

Abstract
Keratinocyte growth factor (KGF) improves cyclophosphamide-induced bladder injury. To understand the mechanisms, we subcutaneously administered KGF to mice 24 hours before i.p. cyclophosphamide administration, followed by histologic assays and immunostaining. In vehicle (phosphate-buffered saline)–pretreated mice, nonapoptotic superficial cell death from 2 to 6 hours and apoptosis in intermediate and basal cells from 4 to 24 hours was observed after cyclophosphamide. Despite superficial cell loss, KGF suppressed intermediate and basal cell apoptosis, likely via AKT signaling. At 6 and 24 hours after cyclophosphamide, KGF-pretreated mice also had apparent extracellular signal-regulated kinase (ERK)–driven proliferation of mostly keratin 5 (KRT5)+/KRT14− intermediate cells. At 1 to 28 days after cyclophosphamide treatment, mostly KRT14+ basal progenitor cells proliferated in response to injury, peaking at 3 days in both treatment groups; however, proliferation rates were lower in the KGF group at 3 days, consistent with less injury. Three days after injury, unlike controls, KGF-pretreated mice had regenerated superficial cells. At 10 and 28 days after cyclophosphamide treatment, KGF-pretreated mice had little proliferation and marked restoration of urothelial layers, whereas the phosphate-buffered saline group had ongoing regeneration. Administration of KGF to uninjured mice reproduced ERK-driven KRT5+/KRT14− proliferation seen in injured mice; KRT14+ cells were unaffected. KGF pretreatment blocks cyclophosphamide-induced intermediate and basal cell apoptosis, likely by phosphorylated AKT, and drives phosphorylated ERK–mediated KRT5+/KRT14− cell proliferation, leading to early urothelial regeneration.
Cyclophosphamide, an alkylating agent that is used widely for solid tumors, leukemias, lymphomas, and nononcologic diseases, such as lupus, is highly bladder toxic.1 Cyclophosphamide is metabolized to acrolein, which concentrates in the urine, leading to direct damage to bladder urothelium. Clinically, acute toxicity is manifested as hemorrhagic cystitis, which ranges in severity from microscopic hematuria and/or painful lower urinary tract symptoms to severe life-threatening bleeding. Chronic sequelae include bladder fibrosis and contractures. Patients treated with cyclophosphamide for either oncologic or nononcologic disease also have dose-dependent increased risks of developing urothelial cancer,2, 3, 4, 5 with one study finding a nearly 15-fold risk for lymphoma survivors given ≥50 g cumulative doses.6
Current therapies to prevent hemorrhagic cystitis are only partially efficacious.1,7,8 Mesna (sodium-2-mercaptoethanesulfonate), the most commonly used therapy, directly binds acrolein in the urine; however, it may cause hypersensitivity reactions and often does not prevent cystitis. One study found that 66% of patients treated with mesna to prevent injury from ifosfamide (metabolized to acrolein, like cyclophosphamide) had urothelial injury by cystoscopy.9 Hydration, another commonly used therapy to dilute acrolein, is also at best partially efficacious and may be contraindicated in patients who cannot tolerate fluid overload.8 Antioxidants or anti-inflammatory drugs have no superior efficacy to mesna.8 There are also no data that mesna (or any drug) prevents cyclophosphamide-driven urothelial cancer.10
Many preclinical and clinical studies have shown that fibroblast growth factor receptor 2 (FGFR2) signaling reduces injury and drives regeneration in epithelial tissues after noxious stimuli.11, 12, 13, 14, 15, 16, 17 FGFR2 is a receptor tyrosine kinase, which has a IIIb isoform that is found in epithelium, including urothelium, and that binds to ligands, including FGF7 [alias keratinocyte growth factor (KGF)].18 Exogenous recombinant human KGF ameliorates toxin or radiation-mediated injury in oral, retinal, alveolar, and intestinal epithelia.12, 13, 14, 15, 16, 17 In 1997, Ulich et al11 published that one i.v. dose of KGF in rats 24 hours before cyclophosphamide administration led to increased urothelial proliferation versus vehicle-pretreated mice and led to marked improvement in bladder histology. The report did not reveal whether the beneficial effects of KGF were related to the hyperproliferation (ie, accelerated regeneration) and/or cytoprotection and did not explore mechanisms of KGF activity in the bladder. Our study examines a detailed histologic analysis of cyclophosphamide-induced injury, how KGF acts to deliver urothelial cytoprotection and drive proliferation, and the mechanisms underlying the latter actions.
Materials and Methods
Mice
Female FVB/NJ mice (aged 2 to 3 months) were used for all experiments. All the proposed mouse experiments were approved by the University of Pittsburgh (Pittsburgh, PA) Institutional Animal Care and Use Committee in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care.
Cyclophosphamide Injections
Mice were administered either 150 mg/kg i.p. cyclophosphamide (Sigma-Aldrich, St. Louis, MO; catalog number C7397) dissolved in phosphate-buffered saline (PBS) or i.p. PBS alone (sham injury).
KGF Pretreatment
Mice were administered 5 mg/kg s.c. injections of KGF (R&D Systems, Minneapolis, MN; catalog number 251-KG-010) dissolved in PBS or of PBS alone (vehicle), 24 hours before cyclophosphamide (or PBS) injections. In some instances, no cyclophosphamide (or i.p. PBS) was administered after KGF or PBS.
Histology, IF, and TUNEL Assays
For histology, paraffin-embedded tissues were sectioned (6 μm thick) and stained with hematoxylin and eosin (H&E). For immunofluorescence (IF), dewaxed paraffin-embedded sections were subjected to antigen retrieval in a pressure cooker for 15 minutes in Tris-EDTA, pH 9.0, buffer and then blocked with normal donkey serum for 1 hour at room temperature. Sections were then incubated overnight at 4°C with the following primary antibodies: anti–keratin 20 (KRT20) at 1:50 (Agilent Technologies, Santa Clara, CA; catalog number M7019; RRID:AB_2133718), keratin 5 (KRT5) at 1:200 (BioLegend, San Diego, CA; catalog number 905901) (RRID Portal, https://scicrunch.org/resources, last accessed August 2019; RRID:AB_2565054), keratin 14 (KRT14) at 1:200 (BioLegend; catalog number 905301; RRID:AB_2565048), uroplakin 3a (UPK3) at 1:200 (Santa Cruz Biotechnology, Dallas, TX; catalog number sc-33570; RRID:AB_2213486), phosphorylated AKT (pAKT) at 1:100 (Cell Signaling Technology, Beverly, MA; catalog number 4060; RRID:AB_2315049), phosphorylated extracellular signal-regulated kinase (pERK) at 1:50 (Cell Signaling Technology; catalog number 9100; RRID:AB_330741), marker of proliferation Ki-67 (Ki-67) at 1:200 (R&D Systems; catalog number AF7649; RRID:AB_2687500), phosphorylated FGF receptor substrate 2α (pFRS2α) at 1:100 (R&D Systems; catalog number AF5126; RRID:AB_2106234), and/or FGFR2 at 1:200 (Abcam, Cambridge, UK; catalog number ab10648; RRID:AB_297369). After washing in PBS, slides were incubated with the following secondary antibodies: Alexa Fluor 643 (Thermo Fisher Scientific, Waltham, MA; catalog number A-31573; RRID:AB_2536183), Alexa Fluor 594 (Thermo Fisher Scientific; catalog number A-21207; RRID:AB_141637), Alexa Fluor 488 (Thermo Fisher Scientific; catalog number A-21202; RRID:AB_141607), and/or Alexa Flour 647 (Jackson ImmunoResearch Labs, West Grove, PA; catalog number 703-606-155; RRID:AB_2340380), all at 1:500 for 2 hours at room temperature, followed by washes. Nuclei were stained with DAPI. To assay for apoptosis, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assays were performed with ApopTag Plus in situ Apoptosis Fluorescein Detection kit (EMD Millipore, Burlington, MA; catalog number S7111-kit), according to manufacturer's instructions. Slides were imaged with a Leica DM2500 fluorescence microscope (Leica Microsystems, Buffalo Grove, IL) or a Zeiss LSM 710 confocal microscope (Carl Zeiss, Thornwood, NY). Total urothelial proliferation rates were assessed by counting Ki-67+/all DAPI+ cells and individual cell type proliferation rates were assessed by counting numbers of Ki-67+ cells that were KRT5+ and/or UPK3+ or that were KRT14+, KRT5+, and/or not colabeled. Counts (blinded to genotype) were performed in two to three planes encompassing entire rings of urothelium per bladder with three mice of each treatment group per time point.
Statistical Analysis
To test the effect of KGF versus PBS pretreatment on injury or sham on each histologic marker, mixed effect models (with random intercept and restricted maximum likelihood ratios) were applied. These are flexible models to account for repeated measures at different time points, adjusting for correlation of outcomes within each mouse. In these models, the interactions between intervention and time measure the overall effect of injury on each study outcome. After testing the overall effect of injury in each model, predictive margins were used to calculate the mean (SEM) for each histologic marker at different time points for the two pretreatment groups as well as the P value of effect on injury or sham at different time points. A two-sided hypothesis test was used for all analyses. All statistical analyses were performed in Stata 15.0 (StataCorp, College Station, TX).
Results
Cyclophosphamide Induces Early Nonapoptotic Death of Superficial Cells, followed by Apoptotic Death of Deeper Layers
H&E staining was performed over several days to determine the global urothelial injury and regeneration pattern after cyclophosphamide injection. Compared with uninjured mice (Figure 1A), significant sloughing of urothelial cell layers as well as submucosal hemorrhage and inflammation were observed 1 day after cyclophosphamide (Figure 1B). At 3 and 10 days after injury, urothelial regeneration was observed with increasing cellular hyperplasia (Figure 1, C and D). At 28 days after injury, apparent restoration of normal urothelial cell layers (Figure 1E), appearing similar to uninjured bladders, was observed.
Figure 1.

