

Abstract
Microfluidics have been used to create “body-on-chip” systems to mimic in vivo cellular interactions with a high level of control. Most such systems rely on optical observation of cells as a readout. In this work we integrated a cell-cell interaction chip with on-line microchip electrophoresis immunoassay to monitor the effects of the interaction on protein secretion dynamics. The system was used to investigate the effects of adipocytes on insulin secretion. Chips were loaded with 190,000 3T3-L1 adipocytes and a single islet of Langerhans in separate chambers. The chambers were perfused at 300–600 nL/min so that adipocyte secretions flowed over the islets for 3 h. Adipocytes produced 80 µM of non-esterified fatty acids (NEFAs), a factor known to impact insulin secretion, at the islets. After perfusion, islets were challenged with a step change in glucose from 3 to 11 mM while monitoring insulin secretion at 8 s intervals by on-line immunoassay. Adipocyte treatment augmented insulin secretion by 6-fold compared to controls. The effect was far greater than comparable concentrations of NEFA applied to the islets demonstrating that adipocytes release multiple factors that can strongly potentiate insulin secretion. The experiments reveal that integration of chemical analysis with cell-cell interaction can provide valuable insights into cellular functions.
Introduction
Cell-cell interactions are vital to normal cell function.1–3 Indeed, it is known that isolated cells in culture may behave differently from the same cell types in vivo due to loss of normal cellular milieu that is comprised of physical structure and chemical secretions from other cell types in both paracrine and endocrine interactions. Studying cells in vivo however can be challenging experimentally. Furthermore, it is difficult to manipulate the interaction of specific cell types in vivo. In vitro models that mimic the in vivo environment better than isolated cells, e.g. by allowing for cell-cell interactions, are potential alternatives. A simple method to study cell-cell interaction is to co-culture different cell types, e.g. in both sides of a transwell4 or a single compartment.5 Although useful, these methods have unrealistic volume-to-cell ratio and lack fluid dynamics and mass transport that are part of the in vivo environment. They also do not lend themselves to dynamic control over the cellular environment and integration with chemical measurements. In this work, we describe a microfluidic system that allows secretions from one cell type to interact with a second while using an integrated immunoassay to monitor protein release from the target cells. The system is used to investigate adipocyte effects on insulin secretion from islet cells.
Microfluidics has emerged as a powerful way to create in vitro cell systems, sometimes called “organ-on-chip” or “body-on-chip”, which mimic an in vivo environment. Such systems were first developed to replicate in vivo pharmacokinetics and pharmacodynamics.3,6,7 The devices had interconnected compartments containing lung, liver, fat and other tissues in a circulatory system so that drugs introduced to the cells were metabolized similar to in vivo. Subsequent improvements include creation of 3-dimensional environments and long-term culture.8–11 The concept has grown beyond drug metabolism so that microfluidics has also been used to study other types of cell-cell interaction such as platelet adhesion to endothelial tissue12 and neovascularlization.13 These systems have revealed advantages such as precise control of cell culture environment, consumption of small amounts of tissue and media, and high-throughput.7–17 In these organ-on-chip studies, the experimental output was typically microscopic imaging of the cells. For drug metabolism studies, metabolites were collected from the chip and assayed off-line.
Microfluidics also enables development of integrated systems for chemical analysis of cells.18–25 Cell function can be monitored by measuring chemical secretions from cells incubated on microfluidic devices.26–35 For example, we have coupled cell culture chips to mass spectrometry to identify and quantify secretions from adipocytes.36 More relevant to the current work, we have monitored the dynamics of insulin secretion from single islets of Langerhans by perfusing cells and analyzing the perfusate using rapid, on-line electrophoretic immunoassay.37 Capillary electrophoresis used to rapidly separate bound and free insulin was also reported by our group before (See supplemental information for more details).38
Islets are 75–200 µm diameter endocrine microorgans located in the pancreas that contain insulin secreting β-cells.39 β-cells secrete insulin at elevated blood glucose concentration40 to help maintain glucose homeostasis.41 Insulin secretion has complex dynamics. In vitro, step increases in glucose concentration (typically from 3 mM to 11 mM) results in an initial burst (first phase) followed by a lower level of sustained secretion (second phase), often with oscillations with periods of 3–5 min,42 Oscillations, which appear to be related to metabolic oscillations,42 may be important in overall islet function, e.g. in vivo oscillations are disrupted in diabetics;43 and they maybe important in for insulin action.
Insulin secretion has been well-studied in vitro; however, it is known that the extracellular environment in vivo contains factors that modulate insulin secretion. One cell type that is of interest for its effects on islet cells is the adipocyte. Adipocytes are fat-storing cells that secrete non-esterified fatty acids (NEFAs) and adipokines (a group of hormones and cytokines) that can enter the bloodstream to contact islets and possibly impact insulin secretion.41,44,45 NEFA effects have been of the most interest because of their relevance to type 2 diabetes. NEFAs have been shown to have bimodal effects on insulin secretion so that short term (typically less than a few hours) they enhance secretion46 but with chronic exposure (typically 1 – 2 days or longer) they may suppress insulin secretion.47 A variety of adipokines are released from fat cells with competing effects on insulin secretion.48–53
Obesity is a risk factor for diabetes which is characterized by impaired insulin secretion. This observation suggests the possibility that adipocyte secretions might play a role in degrading islet cell function. Investigations of adipocyte secretory products on insulin secretions have mostly relied on exposing islets to isolated components, such as NEFAs or individual adipokines; although, some studies54 have focused on adipokine interactions. Here we demonstrate a microfluidic system that better mimics the in vivo environment by allowing adipocyte secretions to interact with islets similar to in vivo circulation. The significance of allowing such cell-cell interactions is revealed in our finding that adipocytes have a profound impact on insulin secretion that cannot be explained by the effect of just NEFAs. The results show that exposing β-cells to the full range of cellular secretions provides a more integrated view of the impacts of the adipocyte-β-cell interaction.
Experimental
Chemicals and reagents
Cell culture chemicals, Amplex UltraRed, and Hank’s buffered salt solution (HBSS) (Cat.No. 14175) were purchased from Life Technologies (Carlsbad, CA). Monoclonal antibody (Ab) to human insulin was purchased from Meridian Life Science (Memphis, TN). Tricine, electrophoresis grade, was obtained from MP Biomedicals (Aurora, OH). Collagenase P was obtained from Roche Diagnostic. Fluorescein isothiocyanate-labeled insulin (FITC-insulin) Tween 20, ethylenediaminetetraacetic acid (EDTA), insulin and fatty acid free bovine serum albumin (BSA) were obtained from Sigma-Aldrich (St. Louis, MO). The fatty acid assay reagents and standard solution were purchased as a HR Series NEFA-HR (2) kit, from Wako Chemicals USA, Inc. (Richmond, VA). All other chemicals were from Fisher (Pittsburgh, PA). All solutions were prepared with Milli-Q (Millipore, Bedford, MA) 18 MΩ deionized water and filtered with 0.2 µm nylon syringe filters (Fisher) before using. Stock antibody solution was stored at 4 °C in the manufacturer provided phosphate buffer saline. Stock FITC-insulin was diluted to 166 µM in immunoassay reagent buffer and stored at −20 °C.
