

Abstract
Pathogenic bacteria scavenge ferric iron from the host for survival and proliferation using small-molecular chelators, siderophores. Here, we introduce and assess the gallium(III) complex of ciprofloxacin-functionalized desferrichrome (D2) as a potential therapeutic for bacterial infection using an in vitro assay and radiochemical, tracer-based approach. Ga-D2 exhibits a minimum inhibitory concentration of 0.23 μM in Escherichia coli, in line with the parent fluoroquinolone antibiotic. Competitive and mutant strain assays show that Ga-D2 relies on FhuA-mediated transport for internalization. Ga-D2 is potent against Pseudomonas aeruginosa (3.8 μM), Staphylococcus aureus (0.94 μM), and Klebsiella pneumoniae (12.5 μM), while Fe-D2 is inactive in these strains. Radiochemical experiments with E. coli reveal that 67Ga-D2 is taken up more efficiently than 67Ga-citrate. In naive mice, 67Ga-D2 clears renally and is excreted 13% intact in the urine. These pharmacokinetic and bacterial growth inhibitory properties qualify Ga-D2 for future investigations as a diagnosis and treatment tool for infection.
Graphical Abstract
INTRODUCTION
Hospital infections and infection-related deaths are caused by a growing number of resistance cases, as reported by the Center for Disease Control (CDC). The rapid identification, localization, and treatment of bacterial infections are of critical importance for effective treatment and favorable patient outcomes.1,2 To improve diagnosis and treatment outcome, there is a need for faster, accurate, and more sensitive diagnosis of bacterial infections, paired with effective therapies.3
Ideal biological imaging and therapy targets of diseases are markers with high abundance within the target tissue or organ, yet completely absent in healthy tissue. Bacteria produce small natural products, termed siderophores, with extraordinarily high affinity for Fe3+ to sequester iron from their host. Siderophore transmembrane transporters are highly conserved and expressed among Gram-positive and Gram-negative bacterial strains, while completely absent in mammalian host cells.4–6 Considerable substrate tolerance for siderophores functionalized with various payloads has been observed.7 Streptomyces produce the natural antibiotic albomycin, a derivative of the siderophore ferrichrome, which takes advantage of this substrate tolerance. Albomycin links a thioribosyl–pyrimidine moiety to the ferrichrome siderophore (Figure 1), which is efficiently released in the bacterial cytoplasm to inhibit seryl t-RNA synthetase, shutting down bacterial protein synthesis.8–10 Albomycin is active against numerous Gram-negative strains and has been utilized effectively as an antibiotic in mammals, with isolation from bacterial culture as the primary source; recently, the total synthesis of albomycin has been reported.11 Another prominent natural product of the siderophore–antibiotic (sideromycin) family is salmycin. Salmycin is composed of the siderophore danoxamine appended to an amino disaccharide, which is proposed to inhibit protein synthesis but exhibits low in vivo stability and is therefore not suitable as a systemically administered antibiotic.12 In addition to the synthesis and evaluation of these natural products, synthetic “Trojan horse” siderophore–antibiotic conjugates have been developed successfully by Miller, Nolan, and others.13–19 These enterobactin-, tris-catecholato-, and danoxamine-derived conjugates show promising activity in circumventing membrane-permeability associated resistance pathways, or specifically targeting pathogenic strains with upregulated side-rophore-degrading enzymes in the cytoplasmic milieu.20–23
Figure 1.

Chemical structures of structurally related hydroxamate siderophore–antibiotic conjugates synthesized and investigated previously, as well as subject of studies in this work: D1, D2 and albomycin (Fe-D3), a “Trojan horse” conjugate produced by Streptomyces. Black: metal ion, red: siderophore, green: linker, blue: antibiotic.
Hexadentate siderophores exhibit exceptionally high affinity for both Fe3+ and the non-redox active, iron mimetic Ga3+ The log Ka of the gallium complexes of desferrioxamine (DFO)24 and desferrichrome25 have been reported as 28.65 and 27, respectively. Functionalized Ga3+ and Fe3+ siderophore complexes are both efficiently recognized by bacterial siderophore membrane transporters and provide ideal vehicles to deliver diagnostic and therapeutic payloads to the cytoplasm, evidenced by the crystal structure of FhuD co-crystallized with gallium-complexed desferrichrome.26,27 Considerable effort has been dedicated to demonstrate the proposed growth-inhibitory effect of Ga3+-salts as non-redox-active mimics of Fe3+. Promising in vitro and in vivo data of therapeutic activity of Ga–DFO in combination with gentamycin in an ocular infection model in rabbits have been studied.28 Currently, Ga(NO3)3 is under investigation in a phase 2 multicenter, randomized, placebo-controlled trial as a treatment of Pseudomonas aeruginosa lung infections in adults with cystic fibrosis (CF).29 The phase 1 trial concluded that Ga(NO3)3 imparts significant therapeutic effects and indicated synergistic activity when co-administered with certain polymyxins and β-lactams as indicated by measurably improved lung function. Other work has explored the gallium complexes of DFO-conjugates for their antibiotic potency, producing complexes with highly strain-specific activity.30 Efforts to elucidate the exact mechanism of action of Ga(III) in P. aeruginosa have recently been reported using pull-down methods for protein-bound Ga(III) with open sites of coordination.31
In addition to the Ga(III) complexes providing an alternative, medicinal inorganic chemistry approach to the management of bacterial infections, the nuclear complexes utilizing radioactive isotopes 67Ga3+ (t1/2 = 3.3 d, single photon emission computed tomography, SPECT) and 68Ga3+ (t1/2 = 1.1 h, positron emission tomography, PET) also offer opportunities to explore nuclear imaging as a diagnostic tool. Nuclear imaging tracers are ideally suited to detect small quantities of colony forming units (CFU) in vivo. Imaging with SPECT or PET isotopes provides a sensitivity of 10−12 M and a high spatial resolution of ~3 mm with unlimited depth penetration. The SPECT tracers 111In(oxime), 99mTc-MDP, and 67Ga-citrate have been used to detect infection; however, they lack specificity or sensitivity in a clinical setting as they target host response to infection, rendering distinction of inflamed and infected tissue difficult.32 New, alternative probes composed of 11C-, 18F-, and 124I-labeled PET tracers have been developed for bacterial infections, but also exhibit drawbacks, as they cannot predict treatment efficacy.33–36 Recently, Decristoforo and co-workers have successfully explored 68Ga-labeled siderophores fusarinine and pyoverdine as tracers for localized fungal infections in preclinical models; these results indicate that functionalized, radiolabeled siderophores could provide a viable option for the noninvasive nuclear imaging of bacterial infections. Based on these concepts explored in literature, we hypothesized that potent gallium–sideromycins can provide opportunities to explore both diagnostic and therapeutic applications: the direct radio-metallation with 67Ga3+ or 68Ga3+ opens possibilities to study stability, in vitro uptake and pharmacokinetics.37–41 Complexation with gallium in lieu of iron may further enhance antibiotic activity of already potent compounds. The concept of a theranostic siderophore-based approach to infection has thus far not been explored widely; a siderophore-mimetic molecular design was implemented by Brönstrup and co-workers recently where a tetraaza-macrocycle based chelator was employed as a carrier for antibiotic and imaging modality. However, the departure from biologically established siderophore structures can be associated with the risk of diminished affinity to siderophore-specific membrane transporters.42
To fulfill requirements as a theranostic Trojan horse conjugate that is likely to take advantage of specific siderophore-based transport mechanisms, we proposed the following criteria: (1) accessible and scalable via multistep organic synthesis, (2) formation of kinetically inert complexes with Fe and Ga under mild conditions, (3) a minimum inhibitory concentration (MIC) equivalent or lower than ciprofloxacin for both Ga complexes, (4) enhanced uptake by bacteria when compared with 67Ga-citrate, and (5) measurable stability of the corresponding 67Ga-labeled conjugate. Here, we introduce two Trojan horse compounds, Ga-D1 and Ga-D2, as ciprofloxacin-bearing mimics of the salmycin and albomycin natural products and explore their potential for a concerted diagnosis and therapy approach for bacterial infections.
RESULTS AND DISCUSSION
Synthesis of Siderophore Conjugates.
The structural components of sideromycins (siderophore, linker, and antibiotic moiety) lend themselves to a modular approach to design and chemically synthesize new, bioactive compounds that can be produced on a large scale. Albomycin and salmycin are natural products that exhibit exceptional activity in a range of Gram-positive and Gram-negative strains, by gaining access to the bacterial cytoplasm via xenosiderophore internalization pathways.43,44 Albomycin is particularly active in Gram-negatives, while salmycin effectively inhibits growth in Gram-positives and K. pneumoniae.45 This represents a contrast to the strain selectivity of enterobactin conjugates which are almost exclusively active in Gram-negative Enterobacteriaceae. The total syntheses of both natural product sideromycins,11,46 as well as their isolation from their native bacterial strains, have been reported.9,47 Albomycin has found limited, yet very successful clinical application in animals and humans in the 1950s.8
As others before us, we drew direct structural inspiration from the natural products salmycin and albomycin (Figure 1) by implementing a conjugation strategy mirroring the connectivity of the natural products but incorporating an antibiotic that provides synthetic accessibility and flexibility for modification with a cytoplasmic inhibitory target such as ciprofloxacin, a DNA gyrase inhibitor. We hypothesized that the delivery of the construct to the bacterial cytoplasm would also increase the concentration of cytoplasmic, non-redox active Ga(III) and inhibit biosynthesis of Fe(III)-dependent, cytoplasmic proteins. This strategy aims to maximize gallium’s potential to exert bacteriotoxicity, as opposed to delivery of the construct exclusively to the periplasm in the case of β-lactam-containing, cell-wall targeting antibiotic conjugates. For this first series of compounds, we elected to forgo the previously established and effective cleavable linker strategies48 for ciprofloxacin–DFO conjugates in order to minimize variability and instability of synthesized apo and complexed forms of compounds, but note that once activity of a first-generation compound is successfully established, modifications to the linker can be made to further increase efficacy. To synthesize D1, we employed DFO as a commercially attainable surrogate of danoxamine and linked ciprofloxacin with a succinate linker in 2 steps by modification of previously published protocols (Scheme 1).48,49
Scheme 1.

The functionalization of desferrichrome presented a greater synthetic challenge, as the pendant hydroxamates are prone to hydrolysis especially under reaction conditions employed for peptide coupling reactions. To this end, we synthesized the acetylated, amino-protected desferrichrome precursor P5 as previously employed in the synthesis of other desferrichrome-based conjugates and pioneered by Miller and co-workers (Scheme 3);50–53 the synthesis, characterization, and efficacy of a desferrichrome-functionalized β-lactam has already been reported.53 A short, noncleavable linker was introduced to attach P5 to the antibiotic moiety in attempts to further increase compound integrity toward peptidases prior to delivery to the cytoplasm: ciprofloxacin was functionalized with β-alanine using in situ protection of the less reactive aromatic carboxylate with trimethyl silane for each conjugation step (Scheme 2).22,54 Amine-functionalized precursor B2 was directly linked to intermediate P5 via amidation. The final product D2 was attained in a <2% overall yield after 17 steps following final purification using reverse phase chromatography. The chemical synthesis of D2 relies on the assembly of building blocks in analogy to the total synthesis of albomycin,11 indicating that assembly remains modular and can enable introduction of modified linkers at a later stage. D3 (desferri-albomycin) was attained from albomycin by adapting a previously reported transchelation procedure for ferrichrome.55 In brief, transchelation of iron was achieved by a 50-fold excess ethylenediaminetetraacetic acid (EDTA), followed by reverse phase chromatography purification to yield sufficient D3 to carry out all prospective radiochemical studies.
Scheme 3.

Synthesis Scheme for LDFC adapted from ref 51
Scheme 2.

Functionalization of Ciprofloxacin, Subsequent Conjugation to Protected Hydroxamate Fragment P5 Followed by Sequential Deprotection Steps Yields the Conjugate D2
Fe/Ga Complex Formation and Characterization.