Representative images of mouse bladder urothelium after cyclophosphamide injection. A–E: Hematoxylin and eosin (H&E) images from 0 to 28 days after cyclophosphamide administration. A: Uninjured adult mouse bladder with three to four cell layers, including large polyploid superficial (arrowhead) and smaller deeper (arrow) cells. B: Mouse bladder 1 day after cyclophosphamide administration shows significant urothelial cell sloughing (black arrowheads), leaving regions of denuding (arrow) and hemorrhage (green arrowheads). C: Mouse bladder 3 days after cyclophosphamide administration shows urothelial regeneration with cellular hyperplasia (arrow). D: Mouse bladder 10 days after cyclophosphamide administration shows ongoing urothelial cell regeneration with increased cellular hyperplasia (arrow). E: Mouse bladder 28 days after cyclophosphamide administration shows apparent restoration of normal urothelial cell layers, including superficial cells (arrowhead) and smaller deeper cells (arrow). F–J: Images at 2 hours after cyclophosphamide administration. F: H&E-stained bladder shows some sloughing of urothelial cells (arrow). G: Immunofluorescence (IF) for superficial cell marker, KRT20 (red), in a section adjacent to F shows that the sloughing cells (arrow) are KRT20+ superficial cells and that regions of remnant urothelium have lost KRT20+ superficial cells (arrowhead). H: IF for the superficial cell and intermediate cell subset marker, uroplakin 3a (UPK3; white) in a section adjacent to G shows coexpression with KRT20 in sloughing superficial cells (arrow) but preserved expression on luminal urothelial surface, even in regions without KRT20 (arrowhead), consistent with preserved UPK3+ intermediate cells. I: IF for the intermediate cell and basal cell marker, KRT5 (green), in the same section as H shows intact KRT5+ intermediate and basal cells. J: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining (green) in a section adjacent to H and I is negative throughout the urothelium. K–O: Images at 12 hours after cyclophosphamide administration. K: H&E-stained bladder showing detachment of sheets of urothelial cells (arrow). L: KRT20 IF (red) in an adjacent section to K is completely negative, even on detaching urothelial cells (arrowhead), consistent with previous loss of superficial cells. M and N: UPK3 IF (white; M) and KRT5 IF (green; N) in a section adjacent to L show injury and detachment of sheets of UPK3+ and KRT5+ intermediate cells (arrows). O: TUNEL staining (green) in a section adjacent to M and N shows widespread apoptosis throughout the detaching and remnant urothelium (arrowheads). A–E, J, and O: Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. G–J and L–O: Blue indicates DAPI. Scale bars = 50 μm (A–O). L, lumen.
The spatiotemporal injury pattern was then determined within the first 24 hours by H&E staining, immunostaining for cell-specific markers, and TUNEL assays. Two hours after cyclophosphamide, variable sloughing of outer layers of urothelial cells was observed by H&E staining (Figure 1F). Immunostaining identified the sloughing urothelial cells as keratin 20+ superficial cells, although some KRT20+ cells were still attached (Figure 1G). Although all sloughing and attached KRT20+ superficial cells were also UPK3+, UPK3+/KRT20− intermediate cells appeared uninjured (Figure 1H). KRT5+ cells (mostly UPK3− intermediate cells and basal cells) appeared uninjured and attached to the urothelium at 2 hours (Figure 1I). No urothelial apoptosis was detected by TUNEL assays 2 hours after cyclophosphamide (Figure 1J).
Four hours after cyclophosphamide, H&E stains revealed worsening urothelial damage, with more cells sloughing into the lumen (Supplemental Figure S1A). Immunofluorescence confirmed that most KRT20+/UPK3+ superficial cells were either detached or detaching from the bladder (Supplemental Figure S1B). Most of the UPK3+/KRT20− intermediate and KRT5+ intermediate and/or basal cells were still attached, although there were areas of interrupted staining in both (Supplemental Figure S1, C and D). TUNEL staining revealed a small number of apoptotic cells within deeper cell layers (Supplemental Figure S1E).
Twelve hours after cyclophosphamide, H&E stains revealed sheets of urothelial cells detaching from the bladder (Figure 1K). KRT20 staining was negative, consistent with absence of all superficial cells (Figure 1L). Detaching sheets of UPK3+ and KRT5+ cells were also observed (Figure 1, M and N). TUNEL staining revealed large numbers of apoptotic cells within the detaching and remaining intermediate and basal cells (Figure 1O). Together, cyclophosphamide induces nonapoptotic death of superficial cells starting at 2 hours, followed by apoptotic loss of intermediate and basal cells starting at 4 hours.
KGF Protects Deeper Layers from Apoptosis but Does Not Prevent Loss of Superficial Cells
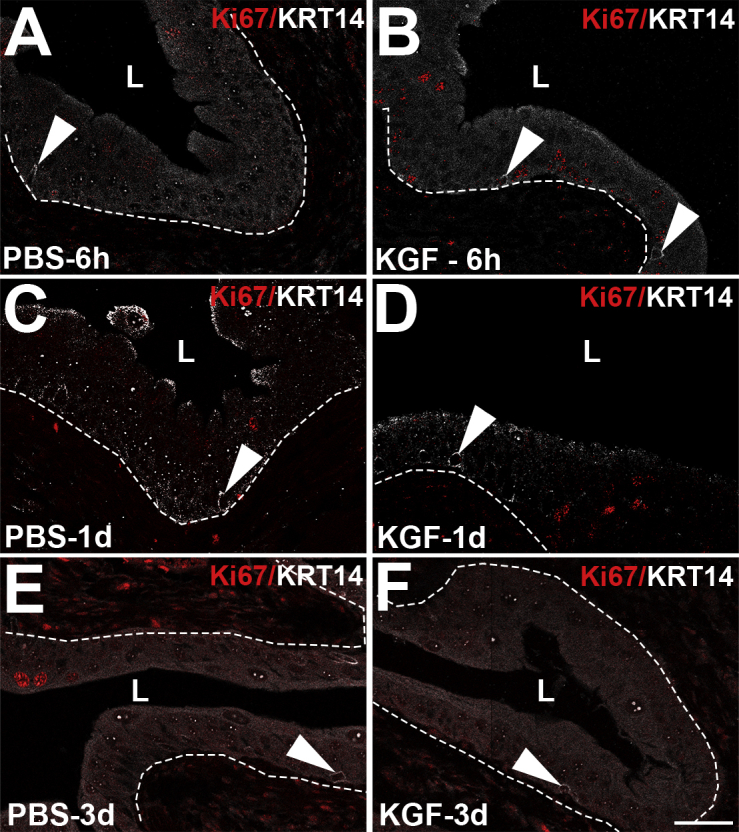
To determine whether exogenous KGF attenuated cyclophosphamide-induced bladder injury, it was first confirmed that its receptor, FGFR2, was expressed throughout uninjured urothelium (Supplemental Figure S2). To confirm that systemically administered KGF directly activates FGFR2 in urothelium, KGF or PBS (vehicle) was administered by s.c. injection and IF was performed for pFRS2α, which is constitutively associated with FGFR2 and phosphorylated on ligand activation of the receptor; at 15 (Supplemental Figure S3) and 30 hours (data not shown) after s.c. KGF administration, pFRS2α membrane staining was seen across multiple layers of urothelium, whereas no signal was observed after PBS. KGF or PBS was then administered; and 24 hours later, cyclophosphamide (or PBS) was injected. Six hours after cyclophosphamide (30 hours after KGF or PBS), H&E staining revealed PBS-treated mice had urothelial injury affecting multiple layers (Figure 2A), whereas KGF-treated bladders appeared to have selective sloughing and injury to superficial cells (Figure 2F). Immunofluorescence confirmed that many KRT20+/UPK3+ superficial cells were sloughing into the lumen after either PBS (Figure 2, B and C) or KGF (Figure 2, G and H) pretreatment. Although PBS-pretreated mice also had large numbers of injured and/or detaching UPK3+/KRT20− and KRT5+ cells (Figure 2, C and D), KGF-pretreated mice had virtually completely intact UPK3+/KRT20− and KRT5+ cells remaining attached to the bladder (Figure 2, H and I). TUNEL staining revealed extensive urothelial apoptosis in PBS-treated mice (Figure 2E), but none in the KGF-treated mice, even in detaching (superficial) cells (Figure 2J).
Figure 2.

Representative images showing effects of keratinocyte growth factor (KGF) pretreatment 6 hours and 1 day after cyclophosphamide injection. A–J: Images from 6 hours after cyclophosphamide administration. A and F: Hematoxylin and eosin (H&E) staining shows PBS-pretreated bladders have extensive urothelial injury, lumenal debris, and cellular sloughing (arrowhead, A) with few remnant urothelial cells attached (arrow, A) versus KGF-pretreated bladders that have loss of outer superficial cells (arrowhead, F) and preservation of deeper urothelial layers (arrow, F). B and G: KRT20 immunofluorescence (IF; red) in sections adjacent to A and F, respectively, shows many of the sloughing cells in PBS-pretreated (B) and KGF-pretreated (G) bladders are KRT20+ (arrowheads). C and H: Uroplakin 3a (UPK3) IF (white) in sections adjacent to B and G, respectively, shows PBS-pretreated bladders have many sloughing UPK3+ cells (arrowhead, C) with few UPK3+ intermediate cells adherent to the bladder (arrow, C); KGF-pretreated bladders also have UPK3+ sloughing cells (arrowhead, H) but many UPK3+ intermediate cells remain attached (arrow, H). D and I: KRT5 IF (green) in the same sections as C and H, respectively, shows that PBS-pretreated bladders have only a single layer of remaining KRT5+ cells that are exposed to the lumen (arrow, D), whereas KGF-pretreated bladders have many layers of apparently uninjured KRT5+ cells (arrow, I). E and J: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining (green) in sections adjacent to D and I, respectively, shows apoptosis in many PBS-pretreated urothelial cells (arrowheads) but none in KGF-pretreated bladders (J), including in sloughing superficial cell nuclei (arrows). B–E and G–J: Blue indicates DAPI. E and J: K–T: Images from 1 day after cyclophosphamide administration. K and P: H&E staining shows that PBS-pretreated bladders (K) have individual urothelial cells (short arrow) and sheets of cells (long arrow) sloughing, leaving regions of denuded bladder (arrowhead) with hemorrhage and inflammation (concave arrows), whereas KGF-pretreated bladders (P) appear to have minimal urothelial damage (concave arrowhead). L and Q: KRT20 IF (green) in sections adjacent to K and P, respectively, is negative (arrowheads) in both PBS-pretreated (L) and KGF-pretreated (Q) bladders, showing ongoing absence of superficial cells in both. M and R: UPK3 IF (white) in sections adjacent to L and Q, respectively, shows that the PBS-pretreated bladders (M) have UPK3+ intermediate cells within the sheet of sloughing cells (long arrow) and few remaining UPK3+ cells remaining attached to the bladder (short arrow), whereas the KGF-pretreated bladders (R) have robust and continuous UPK3 expression across the lumenal surface of the urothelium (arrowhead), consistent with intact UPK3+ intermediate cells. N and S: KRT5 IF (green) in the same sections as M and R, respectively, shows that the PBS-pretreated bladders (N) have KRT5+ intermediate/basal cells within the sheet of sloughing cells (arrow) and at most a single layer of KRT5+ cells remaining attached to the bladder (short arrow), whereas the KGF-pretreated bladders (S) have robust and continuous KRT5 expression across the deeper regions of the urothelium (arrowhead), consistent with intact KRT5+ intermediate and basal cells. O and T: TUNEL staining (green) in sections adjacent to N and S, respectively, shows sheets of apoptotic cells in PBS-pretreated urothelium (arrowheads; O) and no apoptosis in KGF-pretreated (T) bladders. L–O and Q–T: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bars = 50 μm (A–T). L, lumen.
One day after cyclophosphamide (2 days after KGF or PBS), H&E staining revealed worsened injury with hemorrhage, inflammation, and sheets of urothelial sloughing within the PBS-pretreated mice (Figure 2K), whereas KGF-pretreated bladders appeared largely intact (Figure 2P). Immunostaining for KRT20 was also negative in both the PBS- and KGF-pretreated mice, consistent with absence of superficial cells (Figure 2, L and Q). In PBS-pretreated mice, most UPK3+/KRT20− intermediate and many KRT5+ intermediate/basal cells were injured and/or sloughing, leaving the bladder with only KRT5+ cells or no urothelial cells remaining attached (Figure 2, M and N). In contrast, KGF pretreated bladders had intact UPK3+/KRT20− cells at the lumen (Figure 2R) and undamaged deeper KRT5+ cells (Figure 2S). TUNEL staining revealed significant urothelial apoptosis with PBS pretreatment (Figure 2O), but no apoptosis with KGF pretreatment (Figure 2T). By 3 days after cyclophosphamide, minimal apoptosis was observed in the PBS-pretreated bladders (and still none in the KGF-pretreated bladders), consistent with cessation of acute injury in both groups (Supplemental Figure S4). Taken together, although KGF does not mitigate the loss of superficial cells, it largely protects the underlying intermediate and basal layers from apoptotic death within 24 hours of cyclophosphamide administration.
KGF Cytoprotection Is Likely Mediated through AKT Signaling
To determine how KGF suppresses urothelial apoptosis, immunofluorescence was performed 1 day after cyclophosphamide for pAKT and pERK, two major signaling arms of FGFR2. Although PBS-pretreated bladders had minor increases in pAKT staining compared with uninjured (and untreated) bladders (Figure 3, A and B), KGF-pretreated mice had strong pAKT staining throughout most urothelial layers (Figure 3C). In contrast, minimal pERK staining was observed in uninjured, PBS-pretreated, and KGF-pretreated bladders 1 day after cyclophosphamide administration (Figure 3, D–F). Thus, KGF appears to induce AKT signaling to protect against cyclophosphamide-induced apoptosis of intermediate and basal cell layers.
Figure 3.

