

Abstract
Class A flavin-dependent hydroxylases (FdHs) catalyze the hydroxylation of organic compounds in a site and stereoselective manner. In stark contrast, conventional synthetic routes require environmentally hazardous reagents and give modest yields. Thus, understanding the detailed mechanism of this class of enzymes is essential to their rational manipulation for applications in green chemistry and pharmaceutical production. Both electrophilic substitution and radical intermediate mechanisms have been proposed as interpretations of FdH hydroxylation rates and optical spectra. While radical mechanistic steps are often difficult to examine directly, modern quantum chemistry calculations combined with statistical mechanical approaches can yield detailed mechanistic models providing insight that can be used to differentiate reaction pathways. In the current work, we report quantum mechanical/molecular mechanical (QM/MM) calculations on the fungal TropB enzyme that show an alternative reaction pathway in which hydroxylation through a hydroxyl radical coupled electron transfer (HRC-ET) mechanism is significantly favored over electrophilic substitution. Furthermore, QM/MM calculations on several modified flavins provide a more consistent interpretation of the experimental trends in the reaction rates seen experimentally for a related enzyme, para-hydroxybenzoate hydroxylase (PHBH). These calculations should guide future enzyme and substrate design strategies and broaden the scope of biological spin chemistry.
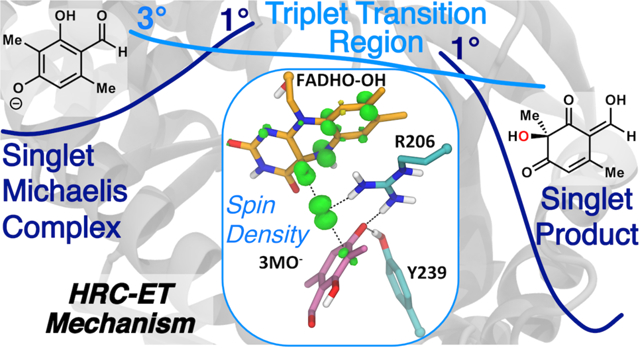
Graphical Abstract
INTRODUCTION
Flavin-dependent hydroxylases (FdHs, or sometimes referred to as monooxygenases) show an incredible range of functional diversity and yet inference from phylogenetic analyses suggest that only a fraction of this diversity has been characterized.1–3 There is immense biotechnological interest in these enzymes, which perform otherwise extremely challenging hydroxylation reactions4–6 with high site and stereoselectivity in a variety of biosynthetic pathways.1, 7 Although these enantioselective transformations are not amenable to the high-throughput evaluations needed for directed evolution approaches8, an in-depth knowledge of the reaction mechanism through computation and/or experiment can facilitate rational enzyme redesign strategies9–12 for the fungal FdHs which to date have not been as well studied as their bacterial counterparts.
Bacterial para-hydroxybenzoate hydroxylase (PHBH) is the most extensively investigated enzyme within class A FdHs.7 The catalytic cycle of both PHBH and TropB13–14, a fungal flavin-dependent hydroxylase, is shown in Figure 1 and includes both reductive and oxidative steps with flavin adenine dinucleotide serving as a cofactor. Since productive substrate binding appears to trigger the steps prior to hydroxylation, an appreciation of the complexity of the catalytic cycle is critical for redesigning substrate selectivity and/or broadening the substrate scope. The catalytic cycle of FdHs (Figure 1) is initiated by substrate binding (i), which (ii) induces a conformational change in FAD to enable formation of a complex with NADPH necessary for hydride transfer from NADPH to FAD. The flavin is reduced to FADH2 and NADP+ is released. The reduced flavin reacts with O2 to form C4a-hydroperoxy-FADH (iii) before a hydroxy group is transferred from the cofactor to the substrate (iv). Finally, (v) the product is released along with one water molecule. In PHBH, the substrate is rearomatized through deprotonation of the penultimate intermediate, while in the TropB reaction, the final product is dearomatized. An important poisonous side reaction can occur at step v if the substrate is not aligned for a productive reaction, leading to formation of hydrogen peroxide.15 Thus, prevention of this reaction by the enzyme is critical to the viability of the cell.
Figure 1.

(Upper) Catalytic cycle of class A Flavin-dependent Monooxygenases (lower) Previously proposed mechanisms for hydroxylation of aromatic substrates using 3-methyl-orcinaldehyde as the example with selected labels for the atoms in the isoalloxazine ring of the FAD cofactor and the 3-methyl-orcinaladehyde (3MO) substrate
A noteworthy feature of these enzymes is that the flavin moiety must swing out to receive the hydride from NAD(P)H before swinging back into the active site to complete the catalytic cycle.16–18 Researchers have dubbed this the “wavin’ flavin”. Substrates that react with FdHs all have a hydroxyl group ortho to the site of hydroxylation and generally para to some other functional group (an aldehydic group in TropB for example).19 Experiments with a variety of substrates suggest that binding of the negatively charged 3-methyl-orcinaldehyde substrate to TropB triggers FAD to transition to the “out” state to receive the hydride from NAD(P)H as in other class A FdHs.13 Thus, electron withdrawing substitutions made to FdH substrates may increase the population of the negatively charged form and thus be more efficient for the reductive steps but perhaps less efficient in the hydroxylation reaction for reasons discussed in this report. Crystal structures of the TropB-FAD complex reveal electron density for the native substrate, 3-methyl-orcinaldehyde, that is consistent with docking calculations of the negatively charged form of the substrate to this receptor. Both the crystal structure and calculations indicate that the substrate atom O4 hydrogen bonds to Arg206 and Tyr239.13
The hydroxylation step of PHBH has been studied via experiment as well as theory. An intermediate, designated Intermediate II7, which has a distinctive optical signature is detected upon oxygen insertion into aromatic substrates of PHBH. The basis of this optical signature has been the subject of intense debate impacting whether the reaction should be classified as an electrophilic aromatic substitution or whether the hydroperoxy Oδ-Op bond breaks homolytically (Figure 1) to form a biradical intermediate pair.7, 20–21 Studies have shown a close correspondence between this optical signature and the combination spectra of hydroxycyclohexadienyl radicals with a C4(a)-hydroxyflavin.20–21 Yet, the experimental data were ultimately interpreted as being consistent with a electrophilic aromatic substitution based on the slope between the calculated pKa value (using the semiempirical AM1 method) of C8-modified hydroxyl-FADs and the hydroxylation reaction rate.22 This study also assumed that these substitutions to FAD would not have an impact on a radical based mechanism. Though electron spin resonance (ESR) spectra at 24 and 280K do not support the existence of a radical pair, several causes can be invoked to explain the absence.20 Following this 1999 paper by Ortiz-Maldonado, Ballou and Massey, several computational studies23–29 were performed assuming the electrophilic aromatic substitution mechanism to be correct, but to the best of our knowledge, alternative reaction mechanisms were never examined. While determination of the most favorable hydroxylation mechanism may not appear to offer any advantage to a biosynthetic chemist (since, in some cases, the products are the same regardless of which reaction path is followed), different mechanisms would require distinct strategies for improving catalysis of nonnative substrates that bind but do not react. Thus, mechanistic insight will be essential for successful enzyme redesign strategies.