Complexation with Fe(III) and Ga(III) salts formed Fe-D1/D2 and Ga-D1/D2 in aqueous, buffered media. Ferric complexes exhibit broad absorption at 420 nm, characteristic of the d–d transition, in addition to the ciprofloxacin-based peak absorptions at 280 and 322 nm (Figure S16). Molar absorptivity and inductively coupled plasma-optical emission spectroscopy (ICP-OES) measurements were employed to produce consistent complex and ligand concentrations for in vitro and in vivo studies. The corresponding complexes were purified using reverse phase chromatography and isolated as orange-red or colorless powders. Conveniently, solubility of the Fe- and Ga-D2 complex remains high in water, with no DMSO required to formulate complexes for bioassays. High-performance liquid chromatography (HPLC) was employed in addition to nuclear magnetic resonance (NMR) and mass spectrometry (MS) methods to characterize conjugates and ascertain appropriate compound purity (Supporting Information Figures S13–S15).
Growth Inhibition in E. coli.
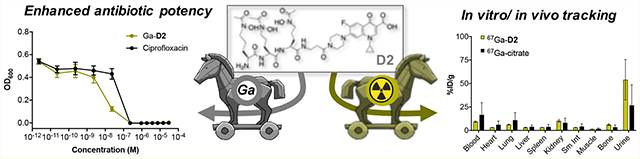
With apo-, Ga, and Fe complexes in hand, we next probed the ability of various constructs to inhibit bacterial growth in a conventional MIC assay. We selected Escherichia coli K12 as a Gram-negative model organism, as it is known to express membrane transporters for both ferrioxamine (FoxA) and ferrichrome (FhuA) in addition to its native enterobactin (FepA).56 Fe-deficient media was prepared by addition of dipyridine (DP).15 Unmodified ciprofloxacin was employed as a control with a MIC of 0.2 μM in our hands, which is consistent with literature values.20 MIC assays showed that the apo and Ga-complexes of DFO and linear desferrichrome (LDFC) show no significant growth inhibition or growth promotion at concentrations tested; the Fe-complexes promote growth at concentrations of 10−5 M and above, indicating that both complexes are efficiently utilized by E. coli as an Fe-source (Figures 2A,B, S24). Next, we probed the ability of the siderophore–antibiotic conjugates D1 and D2 to inhibit growth as their respective apo, Fe and Ga complexes. We found that apo D1 and the corresponding Fe and Ga complexes attenuate the antibiotic activity of the appended ciprofloxacin by 4- to 16-fold (Table 1, Figure 2C). Notably, the apo D1 and the Ga-D1 complex are 4- and 2-fold more active than the Fe-D1 complex, respectively, albeit we note that the 2-fold increased activity observed lies within the error of the MIC assay measurement. Overall, the comparatively high MIC value indicates that the appended DFO–chelator may impact efficient binding of ciprofloxacin to gyrase. This extent of attenuation of target binding was not observed with the LDFC-based siderophore D2. Apo D2 and Ga-D2 show a MIC of 0.23 μM, retaining the growth inhibitory action of ciprofloxacin (Figure 2D). Ga-D2 is one of the most active synthetic ciprofloxacin-conjugated Trojan horse compounds measured to date (Table S7). This is especially notable considering no cleavable linker was employed.
Figure 2.

MIC assays of non-functionalized siderophores (A,B), D1 (C) and D2 (D) in E. coli K12 show that the Ga-D2 complex exhibits greatest potency when compared to ciprofloxacin, while non-functionalized Fe–siderophores act as growth promoters at concentrations above 10−5 M (n = 3) and Fe–sideromycin complexes exhibit attenuated growth inhibition.
Table 1.
Summary of MIC90 Determined in E. coli and in Presence of 100× Fe–Siderophorea
| compound | MIC90 (+DP) (μM) | MIC90 (+DP, +100× Fe siderophore) |
|---|---|---|
| ciprofloxacin | 0.23 | 0.23 μM |
| apo D1 | 0.97 | 1.9 μM |
| Fe-D1 | 3.8 | 7.5 μM |
| Ga-D1 | 1.9 | 30 μM |
| apo D2 | 0.23 | >100 μM |
| Fe-D2 | 0.94 | 15 μM |
| Ga-D2 | 0.23 | 15 μM |
| Fe-D3 (albomycin) | 0.00023 | n.d |
n.d. = not determined.
Furthermore, the Ga-complex exhibits most efficient growth inhibition in the tested strain, which motivates further investigations of the corresponding radiochemical Ga-complex as a true diagnostic equivalent to the most potent therapeutic. Albomycin (Fe-D3) retains exceptional potency as previously reported,11 with a MIC of 0.00023 μM.
Competitive Growth Inhibition.
Siderophore-specific, active transport to the cytoplasm was probed by challenge MIC assay experiments. Ga-D1 and Ga-D2 complexes were challenged with a 100-fold excess of the corresponding Fe and Ga siderophore complexes at each respective concentration. As expected, the presence of excess parent Fe- or Ga–siderophore efficiently attenuates the antibiotic activity of Ga-D1 and D2 (Figure 3, Table 1). Uptake behavior is consistent with active, transporter-mediated delivery. In contrast, Fe–DFO only inhibits the activity of D2 at high concentrations (Figure S27), indicating that Fe–DFO is not directly competing for uptake and is internalized using a different active transporter. This is further supported by lack of attenuation of the MIC of D2 by the corresponding Ga–DFO complex. The presence of 100× Fe–DFO or 100× Fe–LDFC has no impact on the potency of ciprofloxacin alone (Figure S26), further demonstrating that the intracellular transport and the growth-inhibitory activity of Ga-D2 is dependent on siderophore-mediated transport to the cytoplasm.
Figure 3.

(A) MIC challenge assay of Ga-D1 in the presence of 100× excess Fe–DFO and Ga–DFO and (B) Ga-D2 in the presence of 100× excess Fe–LDFC and Ga–LDFC in E. coli K12 (n = 3). Parent siderophore Ga- and Fe-complexes compete effectively for transporter-mediated internalization.
In order to further confirm the involvement of FhuA in the active transport and efficacy of the desferrichrome-derived conjugate complexes, we assessed MIC in E. coli AN193,53 which carries a mutation of the FhuA gene. Figures 4 and S28 summarize results obtained with the AN193 mutant: the previously observed growth inhibitory activity in E. coli K12 of apo D2, Ga-D2 and Fe-D2 as well as Fe-D3 is completely muted, while apo D1, Ga-D1 and Fe-D1 retain comparable MIC values as found with the wt E. coli strain. This further demonstrates that internalization and activity of Ga-D2 is FhuA-dependent.
Figure 4.

MIC assays of apo D1, Fe-D1, Ga-D1 complexes and apo D2, Fe-D2, Ga-D2 in E. coli AN193 (FhuA mutant). Growth inhibitory activity of the LDFC based conjugates was attenuated, whereas the activity of DFO-based conjugates was not significantly affected.
Activity of Ga-D2 against Other Strains.
To probe the antibacterial spectrum of D2 and corresponding complexes, we carried out growth inhibition assays in wild-type Staphylococcus aureus, wild-type P. aeruginosa, and carbapenem resistant K. pneumoniae under iron-limiting conditions. S. aureus and K. pneumoniae possess Fhu transporters for ferrichrome and structurally related sideromycins; they are sensitive to albomycin.45 P. aeruginosa is not susceptible to albomycin, but expresses FiuA and FiuB, a membrane transporter pair capable of binding and transporting ferrichrome to the cytoplasm.57 We found that the Ga-D2 complex exhibited best broad-spectrum activity in all strains, comparing well to the activity of ciprofloxacin (Table 2), followed by apo-D2 and finally the Fe-D2 complex. The growth inhibitory activity of Fe-D2 is muted in P. aeruginosa and Fe-D2 even promotes growth at concentrations >0.01 mM in S. aureus (Figures S29 and S31). Ga-D1 showed enhanced potency when compared with ciprofloxacin in S. aureus but overall remained less active in comparison with Ga-D2. Fe-D3 exhibits exceptional potency in K. pneumoniae, but is inactive in P. aeruginosa, in stark contrast to Ga-D2 (Figure S35). Nonconjugated apo-, Feor Ga-siderophores did not exhibit any growth inhibition, indicating that growth inhibitory activity does not arise from the supplementation of gallium or withholding of iron from the bacteria (Figures S30, S32, S34). The exception is Ga–LDFC, which is inactive in all tested strains except in P. aeruginosa (MIC = 30 μM, Figure S30). This indicates that the growth inhibitory activity of Ga-D2 in P. aeruginosa may also be imparted by the co-administration of the Ga(III)-ion. Further work is needed to determine the individual mechanism of growth inhibitory action of Ga-D2 in each bacterial strain.
Table 2.
Summary of MIC90 Values Determined in S. aureus, P. aeruginosa, and K. pneumoniae for apo D2, Fe-D2, Ga-D2, and Fe-D3
| compound | S. aureus RN4220 (μM) | P. aeruginosa PA01 (μM) | K. pneumoniae CRE-11 (μM) |
|---|---|---|---|
| ciprofloxacin | 1.9 | 0.94 | 3.12 |
| apo D2 | 15 | 7.5 | >100 |
| Fe-D2 | >30 | >30 | >30 |
| Ga-D2 | 0.94 | 3.8 | 12.5 |
| Fe-D3 (albomycin) | 0.94 | >30 | 0.00023 |
Radiolabeling with 67Ga.
Growth inhibition experiments confirmed that Ga-D2 is a potent siderophore–antibiotic conjugate. We next evaluated if a radiochemical tracer approach was feasible to further elucidate and quantitate in vitro and in vivo properties of the siderophores and Trojan horse conjugates synthesized.
We selected a radioisotope that has already shown potential for biomedical imaging and provides a sufficiently long half-life for in vitro uptake experiments: 67Ga-citrate (t1/2 = 79 h) has been extensively used to clinically diagnose inflammation and cancer via SPECT imaging and employed in conjunction with siderophores to image fungal infections.37 The multiday half-life of 67Ga also enables in vitro bacterial uptake studies and pharmacokinetic tracking prior to adapting successful leads for imaging with the short-lived, higher-resolution PET isotope 68Ga (t1/2 = 68 min). 67Ga-citrate is widely available commercially and can be converted to 67GaCl3 as a more convenient reactant for complexation studies. We formed 67Ga-complexes of siderophores, D1, D2, and D3, under mild radiolabeling conditions (pH 7, 10 mM HEPES, 5 min) quantitatively with ligand concentrations of 10−6 M (Figures 5, S17–S20).
Figure 5.

Representative radiolabeling HPLC-trace for the characterization of 67Ga-D2 (Rt = 10.56 min, left axis, 67Ga counts per second), in comparison with HPLC characterization of the apo D2 ligand (Rt = 10.30 min, right axis, absorbance at 280 nm) is shown. Free 67Ga elutes at 0.7 min.
Radiochemical Complex Inertness.
The ability to directly detect the radiochemical species opens possibilities to evaluate compound inertness in various media of interest. We carried out two distinct time-dependent inertness tests: A 10× EDTA transchelation challenge to rank the relative inertness of the corresponding coordination complexes and incubation of complexes in dipyridyl (DP)-treated, Fe-deficient bacterial growth media. The 10× EDTA challenge experiment indicates a relative stability order as follows: 67Ga-DFO > 67Ga-D1 > 67Ga-D3 > 67Ga-LDFC ≈ 67Ga-D2. Incubation of all 67Ga-complexes in growth media indicates that all complexes remain at least 70% intact for at least 2 h (Tables S2 and S3, Figures S22 and S23).
Radiochemical Uptake Experiments in E. coli.