Balance salt solution (BSS) contained 125 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 25 mM Tricine, and 0.7 mg mL−1 BSA. Immunoassay reagent buffer contained 60 mM NaCl, 1 mM EDTA, 20 mM tricine, 0.1% (w/v) Tween 20 and 0.7 mg mL−1 BSA. Electrophoresis buffer was 20 mM NaCl and 150 mM tricine. All buffers were adjusted to pH 7.4.
Adipocyte culture
8 mm diameter glass coverslips (Warner Instruments, Hamden, CT) were sterilized with ethanol and then dried in a sterile culture hood. Three to four coverslips were placed in each 35 mm petri dish prior to seeding preadipocytes. Murine 3T3-L1 preadipocytes were seeded into the 35 mm dishes (200,000 cells per dish), maintained in Dulbecco’s modified Eagle’s medium (Cat. No. 11965–092, Life Technologies) supplemented with 8% v/v bovine calf serum (Denville Scientific, South Plainfield, NJ), 100 units mL−1 penicillin, 100 µg/mL streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate. Cells were incubated at 10% CO2. Two days after the cells became confluent, differentiation (adipogenesis) was induced by adding 500 µM methylisobutylxanthine, 1 µM dexamethasone, and 5 µg/mL insulin.55 Two days post-differentiation, medium was replaced with adipogenic medium consisting of DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate and 5 μg/mL insulin. Every 2 days culture medium was refreshed. Adipocytes were matured to at least 14–20 days post-induction before on-chip experiments. NEFA concentration was determined by collecting perfusates on chip and performing enzyme assays on a multi-plate reader (Perkin Elmer Fusion).29,56
Islet isolation and culture
Pancreatic islets were obtained from 20–30 g male CD-1 mice as previously described.57 The islets were placed in RPMI-1640 cell culture media supplemented with 10% fetal bovine serum, 100 unit mL−1 penicillin, and 100 µg mL−1 streptomycin at 37 °C, 5% CO2, pH 7.4. Islets were used 2–5 days following isolation. Because islet size impacts the amount of secretion, islets were selected for use that had ~100 µm diameter.58
Microfluidic chip fabrication and preparation
Microfluidic devices were fabricated as previously described.30,59 Briefly, the device consisted of 2 etched glass wafers: a bottom wafer for the bottom portion of the adipocyte chamber and a top wafer for the fluidic channels and the top portion of the adipocyte cell chamber (Figure S-1). 1 mm thick Borofloat photomask blanks (3.8 cm X 10.2 cm) coated with a 120 nm layer of chrome and AZ1518 positive photoresist (Telic Company, Valencia, CA) were exposed to UV light for 6 s through a patterned photomask (Fineline Imaging Inc., Colorado Springs, CO). The exposed wafers were developed in AZ 726 MIF developer (Clariant Corp., SummerVille, NJ) and the exposed chrome was removed with CEP-200 etchant (Microchrome Technologies, Inc., San Jose, CA). The exposed glass was etched in 17:96:7 (v/v/v) HNO3/HF/H2O for 57 min to create 250 µm deep cell chamber on the bottom wafer and 17:24:79 (v/v/v) HNO3/HF/H2O for 25 min to create 15 µm deep channels on the top wafer. Fluidic access holes and top portion of the adipocyte cell chamber were drilled with 360 µm diameter (Kyocera Tycom, Costa Mesa, CA) and 9.5 mm diameter (Starlite Industries, Rosemont PA) drill bits, respectively. The remaining photoresist was removed with acetone and the remaining chrome was removed with the CEP-200 chrome etchant. Glass wafers were washed for 20 min in piranha solution (3:1, v/v, H2SO4/H2O2) and then heated RCA solution (5:1:1, v/v/v, H2O/NH4/H2O2) for 40 min. Caution!: piranha solution is aggressive and explosive. Never mix piranha waste with solvents. Check the safety precautions before using it. The wafers were rinsed with water, aligned and bonded at 610 °C for 8 h. Reservoirs and access ports (IDEX Health and Science, Oak Harbor, WA) were glued to the device over drilled access holes with epoxy.
A polydimethylsiloxane (PDMS) plug to seal the adipocyte chamber was made using RTV-615 PDMS (Curbell Plastics, Livonia, MI) with a base to curing agent ratio of 10:1. The plug was made by pouring PDMS into a mold, curing, and then removing the PDMS. A compression frame to enclose the adipocyte portion was built in-house from 2 sheets of acrylic plastic.
The chip was conditioned prior to experiments by flowing 0.1 M NaOH through the channels, followed by deionized water and experimental solutions. All solutions were filtered daily from a stock solution to prevent introduction of particulates to the chip or degradation.
Microfluidic chip operation
For islet experiments, a single islet was placed in the grounded islet reservoir using a pipette. To load adipocytes, 2 coverslips with ~190,000 attached 3T3-L1 adipocytes were removed from a culture dish placed in the cell chamber. The PDMS plug was pressed into place to make a conformal contact. The chip was then placed in the compression frame to seal the plug.
Perfusion and chemical monitoring
During the cell-cell interaction step, BSS buffer was pumped by external syringe pumps (Chemyx, Stafford, TX) onto the chip via fused-silica capillaries (Figure 1A). Flow rates were 0.3 µL min−1 through each adipocyte chamber for a total of 0.6 µL min−1 flowing over the islet. After perfusion for a desired period (typically 3 h), the flow to the islet was switched so that controlled glucose solutions were passed over the islet at 0.6 µL min−1 (Figure 1A, Solid arrow lines). During this time, secretion was measured by electrophoretic immunoassay performed at 8 s intervals. For the immunoassay, negative high voltage (−6 kV) was applied at the waste reservoir of the device and all other reservoirs grounded. Grounding of the islet reservoir allowed the secretions from the islet to be sampled, i.e. continuously pulled by electroosmotic flow into the reaction channel for immunoassay. Likewise, grounding of the antibody (50 nM) and FITC-insulin (100 nM) reservoirs allowed these solutions to be continuously pumped by electroosmosis into the heated reaction channel where they mixed. The thin film resistive heater covers full area of the reaction channel and islet chamber because it is essential to maintain 37 °C for immunoassay reaction and islets.32 Adipocyte chambers are only partially covered because adipocytes secretion is robust in room temperature.29 Repetitive injections of the resulting sample stream were performed as described in detail elsewhere60. Briefly, when the relay was opened, sample entered the separation channel. When gate was returned to ground the sample was flowed to waste and separation is performed. Sample injection time was 0.5 s applied at 7.5 s intervals. During electrophoresis measurements, fresh buffer was continuously pumped into reservoirs via fused-silica capillaries inserted into vials in a reservoir pressurized to 12 psi with helium.32 Continuous buffer supply allowed long-term electrophoresis operation by preventing effects of buffer electrolysis. The separation was monitored 1 cm downstream of the injection cross by laser-induced fluorescence (LIF) using a microscope as described before.30,31
Figure 1.