In order to investigate this mechanism, flexible ligand-receptor docking calculations were performed to establish initial ligand poses, followed by molecular dynamics (MD) simulations on the TropB—C4a-hydroperoxy-FADH—3-methyl-orcinaldehyde complex to construct a model of the Michaelis complex using the X-ray structure of TropB co-crystallized with FAD.13 The reactant and product states were further investigated with third-order Density Functional Tight Binding30/Molecular Mechanical (DFTB3/MM) simulations that demonstrate the importance of explicit inclusion of polarization and charge transfer effects. Both DFT/MM and high-level wavefunction-based quantum chemical methods were used to investigate the mechanism of substrate hydroxylation by C4a-hydroperoxy-FAD and C8-substituted analogs revealing that the hydroxyl radical coupled electron transfer (HRC-ET) mechanism is significantly favored over the electrophilic substitution mechanism.
THEORETICAL CALCULATIONS
Docking of 3-methyl-orcinaldehyde to TropB.
The protocol for ligand docking of 3-methyl-orcinaldehyde (3MO) to the holo-form of TropB with the flavin cofactor in the oxidized form used the CHARMM software package31 and CDOCKER protocol32 and has been detailed in the report on the TropB structure13 and the Supporting Information.
Molecular Dynamics Simulations of the ternary complex.
A number of our low energy docking poses that were also conducive to reaction were used to construct solvated models of the enzyme-substrate system. The final model contained 90761 atoms. MD simulations were performed with the CHARMM/OpenMM interface in version c42b1 using periodic boundary conditions, particle mesh Ewald for long range electrostatic interactions, and a Langevin thermostat (g = friction coefficient = 5) to maintain the temperature at 298 K. Harmonic restraints of 10.0 kcal mol−1 Å−2 were placed on all non-hydrogen atoms of the solute during the first 500 picosecond (ps) of the simulation, which used a 1 femtosecond (fs) timestep. The simulation was continued without restraints for another 24 nanoseconds (ns) with a 2 fs timestep.
Density Functional Tight Binding 3rd Order (DFTB3)/MM Simulations.
These simulations were initiated from a snapshot of the classical MD simulation that showed the substrate poised to react with the hydroperoxyl group. This structure was geometry optimized first with the CHARMM force field then with the DFTB3/MM hybrid potential until the average gradient was less than 0.0001 kcal mol−1 Å−1 using the adopted basis Newton-Raphson algorithm. The model was partitioned into regions represented by the DFTB3 method30 and the CHARMM MM force field.33 The DFTB3 region included the flavin isoalloxazine moiety (plus two carbon centers beyond this towards the adenine moiety), the side chains of Arg206, Tyr239, and Asn248, the 3MO substrate, and 7 proximal water molecules. Three hydrogen link atoms were placed along the Cβ-Cα axis at a distance of 1.09 Å from the Cβ atom for the protein side chains. The fourth link atom was placed along the second C-C bond from N10 in the flavin cofactor. A total of 125 atoms were represented by the DFTB3 method. A potential energy scan for the hydroxylation reaction was performed using the mass-weighted distance difference between the peroxy Oδ-Op bond and the forming Oδ-C3 bond as the reaction coordinate [Reaction coordinate = 0.571(Oδ-Op-distance) – 0.429 (C3-Oδ-distance)]. This reaction coordinate has been used in all past computational studies of a related enzyme model performing the same catalytic reaction. Although a collective reaction coordinate may provide marginally more accurate results, the lack of any significant changes (conformational transitions, intermolecular proton transfers, etc) from the DFTB3/MM simulations and DFT/MM calculations described below associated with progression along the reaction coordinate argues that the reaction coordinate used in this study and all others sufficiently describes the chemical transformation. Atoms further than 32 Å from the substrate (defined in the initial Michaelis complex pose) were fixed. Thirty-three umbrella sampling windows were used and each window was simulated for 50 ps with a 1 fs timestep using a Langevin thermostat (fbeta = 5) at 298K.
Unrestricted DFT/MM and Unrestricted DLPNO-CCSD(T) Potential Energy Scans in Singlet and Triplet States.
Atoms further than 25Å from the Phe56 Cα atom (a central atom) were fixed and water molecules further than 35Å were removed from the DFTB3/MM geometry optimized model of the Michaelis complex. The QM region remained the same as previously described for the DFTB3/MM simulations. The QM region was modeled with the unrestricted generalized gradient approximate (GGA) functional PBE34 using the D3 dispersion correction with Becke-Johnson damping35–36, RI-J approximation, and the def2-SVP basis set with the def2/J (Coulomb fitting) auxiliary basis set37 using pDynamo38/ORCA (Table S1). The potential energy scan was performed on the singlet and triplet surfaces with 50 steps of conjugate-gradient minimization at each reaction coordinate value using a 400 kcal mol−1 Å−2 restraint on the reaction coordinate at consecutive values 0.1 Å.