The radiochemical complexes provide a convenient tool to probe time-dependent compound uptake in bacteria relative to weakly complexed 67Ga-citrate. The relative compound uptake provides complementary information to the MIC assay results. Radiochemical complexes were prepared as outlined previously, with an apparent molar activity of 370 MBq/μmol. E. coli (3.2 × 108 CFU) was incubated with 0.05 MBq of 67Ga-complex at 37 °C. Aliquots were removed at 0.16, 0.33, 0.5, 1, and 2 h following initial incubation. Aliquots were centrifuged to separate pellet from supernatant. Both fractions were isolated and counted for retained radioactivity. Residual activity in the pellet divided by total activity in pellet and supernatant quantified compound uptake. We tested uptake in iron-sufficient and DP-treated, iron-deficient media. Among all siderophores tested, 67Ga-LDFC is taken up most efficiently under iron-limiting conditions with a peak uptake of 41% after 2 h incubation (Tables S4 and S5). Among the sideromycins tested, both 67Ga-D1 and 67Ga-D2 exhibit statistically significant, enhanced compound uptake at the 30 and 60 min time points in comparison to 67Ga-citrate, with 67Ga-D1 displaying almost two-fold enhanced uptake (Figure 6A). These trends are in contrast to trend observed with respect to the growth inhibitory activity of Ga-D1 and Ga-D2, indicating that efficient uptake does not directly correlate to compound efficacy. We also challenged 67Ga-D2 with ferric siderophore in analogy with the MIC challenge experiment. Bacteria were incubated with a mixture of 67Ga-D2 and 120× excess Fe–LDFC. We observed statistically significant depression of 67Ga-D2 uptake within the bacterial pellet (Figure 6B, Table S6), specifically at the 1 h (P < 0.0001) and 2 h (P < 0.01) time point. This result is in agreement with the inhibition of antibiotic activity of Fe-D2 as established with the MIC experiment in presence of excess Fe- and Ga-LDFC.
Figure 6.

(A) Time-dependent, radiochemical bacterial uptake studies in E. coli K12 of 67Ga–siderophore–ciprofloxacin conjugates in iron sufficient (striped bars) and iron-depleted, DP-treated media pH = 7.4 (solid bars) show that uptake is enhanced in comparison with 67Ga-citrate. (B) In the presence of 120× excess Fe–LDFC, the uptake of 67Ga-D2 is attenuated significantly after 1 h (P < 0.0001), corroborating results obtained in challenge MIC experiments.
In Vivo Biodistribution and Pharmacokinetics.
With the 67Ga-compounds validated in vitro using radiochemical stability and bacterial uptake assays, we applied the 67Ga-complexes to the animal model to determine biodistribution and metabolic stability in vivo. Naive CBJ mice (female, 8–10 weeks) were injected with 67Ga-D1, 67Ga-D2, 67Ga-D3, and 67Ga-citrate intravenously, followed by a biodistribution study and analysis of metabolites in the urine using radio-HPLC at 1 h post injection. Biodistribution revealed predominantly renal clearance of all 67Ga-compounds (Figure 7A). No intact 67Ga-D1 or 67Ga-D3 was detected in the urine (Figures S36 and S37), while 67Ga-D2 was detected as 13% intact radiochemical complex in the urine (Figure 7B). A second 67Ga-species is detected, which could correspond to metabolized fragments of the complexed conjugate or alternatively, dechelated 67Ga. Overall, the extent of degradation of the original, radiolabeled 67Ga-D2 is comparable to the degradation of 18F-based infection imaging tracers, which are typically rapidly and fully metabolized in vivo.58
Figure 7.

(A) Comparative biodistribution of 67Ga-D1, 67Ga-D2, 67Ga-D3 and 67Ga-citrate in naive mice (n = 3) shows predominantly renal clearance of all compounds. (B) Metabolite analysis of 67Ga-D2 (open circles) shows detectable intact complex (13%) in the urine 1 h post injection. Radioanalytical HPLC trace of the 67Ga-D2 dose formulation prior to administration is shown as a reference (red line).
The 67Ga-D2 complex remains of interest for further future evaluation for diagnosis and therapy in an animal model of soft tissue infection; the biodistribution profile of 67Ga-D2 compares favorably to previously investigated, radiolabeled small-molecular siderophore complexes, which also generally exhibit rapid renal clearance.39 Furthermore, PET imaging 68Ga-D2 can enable high-quality nuclear imaging superior to SPECT with 67Ga. Strategies to further increase the metabolic stability of the radiochemical Ga-complexes and prevent the loss of any radioactive label in vivo must also be pursued.
CONCLUSIONS
A multitude of approaches is required to identify and eradicate bacteria with high efficiency and specificity. Targeting of the bacterial peri- or cytoplasm using active, siderophore-mediated transport has been established as an effective strategy to enhance the delivery of broad-spectrum antibiotics and increase antibacterial efficacy. Here, we show that the hexacoordinate siderophore desferrichrome can be conjugated to the FDA-approved antibiotic ciprofloxacin and form Fe(III) and Ga(III) complexes. We demonstrate that Ga(III) siderophores alone do not directly impart antibiotic potency; this represents a contrast to previous studies where efficacy was reported for some Ga- salts and Ga-siderophore complexes and hypothesized to provide a broad-spectrum antibacterial approach. Our results indicate that this is not the case. Further work is needed to elucidate the fate of the siderophore and the Ga(III) once the bacterial cytoplasm is reached, to determine the biological target of Ga(III) and if a release of the metal occurs in analogy with Fe(III) -siderophores in the absence of redox chemistry dependent processes accessible under physiological conditions.
In our study, a growth-attenuating effect can only be achieved by enhanced, siderophore-transporter-mediated delivery of the gallium-complexed antibiotic conjugates. The 0.23 μM MIC90 potency in E. coli qualifies Ga-D2 among the most potent, gallium-based antibiotic structures to date. A competitive MIC assay in the presence of excess Fe-bound siderophore attenuates the ability of Ga-D2 to inhibit bacterial growth, and defective FhuA results in no growth inhibition, demonstrating that the delivery of ciprofloxacin as part of D2 depends on active, siderophore-specific membrane transport. Ga-D2 also displays potent growth inhibitory activity in other bacterial strains, namely, S. aureus, P. aeruginosa, and K. pneumoniae with a MIC90 of 1.9, 3.8, and 12.5 μM, respectively, demonstrating broad-spectrum activity which supersedes the activity of the corresponding apo and Fe(III)-complex. At this juncture, we cannot conclude that Ga(III) provides a truly synergistic effect. It is possible that the absence of Fe(III) enhances protein expression of iron-transport mechanisms and thus helps to promote uptake of the Ga(III) -siderophore–antibiotic conjugates. Studies to further enhance antibiotic activity and elucidate the mechanism of action of these compounds are underway.
We also quantified bacterial uptake and measured in vivo pharmacokinetics with a gallium radioisotope. For this, we radiolabeled siderophores and sideromycins with 67Ga. We show that the siderophore–antibiotic conjugates 67Ga-D1 and 67Ga-D2 can be radiochemically synthesized under mild conditions and with high radiochemical yields. The complexes showed enhanced bacterial uptake when compared to 67Ga-citrate. Incubation of bacteria with excess Fe–LDFC efficiently blocks retention of 67Ga-D2, providing a complementary experiment to the attenuation of growth inhibitory activity of Ga-D2 in the presence of excess Fe- and Ga-LDFC. Finally, we show that 67Ga-D1, 67Ga-D2, and 67Ga-D3 complexes are suitable to track integrity and distribution of siderophore-complex conjugates in vivo and evaluate their respective pharmacokinetic stability. Stable 67Ga-D2 was successfully detected in the urine of mice 1 h post injection, motivating future studies to investigate uptake and a combined diagnostic and therapeutic use of the Ga-D2 complex in preclinical models of bacterial infection.
In summary, we have shown that (radio-)gallium complexation provides a promising approach to potentiate the activity of siderophore–antibiotic conjugates and quantify time-dependent in vitro uptake in bacteria and in vivo pharmacokinetics in mice. We note that this dual, medicinal, and radiochemistry approach is broadly applicable to the study of other promising hexacoordinate siderophore-conjugates. A better understanding of in vitro uptake kinetics, metal-dependent metabolism, and in vivo stability can significantly accelerate the clinical translation of synthetic Trojan horse conjugates for the management of bacterial infections.
EXPERIMENTAL SECTION
Materials and Methods.
General Methods.
All starting materials were purchased from Acros Organics, Alfa Aesar, Sigma-Aldrich, or TCI America and used without further purification. Purity analysis of all compounds employed in biological experiments was determined by HPLC analysis of the nonradioactive and/or the radioactive complex species. Percent purity as determined by HPLC is reported individually for each compound, and representative HPLC traces employed for purity analysis are provided within the Supporting Information. Note that the radiochemical 67Ga complex peak retention times exhibit a 0.1–0.15 min delay when compared to the corresponding natGa-complexes due to the sequential detector setup (UV detection prior to gamma detection) of the HPLC. Note that the radiochemical species is present at a 1:1000 complex to the ligand ratio. NMR spectra (1H, 13C) were collected on a 700 MHz AVANCE III Bruker instrument at 25 °C and processed using Topspin 3.5pl7. 19F NMR were collected on a 400 MHz Bruker instrument at 25 °C using TFA as an internal standard (δ: −76 ppm). Chemical shifts are reported as parts per million (ppm). MS: low resolution electrospray ionization (ESI) MS and high-resolution ESI MS were carried out at the Stony Brook University Institute for Chemical Biology and Drug Discovery (ICB&DD) MS Facility with an Agilent LC/MSD and Agilent LCUV-TOF spectrometers, respectively. UV–vis spectra were collected with the NanoDrop 1 C instrument (AZY1706045). Spectra were recorded from 200 to 900 nm in a quartz cuvette with 1 cm path length. HPLC: Preparative HPLC was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient pump, UV–vis detector, manual injector on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed). Method A (preparative purification method): A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B. Method B: A = 10 mM NaOAc (pH = 4.5), B = MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B. Radio HPLC analysis was carried out using a Shimadzu HPLC20AR equipped with a binary gradient pump, UV–vis detector, autoinjector and Laura radio detector on a Gemini-NX C18 column (100 mm × 3 mm, 110 Å, AXIA packed). Method C (analysis of 67Ga complexes): A = 10 mM NaOAc (pH = 4.5), B = MeCN with a flow rate of 0.8 mL/min, UV detection at 254 and 280 nm. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. ICP-OES was carried out using an Agilent 5110 ICP-OES. A 10-point standard with respect to gallium and iron was used and lines of best fit were found with R2 of 0.999. Sample concentration was determined based on this calibration curve. Radiolabeling was carried out using a general protocol for all compounds reported. Preparation of 67Ga-chloride from 67Ga-citrate was carried out as follows: 67Ga-citrate was received from Triad Isotopes, Inc. at an average specific activity of 76.2 MBq/mL. The 67Ga-citrate solution was mixed with two-thirds of its volume of chelex resin-treated water. The solution was then filtered manually over a 100 mg Sep-Pak Vac 1 cc (100 mg) silica cartridge (Waters) with a plastic syringe. The column was washed thrice with 5 mL chelex resin-treated water to remove citrate ions. The retained radioactivity was eluted from the cartridge with 1 mL of 0.1 M HCl to yield 67Ga-chloride. The eluate was collected in 100 μL fractions in Eppendorf tubes, and the fractions with maximum activity were used for radiolabeling. The average specific activity of the resultant 67Ga-chloride solution used for radiolabeling was 27.01 MBq/mL. An aliquot of 67GaCl3 (1.36 MBq, 50 μL) was mixed with a solution of the desired ligand (100 μL, 1 mM) in chelex resin-treated water. The pH of the solution was adjusted with HEPES (50 mM, 350 μL) to 7.4. Complexation was monitored by radio-HPLC, method C. Radiolabeling was found to proceed after 10 min at room temperature. Representative HPLC traces are provided within the Supporting Information. Purity of all metal complexes including radiochemical species was determined using analytical HPLC. All complexes were ≥95% pure.
(S)-2-[(S)-2-(Benzyloxycarbonylamino)-5-(tert-butoxycarbonylamino)valerylamino]-5-(tert-butoxycarbonylamino)valeric Acid, P1.