Representative images of phosphorylated AKT (pAKT) and phosphorylated extracellular signal-regulated kinase (pERK) immunostaining in keratinocyte growth factor (KGF)–pretreated bladders 1 day after cyclophosphamide injection. A–C: Immunostaining for pAKT (red) shows minimal expression in uninjured (and untreated) urothelium (arrows, A), minor increased signal in PBS-pretreated mice 1 day after cyclophosphamide administration (arrows, B), and robust staining throughout the urothelium in KGF-pretreated mice 1 day after cyclophosphamide administration (arrows, C). D–F: Immunofluorescence images for pERK (red) reveal little global signal in uninjured (and untreated) urothelium (D), PBS-pretreated urothelium 1 day after cyclophosphamide administration (E), and KGF-pretreated urothelium 1 day after cyclophosphamide administration (F). Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bars = 50 μm (A–F). L, lumen.
KGF Pretreatment Leads to an Early Proliferative Response in KRT5+/KRT14− Cells
It was next examined if and how KGF pretreatment affected urothelial proliferative/regenerative responses after cyclophosphamide. Coimmunostaining was performed for the proliferation marker Ki-67 with cell-specific markers, UPK3 and KRT5. Six hours after cyclophosphamide, PBS-pretreated mice had few to no Ki-67+ nuclei (Figure 4, A–D), indicating no proliferative response to cyclophosphamide injury. In contrast, KGF-pretreated mice had several Ki-67+ urothelial nuclei (Figure 4E). Moreover, the vast majority of Ki-67+ cells costained exclusively for KRT5, with similarly low numbers that were either KRT5+/UPK3+ or UPK3+ alone (Figure 4, F–H). Quantitatively, an average of 3.3% of KGF-pretreated urothelial cells were Ki-67+ versus 0.3% in PBS-pretreated mice (P < 0.001) (Figure 4I). Moreover, although KGF-pretreated groups had higher percentages of proliferation among each cell type (UPK3+, KRT5+, and UPK3+/KRT5+) than PBS-pretreated groups, most Ki-67+ urothelial cells were KRT5+ (UPK3−) in both pretreatment groups (Figure 4J). Furthermore, the KGF-pretreated group had an even higher percentage of KRT5+ (UPK3−) proliferating cells than the PBS-pretreatment group (76% versus 42%; P < 0.01). Because KRT5+ cells may also express KRT14, which marks bladder progenitor cells that are up-regulated after injury, coimmunostaining was performed for Ki-67, KRT14, and KRT5. Interestingly, both KGF- and PBS-pretreated animals had few KRT14+ cells overall (and virtually all were Ki-67−) (Supplemental Figure S5), similar to uninjured urothelium.19 Thus, KGF drives proliferation of mostly KRT5+/KRT14− (KRT5 only) cells (and a few UPK3+ and UPK3+/KRT5+ cells) 6 hours after cyclophosphamide administration.
Figure 4.

Representative images and graphs of urothelial proliferation and phosphorylated extracellular signal-regulated kinase (pERK) immunostaining in keratinocyte growth factor (KGF)–pretreated bladders 6 hours after cyclophosphamide injection. A–D: Triple-label immunofluorescence (IF) for Ki-67 (red), uroplakin 3a (UPK3; white), and KRT5 (green) shows no Ki-67+ (proliferating) urothelial cells in this bladder section from a PBS-pretreated mouse 6 hours after cyclophosphamide injection. E: Ki-67 staining shows several proliferating urothelial cells in different layers (white arrow, yellow arrow, and arrowheads) in the KGF-pretreated mouse 6 hours after cyclophosphamide injection. A and E: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. F: Ki-67 and UPK3 merged staining shows that although a few of the proliferating urothelial cells in the KGF-pretreated sample are UPK3+ (yellow arrow and white arrow), most are UPK3− (arrowheads). G: Ki-67 and KRT5 merged staining shows that the UPK3− urothelial cells in the KGF-pretreated sample are KRT5+ (arrowheads) and that among the few UPK3+ cells, some are KRT5+ (white arrow) and others are KRT5− (yellow arrow). H: Merged images with all three labels confirm that most of the Ki-67+ urothelial cells in the KGF-pretreated bladder are KRT5+ (arrowheads), whereas a minority are UPK3+ (yellow arrow) or UPK3+/KRT5+ (white arrow). I: Graph showing significantly higher percentages of proliferating urothelial cells (Ki-67+/DAPI+ cells) in KGF- versus PBS-pretreated mice 6 hours after cyclophosphamide injection. J: Graph showing that the percentages of UPK3+, KRT5+, and UPK3+/KRT5+ proliferating (Ki-67+) urothelial cells in KGF-pretreated mice are statistically higher than in the PBS-pretreated mice and that the vast majority of proliferating cells in the KGF group are KRT5+ (UPK3−). K–M: Co-IF for Ki-67 (red) and pERK (white) in a PBS-pretreated mouse bladder 6 hours after cyclophosphamide administration shows one of the rare Ki-67+ urothelial cells (arrowheads) present that does not label for pERK on individual or merged images. N–P: Co-IF for Ki-67 and pERK in a KGF-pretreated mouse bladder 6 hours after cyclophosphamide administration shows that most Ki-67+ cells colabel for pERK (concave arrowheads), although there are rare Ki-67+ cells that do not colabel for pERK (arrowheads) on individual and merged images. Data are expressed as means ± SEM shown (J). N = 9 (3 planes per bladder and 3 bladders per group; J). *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bars: 50 μm (A–H); 10 μm (K–P). L, lumen.
KGF-Induced Proliferation of KRT5+ Cells Is Likely Mediated through ERK Signaling
To determine how KGF drives proliferation in mostly KRT5+/KRT14− cells 6 hours after cyclophosphamide administration, pAKT and pERK staining was examined. Compared with PBS-pretreated bladders that had virtually undetectable urothelial pAKT staining, KGF-pretreated bladders had mildly increased pAKT staining across most urothelial layers (consistent with suspected antiapoptotic effects), without specific increases in Ki-67+ versus Ki-67− cells (Supplemental Figure S6). High-power confocal images also revealed no pERK signal in uninjured (untreated) urothelium (data not shown) and in most PBS-pretreated tissues 6 hours after cyclophosphamide, including in the few Ki-67+ urothelial cells (Figure 4, K–M) (approximately one in five Ki-67+ cells are pERK+ in the PBS group; data not shown). In contrast, we observed nuclear pERK staining in most Ki-67+ cells in KGF-pretreated bladders 6 hours after cyclophosphamide (Figure 4, N–P). Thus, KGF pretreatment induces proliferation of mostly KRT5+ (only) cells, likely via ERK signaling.
At 1 Day after Cyclophosphamide, KGF-Pretreated Mice Have Ongoing Higher Proliferation Rates in KRT5+/KRT14− Cells versus PBS, but Both Groups Have Similar Rates of KRT14+ Cell Proliferation
The effects of KGF on urothelial proliferation/regeneration were determined 1 day after cyclophosphamide injury by performing co-IF for Ki-67 with KRT14 and KRT5 or with UPK3 and KRT5. At 1 day after cyclophosphamide administration, both PBS- and KGF-pretreated mice had apparently higher numbers of Ki-67+ cells than at 6 hours, although the KGF group appeared to still have more proliferating cells than the PBS group (Figure 5, A and D, and Supplemental Figure S7, A and D). Both pretreatment groups now had many KRT14+ cells (approximately half of which were Ki-67+) that had spread across at least half or more of the basal layers, consistent with their activation from injury (Figure 5, B and E). Similar to 6 hours, the KGF group also appeared to have more proliferating KRT5+/KRT14− (KRT5 only) than the PBS group; and both had low numbers of KRT5− (presumably UPK3+) proliferating cells (Figure 5, C and F). Co-IF for Ki-67, UPK3, and KRT5 confirmed that only a few Ki-67+ cells were UPK3+ or UPK3+/KRT5+ (Supplemental Figure S7, A–F). Counts of Ki-67+/DAPI+ cells from all immunostained sections confirmed that, on average, 10.2% of PBS-pretreated urothelium was proliferating versus 18.6% of the KGF-pretreated urothelium (P < 0.01) (Figure 5G). Quantitative analysis of Ki-67/KRT14/KRT5 stained sections revealed similar percentages of proliferating KRT14+ cells or noncolabeled (presumably UPK3+) cells between the PBS and KGF groups, but significantly higher numbers of KRT5+/KRT14− (KRT5-only) proliferating cells in the KGF group (10.1%) versus the PBS group (3.4%; P < 0.01) (Figure 5H). Quantitative analysis of Ki-67/UPK3/KRT5 stained sections revealed no differences in proliferation rates among any cell types, although the KGF group had a trend for a higher percentage of proliferating KRT5+ cells than the PBS group (likely due to higher numbers of KRT5+/KRT14− cells in the former) (Supplemental Figure S7G). Thus, the larger total number of proliferating cells in the KGF-pretreated group 1 day after cyclophosphamide appears to be due to higher numbers of Ki-67+/KRT5+ (KRT5-only) cells in the former than the latter (and not due to more Ki-67+/KRT14+ cells). Furthermore, both groups appeared to have similar numbers of proliferating KRT14+ cells that were likely activated by injury 24 hours after cyclophosphamide.
Figure 5.

Representative images and graphs of urothelial proliferation in keratinocyte growth factor (KGF)–pretreated bladders 1 day after cyclophosphamide injection. A–F: Triple-label immunofluorescence for Ki-67 (red), KRT14 (white), and KRT5 (green) in PBS-pretreated (A–C) and KGF-pretreated (D–F) bladder sections 1 day after cyclophosphamide administration. A and D: Ki-67 staining shows that compared with 6 hours after injury, both the PBS (A) and the KGF (D) groups appear to have more Ki-67+ proliferating urothelial cells 1 day after injury, although the KGF group still appears to have more Ki-67+ cells than the PBS group. Blue indicates DAPI. Dotted lines indicates demarcation between urothelial layer and underlying lamina propria. B and E: Unlike 6 hours after injury, Ki-67 and KRT14 merged staining shows that both PBS-pretreated (B) and KGF-pretreated (E) mice have expansion of KRT14+ cells across the basal layers, many of which are Ki-67+ (arrowheads), although some Ki-67+ cells do not colabel with KRT14 (concave arrowheads). C and F: Ki-67 and KRT5 merged staining shows that most of the Ki-67+ proliferating cells in both the PBS (C) and KGF (F) groups are KRT5+, including in KRT14+ (arrowheads) and KRT14− (concave arrowheads) cells; a rare KRT5− cell is seen in F (arrow). G: Graph confirming higher percentages of proliferating urothelial cells (Ki-67+/DAPI+ cells) in both PBS and KGF groups compared with 6 hours after cyclophosphamide administration and significantly higher percentages of proliferating urothelial cells in the KGF group versus the PBS group 1 day after cyclophosphamide injection (similar to 6 hours after injury). H: Graph showing that the percentages of KRT5+/KRT14− (KRT5 only) proliferating (Ki-67+) urothelial cells are statistically higher in the KGF- versus PBS-pretreated mice, whereas the percentages of KRT14+ and KRT14−/KRT5− (no label) proliferating cells are not statistically different. Data are expressed as means ± SEM (H). N = 9 (3 planes per bladder and 3 bladders per group; H). **P < 0.01. Scale bars = 50 μm (A–F). L, lumen.
KGF Pretreatment Leads to Regeneration of KRT20+ Superficial Cells and Dampened Proliferation 3 Days after Cyclophosphamide versus PBS Pretreatment
It was next examined how KGF- versus PBS-pretreated mouse urothelium responded to injury 3 days after cyclophosphamide. H&E staining showed more urothelial cell layers in PBS-pretreated mice (Figure 6A) than in KGF-pretreated mice (Figure 6E), consistent with more regenerating cells after more injury in the former versus the latter. PBS-pretreated bladders now expressed UPK3 across the lumenal surface (Figure 6B), but were negative for KRT20 (Figure 6C), consistent with regeneration of UPK3+ intermediate cells and/or immature superficial cells. KGF-pretreated bladders had both lumenal UPK3 and KRT20 staining (Figure 6, F and G), consistent with early regeneration of mature superficial cells. Higher-power images revealed that unlike PBS-pretreated mice (Figure 6D), KGF-pretreated mice had KRT20+ cells with large DAPI+ nuclei, consistent with mature polyploid superficial cells (Figure 6H). Thus, KGF pretreatment leads to early regeneration of superficial cells 3 days after cyclophosphamide treatment.
Figure 6.