Using the same reaction coordinate restraints, 40 additional steps of geometry optimization were performed with the unrestricted (U) B3LYP/MM functional using the singlet and triplet structures from the UPBE/MM reaction coordinate driving and minimization. The calculations were performed using the Gaussian definition of B3LYP39–42, the D3 dispersion correction with Becke-Johnson damping35–36, the RIJCOSX approximation43, the def2-TZVPP basis set44 and the def2-TZVPP/C auxiliary basis set.45 The B3LYP method has been shown to give a reasonably accurate description of organic radicals and biradicals.46–47 Some points in the transition region on the singlet surface were spin-contaminated in the unrestricted scheme. For these points, the 40 steps of B3LYP/MM minimization were repeated with a restricted scheme, and the energies were adjusted based on a restricted minimization of the Michaelis complex structure to approximately compare the energy values. To further confirm the accuracy of the UB3LYP method for our system, we performed single-point energy calculations on the singlet and triplet B3LYP-minimized structures with unrestricted domain-based local pair natural orbital coupled-cluster method with single, double, and perturbative triple excitations (DLPNO-CCSD(T))48, which is the most accurate QM method that would be feasible to apply to such a large system. This calculation was also unrestricted and used the def2-TZVP basis set44 and def2-TZVP/C auxiliary basis set.45 The potential energy profiles of the modified flavins were performed in the same manner as the native flavin. See the Supporting Information for further description.
Charge and spin distribution analyses were performed with natural-bond orbital analysis (NBO)49 through the Q-Chem/CHARMM interface.50
Spin Flip.
The Restricted-Active-Space n-Spin Flip method with the double-zeta, polarized 6–31G* basis was used to characterize the electronic structures of the structures at reaction coordinates equal to −0.2, 0.05, and 0.25 Å. This method starts from a high-spin reference state and correlates all electrons within singly occupied molecular orbital (SOMO) at a complete-active-space (CAS) level, which captures the full effect of electronic correlations among the SOMO electrons.51–53 Additional single excitations of the hole, particle type are used to capture the effects of orbital relaxations from this reference. Taken together, the RAS(h,p)-nSF level of theory captures multi-reference correlation effects at relatively low cost.
RESULTS AND DISCUSSION
TropB reactant and product structures from DFTB3/MM simulations.
TropB catalysis is most efficient at pH=8.013–14, 54. At this pH, the native substrate for TropB, 3-methyl-orcinaldehyde, has been shown to be deprotonated.13 The only residues located in the active site that could act as a general base are His235 and His330, though it does not appear that either do so according to results from site-directed mutagenesis studies.13 Thus, our initial atomistic model of the Michaelis complex agrees with the crystal structure and experiments (Figure 2). DFTB3/MM simulations that account for polarization of the active site by the surroundings and charge transfer within the QM region were used to investigate the Michaelis complex and product structure and the fluctuations that may favor reactivity. The reaction coordinate used in these simulations and the potential energy profiles discussed later used the mass-weighted (Op-Oδ) – (Oδ-C3) distance difference. The free energy profile shows energy minima for the Michaelis complex and product state at reaction coordinate values of −0.75 Å and 0.90 Å, respectively (Figure S1). The overall reaction is thermodynamically favorable by 27.2 kcal/mol; consistent with a reaction typically considered irreversible. Although DFTB330 is an excellent method for investigating several types of mechanisms and is efficient enough to facilitate free energy simulations (as opposed to geometry optimizations), the calculated barrier for electrophilic substitution is much lower than the values obtained using more accurate methods.24 In addition, obtaining accurate results for models with unpaired electrons would require further DFTB3 parameterization. Nevertheless, simulations near the reactant and product basins are modeled in the correct electronic state and are further analyzed.
Figure 2.

(left) Representative structure of Michaelis complex between the 3MO substrate and TropB active site from DFTB3/MM simulations. The peroxyl hydrogen atom is hidden from view. The main chain is represented by an aqua ribbon. (right) The product complex from DFTB3/MM simulations
The Michaelis complex (Figure 2) from the DFTB3/MM simulation places the site of hydroxylation (C3) at a distance of 3.8 Å from the distal oxygen of hydroperoxy-FAD (Oδ). Surprisingly, C4 or C5 are always closer than C3 to the reactive Od atom in the Michaelis complex. The deprotonated oxygen of the substrate (O4) accepts hydrogen bonds from Tyr239 and Arg206. Tyr239 is present in other fungal FdHs but is only moderately conserved, with many fungal species containing Phe at this position. Arg206 is strictly conserved (Section III). Two water molecules also contribute a hydrogen bond to O4, and one of these water molecules coordinates with the hydrogen on N5 of the FADHOOH. In accordance with this observation, studies have suggested that hydrogen bonding to this moiety on the hydroperoxyflavin is important for protecting against elimination of hydrogen peroxide.7 The other water molecule donates a hydrogen bond to the substrate O4 while hydrogen bonding to Asn248 (acting as either an acceptor Od or a donor Hδ). Arg206 interacts with Asn248 through this same water molecule. Asn248 is highly conserved in our fungal data set (Supporting Information, Section III) though a few His, Ser, and Ala residues are also present. Asn, His, Ser can all act both as a hydrogen bond donor, acceptor, or both. The hydroperoxyflavin Oδ directly hydrogen bonds with Arg206 while the proximal oxygen (Op) interacts with Arg206 through an intermediate water molecule. The backbone amide oxygen of Pro329, which plays a major role in the hydroxylation reaction calculations of PHBH23–25, hydrogen bonds with a water molecule that is connected to His330 and does not appear to play a direct role in substrate binding (Figure S2).
The product complex from the DFTB3/MM simulations (Figure 2) shows that the substrate carbonyl oxygen, O4, maintains hydrogen bonds with two organized water molecules and Arg206, but loses the hydrogen bond from Tyr239. Instead, O4 directly hydrogen bonds to N5 on the flavin. The newly formed substrate hydroxy group retains the proton originally from the hydroperoxy group. The hydroxyl group accepts a hydrogen bond from a water molecule and donates a hydrogen bond to the oxygen remaining on the flavin (Op). The anionic oxygen remaining on the flavin (Op) accepts 3 additional hydrogen bonds, one from Arg206 directly, one from a water molecule coordinated to Arg206, and one from another, otherwise uncoordinated water molecule. An intramolecular proton transfer between the C2 hydroxyl group and the aldehyde (tautomerization) is required for the formation of the final product. The substrate spontaneously tautomerizes to the final product in 25% of the simulation frames, with higher percentages in windows beyond the product minimum, suggesting that the different tautomers are separated by a very low free energy barrier. Otherwise, a short hydrogen bond of 1.6 Å is observed between the C2 hydroxy group and the aldehydic oxygen. A tabulation of important hydrogen bonding distances is provided in Table S1.