N-tert-Butoxycarbonyl-N′-benzyloxycarbonyl-l-ornithine (1.0 g, 2.7 mmol) was dissolved in tetrahydrofuran (THF) (10 mL), followed by NHS (1.06 equiv, 0.329 g, 2.86 mmol). The solution was placed in an ice bath and cooled to 0 °C. To the solution cooled at 0 °C was added DCC (1.01 equiv, 0.553 g, 2.68 mmol) dissolved in THF (10 mL). The reaction mixture was removed from ice bath and stirred at room temperature for 24 h. After 24 h, the reaction mixture was filtered, and the precipitate was washed with THF, and the filtrate was collected and used without further purification. N-Delta-Boc-l-ornithine (1.2 equiv, 0.752 g, 3.23 mmol) and NaHCO3 (2.61 equiv, 0.591 g, 7.03 mmol) dissolved in H2O/THF (4:3, 35 mL) was mixed with the activated N-tert-butoxycarbonyl-N′-benzyloxycarbonyl-l-ornithine filtrate. The reaction mixture was stirred at room temperature for 48 h. After 48 h, THF was removed from the reaction mixture, and the aqueous solution was diluted with EtOAc (20 mL). The pH of the mixture was adjusted to 2 with 10% citric acid. The aqueous fraction was separated and extracted with EtOAc. The combined organic fractions were washed with brine and dried with NA2SO4. EtOAc was evaporated, and the product was lyophilized, affording P1 (1.252 g, 2.15 mmol, 79%) as a white solid. MS: calcd for C28H44 N4O9, 580.31; found, 581.3 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 12.51 (s, 1H, OH), 8.06−8.04 (d, 1H, NH), 7.32−7.29 (d, 1H, Cbz-NH), 7.38−7.33 (m, 5H, Ar–H), 6.74−6.80 (m, 2H, Boc-NH), 5.02−5.00 (s, 2H, Cbz-CH2), 4.14 (s, 1H, αH) 4.02 (s, 1H, αH), 2.94−2.82 (m, 4H, δH), 1.73−1.34 (m, 26H, βH/γH/tert-butyl-CH3).
13C NMR (DMSO-d6, 175 MHz, ppm): 173.6 (COOH), 171.9−171.3 (2 signals, amide-C=O), 155.7−155.6 (amide-C=O), 137.5 (Ar–C), 128.6−127.0 (3 signals, Ar–C), 77.4 (tert-butyl-C), 65.4 (Cbz-CH2), 54.74−51.76 (2 signals, αC), 33.3 (δC), 28.4 (CH3), 26.3−24.9 (2 signals, βC/γC).
(S)-2-{(S)-2-[(S)-2-(Benzyloxycarbonylamino)-5-(tert-butoxycarbonylamino)valerylamino]-5-(tert-butoxycarbonylamino)valerylamino}−5-(tert-butoxycarbonylamino)valeric Acid, P2.
The dipeptide P1 (1.2526 g, 2.1 mmol) was dissolved in THF (10 mL), followed by NHS (1.06 equiv, 0.263 g, 2.29 mmol). The solution was placed on an ice bath and cooled to 0 °C. Subsequently, DCC (1.01 equiv, 0.430 g, 2.09 mmol) was added dissolved in 10 mL THF. The reaction mixture was removed from ice bath and stirred at room temperature for 24 h. After 24 h, the reaction mixture was filtered, and the precipitate was washed with THF. The filtrate was collected and used without further purification. N-Delta-Boc-l-ornithine (1.2 equiv, 0.585 g, 2.52 mmol) and NaHCO3 (2.61 equiv, 0.4616 g, 5.49 mmol) dissolved in H2O/THF (4:3, 35 mL) were added to the THF solution above. The reaction mixture was stirred at room temperature for 48 h. After 48 h, THF was removed from the reaction mixture and the aqueous solution was diluted with EtOAc (15 mL). The pH of the mixture was adjusted to 2 with 10% citric acid. The aqueous fraction was separated and extracted with EtOAc. The combined organic fractions were washed with brine and dried with NA2SO4. The EtOAc was evaporated, and the product was lyophilized affording P2 (1.250 g, 1.57 mmol, 74%) as a white solid. MS: calcd for C38H62N6O12, 794.44; found, 795.5 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 12.23 (s, 1H, OH), 8.07−8.06 (d, 1H, NH), 7.87−7.86 (d, 1H, NH), 7.37−7.27 (m, 5H, Ar–H), 7.39−7.38 (d, 1H, Cbz-NH), 6.77−6.73 (m, 3H, Boc-NH), 5.02−4.95 (s, 2H, Cbz-CH2), 4.28−4.24 (m, 1H, αH), 4.14−4.10 (m, 1H, αH), 3.99−3.95 (m, 1H, αH), 2.88 (s, 6H, δH), 1.71−1.32 (m, 38H, βH/γH/tert-butyl-CH3). 13C NMR (DMSO-d6, 175 MHz, ppm): 173.8 (COOH), 172.2−171.7 (3 signals, amide-C=O), 159.1−158.4 (2 signals, amide-C=O), 156.4−156.1 (1 signal, amide-C=O), 137.4 (Ar–C), 128.8−127.4 (3 signals, Ar–C), 77.9 (tert-butyl-C), 65.9 (Cbz-CH2), 52.3−52.1 (2 signals, αC), 30.0−29.7 (δC), 28.7 (CH3), 27.3−23.8 (3 signals, βC/γC).
5-Amino-2-{5-amino-2-[5-amino-2(benzyloxycarbonylamino)-valerylamino]valerylamino}valeric Acid, P3.
P2 (1.250 g, 1.57 mmol) was dissolved in TFA/HOAc (7:3, 5 mL). The reaction mixture was stirred at room temperature for 2 h. The volatiles were removed, and the reaction mixture was partitioned between EtOAc (20 mL) and H2O (20 mL). The aqueous fraction was collected, solvent was removed in vacuo, and the product was lyophilized to afford P3 (0.994 g, 1.198 mmol, 75%). MS: calcd for C23H38 N6O6, 494.29; found, 495.2 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 12.72 (s, 1H, OH), 8.31−8.22 (d, 1H, NH), 8.12−8.02 (d, 1H, NH), 7.51−7.48 (d, 1H, Cbz-NH), 7.92−7.81 (s, 6H, NH2), 7.41−7.26 (m, 5H, Ar–H), 5.04−4.99 (m, 2H, Cbz-CH2), 4.33−4.29 (m, 1H, αH), 4.21−4.16 (m, 1H, αH), 4.08−4.02 (m, 1H, αH), 2.78 (s, 6H, δH), 1.83−1.51 (m, 12H, βH/γH). 13C NMR (DMSO-d6, 175 MHz, ppm): 173.5 (COOH), 172.5−171.8, 159.6−158.7, 156.5 (3 signals, amide-C=O), 137.4 (Ar–C), 128.9−128.2 (3 signals, Ar–C), 66.0 (Cbz-CH2), 54.3, 52.0, 51.8 (3 signals, αC), 38.9 (δC), 29.5, 29.3, 28.3 (βC), 24.2, 23.9, 23.8 (γC).
2-{2-[2-(Benzyloxycarbonylamino)-5-(hydroxyamino)valerylamino]-5-(hydroxyamino)valerylamino}−5-(hydroxyamino)-valeric Acid, P4.
P3 (0.992 g, 2.01 mmol) was dissolved in MeOH (10 mL). KOH (0.483 g, 8.63 mmol), benzaldehyde (0.703 g, 0.677 mL, 6.636 mmol), and dry 5 Å molecular sieves were added to the reaction mixture. The reaction mixture was stirred overnight at room temperature. Subsequently, the reaction mixture was filtered, and the precipitate and molecular sieves were washed with MeOH (10 mL). MeOH solution was placed on an ice bath and cooled to 0 mC. mCPBA (1.143 g, 6.626 mmol) dissolved in MeOH (2 mL) was added dropwise to the reaction mixture. The reaction mixture was stirred for an hour at 0 PC. After 1 h, volatiles were removed affording a white solid. To this solid, EtOAc (15 mL) and H2O (15 mL) were added; pH of the solution was lowered to 2 by dropwise addition of HCl (1 M). The layers were separated, and the aqueous layer was washed with EtOAc. Combined organic fractions were collected and washed with brine and dried with NA2SO4. EtOAc was removed to afford a white solid. To this white solid were added TFA (4 mL) and DCM (6 mL). The reaction mixture was stirred at room temperature for 1 h. After an hour, volatiles were removed to yield a white solid. To this white solid were added HCl (1 M, 6 mL) and DCM (6 mL), and the reaction mixture was stirred for additional 1 h. After 1 h, the aqueous fraction was extracted and washed with DCM (10 mL) and hexane (10 mL). Aqueous fraction was isolated, and the solvent was removed in vacuo to afford a glassy white solid, P4 (469.7 g, 0.867 mmol, 43%). MS: calcd for C23H38N6O9, 542.27; found, 543.3 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 11.32 (s, 4H, OH, N–OH), 8.36−8.35 (d, 1H, NH), 8.13−8.12 (d, 1H, NH), 7.49−7.47 (d, 1H, Cbz-NH), 7.40−7.31 (m, 5H, Ar–H), 5.04−5.00 (m, 2H, Cbz-CH2), 4.36−4.30 (m, 1H, αH), 4.18−4.14 (m, 1H, αH), 4.07−4.03 (m, 1H, αH), 3.16−3.01 (s, 9H, δH, δ-NH), 1.76−1.55 (m, 12H, βH/γH). 13C NMR (DMSO-d6, 175 MHz, ppm): 173.6 (COOH), 172.1, 171.8, 156.4 (3 signals, amide-C=O), 137.3 (Ar–C), 128.8, 128.3, 128.2 (3 signals, Ar–C), 66.0 (Cbz-CH2), 54.3, 51.9, 50.1 (3 signals, αC), 40.8 (δC), 29.6, 29.3, 28.3 (βC), 20.3, 20.3, 20.0 (γC).
5-[N-Acetyl(hydroxyamino)]-2-{5-[N-acetyl(hydroxyamino)]-2-{5-[N acetyl(hydroxyamino)]-2-(benzyloxycarbonylamino)valerylamino}valerylamino}valeric Acid, A1.
P5 (0.460 g, 0.85 mmol) was treated with 6% DIPEA in MeOH (6 mL) overnight at room temperature. Subsequently, the volatiles were removed, and the product was lyophilized to afford the product as a yellow oil, A1 (0.287 g, 0.429 mmol, 58%). MS: calcd for C29H44N6O12, 668.30; found, 669.3 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 11.50 (s, 1H, OH), 8.27 (s, 1H, NH), 7.94−7.92 (d, 1H, NH), 7.32−7.28 (d, 1H, Cbz-NH), 7.47−7.32 (m, 5H, Ar–H), 5.06−4.97 (m, 2H, Cbz-CH2), 4.13 (m, 1H, αH), 4.0−3.99 (m, 1H, αH), 3.83 (m, 1H, αH), 3.37 (s, 6H, δH), 1.99−1.89 (m, 9H, CH3), 1.80−1.40 (m, 12H, βH/γH). 13C NMR (DMSO-d6, 175 MHz, ppm): 174.1 (COOH), 172.9, 170.9, 156.6 (3 signals, amide-C=O), 170.7 (N–C=O), 169.6 (O–C=O), 137.5 (Ar–C), 130.3, 129.7, 128.8 (3 signals, Ar–C), 65.8 (Cbz-CH2), 55.0, 53.8, 53.3 (N–CO), 47.7, 47.5, 47.0 (3 signals, αC), 40.8 (δC), 30.1, 29.9, 29.7 (βC), 24.0, 23.4, 23.1 (γC), 21.6, 21.0, 20.8 (CO–CH3).
5-[N-Acetyl(hydroxyamino)]-2-{5-[N-acetyl(hydroxyamino)]-2-{5-[N acetyl(hydroxyamino)]-2-aminovalerylamino}valerylamino}-valeric Acid, A2.
