

Abstract
Hypoxia and apoptosis are involved in the pathogenesis of Alzheimer’s disease (AD). Hypoxia induces the formation of amyloid precursor protein in neurons, leading to the abnormal deposition of β-amyloid protein and hyperphosphorylation of Tau. Such changes increase the risk of AD. In the present study, a cellular model of hypoxia-induced AD was established by exposing HT-22 mouse hippocampal neurons to the chemical hypoxia-mimicking agent cobalt chloride (CoCl2). It was found that hypoxia increased neuronal apoptosis. Hypoxia caused an abnormal increase in the expression of the intracellular calcium channel protein Orai1 and cyclin-dependent kinase 5 (CDK5), resulting in hyperphosphorylation of Tau. Treatment with small-interfering RNA against Orai1 (siOrai1) or an Orai1-overexpression plasmid effectively intervened the CDK5-mediated hyperphosphorylation of Tau. In summary, following hypoxic injury of neuron, the Orai1-induced expression of CDK5 leads to Tau hyperphosphorylation. Tau hyperphosphorylation is an important pathophysiological manifestation in AD patients. These results indicated that hypoxia induces HT-22 cell death by Orai1/CDK5 pathway mediated Tau hyperphosporylation. This study simulated the pathological process associated with AD and proposed that hypoxia of intravascular cells with normal blood oxygen saturation might be one of a pathogenic mechanisms of AD. Therefore, this work may provide a new theoretical basis for AD prevention and treatment.
Keywords: Alzheimer’s disease, hypoxia, hippocampal neuron, tau protein, orai1 protein, cyclin-dependent kinase 5
Introduction
Alzheimer’s disease (AD) is a progressive neurological degenerative disease with an insidious onset. Clinically, AD is characterized by dysmnesia, aphasia, apraxia, agnosia, visuospatial skill impairment, executive dysfunction, personality and behavioral changes, and other manifestations of generalized dementia [1,2]. The pathological features of AD include intraneuronal neurofibrillary tangles in the cerebral cortex and encephalic region, Tau protein hyperphosphorylation-induced neuronal death, and formation of extracellular senile plaques [3].
Hypoxia is a common pathophysiological process, which can be caused by cardiovascular problems, hematological diseases, respiratory dysfunction, poisoning, or environmental conditions. Hypoxia can lead to cell dysfunction, affect the functions of organs and systems, especially the central nervous system [4]. Low oxygen concentrations may cause oxidative stress to induce AD [5]. Kawahara et al. found that hypoxia enhances the conversion of amyloid precursor protein into β-amyloid protein and increases calcium channel expression, resulting in an imbalance of intracellular calcium homeostasis [6]. Sun et al. reported that hypoxia increases the expression of β-secretase and induces the production of β-amyloid protein, thereby causing dementia [7]. Hypoxia experiments, including the two described above, have been performed in either an in vitro hypoxic environment or by ligation of supplying vessels in experimental animals; however, hypoxia simulation experiments have not yet been conducted in vitro by exposing cells to hypoxia-inducing chemicals. Because AD occurs mainly in people with normal blood oxygen saturation, in vitro cellular experiments with chemical hypoxia are necessary to exclude the effects of environmental and vascular factors and explore the correlation between cellular hypoxia and AD.
Changes in intracellular calcium ion concentration ([Ca2+]i) are involved in many cellular physiopathological responses [8,9]. The calcium influx mechanism mediated by the depletion of endoplasmic reticulum (ER) calcium stores, termed store-operated Ca2+ entry (SOCE), is an area of intense focus in calcium influx research [10-13]. The two main proteins in SOCE are stromal interaction molecule 1 (STIM1) and Orai1 (also known as CRACM1). STIM1, which is a calcium sensor located on the ER, senses intracellular calcium release and calcium store depletion. Following translocation to ER-plasma membrane (PM) junctions, STIM1 then interacts with PM molecules to form calcium channels, ultimately triggering calcium influx. Orai1 is scattered on the cell membrane during quiescence but can be recruited by STIM1 to aggregate and bind to the C-terminus of STIM1 to form the calcium channel. The co-expression of STIM1 and Orai1 is therefore the basis of calcium influx channel function [11]. In cardiomyocytes, hypoxia increases Orai1 protein levels, resulting in an increase in apoptosis [12]; however, the effect of hypoxia on Orai1 in neuronal cells remains unclear and requires clarification through further investigation.
The calpain/p35-p25/CDK5 signaling pathway induces retinal cell apoptosis [14]. CDK5 is a member of the CDK family and is located on regions 3 and 6 of chromosome 7 [15,16]. Unlike other CDK family proteins, CDK5 binds to p35 or p39 on the cell membrane to form active dimmers [17]. In the brain, CDK5 is primarily activated by p35. After p35 is proteolytically cleaved to yield p25, CDK5/p25 becomes completely activated, leading to apoptosis [18]. In a study of rat pulmonary microvascular endothelial cells, Kim et al. found that hypoxia induced high expression of CDK5 [19]. In addition, CDK5 may cause Tau hyperphosphorylation, leading to the production of neurotoxic factors, that damage neurons, such factors include β-amyloid deposition [20], hypoxia-ischemia [21,22], oxidative stress [23] and inflammation [24]. It has been shown that Tau protein hyperphosphorylation is closely related to AD [25], hyperphosphory of tau protein in neurofibrillary tangles are neuropathological features of AD, one of the principal [26]. To date, however, there is little research on the correlation between Orai1 protein and CDK5 in AD, supporting the need for clarification through further investigations.
In this study, the mouse hippocampal neuron-derived cell line HT-22 was used as a cellular model of AD. HT-22 cells were treated with the hypoxia-mimicking agent cobalt chloride (CoCl2) to establish an in vitro cellular model of chemical hypoxia-induced neuronal injury. This model excluded environmental and vascular factors and was used to investigate the effect of hypoxia on neuronal proliferation, apoptosis, and production of Orai1 and CDK5. Additionally, the correlation between AD and hypoxia was explored. This study provided insight into the etiology of AD and lays the groundwork for future efforts to improve clinical treatment and prevention of AD.
Materials and methods
Cell culture and treatments
HT-22 cells were obtained from iCell Bioscience Inc. (Shanghai, China) and routinely grown in MEM high-glucose medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco, Waltham, MA, USA), 100 KU/L penicillin and 100 mg/L streptomycin (Beyotime Institute of Biotechnology, Hubei, China) and cultured at 37°C under 5% CO2 in a humidified atmosphere.