Representative images that keratinocyte growth factor (KGF) pretreatment leads to return of superficial cells 3 days after cyclophosphamide injection. A and E: Hematoxylin and eosin staining shows relatively more urothelial cell hyperplasia in the PBS-pretreated mice (arrow, A) than in the KGF-pretreated group (arrow, E). Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. B and F: Immunofluorescence for UPK3 (white) in tissue sections near A and E, respectively, shows robust expression in both the PBS-pretreated (arrowhead, B) and KGF-pretreated (arrowhead, F) mice at the lumenal surfaces. C and G: Immunofluorescence for KRT20 (red) in sections adjacent to B and F, respectively, shows no signal in the PBS-pretreated mice (concave arrowhead), but strong expression in the KGF-pretreated mice (arrowhead). D and H: Magnifications of boxed areas in C and G, respectively, confirm no KRT20 expression in the PBS group (concave arrowhead), but robust KRT20 expression in the KGF group (arrowhead) along with large polyploid nuclei by DAPI staining in the KGF group (arrows). B–D and F–H: Blue indicates DAPI. Scale bars = 50 μm (A–H). L, lumen; UPK3, uroplakin 3a.
Co-IF was performed for Ki-67 along with cell-specific markers to determine effects of KGF versus PBS pretreatment on proliferation rates 3 days after injury. Three days after cyclophosphamide administration, both PBS- and KGF-pretreated mice had more proliferating urothelial cells than earlier time points; however, the PBS group had Ki-67+ cells in multiple layers within the same vertical planes (Figure 7A and Supplemental Figure S7H), whereas KGF mice rarely had greater than one Ki-67+ cell within a vertical plane (Figure 7D and Supplemental Figure S7K). Co-IF with Ki-67, KRT14, and KRT5 revealed that most of the Ki-67+ cells in both groups were KRT14+ and that KRT14+ cells covered the entire basal layers, although the PBS group had approximately two to four urothelial layers of KRT14+ cells (Figure 7B) versus approximately one to three in the KGF group (Figure 7E). Also, almost all KRT14+ cells in the PBS group were Ki-67+ (Figure 7B) versus approximately half in the KGF group (Figure 7E). Most of the remaining proliferating (KRT14−) cells in both groups were KRT5+ only (Figure 7, C and F). Indeed, co-IF for Ki-67, KRT5, and UPK3 confirmed that only a few cells in either group were either UPK3+ or UPK3+/KRT5+ (Supplemental Figure S7, I, J, L, and M). Counts of all Ki-67–stained sections confirmed that an average of 52.9% of PBS-pretreated urothelial cells were proliferating versus 34.2% of the KGF-pretreated urothelium (P < 0.001) (Figure 7G). Quantitative analysis of Ki-67/KRT14/KRT5 coimmunostained sections revealed similar (relatively low) percentages of proliferating KRT5+ only (KRT14−) or noncolabeled (presumably UPK3+) cells in both PBS and KGF groups, but significantly higher numbers of proliferating KRT14+ cells in the PBS group (44.3%) versus the KGF group (30.9%; P < 0.01) (Figure 7H). Quantitative analysis of Ki-67/UPK3/KRT5 coimmunostained sections revealed that the PBS group had higher percentages of proliferating KRT5+ cells (a mix of KRT14+ and KRT14− cells) than the KGF group, with no differences among other cell types (Supplemental Figure S7N). Thus, the larger number of proliferating cells in the PBS-pretreated group 3 days after cyclophosphamide administration appears to be secondary to more Ki-67+/KRT14+ cells than in the KGF group, likely reflecting a more robust regenerative response in the PBS group because of more severe injury. This is also consistent with the KGF-stimulatory effect on KRT5+ cell proliferation being depleted 3 days after cyclophosphamide administration.
Figure 7.

Representative images and graphs of urothelial proliferation in keratinocyte growth factor (KGF)–pretreated bladders 3 days after cyclophosphamide injection. A–F: Triple-label immunofluorescence for Ki-67 (red), KRT14 (white), and KRT5 (green) in PBS-pretreated (A–C) and KGF-pretreated (D–F) bladder sections 3 days after cyclophosphamide administration. A and D: Ki-67 staining shows that compared with 6 hours and 1 day after injury, both the PBS group (A) and the KGF group (D) appear to have more Ki-67+ proliferating nuclei 3 days after injury, although now the PBS group has many more Ki-67+ cells than the KGF group, including three to four rows of proliferating cells in the former versus one to two in the latter. Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. B and E: Ki-67 and KRT5 merged staining shows that both PBS-pretreated mice (B) and KGF-pretreated mice (E) have expansion of KRT14+ cells covering the basal layers and into more superficial layers (two to four total layers in the former and one to three layers in the latter), many of which are Ki-67+ (arrowheads), although some Ki-67+ cells do not colabel with KRT14 (arrows). C and F: Ki-67 and KRT5 merged staining shows that most of the Ki-67+ proliferating urothelial cells from both the PBS group (C) and the KGF group (F) are KRT5+, including in KRT14+ (arrowheads) and the few KRT14− (arrows) cells. G: Graph showing higher percentages of proliferating cells (Ki-67+/DAPI+ cells) in both the PBS and KGF groups compared with 6 hours and 1 day after injury and significantly higher percentages of proliferating urothelial cells in the PBS group versus the KGF group 3 days after cyclophosphamide injection (unlike at 6 hours and 1 day after injury). H: Graph showing that the percentages of KRT14+ proliferating (Ki-67+) urothelial cells are statistically higher in the PBS- versus KGF-pretreated mice, whereas the percentages of KRT5+/KRT14− (KRT5 only) and KRT14−/KRT5− (no label) proliferating cells are not statistically different. Data are expressed as means ± SEM (H). N = 6 (2 planes per bladder and 3 bladders per group; H). **P < 0.01, ***P < 0.001. Scale bars = 50 μm (A–F). L, lumen.
KGF Pretreatment Leads to Faster Resolution of Cyclophosphamide-Induced Urothelial Injury
Ten days after injury, H&E stains revealed that PBS-pretreated mice had ongoing urothelial hypercellularity, similar to nonpretreated/injured mice at 10 days (Figure 1D), consistent with ongoing regeneration (Figure 8A), whereas KGF-pretreated mice had three to four cell layers with polyploid superficial cells (Figure 8F), similar to uninjured urothelium (Figure 1A). IF for KRT20 revealed light and discontinuous staining in PBS-pretreated mice (Figure 8B) versus robust and continuous staining in KGF-pretreated mice (Figure 8G). IF for Ki-67 revealed ongoing proliferation (albeit reduced versus 3 days) in the PBS group (Figure 8C and Supplemental Figure S7O) and few Ki-67+ urothelial cells in KGF-pretreated mice (Figure 8H and Supplemental Figure S7R). Co-IF for Ki-67/KRT14/KRT5 revealed that the vast majority of the proliferating cells in the PBS group were in KRT14+ cells that still lined the basal layer (often two to three layers), followed by fewer cells that were KRT5+ only or that were not colabeled (likely UPK3+) (Figure 8, C–E). In contrast, KGF-pretreated mice had only sparse numbers of KRT14+ cells and low numbers of any proliferating cell type (KRT14+ > KRT5+ only > noncolabeled cells) (Figure 8, H–J). Co-IF for Ki-67/UPK3/KRT5 revealed that the vast majority of proliferating cells in the PBS group were KRT5+ (likely mostly KRT14+) (Supplemental Figure S7, P and Q) and that the KGF group had a more even mix of low numbers of proliferating cells (KRT5+ > the others) (Supplemental Figure S7, S and T). Quantitative analysis of all Ki-67–stained sections confirmed much higher proliferation rates in PBS-pretreated urothelium (20.2%) than in KGF-pretreated urothelium (4.7%; P < 0.001) (Figure 8K). Quantitative analysis of Ki-67/KRT14/KRT5 stained sections revealed that PBS-pretreated mice had significantly higher percentages of proliferating KRT14+ cells than the KGF group, whereas the two groups had similar (relatively low) numbers of proliferating KRT5+ only cells (the PBS group had more proliferating noncolabeled cells than the KGF group, but both were low numbers) (Figure 8L). Quantitative analysis of Ki-67/UPK3/KRT5 stained sections revealed that PBS-pretreated mice had significantly higher mean percentages of proliferating KRT5+ cells (likely from a larger number of Ki-67+/KRT14+ cells) than KGF-pretreated mice, whereas the two groups had similar (low) numbers of UPK3+ cells (the PBS group also had larger numbers of UPK3+/KRT5+ cells versus the KGF group, although percentages were low in both) (Supplemental Figure S7U). Thus, 10 days after injury, PBS-pretreated urothelium was still largely regenerating, led by proliferation of KRT14+ cells, whereas the KGF-pretreated urothelium appeared to be nearly fully regenerated, with regression of many KRT14+ cells.
Figure 8.