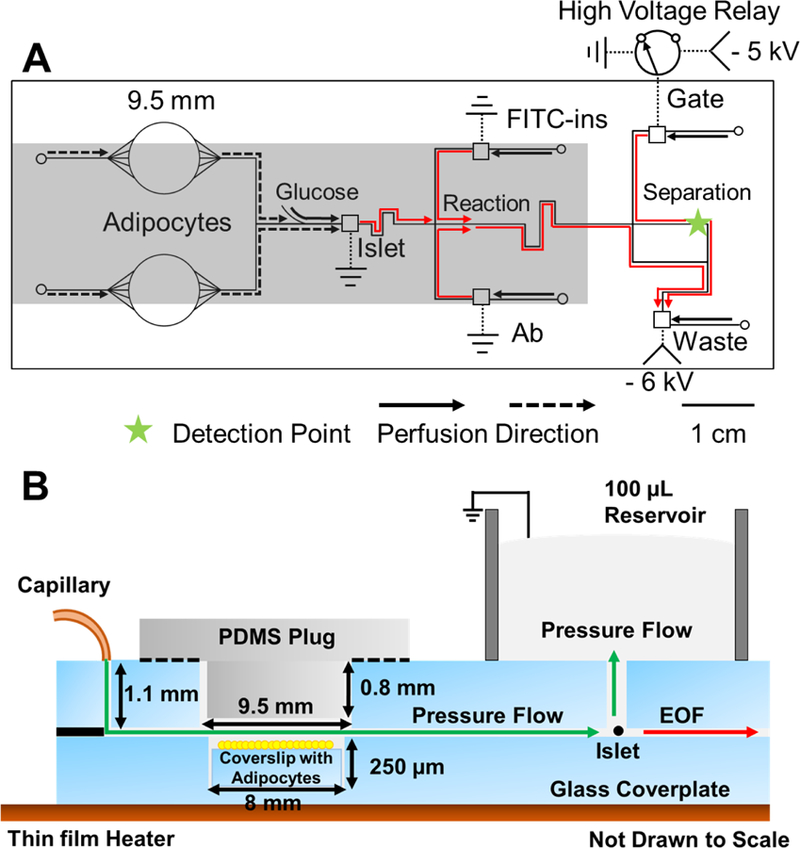
Microfluidic chip layout. (A) Top view of chip illustrating the channel layout, electrical connections, and flow directions for co-culture and monitoring insulin. Solid lines indicate the 15 μm deep and 50 μm wide microfluidic channels. Dotted lines indicate electrical connections. The 9.5 mm circles indicate the adipocyte chambers drilled through the glass. Smaller circles indicate perfusion inlets, and squares indicate reservoirs that are sampled by electroosmotic flow. Electroosmotic flow directions are indicated by the red arrows. All fluidic inlets are 360 µm diameter. Reaction refers to the portion of channel where immunoassay reagents mix, and reaction happens. Arrows indicate perfusion flow direction. Dashed arrow lines indicate direction of flow during 1st step of experiment of perfusion co-culture for 3 h. Islets are co-cultured with adipocytes for 3 h without chemical monitoring. Solid arrow lines indicate flow during 2nd step of experiment of glucose stimulation. Perfusion from adipocytes (dashed lines) is stopped and glucose is delivered from the side channel to stimulate insulin secretion from the islets. Step changes in glucose concentration of 3–11 mM are made using external syringe pumps and valves. The relay is switched to inject sample onto the electrophoresis channel every 8 s. The shaded portion of the chip indicate parts that are heated during experiments with a thin film resistive heater. Laser-induced fluorescence (LIF) detection point occurred 1 cm past injection cross, as indicated by the star. -HV is where high voltage is applied. (B) Side view of adipocyte and islet perfusion culture. Adipocytes and islet are loaded into cell chambers and perfused with pressure-driven flow from the capillary. Perfusate flows into the islet chamber and up into a 100 μL fluidic reservoir. The adipocyte chambers are sealed with a PDMS plug. Solution with insulin from the chamber is sampled by electroosmotic flow (EOF in the drawing) through the sampling channel.
Insulin in the perfusate was quantified by competitive electrophoretic immunoassay. Each electrophoresis separation resulted in a peak due to FITC-insulin bound to antibody (B) and a peak due to free FITC-insulin (F). Concentration of insulin was quantified by comparing B/F peak areas ratios of the electrophoregrams to a calibration curve. Insulin released from the islet competed with FITC-insulin during the on-chip reaction so that higher concentrations of insulin resulted in lower B/F ratios for the FITC-insulin. Calibration was performed daily. High-throughput analysis of collected electropherograms was performed using Cutter software.61
Computational modeling
Perfusion of the cell chambers was modeled in a 3D geometry using COMSOL Muitiphysics (COMSOL, Inc., Burlington, MA). The “laminar flow” and “transport of diluted species” modules were used to model the flow split and theoretical temporal resolution. All simulations assumed water perfusion through the chip, with a density of 998 kg m−3 and a viscosity of 1.002 × 10 −3 Pa ·s’ (20 °C). Oleic acid’s diffusion coefficient, used as a representative of NEFAs, was 5.26 × 10−10 m2 s−1 at 37 °C, adjusted using the Stokes-Einstein relationship.62 The adipocyte and islet chamber were modeled separately to reduce computation times.
Statistical analysis
All data are expressed as means ± standard deviation, unless noted otherwise, and were analyzed with an unpaired two-tailed Student’s t test. Differences were considered significant at P < 0.05.
Results
Microfluidic device overview
Our goal in this work was to produce and test a chip that exposes islets to adipocyte secretions through perfusion and observe the effect on glucose-stimulated insulin secretion (GSIS) as a model of cell-cell interaction with integrated analysis. The microfluidic chip integrated multiple cell chambers and electrophoresis-based immunoassay for insulin monitoring (Figure 1). This chip design builds on previous work aimed at measuring insulin secretion from islets30,31 and NEFAs from adipocytes.28,29 This prior work identified important features such as minimizing the difficulty of loading cells into the chip, prevention of shear stress on the cells during perfusion, and simple regeneration of chips, which are used in this work.