For the control group, HT-22 cells were cultured under the above conditions. For the CoCl2 group, HT-22 cells were treated with the hypoxia-mimicking agent CoCl2 (Invitrogen) to establish an in vitro cellular model of chemical hypoxia-induced neuronal injury. The culture conditions were as described above. For the transfection group, siOrai1 interference fragment (GenePharma, Shanghai, China) or the Orai1 overexpression plasmid (GenePharma) was transfected into HT-22 cells. The transfected cells were cultured in serum-free MEM high-glucose medium at 37°C, 5% CO2, and 100% saturated humidity, while the cells in the transfection+CoCl2 group were transfected and then treated with CoCl2. These cells were cultured at 37°C under 5% CO2, and 100% saturated humidity atmosphere.
Cell transfection
For transfection of HT-22 cells with the siOrai1 or the Orai1 overexpression plasmid, cells in the logarithmic growth phase were plated in 6-well culture plates at a density of 2×105 cells/well and allowed to adhere at 37°C for 12 h. The siOrai1 interference fragment or the Orai1 overexpression plasmid was then transfected into the HT-22 cells using LipofectamineTM2000 (Invitrogen) for 6 h according to the manufacturer’s instructions.
Assessment of cell viability
Viability of cells was assessed using the MTT assay (Beyotime Institute of Biotechnology). HT-22 cells (5×103 cells/well) were seeded in a 96-well culture plate. Cells were pretreated with CoCl2 for 12, 24, or 48 h. After a corresponding 12-, 24-, or 48-h incubation, 20 µl of a 5 mg/ml stock solution of MTT was added into the culture medium, and cells were incubated in the dark for 4 h at 37°C for formazan formation. Then, 100 µl DMSO (Invitrogen) was added into the culture medium to dissolve the formazan crystals, and the OD values were spectrophotometrically determined at 490 nm. The percentage of HT-22 cell viability was measured and normalized to the untreated control value, which was set at 100%.
Apoptosis analysis
Cell apoptosis was analyzed using an Annexin V-FITC/PI apoptosis detection kit (Beyotime Institute of Biotechnology). HT-22 cells (2×105 cells/well) were seeded in a 6-well culture plate. Cells were pretreated with CoCl2 for 24 h, and then incubated with AnnexinV-FITC/PI for 15 min in the dark. The fluorescence of each sample was immediately analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Western blotting
The levels of Orai1 (13130-1-AP, Proteintech, Rosemont, IL, USA), CDK5 (10430-1-AP, Proteintech), Bcl-2 (12789-1-AP, Proteintech), cleaved-caspase-3 (19677-1-AP, Proteintech), total-Tau (T-Tau; 10274-1-AP, Proteintech), and phosphorylated-Tau (P-Tau; Ser396, AF3148, Affinity Biotech, Exeter, UK) were detected in HT-22 cells by western blot analysis. After treatment under various conditions, the protein content of the cells was determined by the bicinchoninic acid (BCA) method (Beyotime Institute of Biotechnology). Equivalent amounts of proteins were separated by SDS-PAGE and subsequently transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The membrane was blocked with blocking solution and incubated with the indicated antibodies. An ECL kit (Beyotime Institute of Biotechnology) was used to visualize the membrane immunoreactivity. The quantification of protein was performed using a computerized imaging program Quantity One (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All statistical analyses were conducted using GraphPad Prism 4.0 (GraphPad Software, La Jolla, CA, USA). Results were expressed as means ± standard deviation of the mean (SD). Student’s t-test was used to evaluate the differences between groups. A value of P<0.05 was considered to be statistically significant.
Results
Hypoxia inhibits cell proliferation and promotes cell apoptosis
In order to explore the effects of chemically induced hypoxia on neuronal cells, HT-22 hippocampal neuronal cells were treated with different concentrations of the hypoxia mimic CoCl2 (0-150 μM) for 0-48 h. The proliferation rate of cells in the CoCl2-treated group was significantly decreased compared with the control group (P<0.05). Cell viability in these CoCl2-treated cells was inhibited in a dose- and time-dependent manner with an IC50 of 100 µM at 24 h (Figure 1A). In addition, apoptosis of HT-22 cells was induced in a concentration-dependent manner by treatment with CoCl2 (0-150 μM), as determined by Annexin V-FITC/PI double-staining flow cytometry (Figure 1B and 1C). Based on these experiments, we selected 100 μM as the concentration of CoCl2 for use in the following experiments.
Figure 1.
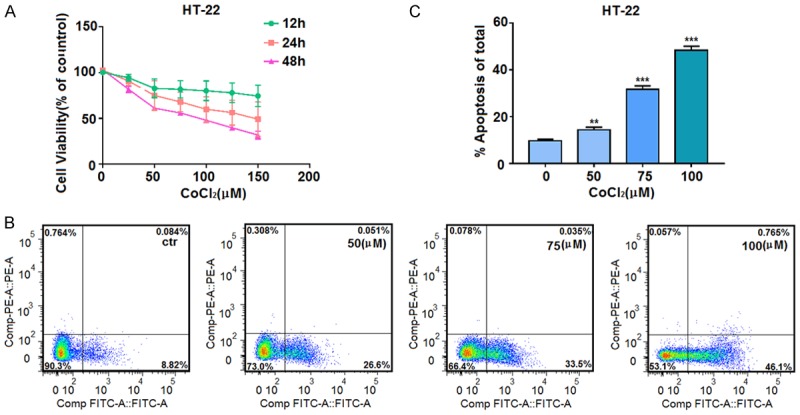
Hypoxia treatment decreases cell viability and induces apoptosis in HT-22 neuronal cells. A. Cells seeded into 96-well plates were treated with the indicated concentrations of CoCl2 for 12, 24, or 48 h. Cell viability was assessed by MTT assay, and the absorbance values of the treated samples are shown as percentages of the absorbance values of the control samples. B, C. After incubation with the indicated concentrations of CoCl2 for 24 h, cells were stained with Annexin V-FITC/PI, and cell apoptosis was then quantified by flow cytometer. Data are expressed as means ± SD from three independent experiments. **P<0.01 and ***P<0.001 vs. Control (0 µm CoCl2).
