circuit reorganization. Neuron. 2017;94:626–641. e4. doi: 10.1016/j.neuron.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen D, Tong J, Yang L, Wei L, Stolz D, Yu J, Zhang J, Zhang L. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci U S A. 2018;115:3930–3935. doi: 10.1073/pnas.1717190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kook S, Zhan X, Cleghorn WM, Benovic JL, Gurevich VV, Gurevich EV. Caspase-cleaved arrestin-2 and BID cooperatively facilitate cytochrome C release and cell death. Cell Death Differ. 2014;21:172–84. doi: 10.1038/cdd.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beauvais DM, Jung O, Yang Y, Sanderson RD, Rapraeger AC. Syndecan-1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: prevention by synstatinIGF1R inhibits tumor growth. Cancer Res. 2016;76:4981–93. doi: 10.1158/0008-5472.CAN-16-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]