A1 (0.097 g, 0.15 mmol) was dissolved in MeOH (3 mL) with Pd/C (0.009 g, 10% w/w). The reaction mixture was stirred for 2 h at room temperature under H2 (1 atm). After 2 h, Pd/C was removed by filtration and washed with MeOH. The filtrate was concentrated and lyophilized to afford a colorless oil, A2 (0.077 g, 0.14 mmol, 99%). MS: calcd for C21H38N6O10, 534.26; found, 535.3 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 9.83 (s, 1H, OH), 8.62 (s, 1H, NH), 7.48−7.41 (m, 1H, NH2), 4.19 (s, 1H, αH), 3.83 (s, 1H, αH), 3.61 (s, 1H, αH), 2.85 (s, 6H, δH), 1.97−1.95 (m, 11H, CH3, NH2), 1.66−1.48 (m, 12H, βH/γH). 13C NMR (DMSO-d6, 175 MHz, ppm): 174.1 (COOH), 172.7, 172.5 (2 signals, amide-C=O), 170.4 (N–C=O), 158.5 (NH2–C), 54.0, 53.4, 51.7 (δN-CO), 47.7, 47.5, 46.9 (3 signals, αC), 40.8 (δC), 30.4, 30.4, 29.9 (βC), 23.3−23.2 (γC), 20.9−20.8 (CO–CH3).
5-(N-Acetyl-N-acetoxyamino)-2-{5-(N-acetyl-N-acetoxy-amino)-2-[5-(N-acetyl-N-acetoxyamino)-2-(benzyloxy-carbonylamino)-valerylamino]valerylamino}valeric Acid, P5.
P4 (0.46 g, 0.85 mmol) was dissolved in NaOAc (10 mM, pH = 4, 6 mL), and the pH was adjusted to 4 by addition of KHCO3 (0.250 g, 2.54 mmol). Acetic anhydride (4.33 g, 4.10 mL, 42.4 mmol) was then added to the reaction mixture dropwise, while maintaining the pH of the solution at 4 by addition of KHCO3. The reaction mixture was stirred for 5 h at room temperature. After 5 h, the volatiles were removed, affording a yellow oil. To this oil, HCl (0.1 M, 6 mL) and EtOAc (6 mL) were added; the layers were separated, and the aqueous layer was washed with EtOAc (3 × 5 mL). The combined organic fractions were collected and washed with brine and dried with NA2SO4. EtOAc was removed, and the product was lyophilized to afford a glassy white solid, P5 (0.31 g, 0.39 mmol, 46%). MS: calcd for C35H50N6O15, 794.33; found, 795.4 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 12.65 (s, 1H, OH), 8.16 (s, 1H, NH), 7.95−7.89 (d, 1H, NH), 7.31−7.30 (d, 1H, Cbz-NH), 7.47−7.33 (m, 5H, Ar–H), 5.02−5.00 (m, 2H, Cbz-CH2), 4.29 (m, 1H, αH), 4.14 (m, 1H, αH), 4.02−4.01 (m, 1H, αH), 3.8 (s, 6H, δH), 2.20 (s, 9H, CH3), 2.01−1.83 (s, 9H, CH3), 1.75−1.46 (m, 12H, βH/γH). 13C NMR (DMSO-d6, 175 MHz, ppm): 173.7 (COOH), 172.5, 171.9, 156.3 (3 signals, amide C=O), 170.7 (N–C=O), 169.6 (O–C=O), 137.47 (Ar–C), 128.8, 128.2, 128.1 (3 signals, Ar–C), 65.8 (Cbz-CH2), 60.31 (NH–CO), 54.5, 52.0, 47.0 (3 signals, αC), 40.4 (δC), 29.8, 29.5, 28.5 (βC), 23.8–22.9 (γC), 20.4 (CO–CH3), 18.6 (Ac-CH3 O).
1-Cyclopropyl-7-(4-{3-[(9H-fluoren-9yl)methoxycarbonylamino]propionyl}−1-piperazinyl)-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, B1.
Ciprofloxacin (0.165 g, 0.499 mmol) and DIPEA (0.368 g, 500 μL, 2.85 mmol) were dissolved in anhydrous DCM (5 mL). To this solution, TMSCl (0.158 g, 185 μL, 1.45 mmol) was added to give a clear yellow solution. Fmoc-β-alanine (0.233 g, 0.748 mmol), PyAOP (0.417 g, 0.8 mmol), and DIPEA (0.260 g, 350 μL, 2.01 mmol) were dissolved in anhydrous DCM (5 mL). The two solutions were combined and stirred overnight at room temperature. Subsequently, the reaction was quenched by addition of MeOH (5 mL). Volatiles were removed, and the crude product was re-dissolved in EtOAc (20 mL). The organic layer was washed with HCl (10 mM, 2 × 10 mL) and saturated NaHCO3 (2 × 10 mL), dried with NA2SO4, and purified using method A to afford a yellow solid, B1 (0.0910 g, 0.145 mmol, 29%). MS: calcd for C35H33FN4O6, 624.24; found, 625.2 [M + H]+.1H NMR (DMSO-d6, 700 MHz, ppm): 8.68 (s, 1H, quinoline-H), 7.97−7.92 (m, 1H, cipro Bn-H), 7.78−7.21 (m, 8H, Fmoc Bn-H), 5.65 (s, 1H, cipro Bn-H), 4.35−4.33 (m, 2H, Cbz-CH2), 4.19−4.16 (m, 1H, Fmoc-H), 3.87 (s, 1H), 3.71 (s, 2H, cipro-H), 3.56−3.54 (m, 2H, NH–CH2), 3.34−3.30 (m, 2H, Cipro-H), 2.65 (s, 2H, CO–CH2), 1.36–1.16 (m, 4H, cyclopropyl-H). 19F (DMSO-d6, 376 MHz, ppm): −121.02.
7-(4-β-Alanyl-1-piperazinyl)-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, B2.
B1 (0.080 g, 0.13 mmol) was dissolved in DMF (6 mL). DEA (0.140 g, 198 μL, 1.92 mmol) was added to the reaction mixture. The reaction was stirred at room temperature for an hour. After 1 h, the volatiles were removed, and the product was lyophilized to afford a yellow solid, B2 (0.051 g, 0.13 mmol, 98 %). MS: calcd for C20H23FN4O4, 402.17; found, 403.1 [M + H]+.
7-(4-{3-[5-(N-Acetyl-N-acetoxyamino)-2-{5-(N-acetyl-N-acetoxyamino)-2-[5-(N-acetyl-N-acetoxyamino)-2-(benzyloxycarbonylamino)valerylamino]valerylamino}valeryl Amino]propionyl}−1-piperazinyl)-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, B3.
B2 (0.010 g, 0.020 mmol) and DIPEA (0.018 g, 0.014 mmol, 25 μL) were dissolved in anhydrous DCM (1 mL). To this solution, TMSCl (0.0086 g, 10 μL, 0.079 mmol) was added to give a clear yellow solution. P5 (0.029 g, 0.037 mmol), PyAOP (0.020 g, 0.039 mmol), and DIPEA (0.013 g, 0.10 mmol, 16 μL) were dissolved in anhydrous DCM (1 mL). The two solutions were combined and stirred overnight at room temperature. Subsequently, the reaction was quenched by addition of MeOH (1 mL). Volatiles were removed, and the crude product was purified using method A on semi-preparative HPLC to afford a yellow solid, B3 (0.010 g, 0.008 mmol, 57%). MS data: calcd for C55H71FN10O18, 1178.49; found, 1179.6 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 8.67 (s, 1H, NH), 8.05−7.80 (d, 1H, cipro Bn-H), 7.59−7.40 (m, 2H, NH), 7.40−7.32 (m, 5H, Ar–H), 7.32−7.27 (s, 1H, NH), 5.02−5.00 (m, 2H, Cbz-CH2), 4.01 (m, 1H, αH), 4.18 (m, 1H, αH), 4.13 (m, 1H, αH), 3.52 (s, 6H, δH), 3.38−3.26 (m, 6H, cipro-αH, CH2), 3.01 (m, 6H, cipro-αH, CH2), 2.19 (s, 9H, Ac-CH3), 1.88 (s, 9H, CO–CH3), 1.41−1.68 (m, 12H, βH/γH), 1.37−1.13 (m, CH2). 19F NMR (DMSO-d6, 376 MHz, ppm): −121.03.
7-(4-{3-[5-(N-Acetyl-N-acetoxyamino)-2-{5-(N-acetyl-N-acetoxyamino)-2-[5-(N-acetyl-N-acetoxya mino)-2-(benzyloxycarbonylamino)valerylamino]valerylamino}-valerylamino]propionyl}−1-piperazinyl)-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, B3.
B2 (0.010 g, 0.024 mmol) and DIPEA (0.001 g, 25 μL, 0.014 mmol) were dissolved in anhydrous DCM (1 mL). To this solution, TMSCl (0.007 g, 10 μL, 0.072 mmol) was added to give a clear yellow solution. S5 (0.029 g, 0.037 mmol), PyAOP (0.020 g, 0.039 mmol), and DIPEA (0.013 g, 16 μL, 0.10 mmol) were dissolved in anhydrous DCM (1 mL). The two solutions were combined and stirred overnight at room temperature. Subsequently, the reaction was quenched by addition of MeOH (1 mL). Volatiles were removed, and the crude product was purified using method A on semi-preparative HPLC to afford a yellow solid, B3 (0.010 g, 0.008 mmol, 57%). MS data: calcd for C55H71FN10O18, 1178.49; found, 1179.6 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 8.67 (s, 1H, NH), 8.05−7.80 (d, 2H, cipro Bn-H), 7.59−7.40 (m, 2H, NH), 7.40−7.32 (m, 5H, Ar–H), 7.32−7.27 (s, 1H, NH), 5.02−5.00 (m, 2H, Cbz-CH2), 4.01 (m, 1H, αH), 4.18 (m, 1H, αH), 4.13 (m, 1H, αH), 3.52 (s, 6H, δH), 3.38−3.26 (m, 6H, cipro-αH, CH2), 3.01 (m, 6H, cipro-αH, CH2), 2.19 (s, 9H, Ac-CH3), 1.88 (s, 6H, CO–CH3), 1.41−1.68 (m, 12H, βH/γH), 1.37−1.13 (m, cyclopropane-CH2). 19F (DMSO-d6, 376 MHz, ppm): −121.03.
7-[4-(3-{5-[N-Acetyl(hydroxyamino)]-2-{5-[N-acetyl(hydroxyamino)]-2-{5-[N-acetyl(hydroxyamino)]-2-aminovalerylamino}-valerylamino}valerylamino}propionyl)-1-piperazinyl]-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, apo-D2.
B3 (0.005 g, 0.004 mmol) was dissolved in MeOH (2 mL) with Pd/C (0.0005 g, 10% w/w). The reaction mixture was stirred for 2 h at room temperature under H2 (1 atm). After 2 h, Pd/C was removed by filtration. The filtrate was concentrated and lyophilized to afford a yellow oil, B4 (0.004 g, 0.003 mmol, 95%). The Cbz-deprotected intermediate was used for the subsequent deacetylation without further purification. B4 was treated with 6% DIPEA in MeOH (2 mL) overnight at room temperature. Subsequently, the volatiles were removed to yield D2 as a DIPEA amine salt, the product was further purified using method A to afford a yellow solid, D2 (0.003 g, 0.004 mmol, 75%). MS: calcd for C41H59FN10O13, 918.42; found, 919.4 [M + H]+. 1H NMR (DMSO-d6, 700 MHz, ppm): 9.78−9.75 (d, 1H, NH), 9.73−9.72 (d, 1H, NH), 8.67 (s, 1H, quinoline-H), 8.09 (s, 1H, cipro Bn-H), 7.93 (d, 1H, NH), 7.59−7.56 (s, 1H, cipro Bn-H), 6.54 (s, 2H, CH2), 4.33 (s, 1H, αH), 4.38 (s, 1H, αH), 3.77−3.65 (m, 2H, αH, CH), 3.62 (s, 6H, δH), 2.59 (s, 2H, COCH2), 2.54−2.50 (m, 2H, cipro-H), 1.67−1.16 (m, 23H, CH3/NH2/βH/γH), 1.14−0.81 (m, 4H, cyclopropyl-H). 19F NMR (DMSO-d6, 376 MHz, ppm): −121.6. Retention time (method C): 10.30 min (purity/peak area: 95.6%).