H2O2 formation and poisoning of the catalyst.
The formation of hydrogen peroxide from the C4a-hydroperoxy-FADH intermediate is thought to occur through intramolecular proton transfer from N5 to the proximal oxygen (Op) breaking the C4a-Op bond. Indeed, in the DFTB3/MM simulations at reaction coordinate values slightly greater or lesser than that of the Michaelis complex, some transient structures are formed that reveal a mechanism for hydrogen peroxide formation. In one instance, the N5 proton transfers to a water molecule in concert with a proton transfer from the water molecule to O4 of the substrate. Loss of the N5 proton causes the C4a-Op bond to break forming a negatively charged peroxide molecule. The peroxide molecule can then take back the proton that was initially transferred from N5 to the water molecule to form hydrogen peroxide as the water molecule abstracts the proton from the substrate (Figure S3). Thus, this minor fluctuation in the simulation supports the notion that exclusion of water molecules from the N5 proton is essential for inhibiting hydrogen peroxide formation. Interestingly, a small percentage of decoupling (H2O2 formation) is seen experimentally in TropB with the native substrate 3-methyl-orcinaldehyde.13
Hydroxylation involves homolytic breaking of the hydroperoxy bond.
Having evaluated the reactant and product structures with DFTB3/MM simulations, DFT/MM calculations were used to investigate the reaction pathways. The calculations revealed an alternative mechanism that is kinetically favored over the closed-shell electrophilic substitution mechanism by ~7 kcal/mol based on DFT and wave-function based methods. This alternative mechanism for hydroxylation of the substrate involves homolytic cleavage of the hydroperoxyl bond and addition of the hydroxyl radical to the substrate followed by electron transfer back to the cofactor. In addition, the mechanism proves to be robust to perturbations of the flavin cofactor as described below.
The UB3LYP/MM potential energy profile reveals that as the reaction coordinate progresses toward the transition region in the singlet state, there is a crossing to the triplet state surface (Figure 3). This crossing places the barrier at 13.8 kcal/mol, which is close to the experimental barrier for the hydroxylation reaction in PHBH at pH 8 (15 kcal/mol55–56). While the spin transition is formally disallowed by nonrelativistic quantum mechanics, there is always some amount of spin-orbital coupling present to cross between states in real systems. In this particular reaction mechanism, the spin switch occurs prior to the singlet transition state, and therefore the system will have sufficient time in the reactant state to undergo crossing to the triplet state. The reaction pathway therefore will be limited by the rate of crossing through the transition state (on the triplet surface) by transition state theory considerations; i.e. the activation barrier remains the relevant quantity of interest. At the triplet geometry nearest the first crossing (reaction coordinate = −0.25), the singlet UKS-DFT/MM energy is 0.2 kcal/mol higher than the triplet and 1.0 kcal/mol more favorable than the UKS-DFT/MM singlet geometry produced by the original scan (referred to as Point 1 in Figure S5). This near equivalence in energy suggests that the minimum energy crossing point is within this narrow region. The potential energy profile was also calculated starting from the product structure and driven along the reaction coordinate back to the Michaelis complex showing only minimal hysteresis (Figure S4). The transition region (where the lowest energy structure is the triplet) is quite broad. The UB3LYP/MM singlet-triplet gap is at least 7.7 kcal/mol at the reaction coordinate corresponding to the singlet transition state; favoring the triplet state. The uncertainty in this energy difference results from the singlet state breaking spin symmetry since it has radical character. Therefore, the singlet-triplet energy gap was calculated with the unrestricted DLPNO-CCSD(T) method48 placing the singlet-triplet gap at 8.0 kcal/mol (Table S1). Thus, the singlet pathway is not the lowest energy pathway based on highly accurate model chemistries, and has a barrier of at least 20.5 kcal/mol with the B3LYP/MM method, which is in poor agreement with the PHBH experimental value. RAS-SF calculations further support the biradical character of the transition structure along the singlet and triplet pathways (Figure S5 and Table S3).
Figure 3.

Unrestricted B3LYP/MM potential energy profile for hydroxylation of 3-methyl-orcinaldehyde showing the spin density in the transition state region. The points for the triplet (light blue) are shown for the two spin crossing regions. The reaction coordinate is the mass-weighted (Op-Oδ) – (Oδ-C3) distance difference described in the Methods. The product structure shows a shared proton between the substrate and cofactor.
A description of the changes in the spin density over the transition region (where the triplet state is lower in potential energy than the singlet, Table S3) helps elucidate the exact mechanism of hydroxylation. Spin-density analysis (Figure 4) reveals that the Oδ-Op bond in hydroperoxy-FADH breaks homolytically, but not in a manner with one unpaired electron for each oxygen atom. Instead, one electron from the FAD π system moves to the proximal oxygen, so that the proximal oxygen and the hydroxyl share a −0.9 charge and only one unpaired electron between them (Scheme 1, Step 1, Table S4). The unpaired electron is located primarily on the transferring hydroxyl (ρ = 0.63 on Od, charge of −0.25e on the hydroxyl) while the negative charge is predominantly on the proximal oxygen (charge of −0.65e, r = 0.37 on Op). As a result of the electron donation, the conjugated rings of FAD contain an average of +0.86 charge and nearly one unpaired electron (ρ = 0.93). This unpaired electron is delocalized through the flavin ring (Figure 4), but the atoms supporting the greatest proportion of the spin density are N5 and C8 (average ρ = 0.31 and 0.25 respectively). Of particular importance, the spin density is delocalized out to C8. Thus, the notion that substituents attached to C8 will have no effect on a radical mechanism22 is not supported by these high-level calculations.
Figure 4. Spin Density plots along the triplet transition state region.

The numbering corresponds to the points shown in Fig. 3. The majority of the density is positive (green) with some minor negative (yellow) contributions. The spin density along this UKS-DFT/MM triplet pathway integrates to 2. Spin distribution remains similar through the early transition region (points 1–4), but as the reaction progresses toward the second crossing, the spin builds up on substrate C3.