Representative images and graphs of urothelial proliferation in keratinocyte growth factor (KGF)–pretreated bladders 10 days after cyclophosphamide injection. A and F: Hematoxylin and eosin staining shows that PBS-pretreated bladders (A) have evidence of ongoing cellular hyperplasia with up to seven to eight layers of smaller cells throughout the urothelium (arrow, A) compared with the KGF-pretreated bladders (F) that have three to four cell layers (arrow, F) and many large superficial cells (arrowheads), similar to uninjured urothelium. B and G: Immunofluorescence (IF) for KRT20 (red) in sections adjacent to A and F, respectively, shows the return of some signal in the outer layers of the PBS-pretreated urothelium (arrowhead, B), although gaps without KRT20 expression remain (concave arrowhead) compared with KGF-pretreated bladders (G) that have robust and continuous KRT20 staining across the lumenal surface (arrowhead, G), consistent with the return of mature superficial cells (arrows in B and G illustrate the larger numbers of DAPI+ urothelial cells in the PBS group versus the KGF group, respectively). C–E and H–J: Triple-label IF for Ki-67 (red), KRT14 (white), and KRT5 (green) in PBS-pretreated (C–E) and KGF-pretreated (H–J) bladder sections adjacent to B and G, respectively. C and H: Ki-67 staining shows that the PBS group (C) continues to have robust numbers of proliferating (Ki-67+) urothelial cells, whereas the KGF group (H) has few proliferating cells. B, C, G, and H: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. D and I: Ki-67 and KRT14 merged staining shows that in PBS-pretreated mice (D), most of the proliferating (Ki-67+) urothelial cells are also KRT14+ cells (arrowheads, D) that remain spread across the urothelium and few Ki-67+ cells are KRT14− (concave arrowhead, D); in contrast, KGF-pretreated bladders (I) have few KRT14+ cells and among the few Ki-67+ cells, similar numbers are KRT14+ (arrowhead, I) and KRT14− (concave arrowhead, I). E and J: Ki-67 and KRT5 merged staining shows that most of Ki-67+ proliferating cells in both the PBS group (E) and the KGF group (J) are KRT5+, including in KRT14+ (arrowheads) and KRT14− (KRT5+ only; concave arrowheads) cells (arrow in J shows a Ki-67+/KRT5− cell with a large nucleus, likely representing a polyploid superficial cell). K: Graph showing significantly higher percentages of proliferating cells (Ki-67+/DAPI+ cells) in the PBS-pretreated urothelium versus KGF-pretreated urothelium 10 days after cyclophosphamide injection. L: Graph showing that the percentages of KRT14+ proliferating (Ki-67+) urothelial cells are much higher in the PBS- versus KGF-pretreated mice [and that the small number of proliferating KRT14/KRT5− (no label) cells are also statistically higher in the PBS group], whereas the percentages of proliferating KRT5+/KRT14− (KRT5 only) cells are not statistically different. Data are expressed as means ± SEM (L). N = 6 (2 planes per bladder and 3 bladders per group; L). **P < 0.01, ***P < 0.001. Scale bars = 50 μm (A–J). L, lumen.
To confirm that KGF pretreatment led to sustained restoration of urothelium after cyclophosphamide administration, IF was performed for Ki-67 and urothelial-specific markers in the KGF and PBS groups 28 days after injury. IF for KRT20 showed that PBS-pretreated mice still had occasional patches of light or absent staining, consistent with incomplete regeneration of superficial cells (Supplemental Figure S8A); KGF pretreated mice had continuous, linear KRT20 staining along the lumen (Supplemental Figure S8F). Triple-label IF for Ki-67, UPK3, and KRT5 showed ongoing patches of proliferating urothelial cells in PBS-pretreated mice (albeit reduced compared with 10 days), which were virtually all KRT5+ and UPK3− (Supplemental Figure S8, B–D). Conversely, KGF-pretreated urothelium had no Ki-67+ urothelial cells that were KRT5+ and UPK3+ (Supplemental Figure S8, G–I). Finally, IF for Ki-67/KRT14 confirmed that PBS-pretreated mice still had many KRT14+ cells present along the basal layer, many of which were proliferating (Supplemental Figure S8E). IF for Ki-67/KRT14 in the KGF-pretreated mice confirmed the absence of urothelial proliferation, although occasional patches of Ki-67−/KRT14+ basal cells were present (more than typically observed in uninjured urothelium) (Supplemental Figure S8J). Thus, KGF pretreatment appears to lead to sustained restitution of the urothelium (with the exception of some remnant KRT14+ cells that had not yet regressed), whereas PBS-pretreated mice continued to have ongoing regeneration with proliferating KRT14+ cells and incomplete superficial cell restoration 28 days after cyclophosphamide administration.
KGF Drives Proliferation of Mostly KRT5+ Cells (and Not KRT14+ Cells) in the Absence of Injury
Given that KGF appears to stimulate KRT5+ (KRT14−) (and to a lesser extent UPK3+ and UPK3+/KRT5+) cell proliferation at 6 and 24 hours after cyclophosphamide administration, proliferation and cell-specific markers were examined in PBS- and KGF-pretreated mice, followed by i.p. PBS (sham injury). PBS-pretreated mice had few Ki-67+ urothelial cells 6 hours or 1 day after sham injury (30 and 48 hours after PBS pretreatment, respectively), whereas KGF-pretreated mice had a small burst of proliferating cells at both time points (Figure 9, A, D, I, and L). Similar to cyclophosphamide-injured mice, co-IF of Ki-67 with UPK3 and KRT5 revealed that the clear majority of the proliferating cells in the sham-injured KGF group at 6 hours and 1 day were KRT5+, with fewer that were UPK3+ or UPK3+/KRT5+. PBS-pretreated mice had low percentages of proliferation among all cell types (Figure 9, B, C, E, F, J, K, M, and N). Quantitative analysis of all Ki-67–stained sections confirmed significantly higher numbers of proliferating cells in the KGF groups versus the PBS groups at 6 hours (7.32% versus 0.15%; P < 0.001) and 1 day (5.09% versus 0.52%; P < 0.001) after sham injury (Figure 9, G and O). Quantitative analysis of Ki-67/UPK3/KRT5 coimmunostaining sections revealed statistically higher numbers of proliferating cells of all types in KGF- versus PBS-pretreated groups at both 6 hours and 1 day after sham injury, although the vast majority of proliferating cells in the KGF groups at both time points were KRT5+ (Figure 9, H and P). Co-IF with Ki-67/KRT14 revealed no expansion of or proliferation of KRT14+ cells (ie, no Ki-67+/KRT14+ cells) in either PBS- or KGF-pretreated mice at 6 hours or 1 day after sham injury (Supplemental Figure S9, A–D). By 3 days after sham injury, both PBS- and KGF-pretreated mice had low numbers of Ki-67+ urothelial cells of any subtype (Supplemental Figure S10) and continued to have low numbers of KRT14+ cells that were not proliferating, similar to the quiescent state (Supplemental Figure S9, E and F). Finally, most of the Ki-67+ cells in the KGF-pretreated groups at 6 hours after sham injury were also pERK+ (data not shown), similar to the injured KGF-pretreated mice 6 hours after injury. Thus, KGF pretreatment drives likely ERK-mediated proliferation of mostly KRT5+ cells, even in the absence of injury. Moreover, KGF does not drive expansion or proliferation of KRT14+ cells in the absence of injury; thus, the robust increase in number and proliferation of KRT14+ cells after injury likely occurs in an FGF-independent manner.
Figure 9.

Representative images and graphs of urothelial proliferation 6 hours and 1 day after sham injury. A–C: Triple-label immunofluorescence (IF) for Ki-67 (red), uroplakin 3a (UPK3; white), and KRT5 (green) 6 hours after sham injury shows minimal proliferation in PBS-pretreated mouse with a single proliferating cell in this section that is UPK3+/KRT5+ (arrows). D: Ki-67 staining shows several proliferating urothelial cells in deeper layers (arrowheads) and one superficial proliferating cell (concave arrowhead) in the keratinocyte growth factor (KGF)–pretreated mouse 6 hours after sham injury. A and D: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. E: Ki-67 and UPK3 merged staining shows many more proliferating cells are UPK3− (arrowheads) than UPK3+ (concave arrowhead). F: Ki-67 and KRT5 merged staining confirms that more of the proliferating cells are KRT5+ (arrowheads) than KRT5− (concave arrowhead). G: Graph showing significantly higher percentages of proliferating urothelial cells (Ki-67+/DAPI+ cells) in KGF- versus PBS-pretreated mice 6 hours after sham injury. H: Graph showing that the percentages UPK3+, KRT5+, and UPK3+/KRT5+ proliferating (Ki-67+) urothelial cells in KGF-pretreated mice are statistically higher than the PBS-pretreated mice after sham injury and that the vast majority of proliferating cells in the KGF group are KRT5+ (UPK3−). I–K: Triple-label IF for Ki-67 (red), UPK3 (white), and KRT5 (green) 1 day after sham injury still shows minimal proliferation in PBS-pretreated mouse with one proliferating cell in this section that is UPK3+/KRT5+ (arrows) and one that is only KRT5+ (arrowheads). L: Ki-67 staining still shows several proliferating urothelial cells in different layers in the KGF-pretreated mouse 1 day after sham injury (arrowheads and arrow). M: Ki-67 and UPK3 merged staining shows many more proliferating cells are UPK3− (arrowheads) than UPK3+ (arrow). N: Ki-67 and KRT5 merged staining confirms that most of the proliferating cells are KRT5+ (arrowheads), with one cell that is UPK3+/KRT5+ (arrow). O: Graph showing ongoing significantly higher percentages of proliferating urothelial cells (Ki-67+/DAPI+ cells) in KGF- versus PBS-pretreated mice 1 day after sham injury. P: Graph showing that the percentages of UPK3+, KRT5+, and UPK3+/KRT5+ proliferating (Ki-67+) urothelial cells in KGF-pretreated mice are statistically higher than in the PBS-pretreated mice after sham injury and that the majority of proliferating cells in the KGF group are KRT5+ (UPK3−). I and L: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Data are expressed as means ± SEM (H and P). N = 9 (3 planes per bladder and 3 bladders per group; H and P). ***P < 0.001. Scale bars = 50 μm (A–F and I–N). L, lumen.
Discussion
Before determining how keratinocyte growth factor affected cyclophosphamide-induced urothelial injury, the injury pattern was first defined after cyclophosphamide administration alone. Between 2 and 6 hours after cyclophosphamide administration, nonapoptotic death and sloughing of superficial cells were observed. From 4 to 24 hours, increased apoptosis of intermediate and possibly basal cells was observed. Although both the apoptotic and nonapoptotic modes of cell death have been reported in many rodent models, few have examined time points earlier than 12 to 24 hours20, 21, 22 and none, to our knowledge, has used cell-specific markers to detail the spatiotemporal injury patterns.23, 24, 25 One report in rats examined multiple time points within 24 hours after cyclophosphamide infusion and without cell-specific labeling and noted that superficial cells underwent necrotic (nonapoptotic) cell death within 6 hours, followed by waves of apoptosis within deeper layers from 12 to 24 hours, similar to our findings.25 Injury severity after cyclophosphamide administration also likely differs among mouse strains, with C57/BL6 mice appearing more resistant than others, such as ICR and DBA/2 mice.26,27 FVB/N mice, which is the strain used in this study, appear to be in the more susceptible group, given the early and extensive injury.
KGF was next administered 24 hours before cyclophosphamide administration. Extensive cytoprotection was noticed within the urothelium. The authors of the previous publication showing beneficial effects of KGF before cyclophosphamide infusion in rats had discussed the possibility of cytoprotective effects, but did not test for any.11 In this study, KGF prevented apoptotic death of deeper urothelial cells (UPK3+ intermediate and KRT5+ intermediate/basal cells), likely via activation of AKT. FGF signaling can suppress apoptosis in cells outside of the bladder.28, 29, 30, 31, 32 Furthermore, many publications have shown that AKT signaling suppresses apoptosis,33 including downstream of FGF signaling in nonbladder tissues.34, 35, 36, 37 Interestingly, one group determined that cyclophosphamide administration led to minor increases in pAKT expression throughout bladder urothelium, mesenchymal tissues, and inflammatory cells within 24 hours (minor increases in urothelium in PBS-pretreated mice were also observed in this study). However, intravesicular administration of an AKT inhibitor enhanced bladder capacity, void volume, and intercontraction void interval, suggesting that increased pAKT signaling was detrimental.38 It is possible that the benefits of inhibiting AKT signaling are specific to blockade in the detrusor or inflammatory cells as opposed to the urothelium. On balance, it appears that the significant up-regulation of AKT signaling in the urothelium via KGF affords cytoprotection against apoptotic injury of deeper urothelial cells after cyclophosphamide administration.
KGF did not protect against nonapoptotic death of superficial cells. This does not appear to be due to the absence of FGFR2 expression in superficial cells, based on immunostaining in uninjured urothelium (Supplemental Figure S2). The staining pattern of pFRS2α after KGF administration 15 hours earlier (indicating FGFR2 activation) does not clarify the issue. Although pFRS2α staining is absent at the apical surfaces of the superficial cells, it may be present at the basolateral surfaces (Supplemental Figure S3). Most likely, KGF/FGFR2 activation simply does not block nonapoptotic cell death (in any cell type). Moreover, the inability of KGF to prevent superficial cell loss is appropriate given that these cells are exposed to high concentrations of acrolein in the urine after cyclophosphamide administration, likely leading to irreversible cellular and DNA damage by reactive oxygen species and nitric oxide that stimulates aberrant NF-κB signaling.1,7,39
In addition to cytoprotection against cyclophosphamide-induced urothelial cell apoptosis, KGF also drove proliferation of urothelial cells. The earlier study in rats showed enhanced numbers of proliferating cell nuclear antigen (PNCA)+ urothelial cells in KGF-pretreated bladders versus vehicle-pretreated bladders 24 hours after cyclophosphamide administration.11 However, that study did not identify specific cell types proliferating after KGF and did not examine effects of KGF on proliferation in uninjured bladder. The source(s) of adult urothelial progenitors (loosely defined as cells with proliferative potential) are likely chiefly KRT14+ basal cells, which normally are relatively sparse in uninjured bladder, but proliferate after injury (as noted in this study).19 Other groups have shown that UPK3+ intermediate cells and KRT20+ superficial cells have potential to proliferate after injury.40,41 Although others showed that KRT5+ basal cells can proliferate after injury, these groups did not perform immunostaining to determine whether they were KRT14+.42,43 This study clearly demonstrates that KRT5+/KRT14− cells have proliferative potential after injury. Moreover, KGF appears to drive proliferation of KRT5+/KRT14− cells and, to a lesser degree, UPK3+ and KRT5+/UPK3+ urothelial cells, both after cyclophosphamide-induced injury and in uninjured bladders. The enhanced urothelial proliferation rates in KGF- versus PBS-pretreated mice at 6 hours and 1 day after cyclophosphamide administration appears to be due to increases in KRT5+ (and to lesser degree UPK3+) cell proliferation. In contrast, the higher proliferation rates in the PBS- versus KGF-pretreated urothelium at 3, 10, and 28 days after cyclophosphamide administration appear to be due to increased numbers of KRT14+ proliferating cells, likely reflecting increased injury in the former. Thus, exogenous KGF does not appear to drive proliferation of KRT14+ cells at all, in either the context of injury or uninjured bladders; the signaling pathway driving KRT14+ cell proliferation after injury remains unclear.
The mechanism by which KGF drives proliferation of (predominantly) KRT5+/KRT14− cells appears to be via ERK, a major effector of FGF signaling.18 KGF has been shown to drive proliferation of nonurothelial cells by ERK activation.44, 45, 46 Furthermore, the nuclear pERK localization in the KGF-treated mice (Figure 4, N–P) is consistent with pERK translocation into the nucleus, leading to a cascade of events resulting in proliferation.47 Whether KGF-driven proliferation impacts on early regeneration of the superficial cells 3 days after cyclophosphamide injury (compared with PBS-pretreated mice) is unclear. KGF-mediated cytoprotection itself likely contributes to the rapid regeneration of superficial cells (versus PBS-pretreated mice that have more extensively injured urothelium). However, the KRT5+/KRT14− (and UPK3+) cells that proliferate in response to KGF are near the lumenal surface (where newly formed superficial cells are located) and, as such, may augment superficial cell regeneration. Interestingly, another group found that ERK was activated in rat bladder urothelium, select nerves, and some inflammatory cells after cyclophosphamide administration (peaking at 48 hours) and that intravesicular infusion of an ERK inhibitor enhanced bladder capacity,48 as with AKT. It is unclear whether the apparent benefits of ERK knockdown are specific to urothelium or other tissues. On balance, activation of ERK by KGF to drive proliferation of select urothelial cells does not appear to do harm and might, in fact, promote regeneration of superficial cells.
It is unclear whether intravesical administration of KGF (ie, directly into the bladder lumen) would lead to cytoprotective and proliferative effects comparable to systemic administration. Ulich et al11 demonstrated that i.v. administration of KGF (same dose as in this study) led to high urinary levels of KGF, peaking at 8 hours after dose. However, the same report showed that mice subjected to bilateral nephrectomy had a similar urothelial proliferative response driven by KGF as those mice with sham nephrectomy.11 Of note, that study did not assess the cytoprotective actions of KGF. In this study, KGF-driven pFRS2α expression in the urothelium (indicating activation of FGFR2) is excluded from apical superficial cell surfaces and is instead expressed along the surfaces of deeper cells throughout the urothelium (and possibly basolateral surfaces of the superficial cells) (Supplemental Figure S3). Thus, topical KGF would need to penetrate the uroplakin barrier to enter these deeper layers to activate FGFR2, which would appear unlikely. Taken together, the topical administration of KGF may not have comparable cytoprotective or proproliferative effects as systemic administration.
Finally, pretreatment with KGF (whether by cytoprotection alone or in combination with proliferation of KRT5+ cells) drives (near) completion of urothelial regeneration at approximately 10 days after cyclophosphamide administration, which persists to 28 days after injury (with the exception of small patches of persistent, nonproliferating KRT14+ cells). In contrast, the PBS-pretreated mice still had ongoing proliferation and regeneration with cellular hyperplasia and incomplete KRT20+ staining 10 days after cyclophosphamide administration, which was still apparent, to a lesser degree, at 28 days after injury. Other rodent models have also shown that regeneration after cyclophosphamide administration can take from 14 days to 2 to 3 months to complete.49,50 Moreover, mice with spinal cord injury–induced bladder dysfunction, a less severe form of urothelial injury than cyclophosphamide injury, did not complete regeneration until approximately 28 days.51 Early completion of regeneration after cyclophosphamide administration may have long-term benefits for patients receiving the drug, including possibly decreasing risks of bladder fibrosis or urothelial cancer. A diagram outlining the apparent urothelial actions of KGF versus PBS after cyclophosphamide administration is shown in Figure 10.
Figure 10.