The chip contains three cell chambers, two to hold adipocytes and one to hold a single islet. Coverslips containing adipocytes were placed at the bottom of the cell chambers, which was reversibly sealed with a PDMS plug, to allow easy loading and re-use of a chip. Although in principle a single adipocyte chamber could be used, pilot experiments revealed that chambers large enough to hold sufficient adipocytes for the experiment were difficult to seal because the PDMS plugs would collapse. Smaller chambers were more robust for sealing. The adipocytes were held in a recessed area that minimized their contact with direct flow and shear stress (Figure 1B). This feature is important because adipocytes are fragile. Single islets were placed in a chamber that was left open to atmosphere (Figure 1B) to allow: 1) easy loading and unloading of the chamber; 2) perfusate from adipocytes to flow out the top of the chamber without causing high flow into the electrophoresis channel; and 3) the potential for placing sensors or electrophysiological probes in the islet for other studies.
For cell-cell interaction experiments, adipocyte perfusate passed into the islet chamber so that islets were exposed to adipocyte secretions. After a 3 h incubation, GSIS was measured at the islets by electrophoretic immunoassay. In principle, the immunoassay could be performed during adipocyte perfusion; however, we passed the glucose only over the islets to avoid any unknown effects of changes in glucose on adipocyte secretions. The perfusion flow rate replaced solution in the islet chamber (approximately 100 nL) in a few seconds to allow high temporal resolution monitoring of insulin secretion from the islet. The islet chamber was continuously sampled at ~2 nL min−1 by electroosmotic flow generated by applied voltage from the chamber to the exit of the electrophoresis system (See Figure 1A). The resulting sample stream was analyzed by electrophoretic immunoassay every 8 s. Figure 2A illustrates sample electropherograms. Using a calibration curve, these data enabled determination of insulin concentrations and secretion rates.
Figure 2.
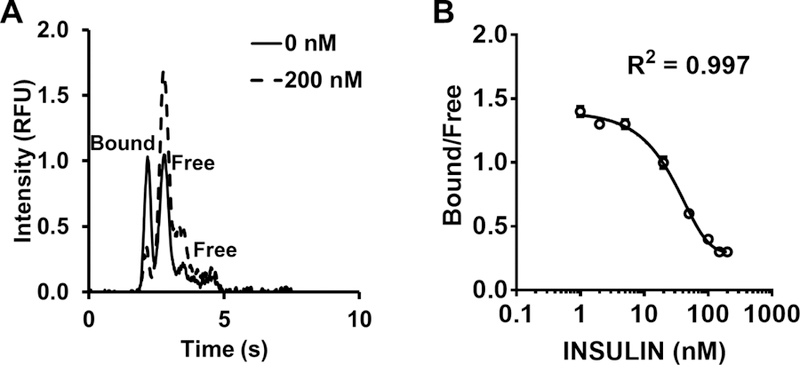
Sample electropherograms and calibration curve. (A) Sample electropherograms at 0 nM and 200 nM insulin standards. Bound and free indicate Ab:Ag* complex and free Ag*, respectively. There are 2 free peaks because FITC-insulin are mono- and double-labeled. We use the first free peak area for B/F calculation. B/F is approximately one at 0 nM insulin. (B) A sample calibration curve. Error bars indicate ±1 standard deviation. The mean and standard deviation were calculated based on 10–15 electropherograms for each data point.
Figure 2B shows calibration data fit to a variable slope sigmoidal dose response function (R2 = 0.997). The calibration curve also shows that the electrophoresis immunoassay produced a dynamic range of 10 to 110 nM, which covers the range expected from islets. The relative standard deviation (RSD) of B/F was less than 5%. The detection limit was 0.5 nM, calculated as the concentration required to give a B/F that was at least 3 standard deviations less than the B/F for 0 nM insulin.
Fluid dynamics of adipocyte and islet chambers
Flow distribution and fluid dynamic response are important performance considerations for the chip. To assist in determining flow properties, COMSOL models of different portions of the chip were made (Figure S-2). Modeling showed that flow across the cell chamber is uniformly distributed, as desired, so that adipocytes at the edge of the chip receive comparable flow to those at the center (Figure S-2A). Modeling also showed that concentration of species entering the islet chamber rapidly reaches comparable concentrations in both the front and rear (relative to the perfusion inlet) of the islet indicating that the islet is exposed to comparable concentrations of adipocyte secretions and glucose around its entire perimeter (Figure S-2D).
Our goal was to expose islets to stable concentrations of adipocyte secretions. As a result of the recessed position for adipocytes (Figure 1B), secretions must diffuse into the flow path to be brought to the islets. Modeling showed that with a stable production of secretions from the floor of the adipocyte chamber, a steady state concentration gradient of secreted chemicals is created in the adipocyte chamber (Figure S-2B). The model also shows that chemicals released from adipocytes tend to stay near the bottom of the chamber as they flow downstream. This flow pattern favors rapid entry into the downstream channels towards the islet. A simulation of the concentration flowing from the chamber (Figure S-2C) had a 10–90% rise time of 4 min. Flow rates from 0.3 to 2.0 μL min−1 had little effect on the concentration reaching the islets so that the effects of adipocytes on islets are relatively immune to flow rate variation.
Fatty acid concentration validation
The NEFA concentration reaching the islet will depend on the number of adipocytes present in the chip and mass transport. Pilot experiments in our laboratory revealed that concentrations of 50–100 µM NEFA produced robust effects on insulin secretion. Based on previous results,29 we estimated that 190,000 adipocytes would produce approximately 80 μM NEFA at the islets. To confirm this expectation, fractions of perfusate were collected from the islet chamber and assayed for NEFA using a fluorescent enzyme assay. We also flowed standard concentrations of 50 µM and 100 µM palmitic acid through the chip to verify transport of known concentrations through the chip. As shown in Figure S-3, standards gave the expected concentration indicating inconsequential NEFA loss through the chip. Adipocytes produced 80 ± 11 μM (n = 3) putting it within the expected range.
Islet pretreatment, co-culture with adipocytes and insulin secretion monitoring
We used the system to co-culture adipocytes and islets for 3 h and record insulin secretion from single islets following 3 to 11 mM glucose step change. Results from adipocytes were compared to perfusion with 0, 50, and 100 μM palmitic acid for the same period as a positive control. Palmitic acid was chosen because it is the most abundant NEFA in serum and released from adipocytes,63,64 and it is widely used to study the effect of fatty acid on insulin secretion in vitro. As shown in Figure 3A, the islet chip records a classical peak of insulin secretion (first phase) followed by a lower rate (second phase) for all conditions. Pretreatment for 3 h with palmitic acid potentiated the first phase of GSIS in a concentration dependent fashion (Figure 3A, Figure 4A, B). Adipocytes producing 80 µM NEFA had an even greater potentiation of secretion resulting in an almost 6-fold increase in insulin compared to controls and 50% more than 100 μM palmitic acid.