Scheme 1.

Hydroxyl radical coupled electron transfer (HRC-ET) mechanism based on DFT/MM calculations
As the Oδ-Op bond is lengthened, the spin density on Oδ slightly increases with concerted decreases in the proximal oxygen through the initial part of the triplet region. As the hydroxyl radical moves closer to the substrate, spin density is induced on C3 (Figure 4 and Scheme 1, Step 2) and decreases on both oxygen atoms of the hydroperoxy group. Spin density is also delocalized through the substrate, primarily on the oxygen atoms. The last point before the triplet state crosses back to the singlet state indicates that one unpaired electron is on the FADHO(−) cofactor (net charge of 0) and one unpaired electron (along with a charge of −0.76) is on the almost fully formed product. Interestingly, at this point the p orbital on the FADHO containing spin density rotates slightly away from being oriented towards the departing hydroxyl radical suggesting that the interaction between these two groups has diminished (Figure 4, point 8).
Electron transfer (ET) from the substrate to the flavin cofactor occurs when the transferring hydroxyl radical is almost equidistant between Op and C3, returning the system back to the singlet state (Scheme 1, Step 3). Natural Bond Orbital (NBO) analysis of the charge distribution along the transition region reveals the details of this ET pathway. As negative charge is depleted on the substrate, particularly near C3, the charges on Oδ, Op and the flavin ring (in order of the electron transfer pathway) become more negative. The ET occurs adiabatically as the substrate C3–C4 double bond and the cofactor C4a-Op bond elongate. The former relaxes the substrate to a position favorable for hydroxyl addition, while the latter positions the cofactor favorably for the negatively charged closed-shell state. Arg206 acts as a “fork” holding the pathway into place or possibly directly involved in an ET pathway. Interestingly, arginine residues are often involved in controlling biomolecular electron transfer pathways.57–59 The overall hydroxylation mechanism proposed here has some similarities with 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase which oxygenates substrates without the need for a cofactor60 and those proposed for bacterial luciferase involving formation of a radical pair and subsequent electron transfer(s).61
Role of surroundings and Arg206 and Tyr239.
Two residues in the TropB active site, Arg206 and Tyr239, are critical for substrate positioning and productive catalysis. In the Michaelis complex Arg206 NH1-H forms a hydrogen bond with Oδ-H that shortens considerably at the beginning of the triplet transition state region to 1.75 Å. The hydrogen bond distance increases somewhat just before the second crossing and the interaction is decreased in the product structure (Table S5). A water molecule also interacts with the Oδ-H group; this hydrogen bond forms tightly at the start of the triplet transition state decreasing to 1.59 Å before the second crossing and remains hydrogen bonded to the product. There are three hydrogen bond donors to the “oxyanionic” oxygen of the substrate (C4-O), Arg206 (NH2-H), Tyr239 and a water molecule. All three form short hydrogen bonds in both the reactant and product structures. The hydrogen bond distances from Arg206 and Tyr239 decrease by 0.03–0.05 Å through the triplet transition region before increasing by the same amount before the second crossing. Clearly, Arg206 and Tyr239 are important in binding the oxyanionic form of the substrate. In the product structure, Arg206 NH1-H hydrogen bonds to the remaining Op-FADH, stabilizing the anionic alkoxyflavin. Ile237, which appears to be important for substrate selectivity, is positioned to direct Tyr239 towards the substrate oxyanion throughout. Here, calculations indicate that the TropB substrate transfers 15% of the negative charge to the surroundings (125 QM atoms in the model). The QM region in all of the PHBH studies contains only the hydroperoxyl isoalloxazine ring and the substrate (49 QM atoms in the model).23–26, 28–29 Previous studies of the hydroxylation reaction in PHBH models is discussed in the Supporting Information, Section VII.
The hydroxylation reaction with C8-substituted flavins.
In order to determine if the hydroxylation reaction rate of flavin C8-substitutions investigated experimentally in PHBH22 could be explained through the HRC-ET mechanism, DFT/MM calculations of these substitutions with the TropB model were performed. Regardless of whether the C8-substituent bonded to FADHOOH was hydrogen, methyl, amino, chloride, or cyano group, a crossing from the singlet to triplet state was calculated to occur in the transition region (Figure S7–S9 & Table S6). Thus, the Od-Op bond is predicted to break homolytically in all cases, which may be a general feature of all flavin-dependent hydroxylases. The UB3LYP/MM calculations clearly indicate that a substituent at C8 can affect the stabilization of the radical. The DFT/MM calculations reveal that resonance electron donors lead to initial stabilization of the delocalized flavin radical leading to lower barriers consistent with the faster reaction rates observed in PHBH. In contrast, resonance electron withdrawing groups favor initial stabilization of the delocalized substrate radical leading to higher barriers relative to native. Thus, the HRC-ET mechanism is consistent with chemical intuition and the experimental data while the proponents of the electrophilic aromatic substitution mechanism must assume that both the cyano (electron withdrawing) substituted flavin and the amino (electron donating) substituted flavin have enhanced hydroxylation reaction rates compared with the native cofactor.22 Further, the calculations indicate that substitutions at C8 more significantly impact the radical mechanism over the electrophilic aromatic substitution mechanism. The results refute the notion that these substitutions would have no effect on a radical based mechanism and further solidify that the hydroxyl radical coupled electron transfer (HRC-ET) is robust to perturbations of this type.