7-[4-(3-Carboxypropionyl)-1-piperazinyl]-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, C1.
Ciprofloxacin (0.331 g, 1.00 mmol) was added to a solution of succinic anhydride (0.100 g, 1.00 mmol) in DMSO (6 mL). The mixture was stirred overnight under reflux. Upon consumption of starting materials as confirmed by MS, the reaction mixture was cooled, and the precipitate was filtered and washed successively with water and Et2O. The solvent was removed under reduced pressure to afford the product as a light-yellow powder (0.2524 g, 0.587 mmol, 59%). MS: calcd for C21H22FN3O6, 431.15; found, 432.1 [M + H]+. 1H NMR (DMSO-d6, 400 MHz, ppm): 8.66 (s, 1H, H1), 7.90 (d, 1H, H2), 7.57 (s, 1H, H3), 3.82 (m, 1H, H4), 3.74−3.64 (t, 4H, H5,6), 3.31−3.24 (t, 4H, H7,8), 2.61 (t, 2H, H9), 2.47 (t, 2H, H10), 1.33 (d, 2H, H11), 1.19 (d, 2H, H12). 13C NMR (DMSO-d6, 175 MHz, ppm): 176.8 (COOH), 174.4 (COOH), 170.2 (C=O), 166.4 (C=O), 152.4 (C=C–F), 148.5 (C=COOH), 145.4 (C=C–N), 139.6 (C=C–N), 119.2 (C=C), 111.6 (C=C), 107.2 (2 signals, C=C aromatic), 50.0 (C–C–N), 49.6 (C–C–N), 44.7 (C–C–N), 40.8 (C–C–N), 36.3 (cyclopropyl), 29.4 (CO–CH3), 27.8 (CO–CH2), 8.0 (2 signals, cyclopropyl).
7-{4-[4-(5-{N-4-[5-(N-4-{5-[N-Acetyl-(hydroxyamino)]pentylamino}−4-oxobutyry(hydroxyamino))pentyl-amino]-4-oxobutyryl(hydroxyamino)}pentylamino)-4-oxobutyryl]-1-piperazinyl}−1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic Acid, apo-D1.
Succinyl β-ciprofloxacin (0.050 g, 0.12 mmol) was synthesized according to literature and added to a solution of N,N-diisopropylethylamine (40.4 μL, 0.232 mmol) in anhydrous DMF (6 mL). The mixture was stirred at room temperature for 10 min. PyAOP (0.73 g, 0.14 mmol) was added to the stirring solution, and the mixture was continued to be stirred at room temperature for another 10 min. DFO mesylate (DFO, 0.0762 g, 0.116 mmol) was dissolved in anhydrous DMF (6 mL) and added to the stirring solution. The mixture was stirred at room temperature overnight. Upon consumption of starting materials as confirmed by MS, the solvent was removed from the mixture under reduced pressure. The yellow solid was dissolved in DMF (1 mL) and purified using Sep-Pak hypersep C18 column (1000 mg) with a linear H2O/MeCN gradient. The pure product was collected from the 50:50 (H2O/MeCN) fraction. The solvent was removed under reduced pressure to afford the product as a white powder (0.048 g, 0.049 mmol, 43%). MS: calcd for C46H68FN9O13, 973.50; found, 974.5 [M + H]+. 1H NMR (DMSO-d6, 400 MHz, ppm): 9.63 (s, 1H, H1), 9.59 (s, 2H, H2), 8.67 (s, 1H, H3), 7.96 (d, 1H, H4), 7.80−7.64 (m, 3H, H5), 7.59 (d, 1H, H6), 3.82 (m, 3H, H7), 3.69 (m, 4H, H8), 3.46−3.43 (m, 6H, H9,10), 3.26 (m, 2H, H11), 3.00−2.99 (m, 6H, H12), 2.67−2.57 (m, 6H, H13,14), 2.32 (t, 2H, H15), 2.26 (t, 4H, H16), 1.96 (s, 3H, H17), 1.49 (s, 6H, H18), 1.38−1.31 (m, 8H, H19,20), 1.23−1.19 (m, 8H, H21,22). 13C NMR (DMSO-d6, 175 MHz, ppm): 176.6, 174.7, 172.1, 171.8, 170.5, 166.5, 152.3, 148.5, 145.2, 139.5, 119.5, 111.4, 107.0, 106.8, 50.0, 49.7, 47.5, 44.9, 41.2, 38.8, 36.2, 30.8, 29.2, 28.0, 26.4, 23.9, 20.8, 8.0. 19F NMR (DMSO-d6, 376 MHz, ppm): −73.5. Retention time (method C): 11.5 min (purity/peak area: 99.1%).
Albomycin (Fe-D3).
The albomycin-producing strain Streptomyces sp. ATCC 700974 was cultivated in two 250 mL Erlenmeyer flasks, each filled with 50 mL of medium containing soluble starch (20 g/L), l-ornithine-HCl (5 g/L), KH2PO4 (1.8 g/L), NA2HPO4·2H2O (10.2 g/L), NaCl (2 g/L), (NH4)2SO4 (2 g/L), MgSO4·7H2O (2 g/L), CaCl2·2H2O (0.8 g/L), ZnSO4·7H2O (0.02 g/L), and FeSO4·7H2O (0.28 g/L), pH = 6.8. After 3 days of incubation at 28 °C with 250 rpm agitation, the cultures were used to inoculate multiple flasks (250 mL), each containing 100 mL of the same medium. The fermentation was continued for 4 days at 28 °C with 200 rpm agitation. All culture flasks were combined and mixed with Celite, followed by filtration under vacum to separate the mycelium and water phase. The filtrate was transferred to a column loaded with Amberlite XAD-16 beads with a surface area 800 m2/g, pore diameter ≈ 250 Å, bed volume 1.5 L, and column dimensions 8 cm × 30 cm. The flow rate was maintained at 7.5 L/h. The resin was washed with deionized water until a clear eluate was obtained. Bound fraction was eluted with 2.2 L of 40% acetone (v/v). The eluate was concentrated to dryness, resuspended in water, and loaded onto a column containing Bio-Gel P2 (400 mesh). Using water as the mobile phase, fractions were monitored by UV–vis at λ = 435 nm followed by bioactivity tests. Fractions of interest were pooled, evaporated, and lyophilized to dryness. Samples containing albomycin were confirmed by analytical HPLC (Shimadzu LC10 pumps) using a reversed-phase column (Nucleosil C18, 5 μM, 4 × 250 mm). The gradient elution consisted of 100% 2 mM ammonium acetate to 100% acetonitrile over 20 min with a flow rate of 2 mL/min. Albomycin was further purified using HPLC with a Phenomenex Kinetex XB-C-182.6 column (75 by 4.6 mm). The flow rate was maintained at 1 mL/min. The gradient program from solvent A (H2O in 0.01% formic acid) to solvent B (acetonitrile in 0.01% formic acid) was 0–40% solvent B in 15 min, 40–95% solvent B in 2 min, 95% solvent B for 3 min, 95–0% solvent B in 3 min, and 0% solvent B for 7 min. Albomycin was eluted at 6.5 min and verified by high-resolution MS, UV–vis absorption spectrum, and the corresponding bioactivity, and 10 mg pure δ2 albomycin was obtained per liter of fermentation culture. Sensitivity assays were performed with aqueous albomycin solutions. Separate TY agar plates were overlaid with 3 mL TY-soft agar thoroughly mixed with 20 μL of overnight grown cultures of test strains. TY plates overlaid with test organisms were directly spotted with 5 μL of the antibiotic solution and incubated at 37 °C for 16–18 h. Inhibition zones indicated susceptibility and lack of inhibition resistance. The laboratory strain E. coli SIP 401 was used as positive control for sensitivity to albomycin. For preparation of the apo-D3 compound, the removal of iron was achieved by incubation of Fe-D3 with a 50-fold excess EDTA at pH 5, followed by reverse-phase chromatography purification to yield sufficient D3 to carry out all prospective radiochemical studies. Fe-D3: 7.53 min (purity/peak area: 98.8%). apo-D3: 7.35 min (purity/peak area: 96.4%). 67Ga-D3: Rt = 7.27 min (purity/peak area: >99%).
Ga–DFO.
DFO (5 mg, 0.008 mmol) was dissolved in H2O (1 mL). To this solution was added Ga(NO3)3 (1.9 mg, 0.0076 mmol). The reaction mixture was stirred at room temperature for 15 min. The pH of the reaction mixture was adjusted to 7.4 by adding 0.1 M NaOH. After 15 min, solvent was removed in vacuo to afford the product as a white solid. The product was dissolved in H2O (1 mL) and purified using Sep-Pak hypersep C18 column (1000 mg) with a linear H2O/MeCN gradient. The pure product eluted in the 80:20 (H2O/MeCN) fraction. The solvent was removed under reduced pressure to afford a white product. MS: calcd for C25H45GaN6O8, 627.25; found, 628.2 [M + H]+. Ga–DFO: Rt = 3.72 min (purity/peak area: >99%).
Ga–LDFC.
A2 (2 mg, 0.004 mmol) was dissolved in H2O (1 mL). To this solution was added Ga(NO3)3 (~1 mg, 0.0047 mmol). The pH of the reaction mixture was adjusted to 7.4 by adding 0.1 M NaOH. The reaction mixture was stirred at room temperature for 15 min. After 15 min, solvent was removed in vacuo to afford the product as a white solid. The product was dissolved in H2O (1 mL) and purified using Sep-Pak hypersep C18 column (1000 mg) with a linear H2O/MeCN gradient. The pure product eluted in the 80:20 (H2O/MeCN) fraction. The solvent was removed under reduced pressure to afford a white product. MS: calcd for C21H35GaN6O10, 600.17; found, 601.1 [M + H]+. Ga–LDFC: Rt = 1.65 min (purity/peak area: >99%).
Fe-D1.
D1 (0.007 g, 0.007 mmol) was added to a solution containing FeCl3 (0.0013 g, 0.0079 mmol) in anhydrous DMF (2 mL). The mixture was stirred for 30 min at room temperature. Upon consumption of starting materials as confirmed by MS, the solvent was removed under reduced pressure. The red solid material was dissolved in water and purified using semi-preparative HPLC (method B), with product eluting at 13.4 min. The fractions containing product were pooled, and the solvent was removed under reduced pressure to afford the product as a red-orange solid (0.0062 g, 0.006 mmol, 84%). MS: calcd for C46H65FFeN9O13, 1026.40; found, 1027.2 [M + H]+. 19F NMR (DMSO, 376 MHz, ppm): −73.46. Retention time (method C): 13.28 min (purity/peak area: 95.6%).
Ga-D1.
D1 (0.008 g, 0.008 mmol) was added to a solution containing Ga(NO3)3 (0.0063 g, 0.025 mmol) in anhydrous DMF (2.5 mL). The pH was adjusted to 7 using 0.1 M NaOH. The mixture was stirred at 60 °C for 1 h. The mixture was then cooled to room temperature and stirred overnight. The solvent was removed from the mixture under reduced pressure. The white solid was dissolved in water and purified using semi-preparative HPLC (method B), with product eluting at 14.1 min. The fractions containing product were pooled and the solvent was removed under reduced pressure to afford the product as a white solid (0.0079 g, 0.0076 mmol, 93%). MS: calcd for C46H65FGaN9O13, 1039.3; found, 1040.2 [M + H]+. 1H NMR (DMSO-d6, 500 MHz, ppm): 8.58 (s, 1H), 8.06 (d, 1H), 7.87−7.81 (m, 3H), 7.45 (d, 1H), 3.67−3.62 (m, 7H), 3.25 (m, 6H), 3.18 (m, 2H), 3.03 (m, 6H), 2.60 (m, 6H), 2.36−2.34 (m, 6H), 2.03 (s, 3H), 1.57 (s, 6H), 1.36−1.28 (m, 8H), 1.27−1.23 (m, 8H). 19F NMR (DMSO-d6, 376 MHz, ppm): −73.59. Retention time (method C): 13.73 min (purity: 98.7%). 67Ga-D1: Rt = 13.85 min (purity/peak area: 99%).