Figure 3.
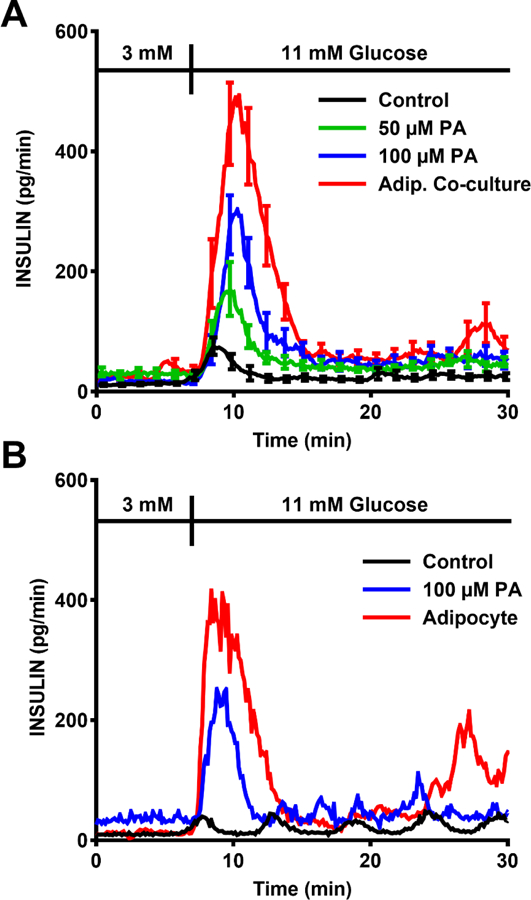
Insulin secretion data is plotted by connecting the insulin concentration data collected every 8 s. (A) Summary of insulin secretion during a 3–11 mM glucose stimulation after 3 h pretreatment with 0 (control), 50 µM, or 100 µM palmitate (PA) compared to similar pretreatment with adipocytes. The error bars indicate ±SEM (n = 5–19 islets from a total of 3–5 mice for each condition). The error bars are shown only on every 10th point for clarity. (B) Representative insulin secretion from single islets for these experiments.
Figure 4.
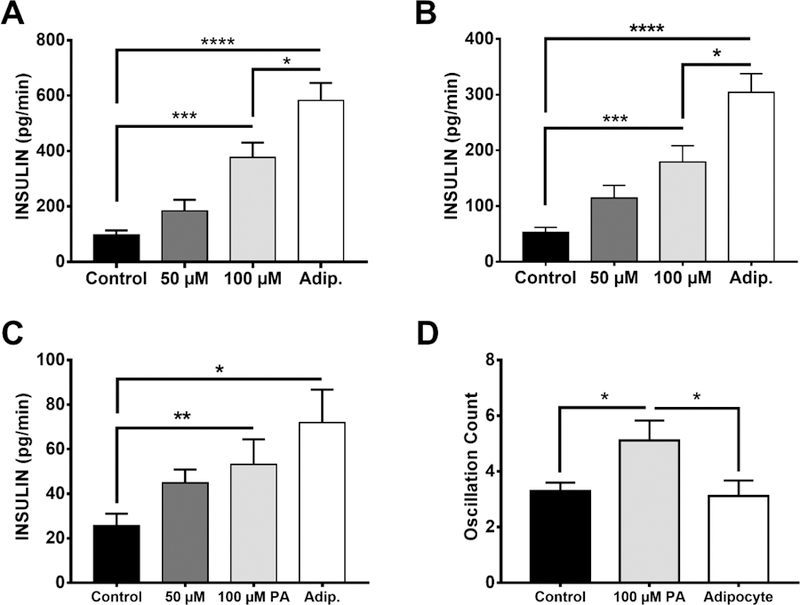
Statistical analysis of insulin secretion. (A) Peak insulin secretion rate, also indicated by the apex value of 1st phase insulin secretion in Figure 5A. (B) Average insulin secretion rate in 1st phase. (C) Average insulin secretion rate in 2nd phase. (D) Quantification of oscillations of control, 100 µM palmitic acid and adipocyte co-cultured islets, occurring from 10 to 30 min after glucose stimulation. Values are averages and error bars indicate ±SEM. **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05, n = 19, 5, 14 and 12 islets in control, 50 µM, 100 µM and adipocyte groups, respectfully.
Second phase insulin secretion, which is typically low in mouse islets in vitro,65,66 was also enhanced by the palmitic acid pretreatment and even more so by adipocyte treatments (Figure 3A, 4C). We also observed oscillations during 2nd phase in many of the islets (see examples in Figure 3B). In summary, we found that 53% (n = 19), 64% (n = 14), and 75% (n = 12) islets had oscillations in control, 100 μM palmitic acid and adipocyte group, respectively (50 μM group is not included in the comparison because of the relatively small n-value). These data suggests that NEFA and adipocytes enhance 2nd phase secretion and possibly induce more oscillations in secretion than glucose alone. As shown in Figure 4D, NEFAs increase the frequency of oscillations but this effect is blunted by adipocytes.