CONCLUSIONS
The HRC-ET mechanism suggests strategies for delocalizing a radical through appropriate substitutions on an aromatic substrate in order to affect the reactivity of additional sites. If successfully employed, the products would serve as a powerful confirmation of the HRC-ET mechanism. Electron transfer pathways from the substrate to surrounding entities (for example Trp or Tyr residues) and ultimately additional substrates could be designed. This scheme is analogous to hypotheses on the basis of avian magnetoreception involving cryptochromes which incidentally also employ a flavin cofactor.62–63
Given that both the electrophilic substitution and HRC-ET hydroxylation mechanisms are predicted to be energetically feasible in FdHs yet mechanistically distinct, interesting evolutionary questions arise such as the following: If the FdH family shows mechanistic promiscuity, how are those environments distinct from one another? Do physical constraints prevent such tinkering64 with the hydroxylation mechanism? Future studies can examine these tradeoffs and trends in the singlet-triplet energy gap across phylogenetic space.65
While fungal TropB and bacterial PHBH are distantly related and share only minimal sequence identity, these two enzymes do have very similar structures and both hydroxylate aromatic substrates. Thus, the HRC-ET mechanism may be shared among a wide variety of flavin dependent hydroxylases. Indeed, hydroperoxyflavin chemistry performed by other flavin-dependent enzymes are highly suggestive of radical mechanisms.66 In addition, the HRC-ET mechanism may also be applicable to peroxyflavin chemistry and thus blur the distinction between hydroperoxyflavin and peroxyflavin reactions; the latter currently characterized as carrying out nucleophilic substitutions.67 The simulations also reveal why sufficient “protection” of the hydroperoxy group by blocking access to the N5 proton is essential for product yields and thus impacts rational redesign efforts of this enzyme for syntheses of alternative products.
Most importantly though, the DFT/MM calculations indicate that delocalization of an unpaired electron over the substrate is energetically unfavorable. Yet, with the appropriate enzyme substitutions, the unpaired electron spin density could readily be altered through intermolecular interactions to facilitate electron transfer and thus explain trends in substrate reactivity. How these same intermolecular interactions would have much impact on the electrophilic mechanism is unclear. In addition, proton abstraction from the substrate either through a general base or increased pH of the solution would undoubtedly create a more electrophilic form of the substrate, whereas this particular activation step appears to be irrelevant to the favorable homolytic breaking of the hydroperoxy O-O bond based on our calculations. Further investigation is needed to determine why some substrates that are very similar to the native substrate bind to TropB but do not react. Finally, determining how substitutions of active site residues that create a positive electrostatic potential68 affect the hydroxylation step in the catalytic cycle would advance FdH enzyme engineering approaches. Understanding of the HRC-ET mechanism may be the critical component of these strategies.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by R35 GM130587-01 (to C.L.B.) and NSF CHE 1506273 (to C.L.B.), funds from the University of Michigan Life Sciences Institute, the National Institutes of Health R35 GM124880 (to A.R.H.N.). A.R.B. was supported by a National Institutes of Health Chemistry Biology Interface Training Grant (T32 GM008597), a Graduate Assistance of Areas in National Need Training Grant (GAANN P200A150164) and a Rackham Merit Fellowship. We thank Itai Palmon for discussions of manuscript clarity and David Braun for computational support.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
The Supporting Information contains additional details about the methods, DFTB3/MM free energy simulations, multiple sequence alignment, spin flip calculations, the mechanism from DFT/MM calculations with the native flavin, and the DFT/MM study of modified flavins, as well as additional DFT/MM analysis of hysteresis, calculations using a minimal QM region, and a comparison of results from the different QM methods used. The following files are available free of charge (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Huijbers MME; Montersino S; Westphal AH; Tischler D; van Berkel WJH Flavin Dependent Monooxygenases. Arch. Biochem. Biophys 2014, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 2.van Berkel WJH; Kamerbeek NM; Fraaije MW Flavoprotein Monooxygenases, a Diverse Class of Oxidative Biocatalysts. J. Biotechnol 2006, 124, 670–689. [DOI] [PubMed] [Google Scholar]
- 3.Mascotti ML; Juri Ayub M; Furnham N; Thornton JM; Laskowski RA Chopping and Changing: The Evolution of the Flavin-Dependent Monooxygenases. J. Mol. Biol 2016, 428, 3131–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu J; Grigoriadis NP; Lee JP; Porco JA Jr. Synthesis of the Azaphilones Using Copper-Mediated Enantioselective Oxidative Dearomatization. J. Am. Chem. Soc 2005, 127, 9342–9343. [DOI] [PubMed] [Google Scholar]
- 5.Sun W; Li G; Hong L; Wang R Asymmetric Dearomatization of Phenols. Org. Biomol. Chem 2016, 14, 2164–2176. [DOI] [PubMed] [Google Scholar]
- 6.Tang M-C; Zou Y; Watanabe K; Walsh CT; Tang Y Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palfey BA; McDonald CA Control of Catalysis in Flavin-Dependent Monooxygenases. Arch. Biochem. Biophys 2010, 493, 26–36. [DOI] [PubMed] [Google Scholar]
- 8.Reetz MT Controlling the Enantioselectivity of Enzymes by Directed Evolution: Practical and Theoretical Ramifications. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5716–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiss G; Çelebi-Ölçüm N; Moretti R; Baker D; Houk KN Computational Enzyme Design. Angew. Chem., Int. Ed. Engl 2013, 52, 5700–5725. [DOI] [PubMed] [Google Scholar]