Fe-D2.
D2 (0.002 g, 0.002 mmol) was dissolved in 200 μL of H2O. To this solution was added FeCl3 (1 mg, 0.002 mmol). The reaction mixture was stirred at room temperature for 15 min. After 15 min, solvent was removed in vacuo to afford the product as a red solid. The product was dissolved in H2O (1 mL) and purified using Sep-Pak hypersep C18 column (1000 mg) with a linear H2O/MeCN gradient. The pure product eluted in the 80:20 (H2O/MeCN) fraction. The solvent was removed under reduced pressure to afford an orange-red product. 19F NMR (DMSO-d6, 376 MHz, ppm): −73.59. Retention time (method C): 10.50 min (purity/peak area: 97.3%).
Ga-D2.
D2 (0.002 g, 0.002 mmol) was dissolved in 200 μL of H2O. To this solution was added Ga(NO3)3 (1 mg, 0.003 mmol). The pH of the reaction mixture was adjusted to 7.4 by adding 0.1 M NaOH. The reaction mixture was stirred at room temperature for 15 min. After 15 min, solvent was removed in vacuo to afford the product as a light yellow solid. The product was dissolved in H2O (1 mL) and purified using Sep-Pak hypersep C18 column (1000 mg) with a linear H2O/MeCN gradient. The pure product eluted in the 80:20 (H2O/MeCN) fraction. The solvent was removed under reduced pressure to afford the product. 1H NMR (DMSO-d6, 700 MHz): 12.05 (s, 1H, OH), 7.8−7.93 (d, 1H, cipro Bn-H), 7.38 (s, 1H, quinoline-H), 7.31 (s, 1H, NH), 7.23 (s, 1H, cipro Bn-H), 4.43 (s, 1H) 4.18−4.10 (m, 4H, αH), under H2O peak ~3.6−3.4 (m, 6H, δH), 3.16−3.15 (m, 4H, cipro-H), 2.90−2.87 (m, 2H, NH–CH2), 2.08 (s, 2H, CO–CH2), 1.31–1.20 (m, 23H, CH3/NH2/βH/γH), 0.87−0.82 (m, 4H, cyclopropyl-H). 19F NMR (DMSO-d6, 376 MHz): −73.59 ppm. Retention time (method C): 10.35 min (purity/peak area: 98.7%). 67Ga-D2: Rt = 10.56 min (purity/peak area: 99%).
Antimicrobial Activity Assays.
Stock solutions of test compounds were prepared and serially diluted to obtain a dilution series of relevant concentration ranges in comparison with ciprofloxacin (control). Bacterial inoculate (wt. E. coli, Mg 1655, E. coli AN193—E. coli Genetic Stock Center Yale University, New Haven, CT) was added to each well, and the plate was analyzed for bacterial growth after overnight incubation at 37 uC. All liquids and media were sterilized by autoclaving (220 °C, 1 h) before use. All aqueous solutions and media were prepared using deionized water. Mueller–Hinton broth (MHB) was purchased from Fisher Scientific. Mueller–Hinton II (MHII) broth (cation adjusted) was prepared by adding sterile aqueous solution of 0.41 mL of 1 M CA2+ and 0.15 mL of 1 M Mg2+ to 250 mL of MHB. Iron-deficient (−Fe) MHII broth was prepared by adding 4.06 mL of a 1 mg/mL sterile aq solution of 2,2′-bipyridine to 250 mL of MHII broth, and 0.3 mM stock solutions of the testing compounds were prepared in sterilized deionized water. Concentrations of siderophore, siderophore–ciprofloxacin conjugate, and corresponding metal complexes were analyzed using ICP-OES and UV–vis spectrophotometry, and 10 μL solution aliquot of test compound (0.3 mM) was added to the first well of the 96-well plate and serial dilutions were performed. Subsequently, 40 μL of growth media and 50 μL of dilute bacterial inoculum were also added to each well, resulting in a total volume of 100 μL and a concentration gradient of 0.3 × 10−4 to 1.56 × 10−12 M. For challenge experiments, serial dilution of test compound was performed, followed by addition of 40 μL of compound at appropriate concentration, 40 μL of growth media, and 50 μL of dilute bacterial inoculum to each well, resulting in a total volume of 100 μL and a concentration gradient of 0.3 × 10−4 to 1.56 × 10−12 M. For challenge experiments, 10 μL of 100-fold excess Fe-, Ga–LDFC or DFO complex was also added to each well, resulting in a total volume of 110 μL/well. The plates were incubated at 37 °C for 18 h, and each plate was examined for bacterial growth using a UV–vis plate reader. The MIC was recorded as the lowest compound concentration (μM) required to inhibit at least 90% of bacterial growth as judged by the absorbance of the culture media relative to a row of wells filled with the negative control.
Radiochemical Complex Challenge Experiments.
To assess the inertness of complexes toward transchelation, the ligand exchange challenge experiment was carried out in duplicates with 10× EDTA, in iron-sufficient and iron-deficient LB Broth (0.8 mL of a 1 mg/mL sterile aq solution of 2,2′-bipyridine added to 49.2 mL of LB). 67Ga complexes were incubated with an equal volume of EDTA (pH = 3), and an aliquot of the reaction mixture was removed at select time points (10 min, 20 min, 30 min, 1 h and 2 h) and analyzed by HPLC, method C.
Bacterial Uptake of 67Ga in wt E. coli.
E. coli (Mg 1655) was grown overnight in 5 mL of LB at 37 °C. The overnight culture was diluted in either iron-sufficient or iron-deficient LB and incubated at 37 eC until the OD100 reached 0.5. Uptake was initiated by adding 67Ga complexes (10 μL, 0.05 MBq) to Eppendorf tubes containing 500 μL bacterial culture and incubating at 37 C. Aliquots (60 μL) were removed after 10 min, 20 min, 30 min, 1 h, and 2 h and centrifuged for 2 min. Pellet and supernatant were separated and collected in individual Eppendorf tubes without washing. All tubes were counted using an automated gamma counter to quantify retained radioactivity. The assay was performed in at least triplicates and in parallel with a 67Ga-citrate control.
To challenge active transporter-mediated uptake, 67Ga complexes (10 μL, 0.05 MBq) and 10 μL of the 120× Fe-LDFC complex were added to each Eppendorf tube containing 500 μL of bacterial culture. Aliquots (60 μL) were removed after 10 min, 20 min, 30 min, 1 h, and 2 h and centrifuged for 2 min. Pellet and supernatant were separated and collected in individual Eppendorf tubes. The assay was performed as described above.
Biodistribution and Metabolite Analysis.
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Stony Brook Medicine in accordance with the approved IACUC protocol number 1104609. 0.8–1.2 MBq of 67Ga conjugates were intravenously injected via tail vein catheter. Mice (female CBJ-mice, 8–10 weeks old, Taconic Bioscience) were sacrificed 1 h p.i., and select organs were harvested. Radioactivity was counted by using a gamma counter, and the radioactivity associated with each organ was expressed as % ID/g. 100 μL of urine was directly injected to the radio HPLC. Eluate was collected in 30 s increments from 0–15 min. Activity in each tube was quantified using a gamma counter. The counts were used to reconstruct the metabolite trace which was then compared to the HPLC traces of the original 67Ga complexes.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Dr. Peter Smith-Jones for gamma counter access. Dr. Peter Tonge is thanked for access to the bacterial strains. We acknowledge Dr. Nicole Sampson for access to a UV-vis plate reader. Dr. Anne-Kathrin Duhme-Klair is acknowledged for her suggestions for bacterial assays. Rajeswari Basu is thanked for her assistance and guidance throughout the bacterial studies.
Funding
E.B. acknowledges funding sources, specifically the NIH for a Pathway to Independence Award (R00HL125728) and Stony Brook University for startup funds. S.G.V.L is supported by NIH (R01AI087849).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01388.
Molecular strings file (CSV)
Spectroscopic data on coordination complexes, representative HPLC traces of ligands and their corresponding complexes, radiochemical characterization and inertness, control MIC experiments, tabulated biodistribution data, and metabolite HPLC traces (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Navarro-San Francisco C; Ruiz-Garbajosa P; Cantón R The what, when and how in performing and interpreting microbiological diagnostic tests in skin and soft tissue infections. Curr. Opin. Infect. Dis 2018, 31, 104–112. [DOI] [PubMed] [Google Scholar]
- (2).Thorpe KE; Joski P; Johnston KJ Antibiotic-resistant infection treatment costs have doubled since 2002, now exceeding $2 billion annually. Health Aff. 2018, 37, 662–669. [DOI] [PubMed] [Google Scholar]
- (3).Laxminarayan R; Duse A; Wattal C; Zaidi AKM; Wertheim HFL; Sumpradit N; Vlieghe E; Hara GL; Gould IM; Goossens H; Greko C; So AD; Bigdeli M; Tomson G; Woodhouse W; Ombaka E; Peralta AQ; Qamar FN; Mir F; Kariuki S; Bhutta ZA; Coates A; Bergstrom R; Wright GD; Brown ED; Cars O Antibiotic resistance-the need for global solutions. Lancet Infect. Dis 2013, 13, 1057–1098. [DOI] [PubMed] [Google Scholar]
- (4).Hider RC; Kong X Chemistry and biology of siderophores. Nat. Prod. Rep 2010, 27, 637–657. [DOI] [PubMed] [Google Scholar]
- (5).Ecker DJ; Matzanke BF; Raymond KN Recognition and transport of ferric enterobactin in Escherichia coli. J. Bacteriol 1986, 167, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Raymond KN; Allred BE; Sia AK Coordination chemistry of microbial iron transport. Acc. Chem. Res 2015, 48, 2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zeng Y; Kulkarni A; Yang Z; Patil PB; Zhou W; Chi X; Van Lanen S; Chen S Biosynthesis of Albomycin δ2 Provides a Template for Assembling Siderophore and Aminoacyl-tRNA Synthetase Inhibitor Conjugates. ACS Chem. Biol 2012, 7, 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gause GF Recent studies on albomycin, a new antibiotic. Br. Med. J 1955, 2, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hartmann A; Fiedler H-P; Braun V Uptake and Conversion of the Antibiotic Albomycin by Escherichia coli K-12. FEBS J 1979, 99, 517–524. [DOI] [PubMed] [Google Scholar]
- (10).Paulsen H; Brieden M; Benz G Verzweigte und kettenverlängerte Zucker, XXXI. Synthese des Sauerstoffanalogons der Desferriform von δ1-Albomycin. Liebigs Ann. Chem 1987, 1987, 565–575. [Google Scholar]
- (11).Lin Z; Xu X; Zhao S; Yang X; Guo J; Zhang Q; Jing C; Chen S; He Y Total synthesis and antimicrobial evaluation of natural albomycins against clinical pathogens. Nat. Commun 2018, 9, 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miller MJ; Malouin F Microbial iron chelators as drug delivery agents: the rational design and synthesis of siderophore-drug conjugates. Acc. Chem. Res 1993, 26, 241–249. [Google Scholar]
- (13).Ghosh M; Miller PA; Möllmann U; Claypool WD; Schroeder VA; Wolter WR; Suckow M; Yu H; Li S; Huang W; Zajicek J; Miller MJ Targeted antibiotic delivery: Selective siderophore conjugation with daptomycin confers potent activity against multidrug resistant Acinetobacter baumannii both in vitro and in vivo. J. Med. Chem 2017, 60, 4577–4583. [DOI] [PubMed] [Google Scholar]