Diagram of proposed actions of keratinocyte growth factor (KGF) versus PBS after cyclophosphamide administration. Left panel: Uninjured: diagram of different cell types in the uninjured urothelium [basal cell (B cell), intermediate cell (I cell), and superficial cell (S cell)]. Middle panel: PBS: cyclophosphamide (CPP) is metabolized to acrolein (Acr), leading to acute injury from 2 hours to 1 day, including superficial cell necrosis and intermediate and basal cell apoptosis. During early regeneration (1 to 3 days), KRT14+ basal cells proliferate to begin reconstituting intermediate cells, but superficial cells remain absent. During late regeneration (10 to 28 days), KRT14+ cells begin to regress, with reduced proliferation, and other urothelial layers are more intact, including partial regeneration of superficial cells. Right panel: KGF: during acute injury (2 hour to 1 day), KGF pretreatment does not prevent superficial cell necrosis, but blocks intermediate and basal cell apoptosis by driving AKT downstream of fibroblast growth factor receptor 2 (FGFR2). During early regeneration, KGF pretreatment leads to an early burst of KRT5+ (only) cell proliferation (starting at 6 hours) and leads to early regeneration of superficial cells (from reduced injury and/or the KRT5+ cell proliferative burst). During late regeneration, KGF pretreatment leads to nearly full restoration of urothelial layers, including the absence of proliferating KRT14+ cells (although small patches of nonproliferating KRT14+ basal cells persist). ERK, extracellular signal-regulated kinase; UPK3, uroplakin 3a.
Acknowledgments
We thank Drs. Rannar Airik, Sunder Sims-Lucas, Jacqueline Ho, and Anthony Kanai for helpful discussions about the experimental design and in interpreting the results.
Footnotes
Supported by NIH grant R01DK104374 (C.M.B.).
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2019.09.015.
Supplemental Data
Supplemental Figure S1.

Representative images of wild-type mouse bladder urothelium 4 hours after cyclophosphamide injection. A: Hematoxylin and eosin–stained bladder shows sloughing of urothelial cells (arrows). B: KRT20 immunofluorescence (IF; red) in a section adjacent to A shows that the sloughing cells are still KRT20+ superficial cells (arrow) and that larger regions of remnant urothelium have attenuated (arrowhead) or no KRT20 expression. C and D: Uroplakin 3a (UPK3) IF (white; C) and KRT5 IF (green; D) in a section adjacent to B shows rare regions of discontinuous urothelial staining for both markers (arrows). E: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining (green) in a section adjacent to C and D is now positive in a few nuclei (arrowheads) consistent with the onset of apoptosis in deeper urothelial; cells. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. B–E: Blue indicates DAPI. Scale bar = 50 μm (A–E). L, lumen.
Supplemental Figure S2.
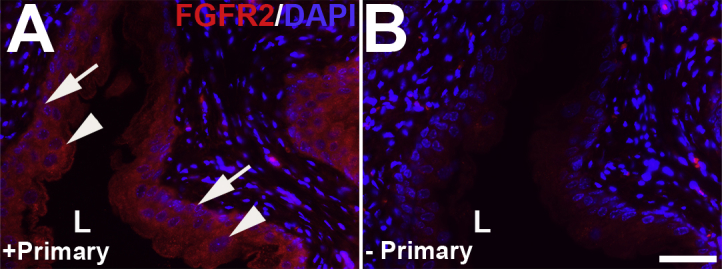
Representative images of fibroblast growth factor receptor 2 (FGFR2) immunostaining in uninjured bladder urothelium. A: Immunofluorescence with primary antibodies (+Primary) against FGFR2 (red) demonstrates strong signal throughout uninjured (and untreated) urothelium, from basal cells (arrows) to polyploid superficial cells (arrowheads). B: Immunofluorescence with no primary antibodies (-Primary) reveals no signal in the urothelium. Blue indicates DAPI. Scale bar = 50 μm (A and B). L, lumen.
Supplemental Figure S3.
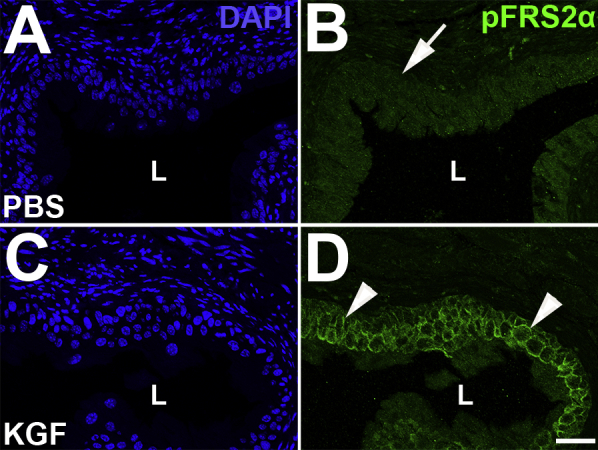
Representative images of pFRS2α urothelial staining 15 hours after s.c. PBS or keratinocyte growth factor (KGF) administration. A–D: Immunofluorescence for pFRS2α (green) reveals no urothelial staining in PBS-pretreated mice (arrow) and linear membrane staining throughout the urothelium in KGF-pretreated mice (arrowheads). A and C: DAPI channels. Scale bar = 50 μm (A–D). L, lumen.
Supplemental Figure S4.
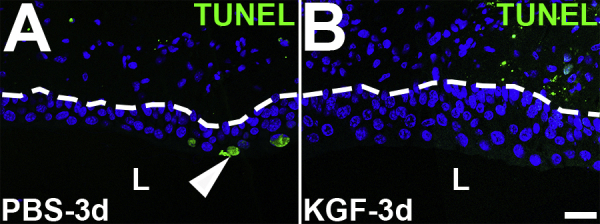
Representative images of terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining in PBS- and keratinocyte growth factor (KGF)–pretreated bladders 3 days after cyclophosphamide injection. A and B: TUNEL staining (green) reveals few apoptotic cells in PBS-pretreated urothelium (arrowhead) and none in KGF-pretreated urothelium (B) 3 days after cyclophosphamide administration. Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bar = 50 μm (A and B). L, lumen.
Supplemental Figure S5.