Detection of Orai1, CDK5, Bcl-2, and cleaved-caspase-3 protein levels by western blotting of HT-22 neuronal cells grown with hypoxia chemical mimics
As apoptosis was increased in the HT-22 cells grown with the hypoxia mimic CoCl2, we sought to examine the effects of this treatment on proteins relevant for apoptosis. HT-22 cells were treated with 50, 75, or 100 μM CoCl2, and levels of proteins of interest were determined by western blot. Compared with the control group, HT-22 cells treated with different concentrations of CoCl2 exhibited decreased expression of the anti-apoptotic protein Bcl-2 and increased expression of the apoptotic protein cleaved-caspase-3 as well as Orai1 (the calcium channel protein) and CDK5 (protein that activates Tau phosphorylation) (Figure 2).
Figure 2.
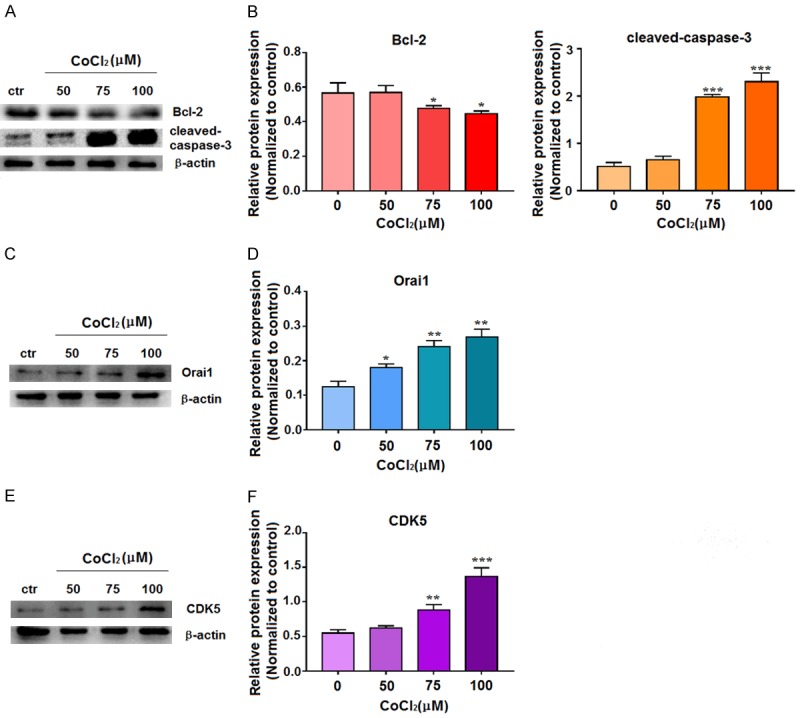
Hypoxia induces expression of apoptosis-related proteins, Orai1, and CDK5 in HT-22 neuronal cells. A, B. Cells were treated with 0, 50, 75, or 100 µM CoCl2 for 72 h, and the extracted proteins were subjected to western blotting with antibodies to Bcl-2 and cleaved-caspase-3. C, D. Cells were treated with 0, 50, 75, or 100 µM CoCl2 for 72 h, and the extracted proteins were subjected to western blotting with Orai1 antibody. E, F. Cells were treated with 0, 50, 75, or 100 µM CoCl2 for 72 h, and the extracted protein was subjected to western blotting with CDK5 antibody. In the western blots, β-actin was used as internal control for normalization of protein loading. For quantitation, data are expressed as means ± SD from three independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs. Control (0 µm CoCl2).
Effects of siOrai1 on neuronal cell proliferation and apoptosis
As Orai1 was upregulated under hypoxic conditions, we next sought to determine the role of the calcium channel protein Orai1 in cell proliferation and apoptosis by examining the effects of knockdown of Orai1 expression in HT-22 cells. We first confirmed knockdown of Orai1 expression following transfection of HT-22 cells with siOrai1 interference fragment for 72 h using western blotting (Figure 3A and 3B). After transfecting HT-22 cells with siOrai1 interference fragment for 6 h, HT-22 cells were treated with 100 μM CoCl2 for 24 h, and then proliferation of HT-22 cells was measured using an MTT assay. Compared with the control untreated cells, proliferation was inhibited in the CoCl2- and siOrai1+CoCl2-treated cells; however, this inhibition was alleviated in the siOra1+CoCl2-treated cells compared to the cells treated with CoCl2 alone (Figure 3C).
Figure 3.
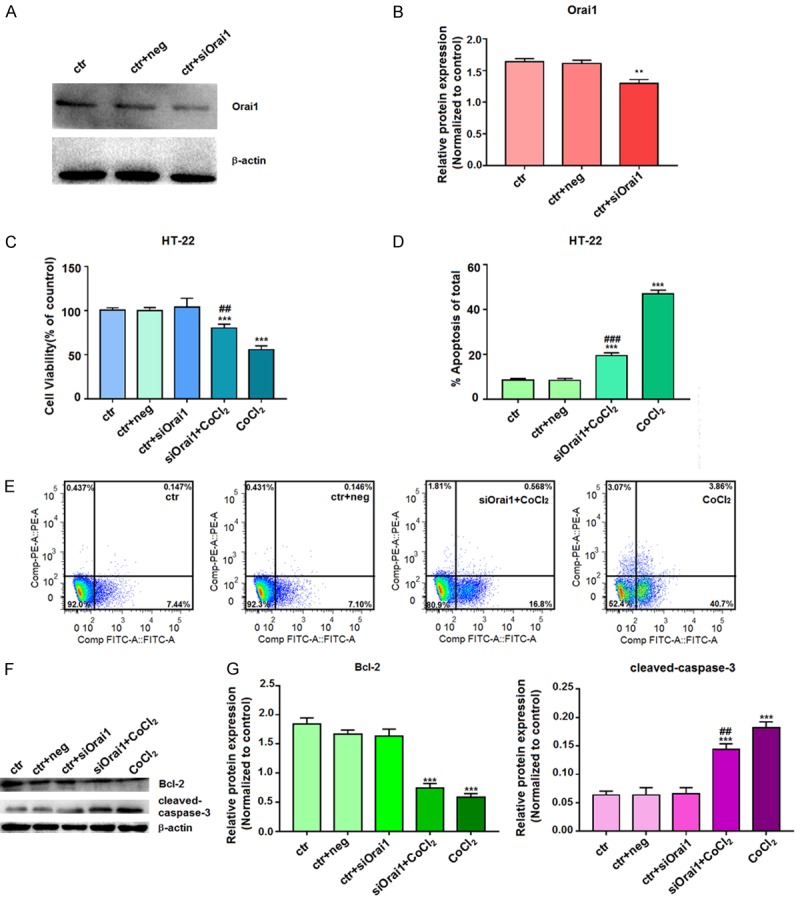
Effects of siOrai1 knockdown on cell proliferation and apoptosis. A, B. After transfection of HT-22 cells with siOrai1 for 72 h, the extracted proteins were subjected to western blotting with the Orai1 antibody. β-actin was used as a control for equal protein loading. Data represent the means ± SD from three independent experiments. C. After transfection of HT-22 cells with siOrai1 for 6 h, cells were then incubated with 100 µM CoCl2 for 24 h. Cell viability was assessed by an MTT assay, and the absorbance values of the treated samples are expressed as percentages of the absorbance values of the control samples. D, E. After transfecting HT-22 cells with siOrai1 for 6 h, cells were incubated with 100 µM CoCl2 for 24 h and then stained with Annexin V-FITC/PI for analysis of cell apoptosis by flow cytometry. F, G. After transfection of HT-22 cells with siOrai1 for 6 h, cells were incubated with 100 µM CoCl2 for 72 h, and the extracted proteins were subjected to western blotting with Bcl-2 and cleaved-caspase-3 antibodies. β-actin was used as a control for equal protein loading. Data are expressed as means ± SD from three independent experiments. **P<0.01 and ***P<0.001 vs. Control; #P<0.05, ##P<0.01, and ###P<0.001 vs. CoCl2 (ctr, untreated cells; ctr+neg, untreated cells incubated with siOrai1 negative control buffer; ctr+siOrai1, untreated cells incubated with siOrai1 buffer; siOrai1+CoCl2, cells were transfected and then treated with CoCl2; CoCl2, cells were treated with CoCl2).