Discussion
Studying isolated cell and tissue types in culture has been a valuable way to dissect cell function. Better mimics of the in vivo environment are likely to be important in fully understanding cell behavior. A significant aspect of the in vivo environment is endocrine interactions wherein secretions from one cell type can enter the bloodstream and then influence other cells. The chip described here advances this study of these effects by integrating cellular interactions through sequential perfusion with an electrophoretic immunoassay to monitor secretory function of the downstream cell, thus mimicking the role of circulation in allowing chemical communication between cell types. The use of modeling and measurements allowed for good understanding and control of the endocrine interaction that was probed. At present the system is limited to allowing 2-cell types to interact. It would be of interest to use added compartments for other cells such as liver and muscle, which also interact with islets and may alter their function. Use of more complex perfusion fluid, perhaps containing blood cell components, would also be of interest. Finally allowing reciprocal interaction, where the release of islets also circulates back to the adipocytes would be useful. Although we used an electrophoretic immunoassay to monitor secretory output, other assays may also be readily coupled to the chip to monitor secretory products such as fluorescence67,68or light-scattering.69
The utility of this new microfluidic system was illustrated by novel observations on the effect of adipocytes on islet function. It is believed that acute exposure to NEFAs will enhance GSIS from islets;70 however, strong data for this effect in mouse islets is scant. Different attempts to test the effect of NEFA have been made by adding and removing fatty acid during low or high glucose treatment.46,71–74 These experiments have resulted in no consensus regarding the actual effect of NEFAs on islets. Our protocol of a 3 h pretreatment with micromolar concentrations of palmitate resulted in robust enhancement of first and second phase GSIS in mouse islets. Oscillatory secretion was also altered suggesting shifts in the metabolism required to generate oscillations.42 Mechanisms for the potentiating effect of NEFAs on GSIS are still being elucidated but metabolic studies suggest that activation of GPR40 receptor, increased glucose metabolism, and glycerolipid formation all play a role.75–78
Although NEFAs enhanced GSIS, the adipocyte pretreatment resulted in much greater enhancement, especially of first and second phase release, but little effect on oscillatory frequency. This result is not due to the amount of NEFAs as the concentrations of NEFA from the adipocytes were bracketed by the control concentrations of palmitate tested. One possible reason for this effect could be that the mix of NEFAs released by adipocytes has a different effect than palmitic acid alone. Even though palmitic acid is the main NEFA released from these adipocytes, several other NEFAs are also released including palmitoleic acid, stearic acid and oleic acid.36 It is possible that this mix has a greater effect than palmitate alone. It is also likely that released adipokines contribute to the effect. Studies have suggested that adiponectin,48 visfatin50 and Interleukin-649,50 can all augment insulin secretion while leptin,51 apelin52 and tumour necrosis factor α (TNFα)53 inhibit insulin secretion. Because all of these adipokines and NEFAs are released by adipocytes, it would be difficult to predict the net effect of adipocytes from prior in vitro studies that examined only individual components in isolation. Our results show the benefit of isolating cell-cell interactions and integrating chemical measurements.
The experiments demonstrate intriguing effects of NEFAs and adipocytes on islets and suggest possible further experiments. It would be of interest to better mimic physiological concentrations. Typical plasma concentrations of NEFA are ~450 µM;64 however, most NEFA are bound to serum albumin, which is present at 500–700 µM and has several NEFA binding sites, so that plasma concentrations of free NEFA are low nanomolar.79 Our conditions have lower total NEFA but higher free NEFA, because of the low albumin concentration used (11 µM), relative to plasma. Using high albumin concentrations in the glass chip was limited by detrimental effects on electroosmotic flow and the electrophoresis assay; therefore, creating a more physiological NEFA/albumin concentration will require overcoming this effect.
Another interesting experiment would be to vary the amount of adipocytes and their condition to mimic different levels of obesity and their effect on islets. Also, the system has potential for chronic measurements that could result in impaired insulin secretion providing a useful in vitro model of the insulin secretion dysfunction in type 2 diabetes.
Conclusion
We have developed a microfluidic chip capable of controlling a cell-cell interaction with integrated continuous monitoring of cell function by microchip electrophoresis immunoassay. With this device we were able to observe a strong potentiating effect of adipocytes on insulin secretion from islets for the first time. Although different chemicals secreted from adipocytes are known to impact insulin secretion in isolation, the net effect of adipocytes would be difficult to predict without such cell-cell interaction measurements. The results illustrate the importance of using live cell interactions to modulate effects of one cell upon the other. The value of integrated chemical analysis is also illustrated as it would be exceedingly difficult to obtain comparable dynamic information on single islets by other means.
Supplementary Material
Figure S-1 Microfluidic chip channel layout. (A) Top piece layout as illustrated in Figure 1A. (B) Bottom piece: The 2 circles with diameter of 8 mm indicate the bottom part of the adipocyte chambers. They are etched to a depth of 250 μm with HF. Before bonded with top piece, they are aligned with the 9.5 mm circles on top layer.
Figure S-2. COMSOL model of NEFA in adipocyte chamber, islet chamber and cellular release. Color-concentration relation is indicated on the bar on the right. (A) A concentration gradient of NEFA moving through the adipocyte chamber to show the profile of flow across the width of the chamber. (B) and (C) Side view of the adipocyte chamber showing a simulation of the concentration flowing from the bottom of chamber assuming a constant release of chemicals from the cells below. The concentration reaching the outlet of the chamber had a 10–90 % rise time of 4 min. (D) Islet chamber and perfusion channel with single islet in it. The average concentration changes of the sphere (mimicking islet) sides facing and away from the perfusion channel were probed. The back side reaches 90% increase at 12 s.
Figure S-3. NEFA concentration measured with off-line enzyme assay. Palmitic acid (PA) was pumped through the chip at the concentrations indicated on the x-axis and collected from the islet chamber to verify that the input concentration was recorded at the islet. During adipocyte perfusion, the same measurement was performed to determine the amount of NEFA reaching the islets. Error bars indicate ±SEM. n = 3.
Acknowledgements
This work was supported by NIH RO1 DK046960. We thank Ting Zhang and Ormond MacDougald for helpful discussions.