- 10.Frushicheva MP; Mills MJL; Schopf P; Singh MK; Prasad RB; Warshel A Computer Aided Enzyme Design and Catalytic Concepts. Curr. Opin. Chem. Biol 2014, 21, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleishman SJ; Baker D Role of the Biomolecular Energy Gap in Protein Design, Structure, and Evolution. Cell 2012, 149, 262–273. [DOI] [PubMed] [Google Scholar]
- 12.Renata H; Wang ZJ; Arnold FH Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem., Int. Ed. Engl 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodríguez Benítez A; Tweedy SE; Baker Dockrey SA; Lukowski AL; Wymore T; Khare D; Brooks CL; Palfey BA; Smith JL; Narayan ARH Structural Basis for Selectivity in Flavin-Dependent Monooxygenase-Catalyzed Oxidative Dearomatization. ACS Catal. 2019, 3633–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abood A; Al-Fahad A; Scott A; Hosny AE-DMS; Hashem AM; Fattah AMA; Race PR; Simpson TJ; Cox RJ Kinetic Characterisation of the Fad Dependent Monooxygenase Tropb and Investigation of Its Biotransformation Potential. RSC Adv. 2015, 5, 49987–49995. [Google Scholar]
- 15.Holtmann D; Hollmann F The Oxygen Dilemma: A Severe Challenge for the Application of Monooxygenases? ChemBioChem 2016, 17, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gatti DL; Palfey BA; Lah MS; Entsch B; Massey V; Ballou DP; Ludwig ML The Mobile Flavin of 4-Oh Benzoate Hydroxylase. Science 1994, 266, 110–114. [DOI] [PubMed] [Google Scholar]
- 17.Wang J; Ortiz-Maldonado M; Entsch B; Massey V; Ballou D; Gatti DL Protein and Ligand Dynamics in 4-Hydroxybenzoate Hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan KS; Chakraborty S; Howard-Jones AR; Walsh CT; Ballou DP; Drennan CL The Fad Cofactor of Rebc Shifts to an in Conformation Upon Flavin Reduction. Biochemistry 2008, 47, 13506–13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chenprakhon P; Wongnate T; Chaiyen P Monooxygenation of Aromatic Compounds by Flavin-Dependent Monooxygenases. Protein Sci. 2019, 28, 8–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson RF; Patel KB; Vojnovic B Absorption Spectra of Radical Forms of 2,4-Dihydroxybenzoic Acid, a Substrate for P-Hydroxybenzoate Hydroxylase. J. Biol. Chem 1991, 266, 13086–13090. [PubMed] [Google Scholar]
- 21.Anderson RF; Patel KB; Stratford MR Absorption Spectra of Radicals of Substrates for P-Hydroxybenzoate Hydroxylase Following Electrophilic Attack of The Oh Radical in the 3 Position. J. Biol. Chem 1987, 262, 17475–17479. [PubMed] [Google Scholar]
- 22.Ortiz-Maldonado M; Ballou DP; Massey V Use of Free Energy Relationships to Probe the Individual Steps of Hydroxylation of P-Hydroxybenzoate Hydroxylase: Studies with a Series of 8-Substituted Flavins. Biochemistry 1999, 38, 8124–8137. [DOI] [PubMed] [Google Scholar]
- 23.Ridder L; Palfey BA; Vervoort J; Rietjens IM Modelling Flavin and Substrate Substituent Effects on the Activation Barrier and Rate of Oxygen Transfer by P-Hydroxybenzoate Hydroxylase. FEBS Lett. 2000, 478, 197–201. [DOI] [PubMed] [Google Scholar]
- 24.Claeyssens F; Harvey JN; Manby FR; Mata RA; Mulholland AJ; Ranaghan KE; Schütz M; Thiel S; Thiel W; Werner H-J High-Accuracy Computation of Reaction Barriers in Enzymes. Angew. Chem., Int. Ed. Engl 2006, 45, 6856–6859. [DOI] [PubMed] [Google Scholar]
- 25.Ridder L; Harvey JN; Rietjens IMCM; Vervoort J; Mulholland AJ Ab Initio Qm/Mm Modeling of the Hydroxylation Step in P-Hydroxybenzoate Hydroxylase. J. Phys. Chem. B 2003, 107, 2118–2126. [Google Scholar]
- 26.Senn HM; Thiel S; Thiel W Enzymatic Hydroxylation in P-Hydroxybenzoate Hydroxylase: A Case Study for Qm/Mm Molecular Dynamics. J. Chem. Theory Comput. 2005, 1, 494–505. [DOI] [PubMed] [Google Scholar]
- 27.Ridder L; Mulholland AJ; Rietjens IMCM; Vervoort J Combined Quantum Mechanical and Molecular Mechanical Reaction Pathway Calculation for Aromatic Hydroxylation by P-Hydroxybenzoate-3-Hydroxylase. J. Mol. Graphics Modell. 1999, 17, 163–175. [DOI] [PubMed] [Google Scholar]
- 28.Mata RA; Werner H-J; Thiel S; Thiel W Toward Accurate Barriers for Enzymatic Reactions: Qm/Mm Case Study on P-Hydroxybenzoate Hydroxylase. J. Chem. Phys 2008, 128, 025104. [DOI] [PubMed] [Google Scholar]
- 29.Bistoni G; Polyak I; Sparta M; Thiel W; Neese F Toward Accurate Qm/Mm Reaction Barriers with Large Qm Regions Using Domain Based Pair Natural Orbital Coupled Cluster Theory. J. Chem. Theory Comput. 2018, 14, 3524–3531. [DOI] [PubMed] [Google Scholar]
- 30.Gaus M; Cui Q; Elstner M, Dftb3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (Scc-Dftb). J. Chem. Theory Comput. 2012, 7, 931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S, et al. Charmm: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagnon JK; Law SM; Brooks CL Flexible Cdocker: Development and Application of a Pseudo-Explicit Structure-Based Docking Method within Charmm. J. Comput. Chem 2016, 37, 753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanommeslaeghe K; Mackerell AD Charmm Additive and Polarizable Force Fields for Biophysics and Computer-Aided Drug Design. Biochim. Biophys. Acta 2015, 1850, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perdew JP; Burke K; Ernzerhof M Generalized Gradient Approximation Made Simple. Phys. Rev. Lett 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 35.Grimme S; Ehrlich S; Goerigk L Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 36.Grimme S; Antony J; Ehrlich S; Krieg H A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (Dft-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 37.Weigend F Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 38.Field MJ The Pdynamo Program for Molecular Simulations Using Hybrid Quantum Chemical and Molecular Mechanical Potentials. J. Chem. Theory Comput. 2008, 4, 1151–1161. [DOI] [PubMed] [Google Scholar]