Furthermore, apoptosis of these HT-22 neuronal cells was examined by flow cytometry. Compared with the control group, the early apoptotic rate was increased in the CoCl2- and siOrai1+CoCl2-treated cells, but the apoptotic rate was increased to a lower extent in the siOrai1+CoCl2-treated cells compared to that for cells treated with CoCl2 alone (Figure 3D and 3E). Western blot analysis demonstrated that expression of the anti-apoptotic protein Bcl-2 was reduced in the CoCl2- and siOrai1+CoCl2-treated cells compared to the control cells, but Bcl-2 expression was not reduced as much in the siOrai1+CoCl2-treated cells compared with the CoCl2-treated cells (Figure 3F). Furthermore, expression of the apoptotic protein cleaved-caspase-3 was elevated in the CoCl2- and siOrai1+CoCl2-treated cells; however, the expression of cleaved-caspase-3 was elevated to lower extent in the siOrai1+CoCl2-treated cells than in the CoCl2-treated cells (Figure 3G). Together, these findings indicate that knockdown of Orai1 dampens the effects of hypoxia on proliferation and apoptosis in HT-22 cells.
Effect of Orai1 overexpression on neuronal cell proliferation and apoptosis
We next examined the effects of overexpression of Orai1 on proliferation and apoptosis in HT-22 cells. After transfection of HT-22 cells with an Orai1 overexpression plasmid for 72 h, the increased expression of Orai1 protein was confirmed by western blotting (Figure 4A and 4B). Following transfection with the Orai1 overexpression plasmid for 6 h, HT-22 cells were induced with 100 μM CoCl2 for 24 h, and cell proliferation was assessed by an MTT assay. Compared with the control cells, cell proliferation was inhibited in the CoCl2- and Orai1+CoCl2-treated cells, and greater cell proliferation inhibition was observed in the Orai1+CoCl2-treated cells compared to that in the CoCl2-treated cells (Figure 4C).
Figure 4.
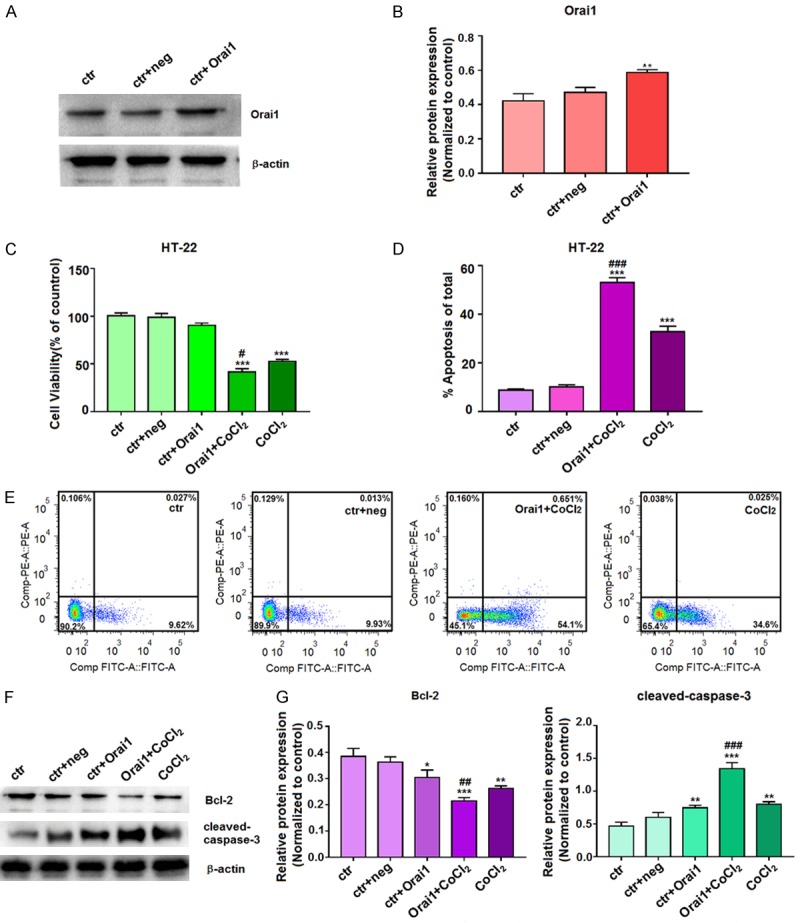
Effects of Orai1 overexpression plasmid on cell proliferation and apoptosis. A, B. After transfection of HT-22 cells with Orai1 overexpression plasmid for 72 h, the extracted proteins were subjected to western blotting with the Orai1 antibody. C. After transfection with Orai1 overexpression plasmid for 6 h, HT-22 cells were incubated with 100 µM CoCl2 for 24 h. Cell viability was assessed by a MTT assay, and the absorbance values of the treated samples are expressed as percentages of the absorbance values of the control samples. D, E. After transfection with Orai1 overexpression plasmid for 6 h, HT-22 cells were incubated with 100 µM CoCl2 for 24 h and then stained with Annexin V-FITC/PI. Cell apoptosis was then quantified by flow cytometry. F, G. After transfection with Orai1 overexpression plasmid for 6 h, HT-22 cells were then incubated with 100 µM CoCl2 for 72 h, and the extracted proteins were subjected to western blotting with antibodies for Bcl-2 and cleaved-caspase-3. For western blotting, β-actin was used as an internal control for equal protein loading. For quantitation, data are expressed as means ± SD from three independent experiments. **P<0.01 and ***P<0.001 vs. Control; #P<0.05, ##P<0.01, and ###P<0.001 vs. CoCl2-treated cells (ctr, untreated cells; ctr+neg, untreated cells incubated with Orai1 negative control buffer; ctr+Orai1, untreated cells incubated with Orai1 buffer; Orai1+CoCl2, cells were transfected and then treated with CoCl2; CoCl2, cells were treated with CoCl2).