- (14).Ji C; Miller PA; Miller MJ Iron Transport-Mediated Drug Delivery: Practical Syntheses and In Vitro Antibacterial Studies of Tris-Catecholate Siderophore-Aminopenicillin Conjugates Reveals Selectively Potent Antipseudomonal Activity. J. Am. Chem. Soc 2012, 134, 9898–9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wencewicz TA; Long TE; Möllmann U; Miller MJ Trihydroxamate Siderophore-Fluoroquinolone Conjugates Are Selective Sideromycin Antibiotics that Target Staphylococcus aureus. Bioconjugate Chem. 2013, 24, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wencewicz TA; Miller MJ Biscatecholate-Monohydroxamate Mixed Ligand Siderophore-Carbacephalosporin Conjugates are Selective Sideromycin Antibiotics that Target Acinetobacter baumannii. J. Med. Chem 2013, 56, 4044–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kong H; Cheng W; Wei H; Yuan Y; Yang Z; Zhang X An overview of recent progress in siderophore-antibiotic conjugates. Eur. J. Med. Chem 2019, 182, 111615. [DOI] [PubMed] [Google Scholar]
- (18).Wencewicz TA; Miller MJ Sideromycins as pathogen-targeted antibiotics. Antibacterials; Springer, 2017; pp 151–183. [Google Scholar]
- (19).Lin Y-M; Ghosh M; Miller PA; Möllmann U; Miller MJ Synthetic sideromycins (skepticism and optimism): selective generation of either broad or narrow spectrum Gram-negative antibiotics. BioMetals 2019, 32, 425–451. [DOI] [PubMed] [Google Scholar]
- (20).Neumann W; Sassone-Corsi M; Raffatellu M; Nolan EM Esterase-Catalyzed Siderophore Hydrolysis Activates an Enterobactin-Ciprofloxacin Conjugate and Confers Targeted Antibacterial Activity. J. Am. Chem. Soc 2018, 140, 5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chairatana P; Zheng T; Nolan EM Targeting virulence: salmochelin modification tunes the antibacterial activity spectrum of β-lactams for pathogen-selective killing of Escherichia coli. Chem. Sci 2015, 6, 4458–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zheng T; Nolan EM Enterobactin-Mediated Delivery of β-Lactam Antibiotics Enhances Antibacterial Activity against PathogenicEscherichia coli. J. Am. Chem. Soc 2014, 136, 9677–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Milner SJ; Snelling AM; Kerr KG; Abd-El-Aziz A; Thomas GH; Hubbard RE; Routledge A; Duhme-Klair A-K Probing linker design in citric acid-ciprofloxacin conjugates. Bioorg. Med. Chem 2014, 22, 4499–4505. [DOI] [PubMed] [Google Scholar]
- (24).Tsionou MI; Knapp CE; Foley CA; Munteanu CR; Cakebread A; Imberti C; Eykyn TR; Young JD; Paterson BM; Blower PJ; Ma MT Comparison of macrocyclic and acyclic chelators for gallium-68 radiolabelling. RSC Adv 2017, 7, 49586–49599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Emery T; Hoffer PB Siderophore-mediated mechanism of gallium uptake demonstrated in the microorganism Ustilago sphaerogena. J. Nucl. Med 1980, 21, 935–939. [PubMed] [Google Scholar]
- (26).Clarke TE; Braun V; Winkelmann G; Tari LW; Vogel HJ X-ray Crystallographic Structures of theEscherichia coliPeriplasmic Protein FhuD Bound to Hydroxamate-type Siderophores and the Antibiotic Albomycin. J. Biol. Chem 2002, 277, 13966–13972. [DOI] [PubMed] [Google Scholar]
- (27).Johnstone TC; Nolan EM Beyond iron: non-classical biological functions of bacterial siderophores. Dalton Trans 2015, 44, 6320–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Banin E; Lozinski A; Brady KM; Berenshtein E; Butterfield PW; Moshe M; Chevion M; Greenberg EP; Banin E The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16761–16766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Goss CH; Kaneko Y; Khuu L; Anderson GD; Ravishankar S; Aitken ML; Lechtzin N; Zhou G; Czyz DM; McLean K; Olakanmi O; Shuman HA; Teresi M; Wilhelm E; Caldwell E; Salipante SJ; Hornick DB; Siehnel RJ; Becker L; Britigan BE; Singh PK Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci. Transl. Med 2018, 10, No. eaat7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Juárez-Hernández RE; Miller PA; Miller MJ Syntheses of Siderophore-Drug Conjugates Using a Convergent Thiol-Maleimide System. ACS Med. Chem. Lett 2012, 3, 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang Y; Han B; Xie Y; Wang H; Wang R; Xia W; Li H; Sun H Combination of gallium (III) with acetate for combating antibiotic resistant Pseudomonas aeruginosa. Chem. Sci 2019, 10, 6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Love C; Palestro CJ Nuclear medicine imaging of bone infections. Clin. Radiol 2016, 71, 632–646. [DOI] [PubMed] [Google Scholar]
- (33).Bettegowda C; Foss CA; Cheong I; Wang Y; Diaz L; Agrawal N; Fox J; Dick J; Dang LH; Zhou S; Kinzler KW; Vogelstein B; Pomper MG Imaging bacterial infections with radiolabeled 1-(2’-deoxy-2’-fluoro- -D-arabinofuranosyl)-5-iodouracil. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 1145–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Diaz LA Jr.; Foss CA; Thornton K; Nimmagadda S; Endres CJ; Uzuner O; Seyler TM; Ulrich SD; Conway J; Bettegowda C; Agrawal N; Cheong I; Zhang X; Ladenson PW; Vogelstein BN; Mont MA; Zhou S; Kinzler KW; Vogelstein B; Pomper MG Imaging of musculoskeletal bacterial infections by [124I] FIAU-PET/CT. PLoS One 2007, 2, No. e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gowrishankar G; Namavari M; Jouannot EB; Hoehne A; Reeves R; Hardy J; Gambhir SS Investigation of 6-[18F]-fluoromaltose as a novel PET tracer for imaging bacterial infection. PLoS One 2014, 9, No. e107951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Weinstein EA; Ordonez AA; DeMarco VP; Murawski AM; Pokkali S; MacDonald EM; Klunk M; Mease RC; Pomper MG; Jain SK Imaging Enterobacteriaceae infection in vivo with 18F-fluorodeoxysorbitol positron emission tomography. Sci. Transl. Med 2014, 6, 259rA146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Petrik M; Haas H; Dobrozemsky G; Lass-Florl C; Helbok A; Blatzer M; Dietrich H; Decristoforo C 68Ga-siderophores for PET imaging of invasive pulmonary aspergillosis: proof of principle. J. Nucl. Med 2010, 51, 639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Petrik M; Haas H; Schrettl M; Helbok A; Blatzer M; Decristoforo C In vitro and in vivo evaluation of selected 68Gasiderophores for infection imaging. Nucl. Med. Biol 2012, 39, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Szebesczyk A; Olshvang E; Shanzer A; Carver PL; Gumienna-Kontecka E Harnessing the power of fungal siderophores for the imaging and treatment of human diseases. Coord. Chem. Rev 2016, 327–328, 84–109. [Google Scholar]
- (40).Petrik M; Zhai C; Haas H; Decristoforo C Siderophores for molecular imaging applications. Clin. Transl. Imaging 2017, 5, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kelson AB; Carnevali M; Truong-Le V Gallium-based anti-infectives: targeting microbial iron-uptake mechanisms. Curr. Opin. Pharmacol 2013, 13, 707–716. [DOI] [PubMed] [Google Scholar]
- (42).Ferreira K; Hu H-Y; Fetz V; Prochnow H; Rais B; Müller PP; Brönstrup M Multivalent Siderophore-DOTAM Conjugates as Theranostics for Imaging and Treatment of Bacterial Infections. Angew. Chem., Int. Ed 2017, 56, 8272–8276. [DOI] [PubMed] [Google Scholar]
- (43).Endicott NP; Lee E; Wencewicz TA Structural basis for xenosiderophore utilization by the human pathogen Staphylococcus aureus. ACS Infect. Dis 2017, 3, 542–553. [DOI] [PubMed] [Google Scholar]
- (44).Mielcarek A; Blauenburg B; Miethke M; Marahiel MA Molecular insights into frataxin-mediated iron supply for heme biosynthesis in Bacillus subtilis. PLoS One 2015, 10, No. e0122538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Braun V; Pramanik A; Gwinner T; Köberle M; Bohn E Sideromycins: tools and antibiotics. BioMetals 2009, 22, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dong L; Roosenberg JM; Miller MJ Total synthesis of desferrisalmycin B. J. Am. Chem. Soc 2002, 124, 15001–15005. [DOI] [PubMed] [Google Scholar]
- (47).Rivera GSM; Beamish CR; Wencewicz TA Immobilized FhuD2 siderophore-binding protein enables purification of salmycin sideromycins from Streptomyces violaceus DSM 8286. ACS Infect. Dis 2018, 4, 845–859. [DOI] [PubMed] [Google Scholar]
- (48).Ji C; Miller MJ Chemical syntheses and in vitro antibacterial activity of two desferrioxamine B-ciprofloxacin conjugates with potential esterase and phosphatase triggered drug release linkers. Bioorg. Med. Chem 2012, 20, 3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ioppolo JA; Caldwell D; Beiraghi O; Llano L; Blacker M; Valliant JF; Berti PJ 67Ga-labeled deferoxamine derivatives for imaging bacterial infection: Preparation and screening of functionalized siderophore complexes. Nucl. Med. Biol 2017, 52, 32–41. [DOI] [PubMed] [Google Scholar]
- (50).Adams CJ; Wilson JJ; Boros E Multifunctional desferrichrome analogues as versatile 89Zr (IV) chelators for immunoPET probe development. Mol. Pharmaceutics 2017, 14, 2831–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ahn SH; Thach D; Vaughn BA; Alford VM; Preston AN; Laughlin ST; Boros E Linear desferrichrome-linked siliconrhodamine antibody conjugate enables targeted multimodal imaging of HER2 in vitro and in vivo. Mol. Pharmaceutics 2019, 16, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vondenhoff GH; Gadakh B; Severinov K; Van Aerschot A Microcin C and Albomycin Analogues with Aryl-tetrazole Substituents as Nucleobase Isosters Are Selective Inhibitors of Bacterial Aminoacyl tRNA Synthetases but Lack Efficient Uptake. Chem-BioChem 2012, 13, 1959–1969. [DOI] [PubMed] [Google Scholar]
- (53).Dolence EK; Minnick AA; Lin CE; Miller MJ; Payne SM Synthesis and siderophore and antibacterial activity of N5-acetyl-N5-hydroxyl-L-ornithine derived siderophore-beta-lactam conjugates: iron transport mediated drug delivery. J. Med. Chem 1991, 34, 968–978. [DOI] [PubMed] [Google Scholar]
- (54).Zheng T; Bullock JL; Nolan EM Siderophore-Mediated Cargo Delivery to the Cytoplasm ofEscherichia coliandPseudomonas aeruginosa: Syntheses of Monofunctionalized Enterobactin Scaffolds and Evaluation of Enterobactin-Cargo Conjugate Uptake. J. Am. Chem. Soc 2012, 134, 18388–18400. [DOI] [PubMed] [Google Scholar]
- (55).Tufano TP; Raymond KN Coordination chemistry of microbial iron transport compounds. 21. Kinetics and mechanism of iron exchange in hydroxamate siderophore complexes. J. Am. Chem. Soc 1981, 103, 6617–6624. [Google Scholar]
- (56).Hannauer M; Barda Y; Mislin GLA; Shanzer A; Schalk IJ The ferrichrome uptake pathway in Pseudomonas aeruginosa involves an iron release mechanism with acylation of the siderophore and recycling of the modified desferrichrome. J. Bacteriol 2010, 192, 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Mislin GLA; Schalk IJ Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Metallomics 2014, 6, 408–420. [DOI] [PubMed] [Google Scholar]
- (58).Zhang Z; Ordonez AA; Wang H; Li Y; Gogarty KR; Weinstein EA; Daryaee F; Merino J; Yoon GE; Kalinda AS; Mease RC; Iuliano JN; Smith-Jones PM; Jain SK; Tonge PJ Positron emission tomography imaging with 2-[18F] F-p-amino-benzoic acid detects Staphylococcus aureus infections and monitors drug response. ACS Infect. Dis 2018, 4, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.