Literature Cited
- (1).Huh D; Torisawa Y; Hamilton GA; Kim HJ; Ingber DE Lab Chip 2012, 12, 2156–2164. [DOI] [PubMed] [Google Scholar]
- (2).Sung JH; Shuler ML Bioprocess Biosyst. Eng 2010, 33, 5–19. [DOI] [PubMed] [Google Scholar]
- (3).Huh D; Hamilton GA; Ingber DE Trends Cell Biol 2011, 21, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lau YY; Chen Y-H; Liu T; Li C; Cui X; White RE; Cheng K-C Drug Metab. Dispos 2004, 32, 937–942. [PubMed] [Google Scholar]
- (5).Li AP; Bode C; Sakai Y Chem. Biol. Interact 2004, 150, 129–136. [DOI] [PubMed] [Google Scholar]
- (6).Esch MB; King TL; Shuler ML Annu. Rev. Biomed. Eng 2011, 13, 55–72. [DOI] [PubMed] [Google Scholar]
- (7).Viravaidya K; Sin A; Shuler ML Biotechnol. Prog 2004, 20, 316–323. [DOI] [PubMed] [Google Scholar]
- (8).Sung JH; Shuler ML Lab Chip 2009, 9, 1385–1394. [DOI] [PubMed] [Google Scholar]
- (9).Zhang C; Zhao Z; Abdul Rahim NA; van Noort D; Yu H Lab Chip 2009, 9, 3185–3192. [DOI] [PubMed] [Google Scholar]
- (10).Maschmeyer I; Lorenz AK; Schimek K; Hasenberg T; Ramme AP; Hübner J; Lindner M; Drewell C; Bauer S; Thomas A; Sambo NS; Sonntag F; Lauster R; Marx U Lab Chip 2015, 15, 2688–2699. [DOI] [PubMed] [Google Scholar]
- (11).van Midwoud PM; Merema MT; Verpoorte E; Groothuis GMM. Lab Chip 2010, 10, 2778–2786. [DOI] [PubMed] [Google Scholar]
- (12).Ku CJ; Oblak TDA; Spence DM Anal. Chem 2008, 80, 7543–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hsu Y-H; Moya ML; Hughes CCW; Georgea SC; Lee AP Lab Chip 2013, 13, 2990–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Frank T; Tay S Lab Chip 2015, 15, 2192–2200. [DOI] [PubMed] [Google Scholar]
- (15).Hirano K; Konagaya S; Turner A; Noda Y; Kitamura S; Kotera H; Iwata H Biochem. Biophys. Res. Commun 2017, 487, 344–350. [DOI] [PubMed] [Google Scholar]
- (16).Sun Y; Yao Z; Lin P; Hou X; Chen L Cell Biol. Int 2014, 38, 647–654. [DOI] [PubMed] [Google Scholar]
- (17).Nahavandi S; Tang SY; Baratchi S; Soffe R; Nahavandi S; Kalantar-Zadeh K; Mitchell A; Khoshmanesh K Small 2014, 10, 4810–4826. [DOI] [PubMed] [Google Scholar]
- (18).Schrum DP; Culbertson CT; Jacobson SC; Ramsey JM Anal. Chem 1999, 71, 4173–4177. [DOI] [PubMed] [Google Scholar]
- (19).Zhao M; Schiro PG; Kuo JS; Koehler KM; Sabath DE; Popov V; Feng Q; Chiu DT Anal. Chem 2013, 85, 2465–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fiedler S; Shirley SG; Schnelle T; Fuhr G Anal. Chem 1998, 70, 1909–1915. [DOI] [PubMed] [Google Scholar]
- (21).Fu AY; Spence C; Scherer A; Arnold FH; Quake SR Nat. Biotechnol 1999, 17, 1109–1111. [DOI] [PubMed] [Google Scholar]
- (22).McClain MA; Culbertson CT; Jacobson SC; Allbritton NL; Sims CE; Ramsey JM Anal. Chem 2003, 75, 5646–5655. [DOI] [PubMed] [Google Scholar]
- (23).Zare RN; Kim S Annu. Rev. Biomed. Eng 2010, 12, 187–201. [DOI] [PubMed] [Google Scholar]
- (24).Roper MG Anal. Chem 2016, 88, 381–394. [DOI] [PubMed] [Google Scholar]
- (25).Rocheleau JV; Piston DW Methods Cell Biol 2008, 89, 71–92. [DOI] [PubMed] [Google Scholar]
- (26).Huang WH; Cheng W; Zhang Z; Pang DW; Wang ZL; Cheng JK; Cui DF Anal. Chem 2004, 76, 483–488. [DOI] [PubMed] [Google Scholar]
- (27).Ma C; Fan R; Ahmad H; Shi Q; Comin-Anduix B; Chodon T; Koya RC; Liu C-C; Kwong GA; Radu CG; Ribas A; Heath JR Nat. Med 2011, 17, 738–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Clark AM; Sousa KM; Jennings C; MacDougald OA; Kennedy RT Anal. Chem 2009, 81, 2350–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dugan CE; Cawthorn WP; MacDougald OA; Kennedy RT Anal. Bioanal. Chem 2014, 406, 4851–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Roper MG; Shackman JG; Dahlgren GM; Kennedy RT Anal. Chem 2003, 75, 4711–4717. [DOI] [PubMed] [Google Scholar]
- (31).Shackman JG; Dahlgren GM; Peters JL; Kennedy RT Lab Chip 2005, 5, 56–63. [DOI] [PubMed] [Google Scholar]
- (32).Reid KR; Kennedy RT Anal. Chem 2009, 81, 6837–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dishinger JF; Reid KR; Kennedy RT Anal. Chem 2009, 81, 3119–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Godwin LA; Pilkerton ME; Deal KS; Wanders D; Judd RL; Easley CJ Anal. Chem 2011, 83, 7166–7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mohammed JS; Wang Y; Harvat TA; Oberholzer J; Eddington DT Lab Chip 2009, 9, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dugan CE; Grinias JP; Parlee SD; El-Azzouny M; Evans CR; Kennedy RT Anal. Bioanal. Chem 2017, 409, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schultz NM; Huang L; Kennedy RT Anal. Chem 1995, 67, 924–929. [DOI] [PubMed] [Google Scholar]
- (38).Tao L; Kennedy RT Electrophoresis 1997, 18, 112–117. [DOI] [PubMed] [Google Scholar]
- (39).Kulkarni RN Int. J. Biochem. Cell Biol 2004, 36, 365–371. [DOI] [PubMed] [Google Scholar]
- (40).Henquin JC Diabetes 2000, 49, 1751–1760. [DOI] [PubMed] [Google Scholar]
- (41).Muoio DM; Newgard CB Nat. Rev. Mol. Cell Biol 2008, 9, 193–205. [DOI] [PubMed] [Google Scholar]
- (42).Kennedy RT; Kauri LM; Dahlgren GM; Jung S-K Diabetes 2002, 51, S152–S161. [DOI] [PubMed] [Google Scholar]
- (43).Hollingdal M; Juhl CB; Pincus SM; Sturis J; Veldhuis JD; Polonsky KS; Pørksen N; Schmitz O Diabetes 2000, 49, 1334–1340. [DOI] [PubMed] [Google Scholar]
- (44).Reshef L; Olswang Y; Cassuto H; Blum B; Croniger CM; Kalhan SC; Tilghman SM; Hanson RW J. Biol. Chem 2003, 278, 30413–30416. [DOI] [PubMed] [Google Scholar]
- (45).Dunmore SJ; Brown JEP J. Endocrinol 2013, 216, T37–T45. [DOI] [PubMed] [Google Scholar]
- (46).Thams P; Capito K Diabetologia 2001, 44, 738–746. [DOI] [PubMed] [Google Scholar]
- (47).Newsholme P; Keane D; Welters HJ; Morgan NG Clin. Sci 2007, 112, 27–42. [DOI] [PubMed] [Google Scholar]
- (48).Gu W; Li X; Liu C; Yang J; Ye L; Tang J; Gu Y; Yang Y; Hong J; Zhang Y; Chen M; Ning G Endocrine 2006, 30, 217–221. [DOI] [PubMed] [Google Scholar]