Apoptosis in these groups of cells was then detected by flow cytometry, indicating that compared with the control cells, the early apoptotic rate was increased in both CoCl2- and Orai1+CoCl2-treated cells. Furthermore, the early apoptotic rate was increased to a greater extent in the Orai1+CoCl2-treated cells than in the CoCl2-treated (Figure 4D and 4E). Western blotting demonstrated reduced expression of the anti-apoptotic protein Bcl-2 following treatment of cells CoCl2 alone or Orai1+CoCl2 compared to levels in control cells (Figure 4F). Moreover, Orai1 overexpression aggravated the reduction of Bcl-2 expression in the presence of CoCl2, as Bcl-2 was reduced to a higher extent in the Orai1+CoCl2-treated cells than in the CoCl2-treated cells. Expression of the apoptotic protein cleaved-caspase-3 was increased in the CoCl2- and the Orai1+CoCl2-treated cells compared with the control cells, and cleaved-caspase-3 expression was increased to a greater extent in the Orai1+CoCl2-treated cells than in the CoCl2-treated (Figure 4G). Thus, overexpression of Orai1 magnifies the effects of hypoxia on cell proliferation and apoptosis in HT-22 cells.
The levels of Orai1 and CDK5 protein in HT-22 cells in the presence of siOrai1 and Orai1 overexpression
Based on our observations that manipulation of Orai1 expression alters the hypoxia-induced effects on apoptosis, we sought to examine the effects of Orai1 (the calcium channel protein) on CDK5 (protein that activates Tau phosphorylation). Western blotting assay showed that the expression of Orai1 was higher in siOrai1+CoCl2-, CoCl2-, and Orai1+CoCl2-treated cells compared to levels in the control cells. Furthermore, Orai1 expression in the CoCl2-treated cells was higher than that in the siOrai1+CoCl2-treated cells, while Orai1 expression was higher in the Orai1+CoCl2-treated cells than in cells treated with CoCl2 alone (Figure 5). Similarly, the expression of CDK5 as higher in the siOrai1+CoCl2-, CoCl2-, and Orai1+CoCl2-treated cells than in the control cells. In addition, CDK5 expression in the CoCl2-treated cells was higher than that in the siOrai1+CoCl2-treated cells but lower than that in the Orai1+CoCl2-treated cells (Figure 5). The above experiments indicate that there is a correlation between Orai1 and CDK5. The overexpression of Orai1 may induce high expression of CDK5. CDK5 might be a downstream molecule of Orai1 and be regulated by Orai1.
Figure 5.
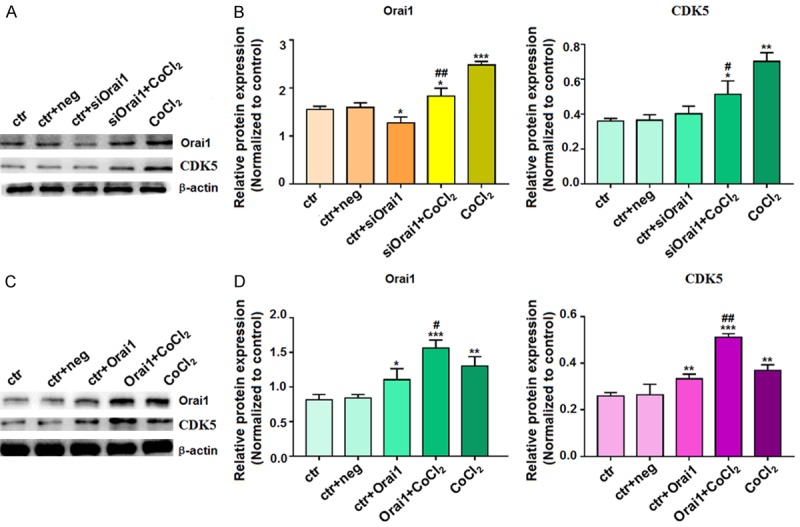
Changes in Orai1 and CDK5 protein expression after transfection of HT-22 cells with siOrai1 and Orai1 overexpression construct. After transfection of HT-22 cells with (A, B) siOrai1 or (C, D) Orai1 overexpression plasmid for 72 h, the extracted proteins were subjected to western blotting with antibodies to (A and C) Orai1 or (B and D) CDK5. β-actin was used as an internal control for equal protein loading. Data are expressed as means ± SD from three independent experiments. *P<0.05, **P<0.01, and ***P<0.001 vs. control; #P<0.05 and ##P<0.01 vs. CoCl2.
Changes in T-Tau and P-Tau protein expression in HT-22 cells grown with hypoxia mimics
As hyperphosphorylation of Tau protein induced by CDK5 in neurons leads to neuronal death and has been implicated in AD, we examined the levels of T-Tau and P-Tau in our cell culture system. Western blotting revealed that the expression of T-Tau and P-Tau increased after hypoxia treatment (Figure 6A and 6B). T-Tau and P-Tau expression was increased in siOrai1+CoCl2- and CoCl2-treated cells compared to control cells, and the levels of T-Tau and P-Tau were higher in the CoCl2-treated cells than in the siOrai1+CoCl2-treated cells (Figure 6C and 6D). In addition, T-Tau and P-Tau levels were increased in both the Orai1+CoCl2- and CoCl2-treated cells compared with the control cells, and levels of T-Tau and P-Tau were higher in the Orai1+CoCl2-treated cells than in the CoCl2-treated cells (Figure 6E and 6F). These results suggest that there is a correlation between Orai1 and Tau (T-Tau and P-Tau). High expression of Orai1 after hypoxia can induce hyperphosphorylation of Tau protein.