Representative images of Ki-67, KRT14, and KRT5 coimmunostaining in keratinocyte growth factor (KGF)–pretreated bladders 6 hours after cyclophosphamide injection. Triple-label immunofluorescence for Ki-67 (red), KRT14 (white), and KRT5 (green). A and D: Ki-67 staining shows no Ki-67+ (proliferating) cells in PBS-pretreated urothelium (A), but several Ki-67+ proliferating cells in the KGF-pretreated urothelium (arrow) 6 hours after cyclophosphamide injection. A, B, D, and E: Ki-67 and KRT14 merged staining shows rare KRT14+ cells in the basal layers of both PBS-pretreated mice (arrowheads in A and B) and KGF-pretreated mice (arrowheads in D and E), none of which are Ki-67+. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. C, E, and F: Ki-67 and KRT5 merged staining shows that the rare KRT14+ cells are also KRT5+ in both groups (arrowheads; C and F) and that the Ki-67+ proliferating cells in the KGF group are all KRT5+/KRT14− (arrows; E and F). A and D: Blue indicates DAPI. Scale bar = 50 μm (A–F). L, lumen.
Supplemental Figure S6.
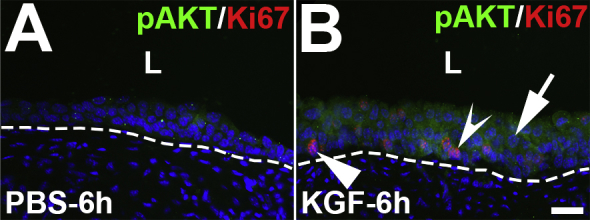
Representative images of phosphorylated AKT (pAKT) and Ki-67 coimmunostaining in keratinocyte growth factor (KGF)–pretreated bladders 6 hours after cyclophosphamide injection. A: Immunostaining for pAKT (green) and Ki-67 (red) reveals limited expression in PBS-pretreated bladders 6 hours after cyclophosphamide administration. B: In comparison to PBS-pretreated/injured mice, KGF-pretreated/injured bladders have increased and diffuse pAKT staining across most of the urothelium, including in most of the predominantly Ki-67− (arrow) and in some of the Ki-67+ (concave arrowhead) cells, although some Ki-67+ cells do not express pAKT (arrowhead). Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bar = 50 μm (A and B). L, lumen.
Supplemental Figure S7.

Representative images and graphs of Ki-67, uroplakin 3a (UPK3), and KRT5 staining in keratinocyte growth factor (KGF)–pretreated bladders 1, 3, and 10 days after cyclophosphamide injection. A–F: Triple-label immunofluorescence (IF) for Ki-67 (red), UPK3 (white), and KRT5 (green) in PBS-pretreated (A–C) and KGF-pretreated (D–F) bladder sections 1 day after cyclophosphamide. A and D: Ki-67 staining shows that compared with 6 hours after injury, both the PBS (A) and the KGF (D) groups appear to have more Ki-67+ proliferating nuclei 1 day after injury, although the KGF group still appears to have more Ki-67+ cells than the PBS group. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. B and E: Ki-67 and UPK3 merged staining shows that both PBS-pretreated mice (B) and KGF-pretreated mice (E) have Ki-67+ proliferating cells that are mostly UPK3− (arrowheads), although a few Ki-67+ cells are UPK3+ (arrow). C and F: Ki-67 and KRT5 merged staining shows that both PBS-pretreated (C) and KGF-pretreated (F) mice have Ki-67+ cells that are mostly KRT5+ (arrowheads), including some UPK3+ cells, as seen in B (arrow). A and D: Blue indicates DAPI. G: Graph showing that the percentages of UPK3+, KRT5+, and UPK3+/KRT5+ proliferating (Ki-67+) urothelial cells are not statistically different, although the percentages of KRT5+ cells may be trending higher in the KGF group versus the PBS group. H–M: Triple-label IF for Ki-67 (red), UPK3 (white), and KRT5 (green) in PBS-pretreated (H–J) and KGF-pretreated (K–M) bladder sections 3 days after cyclophosphamide administration. H and K: Ki-67 staining shows that compared with 6 hours and 1 day after injury, both the PBS group (H) and the KGF group (K) appear to have more Ki-67+ proliferating nuclei 3 days after injury, although the PBS-pretreated group has more Ki-67+ cells than the KGF group, including three to four rows of proliferating cells in the former versus one to two in the latter. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. I and L: Ki-67 and UPK3 merged staining shows that both PBS-pretreated mice (I) and KGF-pretreated mice (L) have Ki-67+ proliferating cells that are mostly UPK3− (arrowheads), although rare Ki-67+ cells are UPK3+ (arrow). J and M: Ki-67 and KRT5 merged staining shows that both PBS-pretreated (J) and KGF-pretreated (M) mice have Ki-67+ cells that are mostly KRT5+ (arrowheads), except for the UPK3+ cell seen in I (arrow). H and K: Blue indicates DAPI. N: Graph showing that the percentages of KRT5+ proliferating (Ki-67+) cells in the PBS group are statistically higher than the KGF group, whereas the percentages of UPK3+ and UPK3+/KRT5+ proliferating urothelial cells are not statistically different. O–T: Triple-label IF for Ki-67 (red), UPK3 (white), and KRT5 (green) in PBS-pretreated (O–Q) and KGF-pretreated (R–T) bladder sections 10 days after cyclophosphamide administration. O and R: Ki-67 staining shows that the PBS group (O) continues to have robust numbers of proliferating (Ki-67+) urothelial cells, whereas the KGF group (R) has few proliferating cells. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. P, Q, S, and T: Most of the Ki-67+ cells in both the PBS-pretreated bladders (P and Q) and the KGF-pretreated bladders (S and T) are UPK3− and KRT5+ (arrowheads). U: Graph showing that the percentages of KRT5+ proliferating (Ki-67+) cells in the PBS group are statistically higher than in the KGF group (as is true for the small number of proliferating UPK3+/KRT5+ cells), whereas the percentages of UPK3+ proliferating urothelial cells are not statistically different. O and R: Blue indicates DAPI. Data are expressed as means ± SEM (G, N, and U). N = 9 (3 planes per bladder and 3 bladders per group; G); N = 6 (2 planes per bladder and 3 bladders per group; N and U). **P < 0.01, ***P < 0.001. Scale bars: 50 μm (A–F, H–M, and O–T). L, lumen.
Supplemental Figure S8.

Representative images of Ki-67 and urothelial marker staining in keratinocyte growth factor (KGF)–pretreated bladders from 28 days after cyclophosphamide injection. A and F: Immunofluorescence (IF) for KRT20 (red) shows regions of incomplete staining along the lumenal surface of PBS-pretreated mice (arrowhead), whereas KGF-pretreated mice have strong, continuous signal (F) 28 days after injury. B–D: Triple-label IF for Ki-67 (red), uroplakin 3a (UPK3; white), and KRT5 (green) in a section adjacent to A shows that PBS-pretreated mice still have proliferating cells along the basal layer that colabel for KRT5 and not UPK3 (arrowheads), 28 days after injury. E: Co-IF for Ki-67 (red) and KRT14 (white) in a section adjacent to D shows that PBS-pretreated mice continue to have many KRT14+ cells along the basal layer, many of which are proliferating (concave arrowheads) 28 days after injury. G–I: Triple-label IF for Ki-67 (red), UPK3 (white), and KRT5 (green) in a section adjacent to F shows that KGF-pretreated mice have no proliferating urothelial cells, including in UPK3+ and KRT5+ cells. J: Co-IF for Ki-67 (red) and KRT14 (white) in a section adjacent to I confirms that KGF-pretreated urothelial cells are not proliferating, although small patches of persistent KRT14+ cells are apparent along the basal layer (arrows) 28 days after injury. A, B, F, and G: Blue indicates DAPI. Scale bar = 50 μm (A–J). L, lumen.
Supplemental Figure S9.
Representative images of Ki-67 and KRT14 coimmunostaining in keratinocyte growth factor (KGF)–pretreated bladders at 6 hours, 1 day, and 3 days after sham injury. Merged images of double-label immunofluorescence for Ki-67 (red) and KRT14 (white) show the presence of few KRT14+ cells (arrowheads) in the basal layer with either PBS pretreatment (A, C, and E) or KGF pretreatment (B, D, and F) at 6 hours (A and B), 1 day (C and D), or 3 days (E and F) after sham injury. None of the KRT14+ cells in any group is Ki-67+. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bar = 50 μm (A–F). L, lumen.
Supplemental Figure S10.