- (49).Suzuki T; Imai J; Yamada T; Ishigaki Y; Kaneko K; Uno K; Hasegawa Y; Ishihara H; Oka Y; Katagiri H Diabetes 2011, 60, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Brown JEP; Onyango DJ; Ramanjaneya M; Conner AC; Patel ST; Dunmore SJ; Randeva HS J. Mol. Endocrinol 2010, 44, 171–178. [DOI] [PubMed] [Google Scholar]
- (51).Brown JEP; Thomas S; Digby JE; Dunmore SJ FEBS Lett 2002, 513, 189–192. [DOI] [PubMed] [Google Scholar]
- (52).Guo L; Li Q; Wang W; Yu P; Pan H; Li P; Sun Y; Zhang J Endocr Res 2009, 34, 142–154. [DOI] [PubMed] [Google Scholar]
- (53).Zhang S; Kim K FEBS Lett 1995, 377, 237–239. [DOI] [PubMed] [Google Scholar]
- (54).Brown JEP; Conner AC; Digby JE; Ward KL; Ramanjaneya M; Randeva HS; Dunmore SJ Peptides 2010, 31, 944–949. [DOI] [PubMed] [Google Scholar]
- (55).Cawthorn WP; Bree AJ; Yao Y; Du B; Hemati N; Martinez-Santibañez G; Macdougald OA Bone 2012, 50, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Clark AM; Sousa KM; Chisolm CN; MacDougald OA; Kennedy RT Anal. Bioanal. Chem 2010, 397, 2939–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Pralong WF; Bartley C; Wollheim CB EMBO J 1990, 9, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Nam K-H; Yong W; Harvat T; Adewola A; Wang S; Oberholzer J; Eddington DT Biomed. Microdevices 2010, 12, 865–874. [DOI] [PubMed] [Google Scholar]
- (59).Manz A; Harrison DJ; Verpoorte EMJ; Fettinger JC; Paulus A; Lüdi H; Widmer HM J. Chromatogr. A 1992, 593, 253–258. [Google Scholar]
- (60).Jacobson SC; Ermakov SV; Ramsey JM Anal. Chem 1999, 71, 3273–3276. [DOI] [PubMed] [Google Scholar]
- (61).Shackman JG; Watson CJ; Kennedy RT J. Chromatogr. A 2004, 1040, 273–282. [DOI] [PubMed] [Google Scholar]
- (62).Stewart JM; Driedzic WR; Berkelaar JA Biochem. J 1991, 275, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Sera RK; McBride JM; Higgins SA; Rodgerson D. J. Clin. Lab. Anal 1994, 8, 81–85. [DOI] [PubMed] [Google Scholar]
- (64).Abdelmagid SA; Clarke SE; Nielsen DE; Badawi A; El-Sohemy A; Mutch DM; Ma DWL PLoS One 2015, 10, e0116195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Ma YH; Wang J; Rodd GG; Bolaffi JL; Grodsky GM Eur. J. Endocrinol 1995, 132, 370–376. [DOI] [PubMed] [Google Scholar]
- (66).Nunemaker CS; Wasserman DH; McGuinness OP; Sweet IR; Teague JC; Satin LS Am. J. Physiol. Endocrinol. Metab 2006, 290, E523–E529. [DOI] [PubMed] [Google Scholar]
- (67).Qian WJ; Gee KR; Kennedy RT Anal. Chem 2003, 75, 3468–3475. [DOI] [PubMed] [Google Scholar]
- (68).Shirasaki Y; Yamagishi M; Suzuki N; Izawa K; Nakahara A; Mizuno J; Shoji S; Heike T; Harada Y; Nishikomori R; Ohara O Sci. Rep 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).McDonald MP; Gemeinhardt A; König K; Piliarik M; Schaffer S; Völkl S; Aigner M; Mackensen A; Sandoghdar V Nano Lett 2018, 18, 513–519. [DOI] [PubMed] [Google Scholar]
- (70).Gravena C; Mathias PC; Ashcroft SJH J. Endocrinol 2002, 173, 73–80. [DOI] [PubMed] [Google Scholar]
- (71).Tian G; Maria Sol ER; Xu Y; Shuai H; Tengholm A Diabetes 2015, 64, 904–915. [DOI] [PubMed] [Google Scholar]
- (72).Alstrup KK; Gregersen S; Jensen HM; Thomsen JL; Hermansen K Metabolism 1999, 48, 22–29. [DOI] [PubMed] [Google Scholar]
- (73).Warnotte C; Gilon P; Nenquin M; Henquin JC Diabetes 1994, 43, 703–711. [DOI] [PubMed] [Google Scholar]
- (74).Doliba NM; Qin W; Vinogradov SA; Wilson DF; Matschinsky FM Am. J. Physiol. Metab 2010, 299, E475–E485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Prentki M; Madiraju SRM Mol. Cell. Endocrinol 2012, 353, 88–100. [DOI] [PubMed] [Google Scholar]
- (76).Latour MG; Alquier T; Oseid E; Tremblay C; Jetton TL; Luo J; Lin DC-H; Poitout V Diabetes 2007, 56, 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Ferdaoussi M; Bergeron V; Zarrouki B; Kolic J; Cantley J; Fielitz J; Olson EN; Prentki M; Biden T; MacDonald PE; Poitout V Diabetologia 2012, 55, 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).El-Azzouny M; Evans CR; Treutelaar MK; Kennedy RT; Burant CF J. Biol. Chem 2014, 289, 13575–13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Richieri GV; Kleinfeld AM J. Lipid Res 1995, 36, 229–240. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S-1 Microfluidic chip channel layout. (A) Top piece layout as illustrated in Figure 1A. (B) Bottom piece: The 2 circles with diameter of 8 mm indicate the bottom part of the adipocyte chambers. They are etched to a depth of 250 μm with HF. Before bonded with top piece, they are aligned with the 9.5 mm circles on top layer.
Figure S-2. COMSOL model of NEFA in adipocyte chamber, islet chamber and cellular release. Color-concentration relation is indicated on the bar on the right. (A) A concentration gradient of NEFA moving through the adipocyte chamber to show the profile of flow across the width of the chamber. (B) and (C) Side view of the adipocyte chamber showing a simulation of the concentration flowing from the bottom of chamber assuming a constant release of chemicals from the cells below. The concentration reaching the outlet of the chamber had a 10–90 % rise time of 4 min. (D) Islet chamber and perfusion channel with single islet in it. The average concentration changes of the sphere (mimicking islet) sides facing and away from the perfusion channel were probed. The back side reaches 90% increase at 12 s.
Figure S-3. NEFA concentration measured with off-line enzyme assay. Palmitic acid (PA) was pumped through the chip at the concentrations indicated on the x-axis and collected from the islet chamber to verify that the input concentration was recorded at the islet. During adipocyte perfusion, the same measurement was performed to determine the amount of NEFA reaching the islets. Error bars indicate ±SEM. n = 3.