Figure 6.
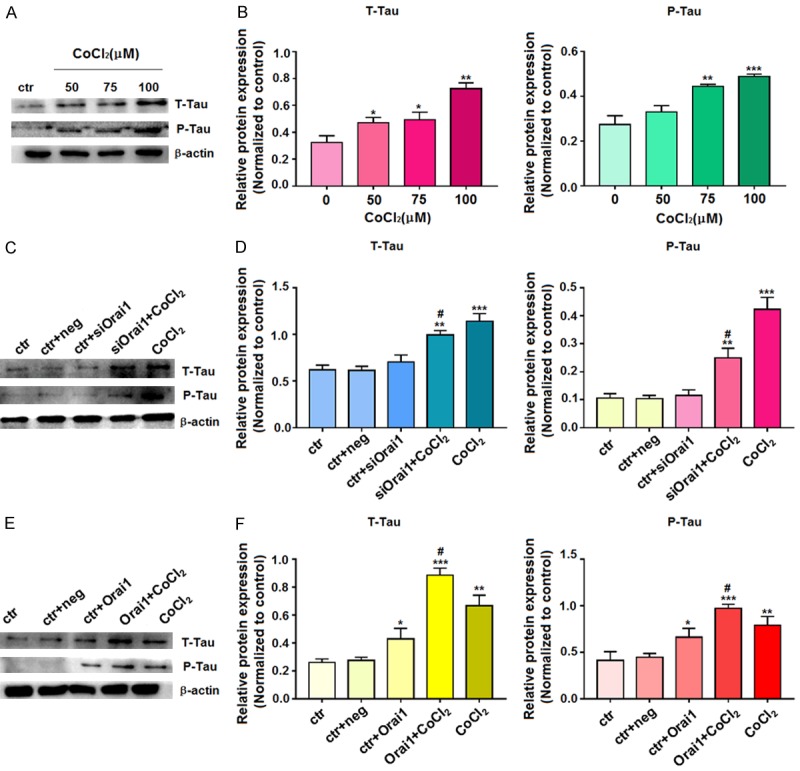
Western blot detection of T-Tau and P-Tau protein expression. A, B. Cells were treated with 0, 50, 75, or 100 µM CoCl2 for 72 h, and then extracted proteins were subjected to western blotting with T-Tau and P-Tau antibodies. C, D. After transfection of HT-22 cells with siOrai1 for 72 h, the extracted proteins were subjected to western blotting with T-Tau and P-Tau antibodies. E, F. After transfection of HT-22 cells with Orai1 overexpression plasmid for 72 h, the extracted proteins were subjected to western blotting with antibodies to T-Tau and P-Tau. For the western blots, β-actin was used as an internal control for equal protein loading. Data are expressed as means ± SD from three independent experiments. *P<0.05, **P<0.01, and ***P<0.001 vs. Control; #P<0.05 vs. CoCl2.
Discussion
The etiology of AD remains largely unknown. Due to the slow clinical progression of AD, investigation of early disease has failed to clearly reveal the pathological mechanism, and pathological changes have mostly been discovered during autopsy of abnormal brain tissue [27]. With respect to neuropathology, abundant extracellular deposition of amyloid, intracellular neurofibrillary tangles, and intracellular Tau protein hyperphosphorylation in the relevant brain regions of AD patients are considered to be the main causes of neuronal cell injury [28,29].
In this study, we found that hypoxia, induced by CoCl2 treatment, in mouse hippocampal neuronal cells leads to elevated intracellular Orai1 protein levels, which result in increased expression of CDK5, which in turn triggers Tau hyperphosphorylation and induces early apoptosis in neurons. Thus, after exclusion of the influence of environmental and vascular factors, these studies indicate that hypoxic injury in hippocampal neurons and the series of resulting pathophysiological manifestations offer insight into prevention and treatment of AD.
Hypoxia has been extensively studied as a mechanism of injury in neurodegenerative diseases. Furthermore, its ability to promote apoptosis has been widely recognized. Our study demonstrated that hypoxia inhibits the growth and proliferation of HT-22 neuronal cells and induces cell damage and early apoptosis. While AD diagnosis involves ruling out a number of diseases at the time of diagnosis, limited studies of whether hypoxic damage exists in the involved brain cells in patients with normal blood oxygen saturation have been performed. We conducted related experiments using HT-22 cells as an AD model with the hypoxia-mimicking chemical agent CoCl2 to simulate the hypoxic environment of cells. In these cells, proliferation was inhibited in a time- and concentration-dependent manner and early apoptosis was initiated following induction of hypoxia. Moreover, the early apoptotic rate increased significantly with CoCl2 incubation in a time- and concentration-dependent manner. Furthermore, expression of the anti-apoptotic protein Bcl-2 decreased following induction of HT-22 cells with different concentrations of CoCl2, while the expression of the apoptotic protein cleaved-caspase-3 protein significantly increased. The Orai1 protein level also increased. The changes in these proteins were CoCl2 concentration-dependent. Additionally, the level of P-Tau increased in cells after hypoxia, suggesting that T-Tau is increasingly phosphorylated. CDK5, a protein that activates Tau phosphorylation, was also increased following induction of hypoxia. It is suggested that hypoxia induces CDK5 expression via interactions between certain proteins and thus impacts T-Tau protein phosphorylation.
To further investigate the mechanism(s) underlying hypoxia-induced neuronal injury, we constructed a small interfering RNA targeting Orai1 (siOrai1) and transfected it into HT-22 cells, and then treated the transfected cells with hypoxia. We found that Orai1’s silencing prevented cell proliferation inhibition and counteracted early apoptosis. Western blotting experiments of Bcl-2 and cleaved-caspase-3 confirmed the above conclusions. At the same time, the siOrai1+CoCl2 treated cells expressed T-Tau, P-Tau and CDK5 at a level higher than the control group but lower than the CoCl2 group. The results suggest that CDK5 may be related to Orai1 and further experiments are needed to clarify this correlation.
To further verify the above results, we constructed an Orai1 overexpression plasmid and transfected the plasmid into HT-22 cells, which were then treated with hypoxia. We found that overexpression of Orai1 gene promoted cell proliferation inhibition and increased early apoptosis rate. Western blotting experiments of Bcl-2 and cleaved-caspase-3 confirmed the above conclusions. The expression levels of T-Tau, P-Tau and CDK5 were increased in the CoCl2- and Orai1+CoCl2 treated cells compared with the control group. The levels of T-Tau, P-Tau and CDK5 in the Orai1+CoCl2 treated cells were higher than those in the CoCl2 group. The results demonstrate that Orai1 overexpression may induce high expression of CDK5.