Representative images of Ki-67, uroplakin 3a (UPK3), and KRT5 coimmunostaining 3 days after sham injury. Triple-label immunofluorescence for Ki-67 (red), UPK3 (white), and KRT5 (green) 3 days after sham injury shows few Ki-67+ cells in either PBS-pretreated (A–C) or keratinocyte growth factor (KGF)–pretreated (D–F) mice; the single Ki-67+ cells in the sections from each group are UPK3+ and KRT5− (arrowheads). A and D: Blue indicates DAPI. Dotted lines indicate demarcation between urothelial layer and underlying lamina propria. Scale bar = 50 μm (A–F). L, lumen.
References
- 1.Korkmaz A., Topal T., Oter S. Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis: implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol Toxicol. 2007;23:303–312. doi: 10.1007/s10565-006-0078-0. [DOI] [PubMed] [Google Scholar]
- 2.Baker G.L., Kahl L.E., Zee B.C., Stolzer B.L., Agarwal A.K., Medsger T.A., Jr. Malignancy following treatment of rheumatoid arthritis with cyclophosphamide: long-term case-control follow-up study. Am J Med. 1987;83:1–9. doi: 10.1016/0002-9343(87)90490-6. [DOI] [PubMed] [Google Scholar]
- 3.Kaldor J.M., Day N.E., Kittelmann B., Pettersson F., Langmark F., Pedersen D., Prior P., Neal F., Karjalainen S., Bell J., Choi W., Koch M., Band P., Pompe-Kirn V., Garton C., Staneczek S., Zaren B., Stovall A., Boffetta P. Bladder tumours following chemotherapy and radiotherapy for ovarian cancer: a case-control study. Int J Cancer. 1995;63:1–6. doi: 10.1002/ijc.2910630102. [DOI] [PubMed] [Google Scholar]
- 4.Shirai T. Etiology of bladder cancer. Semin Urol. 1993;11:113–126. [PubMed] [Google Scholar]
- 5.Vlaovic P., Jewett M.A. Cyclophosphamide-induced bladder cancer. Can J Urol. 1999;6:745–748. [PubMed] [Google Scholar]
- 6.Travis L.B., Curtis R.E., Glimelius B., Holowaty E.J., Van Leeuwen F.E., Lynch C.F., Hagenbeek A., Stovall M., Banks P.M., Adami J., Gospodarowicz M.K., Wacholder S., Inskip P.D., Tucker M.A., Boice J.D. Bladder and kidney cancer following cyclophosphamide therapy for non-Hodgkin's lymphoma. J Natl Cancer Inst. 1995;87:524–530. doi: 10.1093/jnci/87.7.524. [DOI] [PubMed] [Google Scholar]
- 7.Moghe A., Ghare S., Lamoreau B., Mohammad M., Barve S., McClain C., Joshi-Barve S. Molecular mechanisms of acrolein toxicity: relevance to human disease. Toxicol Sci. 2015;143:242–255. doi: 10.1093/toxsci/kfu233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matz E.L., Hsieh M.H. Review of advances in uroprotective agents for cyclophosphamide- and ifosfamide-induced hemorrhagic cystitis. Urology. 2017;100:16–19. doi: 10.1016/j.urology.2016.07.030. [DOI] [PubMed] [Google Scholar]
- 9.Lima M.V., Ferreira F.V., Macedo F.Y., de Castro Brito G.A., Ribeiro R.A. Histological changes in bladders of patients submitted to ifosfamide chemotherapy even with mesna prophylaxis. Cancer Chemother Pharmacol. 2007;59:643–650. doi: 10.1007/s00280-006-0307-5. [DOI] [PubMed] [Google Scholar]
- 10.Monach P.A., Arnold L.M., Merkel P.A. Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: a data-driven review. Arthritis Rheum. 2010;62:9–21. doi: 10.1002/art.25061. [DOI] [PubMed] [Google Scholar]
- 11.Ulich T.R., Whitcomb L., Tang W., O'Conner Tressel P., Tarpley J., Yi E.S., Lacey D. Keratinocyte growth factor ameliorates cyclophosphamide-induced ulcerative hemorrhagic cystitis. Cancer Res. 1997;57:472–475. [PubMed] [Google Scholar]
- 12.Dorr W., Spekl K., Farrell C.L. The effect of keratinocyte growth factor on healing of manifest radiation ulcers in mouse tongue epithelium. Cell Prolif. 2002;35 Suppl 1:86–92. doi: 10.1046/j.1365-2184.35.s1.9.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrell C.L., Rex K.L., Chen J.N., Bready J.V., DiPalma C.R., Kaufman S.A., Rattan A., Scully S., Lacey D.L. The effects of keratinocyte growth factor in preclinical models of mucositis. Cell Prolif. 2002;35 Suppl 1:78–85. doi: 10.1046/j.1365-2184.35.s1.8.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu H., Hao L., Tang C., Zhu Y., Jiang Q., Yao J. Activation of KGFR-Akt-mTOR-Nrf2 signaling protects human retinal pigment epithelium cells from ultra-violet. Biochem Biophys Res Commun. 2018;495:2171–2177. doi: 10.1016/j.bbrc.2017.12.078. [DOI] [PubMed] [Google Scholar]
- 15.Khan W.B., Shui C., Ning S., Knox S.J. Enhancement of murine intestinal stem cell survival after irradiation by keratinocyte growth factor. Radiat Res. 1997;148:248–253. [PubMed] [Google Scholar]
- 16.Wu K.I., Pollack N., Panos R.J., Sporn P.H., Kamp D.W. Keratinocyte growth factor promotes alveolar epithelial cell DNA repair after H2O2 exposure. Am J Physiol. 1998;275:L780–L787. doi: 10.1152/ajplung.1998.275.4.L780. [DOI] [PubMed] [Google Scholar]
- 17.Takeoka M., Ward W.F., Pollack H., Kamp D.W., Panos R.J. KGF facilitates repair of radiation-induced DNA damage in alveolar epithelial cells. Am J Physiol. 1997;272:L1174–L1180. doi: 10.1152/ajplung.1997.272.6.L1174. [DOI] [PubMed] [Google Scholar]
- 18.Ornitz D.M., Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4:215–266. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papafotiou G., Paraskevopoulou V., Vasilaki E., Kanaki Z., Paschalidis N., Klinakis A. KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat Commun. 2016;7:11914. doi: 10.1038/ncomms11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi S.H., Byun Y., Lee G. Expressions of uroplakins in the mouse urinary bladder with cyclophosphamide-induced cystitis. J Korean Med Sci. 2009;24:684–689. doi: 10.3346/jkms.2009.24.4.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park H., Kyung Y.S., Lee G. Urinary uroplakin expression in cyclophosphamide-induced rat cystitis model. Hum Exp Toxicol. 2016;35:613–622. doi: 10.1177/0960327115597466. [DOI] [PubMed] [Google Scholar]
- 22.Veranic P., Romih R., Jezernik K. What determines differentiation of urothelial umbrella cells? Eur J Cell Biol. 2004;83:27–34. doi: 10.1078/0171-9335-00351. [DOI] [PubMed] [Google Scholar]
- 23.Haldar S., Dru C., Bhowmick N.A. Mechanisms of hemorrhagic cystitis. Am J Clin Exp Urol. 2014;2:199–208. [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes F.M., Corn A.G., Nimmich A.R., Pratt-Thomas J.D., Purves J.T. Cyclophosphamide induces an early wave of acrolein-independent apoptosis in the urothelium. Adv Biosci Biotechnol. 2013;4:9–14. doi: 10.4236/abb.2013.48A2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jezernik K., Romih R., Mannherz H.G., Koprivec D. Immunohistochemical detection of apoptosis, proliferation and inducible nitric oxide synthase in rat urothelium damaged by cyclophosphamide treatment. Cell Biol Int. 2003;27:863–869. doi: 10.1016/s1065-6995(03)00175-6. [DOI] [PubMed] [Google Scholar]
- 26.Anton E. Delayed toxicity of cyclophosphamide on the bladder of DBA/2 and C57BL/6 female mouse. Int J Exp Pathol. 2002;83:47–53. doi: 10.1046/j.1365-2613.2002.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraiser L., Kehrer J.P. Murine strain differences in metabolism and bladder toxicity of cyclophosphamide. Toxicology. 1992;75:257–272. doi: 10.1016/0300-483x(92)90007-2. [DOI] [PubMed] [Google Scholar]
- 28.Crescioli C., Maggi M., Luconi M., Vannelli G.B., Salerno R., Sinisi A.A., Bonaccorsi L., Ferruzzi P., Barni T., Forti G., Serio M. Vitamin D3 analogue inhibits keratinocyte growth factor signaling and induces apoptosis in human prostate cancer cells. Prostate. 2002;50:15–26. doi: 10.1002/pros.10028. [DOI] [PubMed] [Google Scholar]
- 29.Fehrenbach H., Kasper M., Koslowski R., Pan T., Schuh D., Muller M., Mason R.J. Alveolar epithelial type II cell apoptosis in vivo during resolution of keratinocyte growth factor-induced hyperplasia in the rat. Histochem Cell Biol. 2000;114:49–61. doi: 10.1007/s004180000157. [DOI] [PubMed] [Google Scholar]
- 30.Firth J.D., Putnins E.E. Keratinocyte growth factor 1 inhibits wound edge epithelial cell apoptosis in vitro. J Invest Dermatol. 2004;122:222–231. doi: 10.1046/j.0022-202X.2003.22124.x. [DOI] [PubMed] [Google Scholar]
- 31.Hines M.D., Allen-Hoffmann B.L. Keratinocyte growth factor inhibits cross-linked envelope formation and nucleosomal fragmentation in cultured human keratinocytes. J Biol Chem. 1996;271:6245–6251. doi: 10.1074/jbc.271.11.6245. [DOI] [PubMed] [Google Scholar]
- 32.Slayden O.D., Rubin J.S., Lacey D.L., Brenner R.M. Effects of keratinocyte growth factor in the endometrium of rhesus macaques during the luteal-follicular transition. J Clin Endocrinol Metab. 2000;85:275–285. doi: 10.1210/jcem.85.1.6251. [DOI] [PubMed] [Google Scholar]
- 33.Song G., Ouyang G., Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang H.L., Sugimoto Y., Liu S., Wang L.S., Huang Y.W., Ye W., Lin Y.C. Keratinocyte growth factor (KGF) regulates estrogen receptor-alpha (ER-alpha) expression and cell apoptosis via phosphatidylinositol 3-kinase (PI3K)/Akt pathway in human breast cancer cells. Anticancer Res. 2009;29:3195–3205. [PubMed] [Google Scholar]
- 35.Qiu W., Leibowitz B., Zhang L., Yu J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene. 2010;29:1622–1632. doi: 10.1038/onc.2009.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao S., Wang Y., Sweeney P., Chaudhuri A., Doseff A.I., Marsh C.B., Knoell D.L. Keratinocyte growth factor induces Akt kinase activity and inhibits Fas-mediated apoptosis in A549 lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288:L36–L42. doi: 10.1152/ajplung.00309.2003. [DOI] [PubMed] [Google Scholar]
- 37.Cai Y., Wang W., Qiu Y., Yu M., Yin J., Yang H., Mei J. KGF inhibits hypoxia-induced intestinal epithelial cell apoptosis by upregulating AKT/ERK pathway-dependent E-cadherin expression. Biomed Pharmacother. 2018;105:1318–1324. doi: 10.1016/j.biopha.2018.06.091. [DOI] [PubMed] [Google Scholar]
- 38.Arms L., Vizzard M.A. Role for pAKT in rat urinary bladder with cyclophosphamide (CYP)-induced cystitis. Am J Physiol Renal Physiol. 2011;301:F252–F262. doi: 10.1152/ajprenal.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang M.S., Wang H.T., Hu Y., Chen W.S., Akao M., Feng Z., Hu W. Acrolein induced DNA damage, mutagenicity and effect on DNA repair. Mol Nutr Food Res. 2011;55:1291–1300. doi: 10.1002/mnfr.201100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gandhi D., Molotkov A., Batourina E., Schneider K., Dan H., Reiley M., Laufer E., Metzger D., Liang F., Liao Y., Sun T.T., Aronow B., Rosen R., Mauney J., Adam R., Rosselot C., Van Batavia J., McMahon A., McMahon J., Guo J.J., Mendelsohn C. Retinoid signaling in progenitors controls specification and regeneration of the urothelium. Dev Cell. 2013;26:469–482. doi: 10.1016/j.devcel.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun W., Wilhelmina Aalders T., Oosterwijk E. Identification of potential bladder progenitor cells in the trigone. Dev Biol. 2014;393:84–92. doi: 10.1016/j.ydbio.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 42.Colopy S.A., Bjorling D.E., Mulligan W.A., Bushman W. A population of progenitor cells in the basal and intermediate layers of the murine bladder urothelium contributes to urothelial development and regeneration. Dev Dyn. 2014;243:988–998. doi: 10.1002/dvdy.24143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schafer F.M., Algarrahi K., Savarino A., Yang X., Seager C., Franck D., Costa K., Liu S., Logvinenko T., Adam R., Mauney J.R. Mode of surgical injury influences the source of urothelial progenitors during bladder defect repair. Stem Cell Reports. 2017;9:2005–2017. doi: 10.1016/j.stemcr.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uzan B., Figeac F., Portha B., Movassat J. Mechanisms of KGF mediated signaling in pancreatic duct cell proliferation and differentiation. PLoS One. 2009;4:e4734. doi: 10.1371/journal.pone.0004734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu J., Han W., Yang W., Liu H., Li C., Guo L., Jin Y., Zhang R., Chen H. Keratinocyte growth factor binding to fibroblast growth factor receptor 2-IIIb promotes epithelial ovarian cancer cell proliferation and invasion. J Cancer Res Ther. 2018;14:S347–S353. doi: 10.4103/0973-1482.235353. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto T., Matsuda Y., Kawahara K., Naito Z., Ishiwata T. Keratinocyte growth factor stimulates growth of MIA PaCa-2 cells through extracellular signal-regulated kinase phosphorylation. Oncol Lett. 2012;3:307–310. doi: 10.3892/ol.2011.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mebratu Y., Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corrow K.A., Vizzard M.A. Phosphorylation of extracellular signal-regulated kinases in urinary bladder in rats with cyclophosphamide-induced cystitis. Am J Physiol Regul Integr Comp Physiol. 2007;293:R125–R134. doi: 10.1152/ajpregu.00857.2006. [DOI] [PubMed] [Google Scholar]
- 49.Romih R., Koprivec D., Martincic D.S., Jezernik K. Restoration of the rat urothelium after cyclophosphamide treatment. Cell Biol Int. 2001;25:531–537. doi: 10.1006/cbir.2000.0658. [DOI] [PubMed] [Google Scholar]
- 50.Stewart F.A. Mechanism of bladder damage and repair after treatment with radiation and cytostatic drugs. Br J Cancer Suppl. 1986;7:280–291. [PMC free article] [PubMed] [Google Scholar]
- 51.Kullmann F.A., Clayton D.R., Ruiz W.G., Wolf-Johnston A., Gauthier C., Kanai A., Birder L.A., Apodaca G. Urothelial proliferation and regeneration after spinal cord injury. Am J Physiol Renal Physiol. 2017;313:F85–F102. doi: 10.1152/ajprenal.00592.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]