- 39.Becke AD Density-Functional Thermochemistry .3. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- 40.Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 41.Vosko SH; Wilk L; Nusair M Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin-Density Calculations - a Critical Analysis. Can. J. Phys 1980, 58, 1200–1211. [Google Scholar]
- 42.Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- 43.Neese F; Wennmohs F; Hansen A; Becker U Efficient, Approximate and Parallel Hartree-Fock and Hybrid Dft Calculations. A ‘Chain-of-Spheres’ Algorithm for the Hartree-Fock Exchange. Chem. Phys 2009, 356, 98–109. [Google Scholar]
- 44.Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 45.Hellweg A; Hattig C; Hofener S; Klopper W Optimized Accurate Auxiliary Basis Sets for Ri-Mp2 and Ri-Cc2 Calculations for the Atoms Rb to Rn. Theor. Chem. Acc 2007, 117, 587–597. [Google Scholar]
- 46.Hait D; Rettig A; Head-Gordon M Well-Behaved Versus Ill-Behaved Density Functionals for Single Bond Dissociation: Separating Success from Disaster Functional by Functional for Stretched H2. J. Chem. Phys 2019, 150, 094115. [DOI] [PubMed] [Google Scholar]
- 47.Riley KE; Op’t Holt BT; Merz KM Jr. Critical Assessment of the Performance of Density Functional Methods for Several Atomic and Molecular Properties. J. Chem. Theory Comput. 2007, 3, 407–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liakos DG; Neese F Is It Possible to Obtain Coupled Cluster Quality Energies at near Density Functional Theory Cost? Domain-Based Local Pair Natural Orbital Coupled Cluster Vs Modern Density Functional Theory. J. Chem. Theory Comput. 2015, 11, 4054–4063. [DOI] [PubMed] [Google Scholar]
- 49.Glendening ED; Landis CR; Weinhold F Nbo 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem 2013, 34, 1429–1437. [DOI] [PubMed] [Google Scholar]
- 50.Woodcock HL; Hodoscek M; Gilbert ATB; Gill PMW; Schaefer HF; Brooks BR Interfacing Q-Chem and Charmm to Perform Qm/Mm Reaction Path Calculations. J. Comput. Chem 2007, 28, 1485–1502. [DOI] [PubMed] [Google Scholar]
- 51.Zimmerman PM; Bell F; Goldey M; Bell AT; Head-Gordon M Restricted Active Space Spin-Flip Configuration Interaction: Theory and Examples for Multiple Spin Flips with Odd Numbers of Electrons. J. Chem. Phys 2012, 137, 164110. [DOI] [PubMed] [Google Scholar]
- 52.Bell F; Zimmerman PM; Casanova D; Goldey M; Head-Gordon M Restricted Active Space Spin-Flip (Ras-Sf) with Arbitrary Number of Spin-Flips. Phys. Chem. Chem. Phys 2013, 15, 358–366. [DOI] [PubMed] [Google Scholar]
- 53.Chien AD; Zimmerman PM Recovering Dynamic Correlation in Spin Flip Configuration Interaction through a Difference Dedicated Approach. J. Chem. Phys 2017, 146, 014103. [DOI] [PubMed] [Google Scholar]
- 54.Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH Biocatalytic Site-and Enantioselective Oxidative Dearomatization of Phenols. Nat. Chem 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Husain M; Entsch B; Ballou DP; Massey V; Chapman PJ Fluoride Elimination from Substrates in Hydroxylation Reactions Catalyzed by P-Hydroxybenzoate Hydroxylase. J. Biol. Chem 1980, 255, 4189–4197. [PubMed] [Google Scholar]
- 56.Entsch B; Palfey BA; Ballou DP; Massey V Catalytic Function of Tyrosine Residues in Para-Hydroxybenzoate Hydroxylase as Determined by the Study of Site-Directed Mutants. J. Biol. Chem 1991, 266, 17341–17349. [PubMed] [Google Scholar]
- 57.Hsiao JC; McGrath AP; Keilmann L; Kalimuthu P; Darain F; Bernhardt PV; Harmer J; Lee M; Meyers K; Maher MJ, et al. The Central Active Site Arginine in Sulfite Oxidizing Enzymes Alters Kinetic Properties by Controlling Electron Transfer and Redox Interactions. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 19–27. [DOI] [PubMed] [Google Scholar]
- 58.Lucas MF; Rousseau DL; Guallar V Electron Transfer Pathways in Cytochrome C Oxidase. Biochim. Biophys. Acta 2011, 1807, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang ZQ; Tejero J; Wei CC; Haque MM; Santolini J; Fadlalla M; Biswas A; Stuehr DJ Arg375 Tunes Tetrahydrobiopterin Functions and Modulates Catalysis by Inducible Nitric Oxide Synthase. J. Inorg. Biochem 2012, 108, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hernandez-Ortega A; Quesne MG; Bui S; Heyes DJ; Steiner RA; Scrutton NS; de Visser SP Catalytic Mechanism of Cofactor-Free Dioxygenases and How They Circumvent Spin-Forbidden Oxygenation of Their Substrates. J. Am. Chem. Soc 2015, 137, 7474–7487. [DOI] [PubMed] [Google Scholar]
- 61.Francisco WA; Abu-Soud HM; DelMonte AJ; Singleton DA; Baldwin TO; Raushel FM Deuterium Kinetic Isotope Effects and the Mechanism of the Bacterial Luciferase Reaction. Biochemistry 1998, 37, 2596–2606. [DOI] [PubMed] [Google Scholar]
- 62.Rodgers CT; Hore PJ, Chemical Magnetoreception in Birds: The Radical Pair Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hore PJ; Mouritsen H The Radical-Pair Mechanism of Magnetoreception. Annu. Rev. Biophys 2016, 45, 299–344. [DOI] [PubMed] [Google Scholar]
- 64.Jacob F, Evolution and Tinkering. Science 1977, 196, 1161–1166. [DOI] [PubMed] [Google Scholar]
- 65.Wymore T; Brooks III C From Molecular Phylogenetics to Quantum Chemistry: Discovering Enzyme Design Principles through Computation. Comput. Struct. Biotechnol. J 2012, 2, e201209018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li L; Liu X; Yang W; Xu F; Wang W; Feng L; Bartlam M; Wang L; Rao Z Crystal Structure of Long-Chain Alkane Monooxygenase (Lada) in Complex with Coenzyme Fmn: Unveiling the Long-Chain Alkane Hydroxylase. J. Mol. Biol 2008, 376, 453–465. [DOI] [PubMed] [Google Scholar]
- 67.Ellis HR The Fmn-Dependent Two-Component Monooxygenase Systems. Arch. Biochem. Biophys 2010, 497, 1–12. [DOI] [PubMed] [Google Scholar]
- 68.Ortiz-Maldonado M; Cole LJ; Dumas SM; Entsch B; Ballou DP, Increased Positive Electrostatic Potential in P-Hydroxybenzoate Hydroxylase Accelerates Hydroxylation but Slows Turnover. Biochemistry 2004, 43, 1569–1579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.