To clarify the correlation between Orai1 protein and CDK5, we observed changes in the expression levels of Orai1 protein and CDK5 in multiple experimental groups. The results showed that compared with the control group, the expression of Orai1 protein in the siOrai1+CoCl2 treated cells, CoCl2- and Orai1+CoCl2 treated cells increased. The expression of Orai1 protein in CoCl2- was higher than that in siOrai1+CoCl2 treated cells, the Orai1+CoCl2 treated cells had higher Orai1 protein expression than the CoCl2 group. At the same time, the same results were obtained for the CDK5 expression levels of the above groups. The results were statistically significant. However, after the Orai1 interference fragment was only transfected into HT-22 cells, although the expression of Orai1 protein was decreased in the siOrai1+ctr treated cells, it did not cause statistically significant changes in CDK5, T-Tau, and P-Tau values compared with the control group. We believe that the reason might be related to the low expression of Orai1 protein in the siOrai1+ctr treated cells. The above results indicated that CDK5, T-Tau, and P-Tau changed significantly when Orai1 expression was increased.
The combined results of the hypoxia experiment, Orai1-interference experiment, and Orai1-overexpression experiment indicate that there is a correlation between Orai1 and CDK5 levels. Compared with the CoCl2-, the siOrai1+CoCl2 treated cells reduced the expression of Orai1, CDK5 decreased, Orai1+CoCl2 treated cells increased the expression of Orai1, and CDK5 increased. Compared with the control group, the expression of Orai1 protein in the siOrai1+CoCl2 treated cells increased and CDK5 increased. The results of Western blotting indicate that CDK5 might be a downstream molecule of Orai1 and be regulated by Orai1. The expression of Orai1 protein increased, and the expression levels of CDK5, T-Tau and P-Tau increased. The expression of Orai1 protein decreased, and the expression levels of CDK5, T-Tau and P-Tau decreased. CDK5 is considered to be the most important protein kinase in the brain that regulates the phosphorylation of Tau and the hyperphosphorylation of Tau protein induces the typical pathological changes of AD [30,31].
In summary, our results showed that Orai1 expression is elevated in HT-22 cells after hypoxic induction, Orai1 regulates the expression of the downstream gene CDK5, high CDK5 expression leads to hyperphosphorylation of Tau, which promotes early apoptosis of the cells, Tau hyperphosphorylation might be related to the Orai1-induced CDK5 expression. P-Tau protein expression increases in HT-22 cells after hypoxic injury. Tau hyperphosphorylation in neuronal cells of specific brain regions is an important pathophysiological manifestation in AD patients. The results of the in vitro post-hypoxic injury simulation are highly similar to the pathophysiological change of related neurons in AD patients, and the results of the experimental group were significantly different from those of the control group. Therefore, we believe brain cells involved in AD patients might experience a pathological process similar to hypoxic injury. Due to the long-term course of AD, this type of hypoxic injury has not drawn attention in early disease stages. Eventually, cellular respiratory activity and energy metabolism are significantly impaired, and organelle function is considerably diminished in these neurons, the apoptotic rate of neurons increases, AD patients develop related symptoms.
The HT-22 cell-based hypoxia simulation experiments suggest that some brain cells in AD patients might experience similar hypoxic injury, however, the specific underlying mechanism remains to be further elucidated. We think that in-depth study on intracellular oxygen utilization in AD-related brain cells should be conducted. Classic hypoxic injury can be caused by insufficient blood supply, poisoning, and declined pulmonary function, during the diagnosis of AD, a variety of diseases are excluded. We think that the hypoxic injury that AD patients might have suffered from is a special type of hypoxic injury, this type of hypoxic injury occurs in brain cells, the patients have normal blood oxygen saturation levels in blood vessel and this hypoxic injury is not caused by poisoning. This provides us with a new understanding of hypoxic injuries and elicits new ideas for the diagnosis and treatment of AD patients. Additional studies are needed to deeperly understand the correlation between hypoxic injury and AD. For elderly non-AD individuals without cognitive impairment, related protective proteins and neurotrophic factors may have protective mechanisms representing new therapeutic strategies for AD patients.
Acknowledgements
This study was supported by the Wuxi Municipal Health Commission Fund for Youth Research Project (Q201755), the Science and Technology Development Fund Project of Nanjing Medical University (2015NJMU134), the training of the “Echelon Talents” eagle plan of the affiliated hospital of Jiangnan University and a grant from Affiliated Hospital of Jiangnan University (FYYB201801).
Disclosure of conflict of interest
None.
References
- 1.Vinothkumar G, Kedharnath C, Krishnakumar S, Sreedhar S, Preethikrishnan K, Dinesh S, Sundaram A, Balakrishnan D, Shivashekar G, Sureshkumar , Venkataraman P. Abnormal amyloid β expression and increased oxidative stress in plasma of CKD patients with cognitive dysfunction: a small scale case control study comparison with Alzheimer’s disease. BBA Clin. 2017;8:20–27. doi: 10.1016/j.bbacli.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haider L, Zrzavy T, Hametner S, Höftberger R, Bagnato F, Grabner G, Trattnig S, Pfeifenbring S, Brück W, Lassmann H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. 2016;139:807–815. doi: 10.1093/brain/awv398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L, de Silva R, Di Giovanni G, Wischik C, Hof PR. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6:6. doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manukhina EB, Downey HF, Shi X, Mallet RT. Intermittent hypoxia training protects cerebrovascular function in Alzheimer’s disease. Exp Biol Med (Maywood) 2016;241:1351–1363. doi: 10.1177/1535370216649060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Niu L, Li S, Le W. Pathological impacts of chronic hypoxia on alzheimer’s disease. ACS Chem Neurosci. 2019;10:902–909. doi: 10.1021/acschemneuro.8b00442. [DOI] [PubMed] [Google Scholar]
- 6.Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer’s disease amyloid beta-protein forms Zn(2+)-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys J. 1997;73:67–75. doi: 10.1016/S0006-3495(97)78048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M, Zhou T, Zhou L, Chen Q, Yu Y, Yang H, Zhong K, Zhang X, Xu F, Cai S, Yu A, Zhang H, Xiao R, Xiao D, Chui D. Formononetin protects neurons against hypoxia-induced cytotoxicity through upregulation of ADAM10 and sAβPPα. J Alzheimers Dis. 2012;28:795–808. doi: 10.3233/JAD-2011-110506. [DOI] [PubMed] [Google Scholar]
- 8.Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C. The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol. 2012;81:510–26. doi: 10.1124/mol.111.074658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groten CJ, Rebane JT, Hodgson HM, Chauhan AK, Blohm G, Magoski NS. Ca2+ removal by the plasma membrane Ca2+-ATPase influences the contribution of mitochondria to activity-dependent Ca2+ dynamics in Aplysia neuroendocrine cells. J Neurophysiol. 2016;115:2615–2634. doi: 10.1152/jn.00494.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakraborty S, Deb BK, Chorna T, Konieczny V, Taylor CW, Hasan G. Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in drosophila neurons. J Cell Sci. 2016;129:3903–3910. doi: 10.1242/jcs.191585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moccia F, Zuccolo E, Soda T, Tanzi F, Guerra G, Mapelli L, Lodola F, D’Angelo E. Stim and orai proteins in neuronal Ca(2+) signaling and excitability. Front Cell Neurosci. 2015;9:153. doi: 10.3389/fncel.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He F, Wu Q, Xu B, Wang X, Wu J, Huang L, Cheng J. Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/reoxygenation induced apoptosis in H9C2 cells. Biosci Rep. 2017;37 doi: 10.1042/BSR20171249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hendron E, Wang X, Zhou Y, Cai X, Goto J, Mikoshiba K, Baba Y, Kurosaki T, Wang Y, Gill DL. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium. 2014;56:482–492. doi: 10.1016/j.ceca.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao Y, Dong LD, Chen J, Hu XC, Yang XL, Wang Z. Involvement of calpain/p35-p25/Cdk5/NMDAR signaling pathway in glutamate-induced neurotoxicity in cultured rat retinal neurons. PLoS One. 2012;7:e42318. doi: 10.1371/journal.pone.0042318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi H, Saito T, Sato K, Furusawa K, Hosokawa T, Tsutsumi K, Asada A, Kamada S, Ohshima T, Hisanaga S. Phosphorylation of cyclin-dependent kinase 5 (Cdk5) at Tyr-15 is inhibited by Cdk5 activators and does not contribute to the activation of Cdk5. J Biol Chem. 2014;289:19627–19636. doi: 10.1074/jbc.M113.501148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 17.Saito T, Yano M, Kawai Y, Asada A, Wada M, Doi H, Hisanaga S. Structural basis for the different stability and activity between the Cdk5 complexes with p35 and p39 activators. J Biol Chem. 2013;288:32433–32439. doi: 10.1074/jbc.M113.512293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amin ND, Zheng Y, Bk B, Shukla V, Skuntz S, Grant P, Steiner J, Bhaskar M, Pant HC. The interaction of Munc 18 (p67) with the p10 domain of p35 protects in vivo Cdk5/p35 activity from inhibition by TFP5, a peptide derived from p35. Mol Biol Cell. 2016;27:3221–3232. doi: 10.1091/mbc.E15-12-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim BS, Serebreni L, Fallica J, Hamdan O, Wang L, Johnston L, Kolb T, Damarla M, Damico R, Hassoun PM. Cyclin-dependent kinase five mediates activation of lung xanthine oxidoreductase in response to hypoxia. PLoS One. 2015;10:e0124189. doi: 10.1371/journal.pone.0124189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ando K, Maruko-Otake A, Ohtake Y, Hayashishita M, Sekiya M, Iijima KM. Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLoS Genet. 2016;12:e1005917. doi: 10.1371/journal.pgen.1005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagishita S, Suzuki S, Yoshikawa K, Iida K, Hirata A, Suzuki M, Takashima A, Maruyama K, Hirasawa A, Awaji T. Treatment of intermittent hypoxia increases phosphorylated tau in the hippocampus via biological processes common to aging. Mol Brain. 2017;10:2. doi: 10.1186/s13041-016-0282-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majd S, Power JH, Koblar SA, Grantham HJ. Early glycogen synthase kinase-3β and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur J Neurosci. 2016;44:1987–1997. doi: 10.1111/ejn.13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, Tyagi N. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of alzheimer’s disease: understanding the therapeutics strategies. Mol Neurobiol. 2016;53:648–661. doi: 10.1007/s12035-014-9053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, Kayed R. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J Alzheimers Dis. 2017;55:1083–1099. doi: 10.3233/JAD-160912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guadagna S, Esiri MM, Williams RJ, Francis PT. Tau phosphorylation in human brain: relationship to behavioral disturbance in dementia. Neurobiol Aging. 2012;33:2798–2806. doi: 10.1016/j.neurobiolaging.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adler DH, Wisse LEM, Ittyerah R, Pluta JB, Ding SL, Xie L, Wang J, Kadivar S, Robinson JL, Schuck T, Trojanowski JQ, Grossman M, Detre JA, Elliott MA, Toledo JB, Liu W, Pickup S, Miller MI, Das SR, Wolk DA, Yushkevich PA. Characterizing the human hippocampus in aging and alzheimer’s disease using a computational atlas derived from ex vivo MRI and histology. Proc Natl Acad Sci U S A. 2018;115:4252–4257. doi: 10.1073/pnas.1801093115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savelieff MG, Lee S, Liu Y, Lim MH. Untangling amyloid-β, tau, and metals in alzheimer’s disease. ACS Chem Biol. 2013;8:856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- 29.Mariano M, Schmitt C, Miralinaghi P, Catto M, Hartmann RW, Carotti A, Engel M. First selective dual inhibitors of tau phosphorylation and beta-amyloid aggregation, two major pathogenic mechanisms in alzheimer’s disease. ACS Chem Neurosci. 2014;5:1198–1202. doi: 10.1021/cn5001815. [DOI] [PubMed] [Google Scholar]
- 30.Mateo I, Vázquez-Higuera JL, Sánchez-Juan P, Rodríguez-Rodríguez E, Infante J, García-Gorostiaga I, Berciano J, Combarros O. Epistasis between tau phosphorylation regulating genes (CDK5R1 and GSK-3beta) and alzheimer’s disease risk. Acta Neurol Scand. 2009;120:130–133. doi: 10.1111/j.1600-0404.2008.01128.x. [DOI] [PubMed] [Google Scholar]
- 31.Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in alzheimer’s disease and PART. Acta Neuropathol. 2018;136:57–67. doi: 10.1007/s00401-018-1855-6. [DOI] [PMC free article] [PubMed] [Google Scholar]